

Abstract
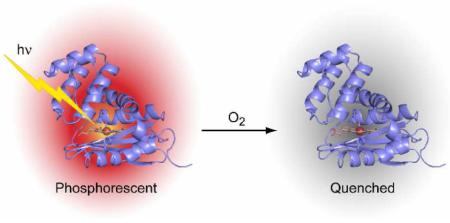
Hemoprotein-based scaffolds containing phosphorescent ruthenium(II) CO mesoporphyrin IX (RuMP) are reported here for oxygen (O2) sensing in biological contexts. RuMP was incorporated into the protein scaffolds during protein expression utilizing a novel method that we have described previously. A high-resolution (2.00 Å) crystal structure revealed that the unnatural porphyrin binds to the proteins in a manner similar to the native heme and does not perturb the protein fold. The protein scaffolds were found to provide unique coordination environments for RuMP and modulate the porphyrin emission properties. Emission lifetime measurements demonstrate a linear O2 response within the physiological range and precision comparable to commercial O2 sensors. The RuMP proteins are robust, readily-modifiable platforms and display promising O2 sensing properties for future in vivo applications.
Proteins that natively bind heme are under-utilized scaffolds for porphyrin-based tools in biology.1,2 Porphyrins have broad applications, ranging from dyes in solar cells3 to sensitizers for radiotherapy4 to frameworks for catalysis.5 Traditional methods for unnatural porphyrin incorporation into hemoproteins have limited their utility as biological tools. Harsh, denaturing conditions are typically required to remove native heme from proteins,6,7 dramatically decreasing the number of viable protein constructs. We recently reported a novel method for incorporation of unnatural porphyrins into hemoproteins during protein expression.8 Here we demonstrate the versatility of this expression-based method through the development of protein-based sensors, in which the native heme cofactor of different hemoprotein scaffolds has been substituted with an unnatural porphyrin for oxygen (O2) sensing within biological contexts.
O2 is a key metabolic indicator for profiling the physiology of tissues and cells.9 Quenching of small molecule luminescence by O2 is a simple, non-invasive method for imaging in vivo O2 levels.10 However, the utility of small molecules for this application is hampered by lack of targetable delivery, non-specific binding, self-aggregation, and limitations in photophysical properties.11,12 Incorporation of luminescent porphyrins into hemoprotein scaffolds provides a novel platform to address these issues.
Ruthenium(II) CO mesoporphyrin IX (RuMP) (Figure S1) is an ideal cofactor for protein-based sensors because it exhibits O2-senstive phosphorescence13 and presents a proximal axial ligation site14 to facilitate binding to the protein scaffold. Myoglobin (Mb) and the H-NOX (Heme Nitric oxide / OXygen binding) domain from the thermophilic bacterium Thermoanaerobacter tengcongensis (Tt H-NOX) are robust proteins for RuMP sensors, as they can be readily modified with genetically-encoded affinity tags and site-directed mutagenesis.1,15 In addition, Tt H-NOX is stable under extreme temperatures (>70 °C).15
Experimental details for preparation and characterization of RuMP-substituted Mb (Ru Mb) and Tt H-NOX (Ru Tt H-NOX) are described in Supporting Information. Briefly, RuMP was synthesized in a manner similar to published methods14 and incorporated into Mb and Tt H-NOX during anaerobic protein expression. The RuMP-substituted proteins were isolated containing a stochiometric amount of porphyrin (Table S1). Indeed, further evaluation of the stability of Ru Tt H-NOX indicated no detectible porphyrin loss for >24 hours under biological conditions (mouse plasma at 37 °C, Figure S2).
Purified Ru Tt H-NOX was crystallized to verify proper porphyrin insertion and preservation of the protein fold. The high-resolution (2.00 Å) structure of Ru Tt H-NOX (Figure 1, Table S2) is the first crystal structure of a Ru porphyrin bound to a protein and demonstrates that the unnatural porphyrin maintains key contacts with surrounding heme pocket residues. These contacts include coordination of the proximal histidine to Ru and hydrogen bonding between the distal porphyrin ligand and a tyrosine residue (Figure 1, Figure S3). In fact, comparison of heme-bound Tt H-NOX with its Ru analogue indicates little perturbation of the protein fold (overall rmsd 1.3 Å, Figure S4).
Figure 1.
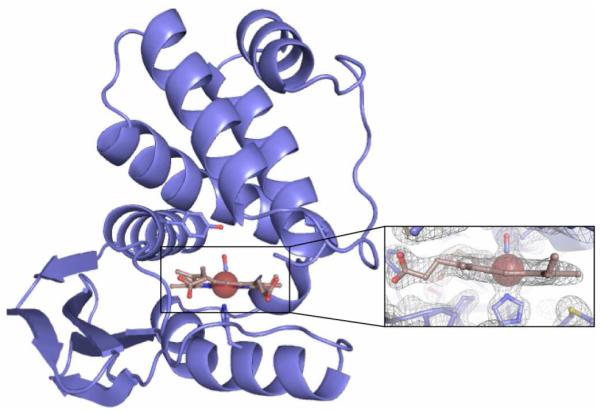
Crystal structure of Tt H-NOX containing RuMP solved at 2.00 Å resolution. Inset: 2(Fo-Fc) electron density map calculated by omitting RuMP and the proximal histidine side chain.
Steady-state and time-resolved spectroscopies were employed to examine the spectral properties of RuMP bound to the protein scaffolds. UV-visible spectra for Ru Tt H-NOX and Ru Mb show similar Soret band features at 400 nm and 397 nm, respectively (Figure 2, Table 1). However, the α band at ~550 nm for Ru Mb is split, as observed previously.14 Steady-state emission spectra reveal a blue-shifted emission band and decreased emission quantum yield for Ru Mb as compared to Ru Tt H-NOX (Table 1). Time-resolved emission spectroscopy conducted to further probe the spectral features of the porphyrin-protein complexes yielded single-exponential emission decays (following 550 nm laser excitation) under anaerobic conditions that vary widely between the two proteins (τo = 7.7 μs for Ru Tt H-NOX vs. 37.3 μs for Ru Mb, respectively). Taken together, these data indicate a substantially different conformation and/or chemical environment for RuMP in Mb and Tt H-NOX. Indeed, the crystal structure of Mb reveals that the heme is partially exposed to solvent,1 whereas the heme in Tt H-NOX is buried within the protein matrix (Figure 1).
Figure 2.
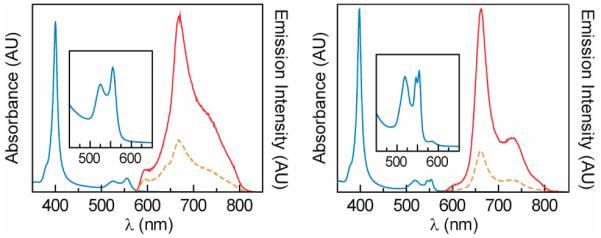
Steady-state absorbance (▬) and emission spectra of Ru Tt H-NOX (left) and Ru Mb (right) in aqueous HEPES/NaCl buffer. Emission spectra were acquired following excitation at 550 nm in the presence (----, 256 μM) and absence (▬) of O2.
Table 1.
Spectroscopic and photophysical properties of Ru proteins
| Ru Protein | λabs (nm) / (ε) | λema (nm) | Φ emc | τemd (μs) |
|---|---|---|---|---|
| Tt H-NOX | 400 (173) | 668 | 1.7 × 10−4 | 7.7 (−O2) |
| 524 (10.7) | ~734b | 2.9 (+O2) | ||
| 555 (13.9) | ||||
| Mb | 397 (197) | 663 | 4.8 × 10−5 | 37.3 (−O2) |
| 518 (12.2) | ~733b | 12.2 (+O2) | ||
| 553 (13.2) |
λex = 550 nm.
shoulder.
λex = 550 nm, no O2.
λex = 550 nm, λdet = 640 nm, no O2 and 256 μM O2.
The ability of phosphorescent molecules to sense O2 is determined by the degree of emission quenching in the presence of O2. Comparison of the steady-state emission spectra of Ru Tt H-NOX and Ru Mb measured under aerobic and anaerobic conditions reveals that O2 appreciably quenches the emission of both proteins (Figure 2). To further evaluate the highly stable Ru Tt H-NOX protein as an O2 sensor, its excited state lifetime was measured at several O2 concentrations (Figure 3). The data were analyzed according to the Stern-Volmer (SV) equation for O2 quenching (Supporting Information) and yielded a bimolecular quenching constant, kq, of 1350 mmHg−1s−1 (8.2 × 108 M−1s−1). In addition to intrinsic emission properties, the precision of lifetime-based O2 sensors is governed by the instrument error associated with the lifetime measurement. Taking our instrument error of 2.5% into account, Ru Tt H-NOX can be used to determine O2 concentrations to within ±2.5 mmHg (4.2 μM) in spite of its low quantum yield. This precision is comparable to that reported for commercial O2 sensors (Table 2) and is ideally suited for determining O2 concentrations in biology. Indeed, emission quenching was observed to be linear across the biologically relevant range of O2 concentrations (Figure 3).9
Figure 3.
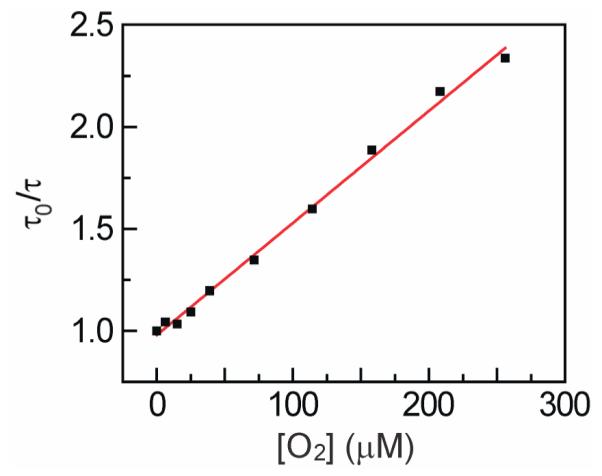
Stern-Volmer plot of the excited state lifetime of Ru Tt H-NOX vs. [O2] showing linear phosphorescence quenching by O2 from 0 to 256 μM O2 (R2 = 0.9957).
Table 2.
Comparison of Ru Tt H-NOX with select O2 sensors
| Complex |
kq (mmHg−1s−1 |
τ0 (μs) |
Precisiona (mmHg) |
Ref. |
|---|---|---|---|---|
| Ru Tt H-NOX | 1350 | 7.7 | 5.0 | this work |
| RuII(bpy)32+ | 4300 | 0.58 | 21 | 16 |
| Oxyphor | 293 | 707 | 0.25 | 17 |
| PtP-C343 | 150 | 60 | 5.9 | 12 |
Determined assuming an error of 2.5% in τ0 measurement
The RuMP proteins described here represent a new class of sensors for detection of dissolved O2. The sensors are readily expressed in E. coli, exceptionally robust, and able to detect O2 levels in the biologically relevant range. The photophysical properties may be further modulated with the choice of emissive porphyrin or through modification of the protein scaffold (e.g. via site-directed mutagenesis). In addition, the proteins may be expressed with genetically-encoded tags for targeted delivery in biology and derivatized for enhancing biocompatibility. We anticipate that this new class of sensors will prove useful for monitoring O2 levels in biological contexts. One area of particular interest for sensing O2 is in tumor microenvironments wherein detailed knowledge of local O2 concentrations is key to improving cancer diagnosis and treatment.9
Supplementary Material
Acknowledgment
We thank the Army Research Office (W911NF-06-1-0101) (D.G.N.) and the National Institutes of Health (R01CA126642-02) (D.G.N.) and (GM070671) (M.A.M.) for financial support. We also thank Prof. J. Kuriyan for structural analysis, Prof. C. Drennan and Prof. S. Marqusee for use of equipment, R. Tran for assistance with cloning, Dr. E. Weinert for assistance with plasma stability experiments, and Dr. A. Iavarone for mass spectrometry acquisition.
Footnotes
Supporting Information Available: Synthetic details, porphyrin stochiometry, further photophysical characterization of the RuMP proteins, structural alignments, and x-ray diffraction data.
References
- (1).Dou Y, Maillett DH, Eich RF, Olson JS. Biophys. Chem. 2002;98:127–148. doi: 10.1016/s0301-4622(02)00090-x. [DOI] [PubMed] [Google Scholar]
- (2).Gillam EMJ. Chem. Res. Toxicol. 2008;21:220–231. doi: 10.1021/tx7002849. [DOI] [PubMed] [Google Scholar]
- (3).Imahori H, Fukuzumi S. Adv. Func. Mat. 2004;14:525–536. [Google Scholar]
- (4).Vicente MGH. Curr. Med. Chem.: Anti-Cancer Agents. 2001;1:175–194. doi: 10.2174/1568011013354769. [DOI] [PubMed] [Google Scholar]
- (5).Chang CJ, Chng LL, Nocera DG. J. Am. Chem. Soc. 2003;125:1866–1876. doi: 10.1021/ja028548o. [DOI] [PubMed] [Google Scholar]
- (6).Teale FW. Biochim. Biophys. Acta. 1959;35:543. doi: 10.1016/0006-3002(59)90407-x. [DOI] [PubMed] [Google Scholar]
- (7).Schmidt P, Schramm M, Schröder H, Stasch JP. Protein Expr. Purif. 2003;31:42–46. doi: 10.1016/s1046-5928(03)00142-6. [DOI] [PubMed] [Google Scholar]
- (8).Woodward JJ, Martin NI, Marletta MA. Nat. Methods. 2007;4:43–45. doi: 10.1038/nmeth984. [DOI] [PubMed] [Google Scholar]
- (9).Helmlinger G, Yuan F, Dellian M, Jain RK. Nat. Med. 1997;3:177–182. doi: 10.1038/nm0297-177. [DOI] [PubMed] [Google Scholar]
- (10).Vanderkooi JM, Maniara G, Green TJ, Wilson DF. J. Biol. Chem. 1987;262:5476–5482. [PubMed] [Google Scholar]
- (11).Berg K, Selbo PK, Weyergang A, Dietze A, Prasmickaite L, Bonsted A, Engesaeter BØ, Angell-Petersen E, Warloe T, Frandsen N, Høgset A. J. Microsc. 2005;218:133–147. doi: 10.1111/j.1365-2818.2005.01471.x. [DOI] [PubMed] [Google Scholar]
- (12).Finikova OS, Lebedev AY, Aprelev A, Troxler T, Gao F, Garnacho C, Muro S, Hochstrasser RM, Vinogradov SA. Chem. Phys. Chem. 2008;9:1673–1679. doi: 10.1002/cphc.200800296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Vanderkooi JM, Wright WW, Erecinska M. Biochim. Biophys. Acta. 1994;1207:249–254. doi: 10.1016/0167-4838(94)00073-5. [DOI] [PubMed] [Google Scholar]
- (14).Paulson DR, Addison AW, Dolphin D, James BR. J. Biol. Chem. 1979;254:7002–7006. [PubMed] [Google Scholar]
- (15).Boon EM, Marletta MA. J. Am. Chem. Soc. 2006;128:10022–10023. doi: 10.1021/ja0632714. [DOI] [PubMed] [Google Scholar]
- (16).Oter O, Ribou AC. J. Fluoresc. 2009;19:389–397. doi: 10.1007/s10895-008-0425-z. [DOI] [PubMed] [Google Scholar]
- (17).Dunphy I, Vinogradov SA, Wilson DF. Anal. Biochem. 2002;310:191–198. doi: 10.1016/s0003-2697(02)00384-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.