

Abstract
A Plasmodium falciparumhexose transporter (PfHT) has previously been shown to be a facilitative glucose and fructose transporter. Its expression in Xenopus laevisoocytes and the use of a glucose analogue inhibitor permitted chemical validation of PfHT as a novel drug target. Following recent re-annotations of the P. falciparum genome, other putative sugar transporters have been identified. To investigate further if PfHT is the key supplier of hexose to P. falciparum and to extend studies to different stages of Plasmodium spp., we functionally analysed the hexose transporters of both the human parasite P. falciparum and the rodent parasite Plasmodium berghei using gene targeting strategies. We show here the essential function of pfht for the erythrocytic parasite growth as it was not possible to knockout pfht unless the gene was complemented by an episomal construct. Also, we show that parasites are rescued from the toxic effect of a glucose analogue inhibitor when pfht is overexpressed in these transfectants. We found that the rodent malaria parasite orthologue, P. berghei hexose transporter (PbHT) gene, was similarly refractory to knockout attempts. However, using a single cross-over transfection strategy, we generated transgenic P. berghei parasites expressing a PbHT–GFP fusion protein suggesting that locus is amenable for gene targeting. Analysis of pbht-gfp transgenic parasites showed that PbHT is constitutively expressed through all the stages in the mosquito host in addition to asexual stages. These results provide genetic support for prioritizing PfHT as a target for novel antimalarials that can inhibit glucose uptake and kill parasites, as well as unveiling the expression of this hexose transporter in mosquito stages of the parasite, where it is also likely to be critical for survival.
Introduction
Malaria still afflicts around 500 million people and continues to kill around 1 million children a year. The recent emergence of resistance to artemisinin (Noedl et al., 2008; Dondorp et al., 2009), the last line of defence against multi-resistant parasites in some parts of the world, makes the search for new antimalarial drug targets an urgent one.
Although transport proteins are excellent drug targets in other systems (Imming et al., 2006), they are underexploited as targets in Plasmodium falciparum, the most pathogenic malarial parasite, and other Plasmodium spp. (Cowman and Crabb, 2003).
We first identified the hexose transporter of P. falciparum (PfHT, PFB0210c) as a potential drug target by studying its function after expression in Xenopus laevisoocytes, and demonstrating selective inhibition of PfHT versus mammalian orthologues using an o-3 undecenyl glucose derivative (CM3361) (Joet et al., 2003). These studies were extended to the Plasmodium berghei murine model, to establish that CM3361 can attenuate parasitaemias in vivo. There is also added value (in addition to parasite clearance) in inhibiting glucose uptake by infected erythrocytes, because competition for this substrate between the parasite and the host tissue may be eliminated rapidly. Interfering with metabolic diversion of glucose may be particularly important in cerebral tissues in syndromes like cerebral malaria, where glucose delivery may become rate limiting.
PfHT is a facilitative hexose transporter in the Major Facilitator Superfamily of integral membrane proteins that mediates the uptake of glucose and fructose by the parasite (Woodrow et al., 1999). Pfht is a single copy gene in the P. falciparumgenome with no close paralogues. Three other proteins, PFI0955w, PFI0785c and PFE1455w have been annotated with putative sugar transport function (Gardner et al., 2002; Martin et al., 2005), although they have diverged considerably from typical sugar transporters (21%, 13% and 7% amino acid sequence identity compared with PfHT, respectively). Unlike PfHT, PFI0955w and PFI0785c are expressed only late in the asexual cycle. Together with chemical validation data demonstrating that PfHT is essential for parasite survival, this makes it unlikely that PFI0955w and PFI0785c function in alternative mechanisms for delivery of hexoses as energy substrates for P. falciparum. To test this hypothesis, we performed gene transfection experiments aiming at determining the essentiality (or otherwise) of pfht in P. falciparum and of its orthologue in P. berghei (pbht, PB000562.01.0). We also generated Pbht-gfp transgenic parasites and employed them in the visualization of expression of this transporter during malaria parasite development. In addition, we also attempted to correlate the level of pfht expression in a transfected P. falciparum line with susceptibility to a specific inhibitor, CM3361.
Results
Pfht is indispensable for the erythrocytic development in P. falciparum
To test if pfht can be disrupted during the asexual stages of P. falciparum, two transfection experiments were performed (Fig. 1A). In the first transfection, 3D7 parasites were electroporated with the knockout plasmid, pCAM-BSD-HT. Transfected populations were subjected to blasticidin pressure, and blasticidin-resistant parasites were readily obtained (14 days post transfection). The second transfection consisted of electroporation of 3D7 parasites with both the knockout and the complementation constructs. Double selection with blasticidin and WR99210 was applied 48 h post transfection. Doubly resistant parasites appeared in the culture somewhat later than blasticidin-resistant parasites, 19 days after the electroporation.
Fig. 1.
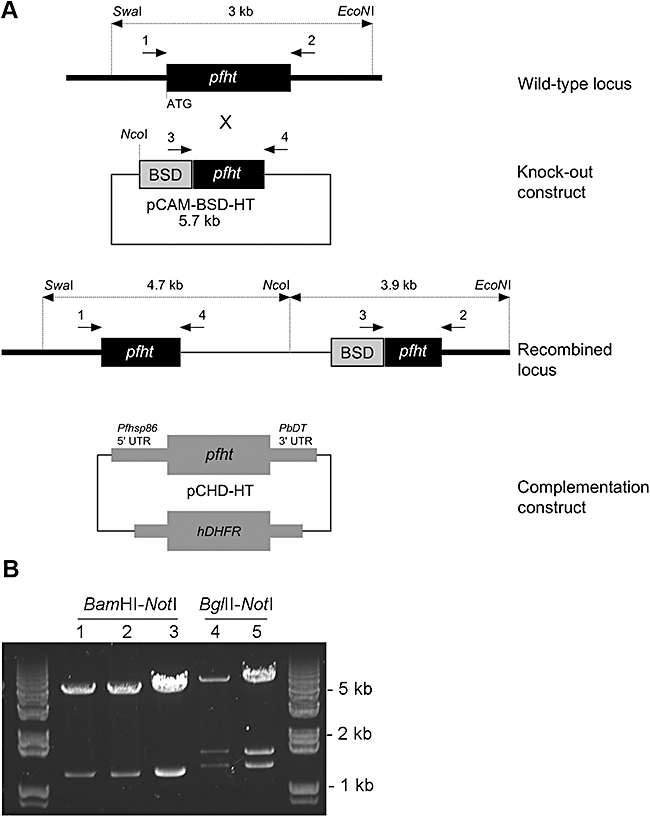
A. Strategy for disruption of the PfHT gene. Single cross-over homologous recombination of the knockout plasmid and the endogenous gene results with two truncated copies of the gene. The location of PCR primers is indicated by numbered arrows. Restriction sites of enzymes used to digest genomic DNA prior to Southern blotting and expected Southern blot fragments are also indicated. The complementation construct (pCHD-HT) allows pfht expression under the Pfhsp86 promoter. B. Plasmid rescue of episomes from transfected parasites. Lane 1: re-isolated episome from parasites transfected with the knockout construct (pCAM-BSD-HT). Lanes 2 and 4: re-isolated episomes from parasites co-transfected with the knockout and the complementation construct (pCHD-HT). Lane 3: pCAM-BSD-HT plasmid. Lane 5: pCHD-HT plasmid. BamHI-NotI digestion releases a 1.2 kb knockout fragment from pCAM-BSD-HT. BglII-NotI digestion releases the PfHT gene (1.5 kb) and an additional 1.2 kb fragment from pCHD-HT.
Episomal plasmids were recovered from all transfected parasites by plasmid rescue to confirm that parasites contained the right constructs (Fig. 1B). Bacterial clones transformed with genomic DNA from blasticidin-resistant parasites all contained pCAM-BSD-HT plasmid while bacterial clones transformed with genomic DNA from doubly resistant parasites carried either the pCAM-BSD-HT or the pCHD-HT plasmid.
Genomic DNA was isolated from both singly and doubly transfected parasites at various stages and was analysed by PCR to establish if integration of the knockout vector and disruption of pfht had occurred.
We found we were not able to obtain a knockout of pfht in parasites transfected only with the knockout vector. PCR detected only the wild-type locus and episomal presence of the knockout construct, but did not show any amplicons diagnostic for integration of the construct into the pfht locus (Fig. 2A). This suggests that the pfht gene is essential for survival of asexual stages of P. falciparum. Co-transfection of 3D7 parasites with both the knockout and the complementation constructs confirms that pfht locus is accessible for homologous recombination. PCR analysis of these parasites detected bands corresponding to the 5′ and 3′ ends of the pCAM-BSD-HT integration event (Fig. 2A). A wild-type locus was still detectable in these parasites, indicative of a genotypically diverse population of parasites. To select for parasites with a disrupted pfht locus, blasticidin selective pressure was removed for 3 weeks. During this time, parasites would tend to lose the pCAM-BSD-HT episome. After 3 weeks, blasticidin was reapplied for another 3 weeks and in this time parasites with pCAM-BSD-HT integrated into their genome would be positively selected (as the PfHT protein was expressed from the complementing plasmid also present in the cells). This cycling selection was repeated twice. PCR analysis on genomic DNA isolated from parasites selected in this way (below referred to as complemented) showed complete loss of the pfht wild-type locus, and presence of the pCAM-BSD-HT integration and episome (Fig. 2A).
Fig. 2.
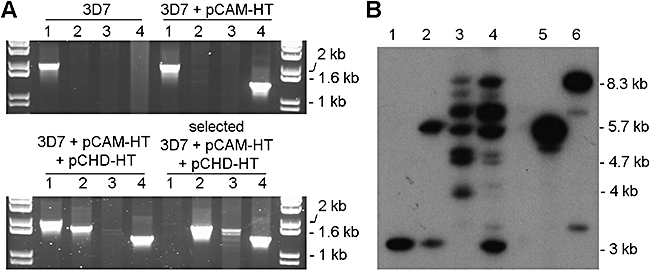
Genotype analysis of wild-type 3D7 parasites and parasites transfected with pCAM-BSD-HT alone or co-transfected with pCAM-BSD-HT and pCHD-HT. A. PCR analysis: lane 1, detection of the wild-type pfht locus 2 kb (primers 1 + 2, see Fig. 1); lane 2, detection of the 5′ integration of pCAM-BSD-HT into the pfhtlocus ∼1.8 kb (primers 1 + 4); lane 3, detection of the 3′ integration of pCAM-BSD-HT ∼1.7 kb (primers 3 + 2); lane 4, detection of the pCAM-BSD-HT episome 1.4 kb (primers 3 + 4). B. Southern blot analysis. Genomic DNA extracted from wild-type 3D7 parasites (lane 1), parasites transfected with pCAM-BSD-HT (lane 2), selected parasites co-transfected with pCAM-BSD-HT and pCHD-HT after blasticidin cycling – complemented parasites (lane 3) and unselected co-transfected parasites prior to blasticidin cycling (lane 4), and plasmid DNA, pCAM-BSD-HT (lane 5) and pCHD-HT (lane 6) were digested with SwaI, NcoI and EcoNI. The blot was probed with the 1.2 kb pfhtfragment that was used as an insert for the pCAM-BSD-HT plasmid; wild-type locus 3 kb, integration of pCAM-BSD-HT 4.7 kb (5′) and 3.9 kb (3′), pCAM-BSD-HT episome 5.7 kb, pCHD-HT episome 8.3 kb.
Southern blot analysis was performed to confirm genotypes (Fig. 2B). When probed with a pfht fragment, DNA from blasticidin-resistant single-transfected parasites yielded two bands corresponding to the wild-type locus and the knockout plasmid (3 kb and 5.7 kb, respectively), while doubly resistant parasites contained pCAM-BSD-HT integrated into the pfht locus (4.7 kb and 3.9 kb) and complete loss of the wild-type locus after cycling with blasticidin pressure. Additionally, complemented parasites showed more complicated Southern blot patterns, suggesting that super-integration events of both plasmids may have occurred in these parasites, as previously observed in similar instances of complementation approaches (Dorin-semblat et al., 2007).
Phenotype of P. falciparum transgenic parasites with an overexpression of pfht
Our complemented transgenic P. falciparum parasites do not express PfHT from the endogenous locus (which is disrupted in these parasites, see Fig. 2), but from the complementing episome, which drives the expression of PfHT from the Pfhsp86 promoter. Complemented parasites have shown a stable phenotype as their removal from the WR99210 selective pressure for a prolonged period did not result in an impaired growth upon the restoration of the selection when compared with complemented parasites continuously cultured exposed to WR99210 (not shown). This finding suggests that parasites were not losing the complementation plasmid in the absence of the selection drug.
Because the Pfhsp86 promoter is highly active throughout the asexual cycle, it can be predicted that PfHT levels are higher in the transgenic parasites than in wild-type cells. To test this hypothesis, we measured the sensitivity of both lines to CM3361, a specific inhibitor of PfHT. When compared with wild-type 3D7, complemented parasites with episomal pfht expression showed a 2.5-fold increase in IC50 value for CM3361 (44.2 ± 8.7 and 109.5 ± 9.6 µM, respectively; P = 0.001 student's t-test, unpaired, two-tailed, n = 5) (Fig. 3A). The sensitivity to chloroquine was the same for these two strains of parasites (wild type: 17.6 ± 4.8 nM, n = 4; complemented transfectants: 15.3 ± 2.6 nM, n = 3; P = 0.7 student's t-test, unpaired, two-tailed). The possibility exists that the 2.5-fold shift in the parasite sensitivity to 3361 is due to an off-target effect as a result of selection with blasticidin and WR99210 although this is less likely given the parasite's unaffected sensitivity to chloroquine, an unrelated compound.
Fig. 3.
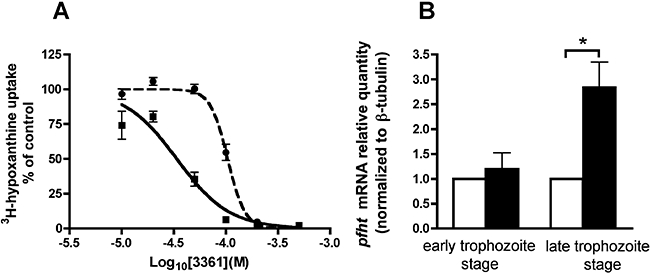
Phenotype analysis of wild-type 3D7 parasites and complemented parasites. A. Effect of compound 3361 on growth of wild-type 3D7 parasites (squares) and complemented parasites (circles). Complemented parasites have disrupted endogenous pfht locus, instead pfht is expressed from pCHD-HT episome under the pfhsp86promoter. Growth inhibition was measured by incorporation of 3H-hypoxanthine with five replicates used per inhibitor concentration. The experiment was repeated five times. Result of a single experiment is shown. Obtained 3361 IC50 values for 3D7 and complemented parasites were 44.2 ± 8.7 and 109.5 ± 9.6 µM respectively (P = 0.001 student's t-test, unpaired, two-tailed, n = 5). B. Real-time PCR analysis of pfhtexpression normalized to β-tubulin in wild-type 3D7 (white bars) and complemented parasites (black bars) (result of 6 experiments with 3 replicates each, one sample t-test, *P = 0.015).
Using quantitative PCR technology, pfht expression was compared at ring (∼10 h post invasion) and trophozoite stages (∼30 h post invasion) in transfected and control cell lines. Significantly increased pfht expression at the trophozoite stage (approximately threefold) was found in complemented parasites compared with 3D7 parasites. In ring-stage parasites there was no difference between the experimental groups (Fig. 3B).
PbHT is essential for erythrocytic stages of P. berghei
Taking into account reported differences between P. falciparum and P. berghei, we were interested to investigate if the P. berghei hexose transporter (PbHT) is also essential for parasite growth similarly to its P. falciparum orthologue. Pbht 5′ and 3′ untranslated regions were cloned upstream and downstream of the dihydrofolate reductase (DHFR) cassette, respectively, in the knockout construct designed for double cross-over homologous recombination. Under pyrimethamine drug pressure, parasites containing the DHFR cassette were selected after three independent transfection experiments. PCR on genomic DNA obtained from all three drug-resistant populations did not detect any integration of the knockout construct or disruption of pbht, suggesting the essential function of the gene (Fig. 4). In contrast, we readily obtained transfected P. berghei parasites containing the integration of the PbHT-GFP construct into the pbht locus. This generated transgenic parasites expressing C-terminally GFP-tagged PbHT, which demonstrates that the locus is accessible for genetic targeting and confirms the essentiality of the hexose transporter gene for completion of the erythrocytic asexual cycle of malaria parasites (Fig. 5).
Fig. 4.
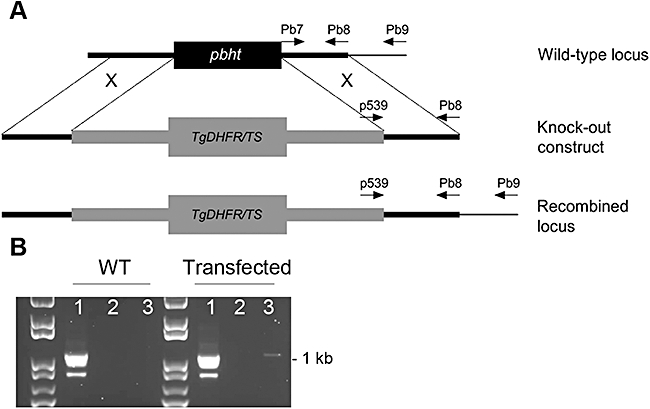
Pbht knockout attempt. A. A double cross-over knockout strategy used to attempt a knockout of pbht and investigate its function; the locations of primers used for PCR analysis of the locus are indicated by arrows. Tg DHFR/TS, Toxoplasma gondii dihydrofolate reductase/thymidylate synthase. B. PCR analysis of the pbht locus in wild type (WT) and parasites transfected with the knockout construct; lane 1, detection of the wild-type locus 1.1 kb (primers Pb7 + Pb9); lane 2, integration detection 1.2 kb (primers p539 + Pb9); lane 3, episome detection 1.1 kb (primers p539 + Pb8).
Fig. 5.
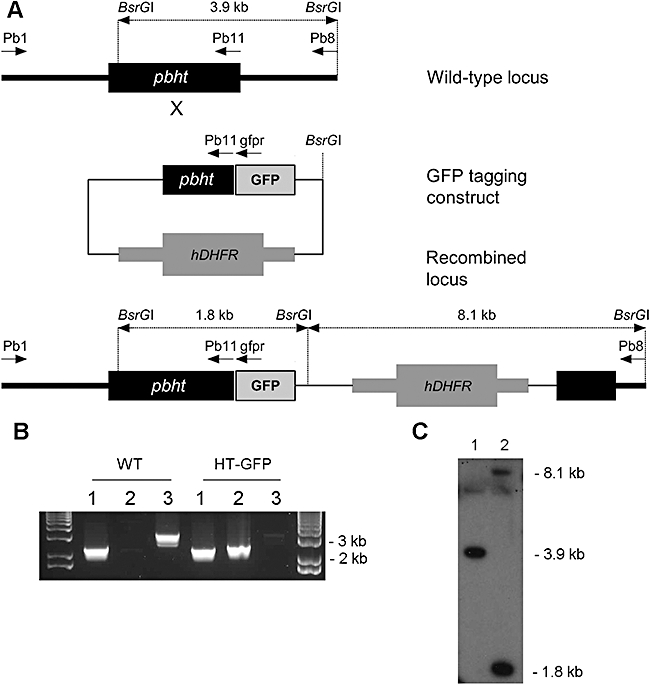
Tagging of the pbht locus with GFP. A. The strategy for GFP-tagging of the pbht locus; arrows indicate the location of PCR primers. B. PCR analysis of the wild type (WT) and a pyrimethamine-resistant pbht-gfp transfected line. Lane 1, positive control 2.3 kb (primers Pb1 + Pb11); lane 2, detection of the gfp-tagged locus 2.4 kb (Pb1 + gfpr); lane 3, detection of the wild-type locus 3.2 kb (Pb1 + Pb8). C. Southern blot analysis of wild-type (1) and pbht-gfp transfected line (2). gDNA was digested with BsrGI, and blot was probed with a pbht fragment used for generation of the tagging construct; wild-type locus 3.9 kb, integration bands 1.8 kb (5′) and 8.1 kb (3′).
Expression profile of pbht-gfp during P. berghei life cycle
Available microarray data (PlasmoDB, Version 6.2) indicate that PfHT mRNA is detectable throughout the erythrocytic stages of development. Direct fluorescence imaging of blood stages of pbht-gfp transgenic parasites reveals fluorescence signal associated with the parasite's plasma membrane present in both early and late blood stages (Fig. 6A and B), demonstrating that PbHT protein is indeed present throughout the asexual cycle. Some of the observed fluorescence signal is internal and may be associated with perinuclear localization as well as the food vacuole, although colocalization studies are needed to ascertain this point. Observed internal PbHT-GFP signal could be associated with developing PbHT in its trafficking pathway or may come from a small proportion of PbHT-GFP protein that has been mislocalized. It is noteworthy that taken together with all our other work supporting the essentiality of this transporter, mislocalization of a greater proportion of the protein would have had functional consequences that have not been observed.
Fig. 6.
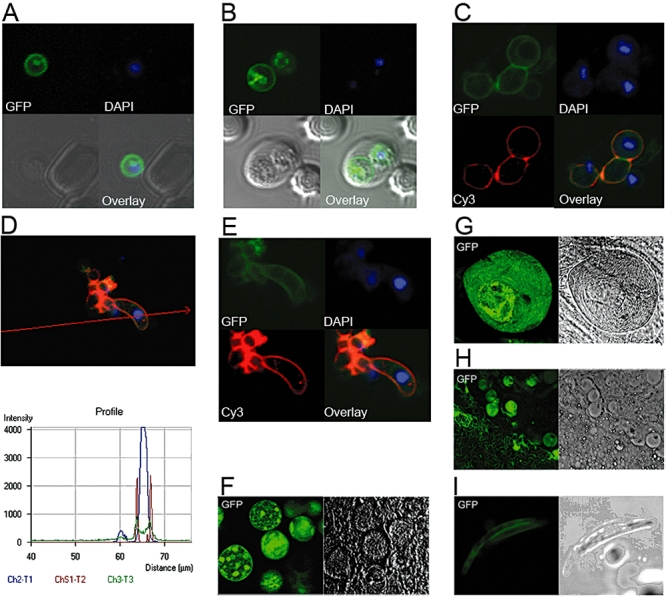
Direct fluorescence imaging of pbht-gfp transgenic line. A. A young, blood-stage pbht-gfp P. berghei parasite. B. Two pbht-gfp P. bergheitrophozoites inside an erythrocyte. C. Zygotes/or female gametes; interestingly a smaller round cell containing surface GFP fluorescence but no P28 staining is also observed. D. Analysis of fluorescence intensities across the ookinete cell. E. Ookinete [C–E: live parasites from 20 to 24 h culture in the ookinete medium, immunostained with a monoclonal antibody against the female gamete/zygote/ookinete marker P28 (red); DAPI was used as a nuclear dye (blue)]. F. A. stephensi midgut 11 days post infection. G. Sporulating midgut oocyst 21 days post infection. H. Sporozoites released from ruptured midgut oocysts. I. Sporozoites released from salivary glands of A. stephensi 21 days post infection.
Very limited information is available regarding glucose uptake during the sexual stages of malaria parasite development especially during its development within the mosquito. We used the pbht-gfp transgenic parasites to investigate the expression of this transporter during the parasite's development in the mosquito host.
We analysed PbHT-GFP expression in live parasites from an ookinete-enriched culture, and showed its expression and colocalization with P28, a surface marker of the ookinete, female gamete and zygote (Fig. 6C–E).
Analysis of mosquito midgut oocysts dissected 11 days post infection showed internal foci of fluorescence of developing sporoblasts and lower intensity membrane fluorescence signal (Fig. 6F). Twenty-one days post infection, strong fluorescence signal was detectable from sporulating oocysts and oocyst-derived sporozoites (Fig. 6G and H). Similarly, sporozoites released from dissected salivary glands 21 days post infection showed expression of PbHT-GFP (Fig. 6I).
Expression of PbHT-GFP in parasites from all analysed developmental stages was confirmed by Western blotting using an anti-GFP monoclonal antibody (Fig. 7). As a positive control for blotting and antibody staining, we used PbGFPCON line, a P. berghei line that constitutively expresses GFP throughout the life cycle (Franke-Fayard et al., 2004). A protein band of ∼82 kDa appeared for all analysed pbht-gfp samples, which corresponds to a predicted mass of PbHT–GFP fusion protein (84.4 kDa).
Fig. 7.
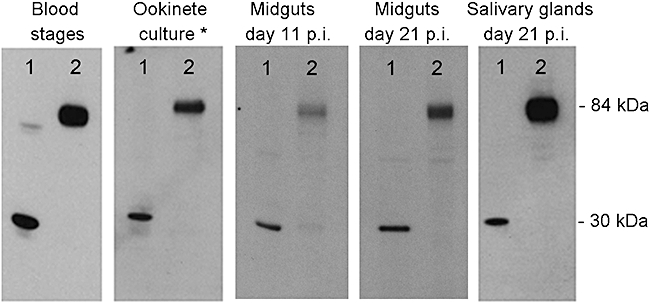
Western blot analysis. Ten micrograms of parasite material from asexual blood stages, ookinete-enriched culture and mosquito midguts and salivary glands stages of PbGFPCON (lane 1) and pbht-gfp line (lane 2) was subjected to SDS-PAGE (10% acrylamide), transferred to a nitrocellulose membrane and probed with 1:1000 diluted anti-GFP antibody (Roche) and 1:3000 diluted HRP-conjugated anti-mouse antibody. *Ookinete-enriched culture obtained by incubation of blood sample of an infected mouse in the ookinete medium overnight at 19°C. p.i., post infection.
Discussion
Identifying inhibitors of the parasite's essential nutrient uptake pathways may lead to the development of novel antimalarials. The P. falciparum hexose transporter, PfHT, has previously been investigated using chemical validation methodologies, which suggested that a functional PfHT molecule is required for parasite survival (Joet et al., 2003). The present study now provides genetic evidence that PfHT is indeed indispensable for erythrocytic stages of P. falciparum. Pfht could not be disrupted in the genome unless its expression was maintained by an exogenously supplied construct. We have further predicted that altered pfht expression would lead to changes in the parasite's sensitivity to a specific inhibitor of PfHT, 3361. We therefore investigated the transfected line containing PfHT expressed only from a complementing episome and lacking endogenous pfht. Quantitative PCR showed significantly increased expression of pfht in these parasites compared with wild-type controls at the trophozoite stage, whereas expression at ring stages was not affected. A previous study showed the peak of pfht expression to be at the early ring stage (8 h post invasion), with falling expression at the late ring stage (16 h) and intermediate levels of expression at the trophozoite stage (Woodrow et al., 1999). This indicates that highly regulated pfht expression in wild-type parasites can be replaced by higher but less regulated pfht expression in transfected lines. In support of this finding, the transfected line was also more resistant to growth inhibition by CM3361 (2.5-fold increase in mean IC50 values compared with controls; P < 0.001). These findings further validate PfHT as a novel target and provide strong evidence that PfHT is the specific target of CM3361 in parasites.
Although there is a growing evidence for a functional TCA cycle in blood stages of P. falciparum, its role is not fully defined. Expression profiling studies of P. falciparum parasites from patients found three metabolically distinct clusters: a glycolytic metabolism state, a starvation response with induction of metabolism of alternative carbon sources and an environmental stress response (Daily et al., 2007). The study by Daily et al. suggests that parasites can undergo a metabolic shift in vivo, switching from predominately glycolytic metabolism to metabolism of alternative carbon sources with induction of gene sets associated with function of mitochondria and apicoplast. Despite the complexity of metabolic patterning that emerges from these types of studies, glucose delivery is nevertheless essential for parasite survival and may also be critical for metabolic diversion of this key substrate from host tissues, thereby exacerbating disease processes. Here, when taken together with previous studies on PfHT, the central role of PfHT in hexose delivery to parasites is confirmed.
We have also extended our analysis of the Plasmodium hexose transporter to a rodent malaria model, P. berghei, which enabled studies with insect stages of the parasite's life cycle. Energy requirements and metabolism during parasite development in the invertebrate host are largely unknown, although it is established that gametocytes display increased expression of TCA cycle and oxidative phosphorylation enzymes compared with asexual stages (Hall et al., 2005). In addition to asexual blood stages, proteome analyses of gametocytes, ookinetes, mosquito midgut oocysts and sporozoites have been performed. Proteomic survey of P. berghei life cycle has detected PbHT expression in asexual blood stages (Hall et al., 2005) and gametocytes (Khan et al., 2005). Similarly, P. falciparum proteome studies identified PfHT expression in asexual stages and gametocytes (Florens et al., 2002; Lasonder et al., 2002). More recently, PfHT has been found expressed in oocyst-derived sporozoites (Lasonder et al., 2008). These findings may reflect in part methodological limitations of applying current proteomic approaches to the detection of integral membrane proteins. They suggest that other membrane proteins should also be examined individually for expression in different stages of the life cycle. Here we show, by direct fluorescence as well as Western blot analysis, that PbHT is expressed in female gametes/or zygotes, ookinetes, midgut oocysts and sporozoites derived from both midgut oocysts and salivary glands.
Furthermore, the importance of glucose transport for a cell may be inferred from the presence of expressed glycolytic enzymes. In the P. falciparum sporozoite proteome, with an exception of 6-phosphofructokinase, all glycolytic enzymes have been detected. However, this enzyme has been detected in the P. berghei sporozoite proteome, indicating that its absence from P. falciparum sporozoite proteome is probably due to the limitations of detection technology rather than true absence. Therefore, available proteomic data and our data presented here together suggest that the sugar uptake and glycolysis are present in sporozoites. Joet et al. (2002) analysed temperature dependence of Plasmodium hexose transporters activity and showed that they are active in a broad temperature range, with approximately 50 % of maximal activity retained at 20°C. This finding shows that these transporters can function in insect stages of development that take place at 22–26°C.
Further studies will evaluate potential of specific PfHT inhibitors in reducing gametocytaemia in culture. Novel drugs that would have activity against both asexual blood stages and gametocytes are necessary if the malaria research community is aiming towards the eradication of the disease.
These studies may help in identifying inhibitors of PfHT that can contribute to the challenging drug discovery process. Indeed, glucose is an essential energy substrate, not only for malaria parasites, but for other unrelated parasites as well, such as the kinetoplastida (Barrett et al., 1998; Burchmore et al., 2003).
Experimental procedures
Ethics statement
All animal work has passed an ethical review process and was approved by the United Kingdom Home Office. Work was carried out in accordance with the United Kingdom ‘Animals (Scientific Procedures) Act 1986’ and in compliance with ‘European Directive 86/609/EEC’ for the protection of animals used for experimental purposes.
Construction of plasmids for transfection of P. falciparum
pCAM-BSD-HT
A 1.2 kb fragment of the central region of pfht[nucleotides 163–1348 of the pfht open reading frame (ORF)] was amplified using primers KOHTf and KOHTr (Table 1). An amplified fragment was inserted into the pCAM-BSD plasmid using BamHI and NotI restriction sites (Sidhu et al., 2005). The resulting plasmid, pCAM-BSD-HT was used in transfections as a gene disruption (knockout) vector.
Table 1.
P. falciparum primer sequences.
| Primer | Sequence | |
|---|---|---|
| Knockout fragment | KOHTf | 5′-CAGGATCCGTTGTAGAATTTGAATGGTGTAAAGG |
| KOHTr | 5′-caGCGGCCGCTAATAATGTCTGATGGGAAGACAAC | |
| Complementation gene | XKBNf | 5′-CCAAGTCGGTTGTATGAGCGGCCGCTTACCACTAAACCAGCC |
| XKBNr | 5′-GGCTGGTTTAGTGGTAAGCGGCCGCTCATACAACCGACTTGG | |
| PCR detection of knockout plasmid integration | 1 | 5′-TATATATAAAATGAGGAATTGGAAAATTTTTC |
| 2 | 5′-AATGGAATAAATGTCGATTGGATAATGTTTG | |
| 3 | 5′-TATTCCTAATCATGTAAATCTTAAA | |
| 4 | 5′-CAATTAACCCTCACTAAAG | |
| Real-time PCR analysis | PfHT-f | 5′-GAAATGTTTCCATCAGAAATAAAAG |
| PfHT-r | 5′-GAATCGAAGGGGATTTCTTAATA | |
| PfHT probe | 5′-CAACAATAATTGCACAAACCCAATTAACTAATGA | |
| Tubulin-f | 5′-TGATGTGCGCAAGTGATCC | |
| Tubulin-r | 5′-TCCTTTGTGGACATTCCTTCCTC | |
| Tubulin probe | 5′-TAGCACATGCCGTTAAATATCTTCCATGTCT | |
pCHD-HT
Xkb.1 plasmid contains the full-length pfht gene flanked by BglII sites and was used as a source of the pfht gene (Woodrow et al., 1999). First, a pair of complementary PCR primers (XKBNf and XKBNr, Table 1) was used in site-directed mutagenesis to convert a 3′ BglII site immediately after the stop codon into a NotI site generating the XkbN plasmid. Subsequently, the pfhtgene was subcloned from XkbN plasmid into a pHGB vector downstream of the pfhsp86 promoter using BglII and NotI restriction sites (Tonkin et al., 2004). The resulting plasmid, pHGB-HT, was used as an entry vector in a LR recombination reaction with the destination vector, pCHD that contains DHFR cassette conferring resistance to WR99210 (Tonkin et al., 2004). After the recombination reaction, the resulting vector, pCHD-HT, contains pfht gene under the control of pfhsp86 promoter and the DHFR resistance cassette, and was used in co-transfections as a complementation plasmid.
All transfection constructs were verified by sequence analysis.
P. falciparum culture and transfection
Plasmodium falciparum 3D7 clone was grown as described previously (Trager and Jensen, 1976) and used for transfections. Parasites were sorbitol-synchronized, and ring-stage parasites were subjected to electroporation in the presence of 100 µg of pCAM-BSD-HT plasmid or 50 µg of both pCAM-BSD-HT and pCHD-HT plasmids, as described previously (Dorin-semblat et al., 2007). Blasticidin (2.5 µg ml−1) and WR99210 (5 nM) were added 48 h after transfection to select for transformed parasites. Blasticidin-resistant parasites appeared in culture 14 days post transfection and parasites resistant to both blasticidin and WR99210 appeared 19 days after transfection.
After the detection of homologous integration of the knockout vector in doubly transfected parasites by PCR, they were subjected to blasticidin cycling to select for parasites with pCAM-BSD-HT integrated into the genome. The cycling consisted of 3 weeks of culturing without blasticidin, followed by 3 weeks with blasticidin pressure. This selection cycle was repeated twice. No decrease in parasiteamia was observed after reapplying the drug pressure.
Genotype characterization
Plasmid rescue of episomes
Parasite pellets were obtained by saponin lysis and genomic DNA was extracted by proteinase K (0.1 mg ml−1) and SDS (2%) treatment for 2 h at 55°C. Genomic DNA extracted from both knockout and co-tranfected parasites was used to transform E. colisupercompetent cells. Plasmid DNA was isolated from obtained bacterial clones using standard methods and digested with BamHI and NotI or BglII and NotI.
PCR
PCR analysis of transfectants was performed on genomic DNA using Taq polymerase (Sigma) and following cycling conditions: 1 cycle at 92°C (2 min); 35 cycles at 92°C (30 s), 54°C (45 s) and 65°C (1.5 min) and 1 cycle at 65°C (5 min). Sequences of primers used are shown in Table 1. PCR primers 1 and 2 anneal to the pfht wild-type locus outside the ORF region inserted in pCHD-HT. Primers 3 and 4 anneal to the pCAM-BSD-HT and flank the insertion site.
Four different combinations of primers were used in order to detect: the wild-type pfht locus (primers 1 and 2), 5′ end of integration event (primers 1 and 4), 3′ end of the integration event (primers 2 and 3) and the presence of episome (primers 3 and 4).
Southern blotting
Genomic DNA extracted from parasite pellet obtained by saponin lysis was digested with SwaI, NcoI and EcoNI. pCAM-BSD-HT and pCHD-HT plasmid DNA were also digested with same enzymes and used as controls. One microgram of digested genomic DNA and 1.5 ng of digested plasmid DNA were separated on 0.8% agarose gel and transferred to positively charged nylon membrane (Hybond-XL, Amersham). Subsequently, the membrane was hybridized overnight at 58°C with 1 kb pfht knockout fragment labelled with 32P-dCTP (Sambrook et al., 1989).
Phenotype characterization
Quantitative PCR
Parasites were sorbitol-synchronized and frozen down in RNA-Latersolution (Ambion) for RNA extraction at rings (∼10 h post invasion) and trophozoite stages (∼30 h post invasion). RNA was isolated with RNA isolation kit (Qiagen) according to manufacturer's instructions. Isolated RNA was treated with DNase I (Promega) and used in reverse transcription reactions (High Capacity cDNA Reverse Transcription Kit with RNase inhibitor, Applied Biosystems).
Quantification of the gene expression was assessed by TaqMan real-time PCR (Stratagene Mx3005P QPCR System). Primers and probes are summarized in Table 1. The pfht probe was FAM-labelled at the 5′ end, and the β-tubulin probe was VIC-labelled. Both probes had a TAMRA quencher at the 3′ end. Amplification reactions were done as duplex PCR in 96-well plates using TaqMan Universal PCR Master Mix (Applied Biosystems). pfhtprimers and probe concentrations were 600 nM and 40 nM respectively. β-tubulin primers and probe were all used at 100 nM (Price et al., 2004). The following cycling conditions were applied: 95°C 10 min, followed by 45 cycles of 95°C (15 s) and 57°C (1 min). Reactions were done in triplicate and experiments repeated three times.
Relative pfht expression in complemented parasites compared with calibrator (3D7 parasites) was calculated using the efficiency-corrected comparative quantification method. β-tubulin was used as a normalizer gene. A standard curve was run together with experimental samples during each run to estimate the efficiencies of target (pfht) and normalizer (β-tubulin) assays. The following equation was used to calculate relative quantity to the calibrator: (1 + EGOI)ΔCtGOI/(1 + Enorm)ΔCtnorm, where EGOI and Enorm are efficiencies of the target and the normalizer assays and ΔCt = (Ctcalibrator – Ctunknown). Primers sequences are in Table 1.
Parasite growth and sensitivity to CM3361
3-O-(undec-10-en)-yl-D-glucose (CM3361) was synthesized as described earlier (Ikekawa et al., 1987). The effect of glucose analogue inhibitor CM3361 on the growth inhibition of the wild-type and complemented parasites was determined using the [3H]-hypoxanthine incorporation assay as previously described. Ring-stage parasites were seeded in the 96-well plates at 0.5% parasitemia and 2% hematocrit in the presence of increasing concentrations of the inhibitor (five replicates per inhibitor concentration). [3H]-hypoxanthine was added after 24 h of incubation and parasites harvested after 48 h. Five independent experiments were performed.
Statistical analysis
For P. falciparum IC50 assays, a sigmoidal dose–response model with the variable slope was fitted to results using GraphPad Prism (Version 4 for Macintosh). IC50 values obtained with the wild-type and the complemented parasite line were compared using Student's t-test (unpaired, two-tailed). For the analysis of pfht mRNA expression, the relative pfht expression in the complemented parasite line was compared with 3D7 parasites using one sample t-test. Statistical significance was set at P ≤ 0.05.
P. berghei culture and transfection
Transfection experiments were performed on P. berghei ANKA strain 2.34 parasites according to a described protocol (Janse et al., 2006). The pbht knockout vectors were constructed for a double cross-over homologous recombination in the pBS-DHFR plasmid that contains a Toxoplasma gondii dhfr/ts cassette conferring resistance to pyrimethamine (Dessens et al., 1999). The knockout construct was generated by inserting 0.8 kb of the pbht 5′ untranslated (UTR) region upstream and 1 kb of the pbht 3′ UTR region downstream of the dhfr cassette (sequences of primers used to amplify fragments from P. berghei genomic DNA are given in Table 2). The final knockout construct was digested with KpnI and NotI to release the fragment for transfection.
Table 2.
P. berghei primers sequences.
| Primer | Sequence | |
|---|---|---|
| PbHT 5′ UTR | Pb1 | 5′-cgaGGTACCGTGTAAAAATTTATCGTTAAGAGAG |
| Pb2 | 5′-CATGGGCCCTTTTTTCGTATTAATACACATATATTTCTTG | |
| PfHT ORF | Pb3 | 5′-AAAGGGCCCATGACGAAAAGTTCGAAAGAT |
| Pb4 | 5′-caaGTCGACTCATACAACCGACTTGGTCATATG | |
| Spacer 3′ UTR | Pb5 | 5′-atcGTCGACATTTTGATACGCATAAATCGTATAG |
| Pb6 | 5′-catAAGCTTTTTGATACATATATATTTGATACATATATATTTG | |
| PbHT 3′ UTR | Pb7 | 5′-gtcGATATCATTTTGATACGCATAAATCGTATAGATATAG |
| Pb8 | 5′-caaGCGGCCGCAAAAAAATAGAAATCAAATGATATATATTTACCC | |
| PbHT-GFP fragment | Pb10 | 5′-catGGTACCTAACATTTGGAATATTTGTTGCAGTTTTATTGGG |
| Pb11 | 5′-caaGGGCCCAACTCTTGATTTGCTTATATGTTTTTGTCTTTCTTC | |
| Integration detection | Pb9 | 5′-CACCATTTTATTCACCATATTTTTAC |
| 539 | 5′-CAATGATTCATAAATAGTTGGACTTG | |
| gfpr | 5′-ACGCTGAACTTGTGGCCG | |
To generate a pbht-gfp construct for a single cross-over homologous recombination, a 1 kb region of the pbht without the stop codon was inserted in frame and upstream of the gfp sequence in the plasmid p277 containing the human dhfr cassette and conveying resistance to pyrimethamine (Liu et al., 2008). Prior to transfection, the final construct was digested with BstXI that cuts the plasmid in the middle of the insert, which is optimal for the homologous recombination event.
Transfected P. berghei parasites were selected with pyrimethamine selection pressure according to a described protocol (Janse et al., 2006). Transfection of P. berghei parasites with the knockout construct was carried out minimum of three times.
Genotype analysis of P. berghei transfectants
PCR
PCR analysis was used to inspect if the transfection construct was integrated into the correct locus in pyrimethamine-resistant parasites. For the analysis of parasites transfected with the pbht knockout construct, combinations of the following four primers were used to explore the pbht locus: primer 539 anneals to the DHFR cassette in the construct, primers Pb7 and Pb8 anneal to the pbht 3′ UTR inserted in the construct, whereas primer Pb9 anneals further downstream from Pb8 in pbht 3′ UTR and does not anneal to the sequences present in the construct. For the analysis of pbht-GFP transgenic parasites, Pb1, Pb8, Pb11 and gfpr primers were used.
Southern blot
Genomic DNA extracted from pbht-GFP parasite pellet obtained by lysis with NH4Cl was digested with BsrGI. One microgram of digested genomic DNA was separated on 0.8% agarose gel and transferred to a positively charged nylon membrane (Hybond-XL, Amersham) that was hybridized overnight at 55°C with 1 kb pbht probe labelled with 32P-dCTP.
Analysis of the HT- GFP localization during malaria life cycle
Images of GFP-expressing parasites were captured with LSM 510 META Confocal Laser Scanning Microscope (Zeiss) and Leica SP5 confocal microscope. Hoechst33342 or DAPI were used for nuclear staining of blood stages parasites and ookinetes. Anopheles stephensi mosquitoes were fed on mice infected with P. berghei pbht-gfp and PbGFPCON parasites as described (Sinden et al., 2002). Eleven days post infection, mosquitoes were dissected and midguts were collected for direct fluorescence imaging and Western blotting analysis (∼30 midguts per parasite strain). Dissection of mosquitoes was repeated at day 21 post infection when midguts and salivary glands were collected for the analysis of pbht expression.
Western blot analysis
For Western blot analysis parasite pellets of pbht-GFP and PbGFPCON (Franke-Fayard et al., 2004). P. berghei blood stages were obtained by erythrocytes lysis with NH4Cl. Pellets of mosquitoes' midguts and salivary glands were ground with pestle and pellets resuspended in PBS containing Complete™ protease inhibitor cocktail tablets (Roche). Approximately 10 µg of each extract were heated at 70°C in NuPAGE® LDS Sample Buffer (Invitrogen), separated on Novex® 10% Bis-Tris gel (Invitrogen) and electro-transferred to a nitrocellulose membrane (Amersham). Membranes were blocked overnight at 4°C in PBS containing 5% skimmed milk and 0.1% Tween 20. Blots were probed with a mouse monoclonal GFP antibody (Roche), diluted 1:1000. Bound antibodies were detected with a HRP-conjugated anti-mouse secondary antibody, diluted 1:3000 (Bio-Rad) and ECL Western Blotting detection reagents (Amersham).
Ookinete culture
Blood was taken from a pbht-gfp infected mouse on day 4 post infection into a heparinized syringe, mixed with the ookinete culture medium (RPMI1640 containing 25 mM HEPES, 25% fetal bovine serum, 10 mM sodium bicarbonate, 50 µM xanthurenic acid, pH 7.6) and cultured at 19°C for a further 21–24 h. For direct immunolabelling cultured cells were pelleted for 2 min at 800 g and then labelled for 10 min on ice in 50 µl of ookinete medium containing Hoechst33342 and Cy3-conjugated mouse monoclonal antibody specific for P28 (Reininger et al., 2009).
Acknowledgments
We thank A. Talman and K. A. Szewczyk for their kind technical help with the confocal microscopy and M. Lanzer for helpful discussions and support. SK and CD are funded by the European Commission projects ANTIMAL (Grant No. 018834) and MALSIG (Grant No. 223044). CD is also supported by the European Commission project BioMalPar Network of Excellence and by the French Institut National de la Santé de la Recherche Médicale (Inserm). RT is funded by the Wellcome trust and MRC grant (through which US is supported).
References
- Barrett MP, Tetaud E, Seyfang A, Bringaud F, Baltz T. Trypanosomeglucose transporters. Mol Biochem Parasitol. 1998;91:195–205. doi: 10.1016/s0166-6851(97)00192-8. [DOI] [PubMed] [Google Scholar]
- Burchmore RJ, Rodriguez-Contreras D, McBride K, Merkel P, Barrett MP, Modi G, et al. Genetic characterization of glucose transporter function in Leishmania mexicana. Proc Natl Acad Sci USA. 2003;100:3901–3906. doi: 10.1073/pnas.0630165100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowman AF, Crabb BS. Functional genomics: identifying drug targets for parasitic diseases. Trends Parasitol. 2003;19:538–543. doi: 10.1016/j.pt.2003.09.006. [DOI] [PubMed] [Google Scholar]
- Daily JP, Scanfeld D, Pochet N, Le Roch K, Plouffe D, Kamal M, et al. Distinct physiological states of Plasmodium falciparum in malaria-infected patients. Nature. 2007;450:1091–1095. doi: 10.1038/nature06311. [DOI] [PubMed] [Google Scholar]
- Dessens JT, Beetsma AL, Dimopoulos G, Wengelnik K, Crisanti A, Kafatos FC, Sinden RE. CTRP is essential for mosquito infection by malaria ookinetes. EMBO J. 1999;18:6221–6227. doi: 10.1093/emboj/18.22.6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorin-Semblat D, Quashie N, Halbert J, Sicard A, Doerig C, Peat E, et al. Functional characterization of both MAP kinases of the human malaria parasite Plasmodium falciparum by reverse genetics. Mol Microbiol. 2007;65:1170–1180. doi: 10.1111/j.1365-2958.2007.05859.x. [DOI] [PubMed] [Google Scholar]
- Florens L, Washburn MP, Raine JD, Anthony RM, Grainger M, Haynes JD, et al. A proteomic view of the Plasmodium falciparum life cycle. Nature. 2002;419:520–526. doi: 10.1038/nature01107. [DOI] [PubMed] [Google Scholar]
- Franke-Fayard B, Trueman H, Ramesar J, Mendoza J, van der Keur M, van der Linden R, et al. A Plasmodium berghei reference line that constitutively expresses GFP at a high level throughout the complete life cycle. Mol Biochem Parasitol. 2004;137:23–33. doi: 10.1016/j.molbiopara.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall N, Karras M, Raine JD, Carlton JM, Kooij TW, Berriman M, et al. A comprehensive survey of the Plasmodium life cycle by genomic, transcriptomic, and proteomic analyses. Science. 2005;307:82–86. doi: 10.1126/science.1103717. [DOI] [PubMed] [Google Scholar]
- Ikekawa T, Irinoda K, Saze K, Katori T, Matsuda H, Ohkawa M, Kosik M. Studies on synthesis of 3-O-alkyl-D-glucose and 3-O-alkyl-D-allose derivatives and their biological activities. Chem Pharm Bull (Tokyo) 1987;35:2894–2899. doi: 10.1248/cpb.35.2894. [DOI] [PubMed] [Google Scholar]
- Imming P, Sinning C, Meyer A. Drugs, their targets and the nature and number of drug targets. Nat Rev Drug Discov. 2006;5:821–834. doi: 10.1038/nrd2132. [DOI] [PubMed] [Google Scholar]
- Janse CJ, Ramesar J, Waters AP. High-efficiency transfection and drug selection of genetically transformed blood stages of the rodent malaria parasite Plasmodium berghei. Nat Protoc. 2006;1:346–356. doi: 10.1038/nprot.2006.53. [DOI] [PubMed] [Google Scholar]
- Joet T, Holterman L, Stedman TT, Kocken CH, Van Der Wel A, Thomas AW, Krishna S. Comparative characterization of hexose transporters of Plasmodium knowlesi, Plasmodium yoeliiand Toxoplasma gondii highlights functional differences within the apicomplexan family. Biochem J. 2002;368:923–929. doi: 10.1042/BJ20021189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joet T, Eckstein-Ludwig U, Morin C, Krishna S. Validation of the hexose transporter of Plasmodium falciparum as a novel drug target. Proc Natl Acad Sci USA. 2003;100:7476–7479. doi: 10.1073/pnas.1330865100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SM, Franke-Fayard B, Mair GR, Lasonder E, Janse CJ, Mann M, Waters AP. Proteome analysis of separated male and female gametocytes reveals novel sex-specific Plasmodium biology. Cell. 2005;121:675–687. doi: 10.1016/j.cell.2005.03.027. [DOI] [PubMed] [Google Scholar]
- Lasonder E, Ishihama Y, Andersen JS, Vermunt AM, Pain A, Sauerwein RW, et al. Analysis of the Plasmodium falciparum proteome by high-accuracy mass spectrometry. Nature. 2002;419:537–542. doi: 10.1038/nature01111. [DOI] [PubMed] [Google Scholar]
- Lasonder E, Janse CJ, van Gemert GJ, Mair GR, Vermunt AM, Douradinha BG, et al. Proteomic profiling of Plasmodium sporozoite maturation identifies new proteins essential for parasite development and infectivity. PLoS Pathog. 2008;4:e1000195. doi: 10.1371/journal.ppat.1000195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Tewari R, Ning J, Blagborough AM, Garbom S, Pei J, et al. The conserved plant sterility gene HAP2 functions after attachment of fusogenic membranes in Chlamydomonasand Plasmodium gametes. Genes Dev. 2008;22:1051–1068. doi: 10.1101/gad.1656508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RE, Henry RI, Abbey JL, Clements JD, Kirk K. The ‘permeome’ of the malaria parasite: an overview of the membrane transport proteins of Plasmodium falciparum. Genome Biol. 2005;6:R26. doi: 10.1186/gb-2005-6-3-r26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noedl H, Se Y, Schaecher K, Smith BL, Socheat D, Fukuda MM. Evidence of artemisinin-resistant malaria in western Cambodia. N Engl J Med. 2008;359:2619–2620. doi: 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]
- Price RN, Uhlemann AC, Brockman A, McGready R, Ashley E, Phaipun L, et al. Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet. 2004;364:438–447. doi: 10.1016/S0140-6736(04)16767-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reininger L, Tewari R, Fennell C, Holland Z, Goldring D, Ranford-Cartwright L, et al. An essential role for the Plasmodium Nek-2 Nima-related protein kinase in the sexual development of malaria parasites. J Biol Chem. 2009;284:20858–20868. doi: 10.1074/jbc.M109.017988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning – A Laboratory Manual. Cold Spring Harbour, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Sidhu AB, Valderramos SG, Fidock DA. pfmdr1 mutations contribute to quinine resistance and enhance mefloquine and artemisinin sensitivity in Plasmodium falciparum. Mol Microbiol. 2005;57:913–926. doi: 10.1111/j.1365-2958.2005.04729.x. [DOI] [PubMed] [Google Scholar]
- Sinden RE, Butcher G, Beetsma A. Maintance of the Plasmodium berghei life cycle. In: Doolan DL, editor. Malaria Methods and Protocols. Totowa, NJ: Humana Press; 2002. pp. 25–40. Vol. 72. [DOI] [PubMed] [Google Scholar]
- Tonkin CJ, van Dooren GG, Spurck TP, Struck NS, Good RT, Handman E, et al. Localization of organellar proteins in Plasmodium falciparum using a novel set of transfection vectors and a new immunofluorescence fixation method. Mol Biochem Parasitol. 2004;137:13–21. doi: 10.1016/j.molbiopara.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- Woodrow CJ, Penny JI, Krishna S. Intraerythrocytic Plasmodium falciparum expresses a high affinity facilitative hexose transporter. J Biol Chem. 1999;274:7272–7277. doi: 10.1074/jbc.274.11.7272. [DOI] [PubMed] [Google Scholar]