

Abstract
Nicotiana tabacum (tobacco) 5-epi-aristolochene synthase (TEAS) serves as an useful model for understanding the enzyme mechanisms of sesquiterpene biosynthesis. Despite extensive bio-chemical and structural characterization of TEAS, a more detailed analysis of the reaction product spectrum is lacking. This study reports the discovery and quantification of several alternative sesquiterpene products generated by recombinant TEAS in the single-vial GC–MS assay. The combined use of chiral and non-polar stationary phases for gas chromatography separations proved critical for resolving the numerous sesquiterpene products of TEAS for mass spectral analysis and identification. Co-injection studies with available authentic standards from both synthetic and natural sources further corroborated the assignment of several compounds, resulting in an annotated reaction mechanism accounting for their biosynthesis. Moreover, a previously undocumented farnesyl trans–cis isomerization pathway was observed.
Keywords: Sesquiterpene, Terpene synthase, Terpene cyclase, GC–MS, Gas chromatography, Biosynthesis, Product identification, Isomerization, Enzyme mechanism, Electrophilic cyclization
Terpenes comprise the most diverse family of natural products known [1], possess a myriad of biological activities in their natural hosts, serve important commercial roles for mankind as therapeutic agents, flavors and fragrances, and insecticides, and continue to represent challenging synthetic targets for chemists due to their stereo-chemical and regio-chemical complexity [2]. Terpene synthases (cyclases) represent a functionally diverse family of structurally conserved enzymes in bacteria, fungi, and plants that often catalyze electrophilic transformations of achiral, acyclic, isoprenoid substrates into multi-cyclic, multi-chiral terpene products [3]. Terpene cyclases can be classified according to their specificity for 10-, 15-, or 20-carbon isoprenoid diphosphate substrates, referred to as mono-, sesqui-, or di-terpene cyclases, respectively. Sesquiterpene cyclases of particular interest here catalyze the transformation of a single 15-carbon substrate (E,E)-farnesyl diphosphate (FPP1) into over 300 distinct hydrocarbon skeletons [4]. Remarkably, the cyclization of acyclic isoprenoid substrates in a terpene cyclase active site proceeds through several highly reactive carbocationic intermediates, which readily undergo dramatic regio- and stereo-chemical rearrangements en route to a product. Several cyclases, however, produce numerous cyclic hydrocarbon products, providing clues as to the cyclization route of the ionized FPP as it transforms into the dominant reaction product [5-7].
Tobacco 5-epi-aristolochene synthase (TEAS), a sesquiterpene cyclase from Nicotiana tabacum, serves as a valuable model system for understanding the chemical mechanisms of sesquiterpene biosynthesis. The major product of this enzyme, (+)-5-epi-aristolochene (13), was first identified as the cyclic precursor of the phytoalexin capsidiol, produced in response to fungal elicitation in tobacco tissue cultures [8-11]. The assigned structure of (+)-5-epi-aristolochene (13) enabled the proposal of a reaction mechanism involving initial formation then protonation of a germacrene A intermediate, followed by sequential 1,2-hydride and methyl migrations on the same face of a eudesmyl carbocation intermediate A, culminating in formation of the final product (13) (Scheme 1).
Scheme 1.
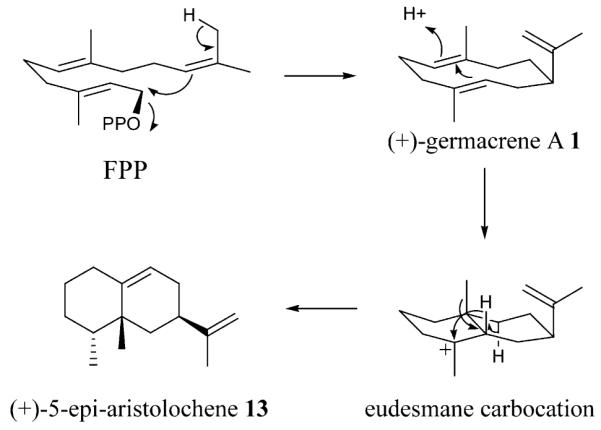
Reaction sequence of TEAS: cyclization of FPP to 5-epi-aristolochene via germacrene A and eudesmyl carbocation intermediates.
Tobacco 5-epi-aristolochene synthase was the first plant sesquiterpene cyclase to be cloned and sequenced [12], and the deduced amino acid sequence has been widely used for sequence comparisons with newly discovered cyclases [13-15]. Robust bacterial expression and high solubility of recombinant TEAS protein has facilitated extensive biochemical characterization [16], including pre-steady-state rapid quench and isotope trapping experiments [17]. Most significantly, TEAS yielded the first high-resolution crystal structure of a plant terpene cyclase, an advance which made it possible to propose a structural basis for the enzymatic conversion of FPP to (+)-5-epi-aristolochene (13) [18]. The structure of TEAS continues to serve as an essential frame-work for understanding stereo-chemical selectivity displayed by terpene cyclases, as exemplified by its use as a template for modeling other cyclases [19-21].
Despite the biochemical and biophysical characterization of TEAS, a more thorough analysis of the reaction product spectrum is lacking. Previously, the wild type enzyme had been reported to make an unknown eremophilane compound (17), later identified as (−)-4-epi-eremophilene (14) [22,23], indicating that TEAS produces at least one alternative reaction product. This report details the identification and quantification of alternative sesquiterpene products produced by TEAS catalysis and presents an annotated reaction mechanism accounting for their formation that is consistent with the known identity of authenticated products. A striking observation made during the course of this study was the presence of a subset of sesquiterpenes derived mechanistically from an initial isomerization of the C2–C3 bond from trans to cis prior to subsequent cyclization. Notably, this feature defines a large class of sesquiterpene cyclases (Scheme 2) [24]. Furthermore, careful quantification of reaction products as a function of temperature reveals changing flux of carbocations through the cyclization pathway consistent with kinetic control of the reaction by TEAS. These findings provide new insights into the energetics of farnesyl cation cyclization and contribute to the continued development of the TEAS model system for understanding the mechanisms and biophysical basis of enzyme-mediated terpene cyclization.
Scheme 2.

Isomerization of (E,E) to the (Z,E)-farnesyl cation.
Materials and methods
Materials
(E,E)-Farnesyl diphosphate (farnesyl-pyrophosphate) was purchased from Echelon Biosciences, as a powder and re-suspended in 25 mM NH4CO3 as a 50 mM stock solution. The sesquiterpene standard (−)-α-cedrene was purchased from Sigma–Aldrich. Synthetic standards for (−)-4-epi-eremophilene and (−)-prezizaene were kindly provided by R.M. Coates and celery oil extract containing α-selinene was provided by T. Kollner.
Protein expression and purification
Tobacco 5-epi-aristolochene synthase, LTC1 (Lactuca sativa germacrene A synthase), and HPS (Hyoscyamus muticus premnaspirodiene synthase) were cloned into the Escherichia coli expression vector pHis9-GW (an in-house Gateway destination vector), and transformed into E. coli BL21(λDE3). Transformed E. coli were grown at 37 °C in terrific broth containing 50 μg/ml kanamycin until an A600 value of 1.0. After induction with 0.1 mM isopropyl 1-thio-β-galactopyranoside, the cultures were grown for 6 h at 22 °C. Cells were harvested by centrifugation and re-suspended in lysis buffer (50 mM Tris–HCl, pH 8.0, 500 mM NaCl, 20 mM imidazole, pH 8.0, 20mM β-mercaptoethanol, 10% [v/v] glycerol, and 1% [v/v] Tween 20) and stirred at 4 °C for 1 h with lysozyme (0.5 mg/ml). After sonication and centrifugation, the supernatant was passed over a column of Ni2+–NTA resin (Qiagen), washed with 10 bed volumes of lysis buffer and 10 bed volumes of wash buffer (50 mM Tris–HCl, pH 8.0, 500 mM NaCl, 20 mM imidazole, pH 8.0, 20 mM β-mercaptoethanol, and 10% [v/v] glycerol), and the His-tagged protein was eluted with elution buffer (50 mM Tris–HCl, pH 8.0, 500 mM NaCl, 250 mM imidazole, pH 8.0, 20 mM β-mercaptoethanol, and 10% [v/v] glycerol). Dialysis for 24 h at 4 °C against 50 mM Tris–HCl, pH 8, 100 mM NaCl, and 1 mM DTT was followed by concentration of protein solutions to greater than or equal to 10 mg/ml by centrifugation using 30,000 molecular weight cut-off concentrators (Millipore, Bedford, MA). An equal volume of glycerol was added (50% (v/v) final concentration) for storage at −20 °C. Proteins were deemed greater than or equal to 95% pure, as judged from SDS–PAGE analysis.
Product identification and quantification by GC–MS
Reaction products were analyzed using a Hewlett–Packard 6890 GC coupled to a 5973 mass selective detector (MSD) outfitted with a 7683 series injector and autosampler, and equipped with either an HP-5MS capillary column (5% diphenyl/95% dimethyl siloxane) for standard separations or an HP-Chiral-20B column (20% β-cyclodextrin) for chiral resolution (0.25 mm i.d. × 30 m with 0.25-μm film dimensions) (Agilent Technologies). Needle sampling depth was adjusted to place the needle tip in the middle of the organic layer (near the 750 μl level in 2 ml glass vials) as previously described for the vial assay [25]. For standard separations, the GC was operated at a He flow rate of 2 ml/min and the MSD was operated at 70 eV. Splitless injections (2 μl) were performed with an injector temperature of 250 °C. The GC was programmed with an initial oven temperature of 50 °C (5-min hold), which was then increased 10 °C/min up to 180 °C (4-min hold), followed by a 100 °C/min ramp until 240 °C (1-min hold).
For chiral separations, the GC was operated at a He flow rate of 1.5 ml/min with an injection port temperature of 200 °C. The GC oven program was identical to that described for standard separations. For all runs, a solvent delay of 8.5 min was allowed prior to acquisition of MS data. Product peaks were quantified by integration of peak areas using Enhanced Chemstation version B.01.00 (Agilent Technologies).
Single-vial assay for product analysis
The vial assay for product characterization of recombinant TEAS was performed according to previously published methods [25]. Reactions were formulated in 0.5 ml volumes in assay buffer (50 mM Bis-Tris propane–HCl, pH 7.5, 20 mM MgCl2) with 1.0 μM enzyme and 500 μM farnesyl diphosphate. Reaction components were mixed in 2 ml screw top glass vials, allowed to equilibrate at room temperature prior to the addition of enzyme, then reactions were overlaid with 0.5 ml ethyl acetate containing 50 μM farnesol as an internal standard, and caps were affixed. After ~1h incubations at room temperature, the hydrocarbon products were extracted by vigorous vortexing for 10–15 s, followed by GC–MS analysis. For temperature dependent studies, glycerol was added to 10% (v/v) and the reaction buffers were adjusted to, pH 7, at the indicated temperature. Reaction components were mixed and allowed to equilibrate to temperature prior to the addition of enzyme, then overlaid with ethyl acetate (with farnesol). Incubations at room temperature or higher were allowed to proceed for ~1 h prior to quenching by vortexing, whereas reactions at lower temperatures were allowed to incubate overnight.
Results
Identification and characterization of TEAS reaction products
In addition to the dominant product (+)-5-epi-aristolochene (13), recombinant TEAS catalyzes the synthesis of a diverse array of sesquiterpene hydrocarbons using FPP as a substrate. Both non-polar (5% diphenyl/95% dimethylsiloxane) and chiral (20% β-cyclodextrin) stationary phases proved complementary for the separation of reaction product mixtures by gas chromatography. Careful analysis of the corresponding mass spectrometry data led to the recognition of 25 distinct compounds numbered according to their order of elution (Fig. 1). Relative retention times of the various compounds were similar from column to column; however, the degree of peak-to-peak separation varied depending on the column’s stationary phase. For instance, (+)-5-epi-aristolochene (13) and (−)-4-epi-eremophilene (14) were easily separated on the non-polar phase whereas, these two products co-elute using the chiral column. Conversely, spirolepechinene (5) and isoprezizaene (8), which are poorly resolved on the non-polar phase, migrate as distinct peaks during chiral separations. Of note, the chiral-phase column was operated at reduced inlet temperatures, as compared to the non-polar column (lower temperature rating for the stationary phase) resulting in a moderate decrease in analyte signal compared to the non-polar separation. This lowered temperature likely accounted for the absence of several of the least abundant minor products during chiral separations.
Fig. 1.

Gas chromatographic resolution of TEAS reaction products. (A) Separation of TEAS reaction products with a non-polar stationary-phase column (in black) was overlaid on a chromatogram from a control reaction with no added enzyme (gray). (B) Separation of TEAS reaction products with a chiral stationary-phase column (in black) was overlaid on a chromatogram from a control reaction with no added enzyme (gray).
Product specificity, defined here as the degree to which a cyclase generates a single product exclusively, was quantified by integration of peak areas from total ion chromatograms and expressed in terms of the percentage of the total sesquiterpene product (Table 1). Under the experimental conditions employed at room temperature, TEAS generates (+)-5-epi-aristolochene (13) as 78.9% hydrocarbon product, however, about one out of every five TEAS-catalyzed reactions results in 1 of 24 possible alternative products. (−)-4-epi-Eremophilene (14) is the most abundant of these at 6.2%, followed by (+)-germacrene A (1) at 3.6%. The remaining 22 hydrocarbons contribute ~12% of TEAS sesquiterpene products. Dominant reaction products were easily identified through matches to library mass spectra, but even minor products that constitute ~0.1% of the total extractable product, such as β-acoradiene ( 9) and β-selinene (19), displayed mass spectra of sufficient integrity to make reasonable matches to library spectra possible (described below).
Table 1.
TEAS reaction products identified by GC–MS
| Peaka | Compound | % abundanceb | Validationc | Retention time (min) |
|
|---|---|---|---|---|---|
| Chiral | Non-polar | ||||
| 13 | (+)-5-epi-Aristolochene | 78.87 | std | 17.89 | 15.59 |
| 14 | (−)-4-epi-Eremophilene | 6.21 | std | ND | 15.67 |
| 1 | (+)-Germacrene Ad | 3.65 | std | 16.42 | 14.38 |
| 11 | Selina-4,11-diene | 1.88 | lib | 17.68 | 15.48 |
| 8 | Isoprezizaene | 1.77 | std | 17.58 | 15.19 |
| 16 | (−)-Premnaspirodiene | 1.66 | std | 18.05 | 15.83 |
| 5 | Spirolepechinene | 1.40 | lib | 17.22 | 15.15 |
| 15 | α-Selinene | 1.06 | std | 17.99 | 15.73 |
| 12 | Unknown-12 | 0.71 | — | 17.83 | ND |
| 20 | Unknown-20 | 0.55 | — | 19.14 | 16.38 |
| 4 | Unknown-4 | 0.48 | — | ND | 14.97 |
| 2 | (−)-α-Cedrene | 0.46 | std | 16.98 | 14.67 |
| 22 | Unknown-22 | 0.24 | — | ND | 17.51 |
| 3 | Unknown-3 | 0.22 | — | 17.15 | 14.76 |
| 9 | β-Acoradiene | 0.18 | lib | ND | 15.35 |
| 7 | Unknown-7 | 0.12 | — | 17.50 | ND |
| 19 | β-Selinene | 0.12 | lib | ND | 16.01 |
| 17 | Unknown-17 | 0.11 | — | 18.18 | ND |
| 10 | Unknown-10 | 0.07 | — | ND | 15.37 |
| 6 | Unknown-6 | 0.06 | — | 17.34 | ND |
| 21 | Unknown-21 | 0.06 | — | 19.46 | 16.67 |
| 24 | Unknown-24 | 0.03 | — | ND | 17.63 |
| 18 | Unknown-18 | 0.03 | — | ND | 15.88 |
| 23 | Unknown-23 | 0.02 | — | ND | 17.57 |
| 25 | Unknown-25 | 0.01 | — | ND | 17.65 |
ND, not detected.
Peak numbers were assigned according to the order in which compounds eluted from the GC.
The percentage abundance was calculated as the total ion count of a given product peak divided by the sum of the total ion counts of all product peaks multiplied by 100.
Products for which authentic standards were used for validation with GC–MS are indicated by the abbreviation std., whereas, product assigned on the basis of retention index and mass spectral matching to library compounds are abbreviated as lib.
(+)-Germacrene A (1) is detected as its thermally induced Cope rearrangement product (−)-β-elemene.
Product identification was accomplished using a two-dimensional search algorithm, taking both mass spectra and retention index into account during computer searches of a mass spectral library of sesquiterpenes [26]. Close matches to several compounds were found in this manner (Supplementary data, Table 1). For example, the mass spectrum of peak 5 matches the library reference spectra of (−)-spirolepechinene (5). The appearance of (−)-β-elemene, initially assigned from a library match, can be rationalized as the Cope rearrangement product of (+)-germacrene A (1) that occurs at high inlet temperatures used in the analysis (Scheme 3) as described previously [27]. No (−)-β-elemene is observed at low inlet temperatures. In fact, reducing the inlet temperature to 150 °C or below allows detection of (+)-germacrene A with a corresponding reduction in the (−)-β-elemene peak (not shown). The mass spectra of numerous other products displayed fragmentation patterns characteristic of sesquiterpenes with parent ions of m/z=204, but either the quality of spectra was insufficient for a reasonable match or the spectral library did not contain a suitable reference spectrum (Supplementary data, Table 2). Interestingly, compounds (22) through (25) were observed with longer retention times and parent ions of m/z=222 consistent with terpene alcohols.
Scheme 3.
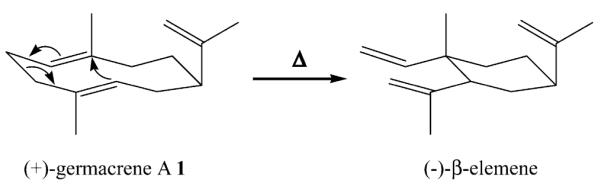
Mechanism of the thermally induced Cope rearrangement of (+)-germacrene A.
Co-injection studies with available authentic standards from both synthetic and natural sources further corroborated the assignment of several compounds (Fig. 2). For example, a commercially available (−)-α-cedrene standard and peak (2) co-elute on both non-polar and chiral phases, and share identical mass spectra. The synthetic standard (−)-prezizaene and peak (8) have identical mass spectra, and co-elute on the non-polar column; however, these products are clearly resolved as separate peaks on the chiral phase. The most abundant alternative product of TEAS, (−)-4-epi-eremophilene (14), was confirmed by comparison to a recently synthesized and characterized source [23]. Whereas, clear separation of peak 14 from (+)-5-epi-aristolochene (13) was observed on the non-polar phase, both products co-elute on the chiral phase. Enzymatically generated products (−)-premnaspirodiene from recombinant Hyoscyamus muticus premnaspirodiene synthase (HPS) and (+)-germacrene A from L. sativa germacrene A synthase (LTC1) provided biosynthetic standards to further support the assigned identity of peaks (1) and (16) in the product spectrum of TEAS.
Fig. 2.

Co-injection of authentic standards for product identification. An overlay of chromatograms from GC separations of TEAS reaction products in the presence (black) or absence (gray) of added authentic standard (as indicated on the top of each panel) is shown. A chiral stationary-phase column was used for the separations and boxes enclose the relevant portion of the chromatogram for each particular co-injection experiment. Peaks are labeled according to the numbering scheme found in Fig. 1.
Mechanistic rationalization of products
The identity of alternative reaction products strengthens the proposed cyclization pathway leading to the major reaction product (+)-5-epi-aristolochene (13), and further suggests the intermediacy of several distinct carbocation species during TEAS-catalyzed FPP turnover (Fig. 3). The TEAS-catalyzed reaction begins with magnesium ion-assisted ionization of FPP to generate the (E,E)-farnesyl cation and diphosphate ion pair. The juxtaposition of the C10–C11 double bond to the nascent carbocation center at C1 primes a facile 1,10 cyclization, which is followed by loss of a proton from the C13 methyl group of the germacren-11-yl cation, resulting in the formation of the neutral intermediate (+)-germacrene A (1). The formation of this proposed neutral intermediate has been clearly demonstrated for the Y520F mutant of TEAS [28]. Subsequent studies report the observation of germacrene A (1) as a product of the wild type enzyme at elevated pH [29,30].
Fig. 3.

Proposed TEAS cyclization mechanism accounting for identified products. All-trans FPP substrate is shown in the center and each major pathway involving cyclization of either the (E,E) or (Z,E)-farnesyl carbocations is highlighted with a large circular arrow terminating at the dominant product of the specific pathway. Branch points leading to multiple products are labeled A–D as referred to in the text. Products are labeled according to the numbering scheme found in Fig. 1.
A key step of fundamental importance to TEAS and related cyclases is the efficient activation of germacrene A (1) via protonation to generate a germacren-8-yl cation to promote further electrophilic cyclizations necessary to generate the complex cyclic products observed (in preparation). Protonation of germacrene A (1) at C6 generates the germacrenyl cation, followed by a 2,7 ring closure to form the eudesmyl cation, a cis-decalin ring system (Fig. 3, step A). Elimination of a proton at positions α, β, or γ to the carbocation center readily accounts for the formation of the alternative products α-selinene (15), β-selinene (19), and selina-4,11-diene (11), respectively. Notably, the presence of these selinenes requires the presence of fully active TEAS. In lieu of elimination, a 1,2 hydride shift from C2 to the Re face of the C3 carbocation results in the R stereochemistry of C3 observed in (+)-5-epi-aristolochene (13) and movement of the carbocation center to C2 (Fig. 3, step B). At this point in the pathway, alkyl shifts from either C6 (red arrow) or C8 (blue arrow) to C2 can occur to generate spirojatamyl or vetispiryl carbocations, which subsequently eliminate to (−)-spirolepechinene (5) or (−)-premnaspirodiene (16), respectively. In TEAS, migration of the C14 methyl from C7 to C2 is the prevailing Wagner–Meerwein rearrangement resulting in formation of an eremophilenyl cation (Fig. 3, step C). Elimination of a proton from C6 leads to (−)-4-epi-eremophilene (14), the most abundant minor product, whereas C8 elimination generates the dominant sesquiterpene product (+)-5-epi-aristolochene (13).
Among the anticipated minor products along the cyclization path of the (E,E)-farnesyl cation to (+)-5-epi-aristolochene was the startling discovery of (−)-α-cedrene (2), isoprezizaene (8), and β-acoradiene (9), products derived from the (Z,E)-farnesyl cation. The route to these products involves an initial isomerization of the C2–C3 bond following ionization and prior to subsequent cyclization events. This isomerization event is a mechanistic feature defining a large family of cyclases. These unexpected sesquiterpene products from TEAS account for ~2.5% of the overall hydrocarbon products (Table 1), and so 1 in every 40 turn-overs by TEAS appears to proceed via an initial isomerization of the farnesyl cation. Work currently in preparation examines in detail the enzymatic formation of isomerization products derived from the (Z,E)-farnesyl cation pathway in TEAS (in preparation).
The cyclization mechanism forming these products is proposed to resemble the athway described for epi-cedrol synthase [5]. In brief, a 1,6 cyclization of the (Z,E)-farnesyl cation, followed by a 1,2 hydride shift leads to a syn-6,10 ring closure which generates an acoradilyl cation (Fig. 3, step D). Elimination of a proton from either C12 or C13 of the isopropylidene tail of the acoradilyl cation readily explains the formation of β-acoradiene (9). Alternatively, a 2,11 ring closure followed by elimination of a proton at C2 is the likely pathway to (−)-α-cedrene (2). A 3,11 ring closure of the acoradilyl cation is the most common event, based on the relative abundance of isomerization products observed, and is followed by Wagner–Meerwein rearrangement and proton elimination from C15 to produce isoprezizaene (8), the dominant (Z,E)-farnesyl cation-derived product.
Temperature dependence of TEAS product specificity
Striking changes in the relative proportions of TEAS-catalyzed products were uncovered through careful quantification of products under conditions of changing temperatures (Table 2). TEAS product specificity drops from 87.6 to 66.2% in going from 0 to 42 °C, the latter temperature at which a third of all TEAS catalyzed reactions results in alternative products. The most dramatic shifts in product distributions were observed for germacrene A (1), which increased from 1.1 to 8.1% of the total reaction products, of equal abundance to 4-epi-eremophilene (14). Nearly every alternative product showed at least a modest increase in relative abundance at higher temperatures with the exception of isoprezizaene (8), which actually decreased over the examined temperature range.
Table 2.
Temperature effects on the relative quantities of TEAS reaction products
| Carbocationa | Peakb | TEAS product | Temperature (°C) | ||||
|---|---|---|---|---|---|---|---|
| 0 | 13.5 | 20 | 33 | 42 | |||
| 1 | (+)-Germacrene Ac | 1.13 | 2.41 | 3.65 | 5.62 | 8.11 | |
| A | 15 | α-Selinene | 0.75 | 0.81 | 0.98 | 0.94 | 1.01 |
| 19 | β-Selinene | 0.03 | 0.11 | 0.12 | 0.08 | 0.11 | |
| 11 | Selina-4,11-diene | 1.13 | 1.63 | 1.88 | 2.01 | 2.75 | |
| Total | 1.91 | 2.56 | 2.98 | 3.03 | 3.86 | ||
| B | 5 | (−)-Spirolepechinene | 0.71 | 1.11 | 1.40 | 1.74 | 2.07 |
| 16 | (−)-Premnaspirodiene | 0.99 | 1.31 | 1.66 | 1.96 | 5.81 | |
| Total | 1.70 | 2.42 | 3.07 | 3.70 | 7.88 | ||
| C | 13 | (+)-5-epi-Aristolochene | 87.66 | 82.20 | 78.86 | 75.21 | 66.28 |
| 14 | (−)-4-epi-Eremophilene | 3.23 | 5.11 | 6.21 | 7.14 | 8.47 | |
| Total | 90.89 | 87.31 | 85.07 | 82.35 | 74.75 | ||
| D | 8 | Isoprezizaene | 1.66 | 1.86 | 1.77 | 1.71 | 1.22 |
| 9 | β-Acoradiene | 0.11 | 0.20 | 0.18 | 0.14 | 0.25 | |
| 2 | (−)-α-Cedrene | 0.48 | 0.50 | 0.46 | 0.46 | 0.90 | |
| Total | 2.26 | 2.56 | 2.41 | 2.31 | 2.38 | ||
| Un-knowns | 2.12 | 2.73 | 2.82 | 3.00 | 3.01 | ||
For simplicity, products can be grouped according to the carbocations (A–D) that putatively give rise to each observed hydrocarbon (Table 2 and Fig. 3). The eremophilenyl carbocation derivatives, (−)-4-epi-eremophilene (14) and (+)-5-epi-aristolochene (13) (group C), decline by ~15% over the temperature range employed. These levels of product (13) and (14) are offset by increased relative amounts of the spiranes, (−)-spirolepechinene (5) and (−)-premnaspirodiene (16) (group B), and the premature release of (+)-germacrene A (1). The degree of farnesyl carbocation isomerization seems unaffected by temperature as reffected by constant relative amounts of (Z,E)-farnesyl cation-derived products (group D).
Discussion
Cyclases face the daunting task of channeling highly reactive carbocation intermediates through regio- and stereo-chemical contortions to arrive at a single unique product. Not surprisingly, numerous competing events like premature quenching of carbocations by proton elimination or water capture, or alternative alkyl and/or hydride shifts can cause derailment of the major reaction pathway. These alternative products provide useful information about the reaction trajectory of ionized FPP along the main mechanistic pathway, and the careful elucidation of these off-branch hydrocarbons have served as the basis for postulating mechanistic transformations of highly reactive ionized and neutral intermediates [5-7].
This report describes the first detailed characterization of the product spectrum of TEAS. This detailed and quantitative analysis is an essential step for constructing an accurate picture of the cyclization pathway to (+)-5-epi-aristolochene (13). The observed formation of selinene compounds, for example, can be readily attributed to elimination of a proton from one of three positions on the eudesmyl carbocation intermediate (Scheme 4A and Fig. 1A). Competing alkyl shifts divert another eudesmyl carbocation (Scheme 4B and Fig. 3, step B) into either (−)-premnaspirodiene (16) or (−)-spirolepechinene (5), the latter of which is a rare skeletal class of spirosesquiterpene found in Lepechinia bullata [31]. The final branch point in the pathway terminates at an eremophilenyl cation (Scheme 4C and Fig. 3, step C), which can undergo elimination at either C6 or C8 to yield the dominant eremophilane products. Taken together, the identification of these alternative products implicate transit through at least three carbocation decision points (A–C), following protonation of (+)-germacrene A (1), on the way to (+)-5-epi-aristolochene (13).
Scheme 4.
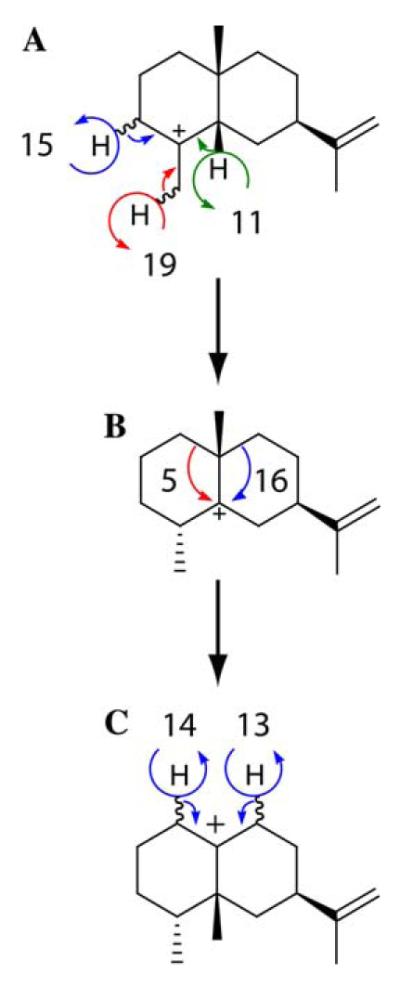
Carbocations from the TEAS-catalyzed cyclization of the (E,E)-farnesyl cation to 5-epi-aristolochene. Arrows are labeled with numbers corresponding to reaction products as defined by elution order from the GC (Fig. 1 and Table 1).
The mechanistic insights gleaned from identification of alternative reaction products underscore the importance of a sensitive and quantitative GC–MS assay for sesquiterpene cyclases [24]. The combined use of chiral and non-polar stationary phases for gas chromatographic separations proved critical for resolving the numerous sesquiterpene products of TEAS for mass spectral analysis. The identification and validation of (−)-α-cedrene (2) as a TEAS-derived reaction product was accomplished in this manner, enabling the discovery of a previously undocumented isomerization activity in this cyclase. The known configuration of (−)-α-cedrene (2) provides a compelling basis for proposing the cyclization mechanism and stereochemistry of the products arising from the (Z,E)-farnesyl cation pathway (Scheme 5). A key step following isomerization is electrophilic attack of C10 on the Si face of the (7R) bisabolyl cation, establishing the S configuration at both C6 and C10 which is shared by all identified isomerization products. The geometry of the resulting acoradilyl cation (Scheme 5D and Fig. 3, step D) directs the subsequent 3,11 closure and Wagner–Meerwein rearrangement to arrive at the proposed configuration of isoprezizaene (8), the major isomerization-derived product. The absolute configuration of the (−)-prezizaene synthetic standard used in this study has been well characterized [32]. This compound shared identical mass spectra with isoprezizaene (8), co-eluted on the non-polar column, but was clearly separated on the chiral column during co-injection GC–MS runs (Fig. 2). Therefore, it is concluded that the novel enzymatic product isoprezizaene (8) is a diastereomer of (−)-prezizaene, based on chromatographic separation and the mechanistic assumption that it is derived from the same acoradilyl ion leading to (−)-α-cedrene (2) (Fig. 3, step D).
Scheme 5.
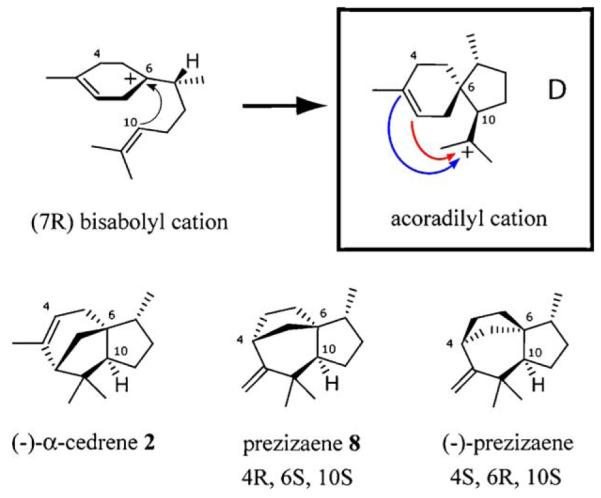
Cyclization of carbocations along the (Z,E)-farnesyl cation pathway. Selected atoms are labeled to illustrate the stereo-chemical rationalization for isoprezizaene (8) biosynthesis.
Although there have been numerous reports on the characterization of terpene synthases (cyclases) which generate multiple products, this work is the first to describe changes in a cyclase’s product profile with temperature. An inverse relationship between temperature and product specificity was observed as decreases of both (+)-5-epi-aristolochene (13) and isoprezizaene (8), the dominant products from the respective (E,E) and (Z,E)-farnesyl cation cyclization pathways, with increasing temperature. This effect is strongly suggestive of kinetic control of the cyclization reaction; as the temperature increases, alternative conformations of the prenyl tail of FPP become populated and the final distribution of conformers reflects the energy differences between them. Electrophilic cyclization is assumed to proceed rapidly following ionization of FPP, such that the conformation of the prenyl tail at the moment of ionization dictates the available cyclization path and hence the final product.
Among the TEAS products formed, (+)-germacrene A (1) production rose substantially at elevated temperatures. This proposed neutral reaction intermediate was first observed as the sole product of the Y520F mutant of TEAS [28], and later detected in the product spectrum of the mechanistically related aristolochene synthase from Penicillium roqueforti [33]. On the basis of crystallographic data, the J–K loop was proposed to close and seal off the active site from bulk solvent [18]. This was evident in the TEAS–substrate analogue complexes and noted in later structural studies of another sesquiterpene cyclase trichodiene synthase from Fusarium sporotrichioides [34]. It is proposed here that increased dynamic fluctuations of the J–K loop accompany temperature increases, in turn allowing germacrene A (1) to escape the active site prior to protonation and further cyclization. Furthermore, global motions of the protein may impinge on the cyclase active site to disrupt germacrene A activation and/or allow additional conformers of FPP to populate the active site of an ensemble of TEAS conformations in solution. Moreover, this population distribution can extend to the conformation of the carbocations formed throughout the TEAS-catalyzed reaction to ultimately influence the distribution of resultant sesquiterpene products.
New insights emerge from monitoring changes in the relative abundance of groups of products according to the carbocation from which they are derived (Fig. 3, steps A–D and Table 2), allowing one to relate changes in product abundance to the net flux of carbocations through the proposed TEAS cyclization pathway (Fig. 4). One immediately obvious observation is the decline of the eremophilanes (group C) and the upsurge of the spiro-sesquiterpenes (group B). Therefore, carbocation B is an important control point of the cyclization cascade for TEAS. The products resulting from the (Z,E)-farnesyl cation pathway (group D) remain constant over the temperature range used in this study, indicating that to a first approximation isomerization is temperature independent.
Fig. 4.
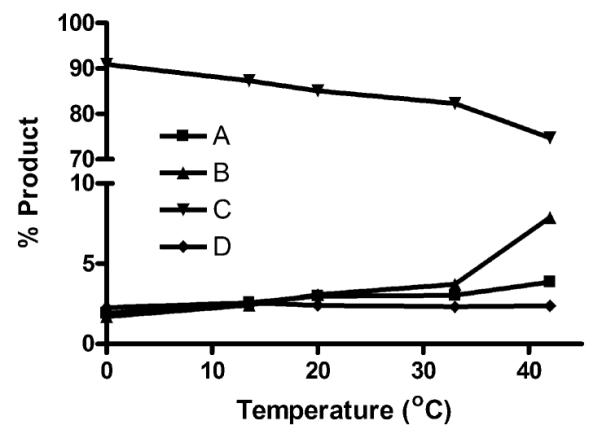
Temperature effects on the product composition of TEAS. The relative abundance of products derived from a particular reaction step involving a carbocation, A–D (Fig. 3), expressed as a sum of the percentage of total product (Table 2), are plotted versus temperature.
Conclusions
The ultimate goal of understanding the biophysical mechanisms responsible for the chemical diversity created by terpene synthases (cyclases) requires a detailed map of the energetic landscapes controlled by these enzymes. The discovery and identification of alternative TEAS reaction products reported in this study reveal carbocations at mechanistic crossroads that signify key energetic reference points along the cyclization pathway to (+)-5-epi-aristolochene (13). This empirical groundwork provides the necessary experimental foundation for rigorous computational investigations of the energetics of enzymemediated terpene-based cyclization, which are currently underway (in preparation). Analytical chemistry, molecular biology, enzymology, structural biology, and comparative bioinformatics will continue to be crucial components of these studies. An important caveat to bear in mind for GC–MS quantification of mixtures of volatiles is that individual compounds may have different extraction efficiencies and instrument responses, thereby requiring the use of authentic standards for accurate quantification of absolute analyte concentrations. The successful integration of analytical and computational chemistry with structural biology and enzyme engineering will provide rational and integrated approaches to harness these biocatalysts for the complex chemo-enzymatic synthesis of rare terpene-based natural products.
Supplementary Material
Acknowledgments
We thank T. Kollner for the celery seed extract, J.W. Mansfield for the LTC1 clone, and R. Coates for synthetic standards of (−)-4-epi-eremophilene and (−)-prezizaene. We appreciate the insightful comments of Robert Coates, Bryan Greenhagen, and Andy Hess on various aspects of the mechanistic work. We are grateful to the National Institutes of Health for a grant that supported this work (GM54029 to Joseph P. Noel/Joseph Chappell). Paul E. O’Maille is an NIH Postdoctoral Research Fellow (GM069056). Joseph P. Noel is an investigator of the Howard Hughes Medical Institute.
Footnotes
- FPP
- (E,E)-farnesyl diphosphate
- TEAS
- tobacco 5-epi-aristolochene synthase
- HPS
- Hyoscyamus muticus premnaspirodiene synthase
- LTC1
- Lactuca sativa germacrene A synthase
Appendix A. Supplementary data Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.abb.2005.10.028.
References
- [1].Dewick PM. Nat. Prod. Rep. 1997;14:111–144. doi: 10.1039/np9971400111. [DOI] [PubMed] [Google Scholar]
- [2].Nicolaou KC, et al. Nature. 1994;367:630–634. doi: 10.1038/367630a0. [DOI] [PubMed] [Google Scholar]
- [3].Cane DE. Chem. Rev. 1990;90:1089–1103. [Google Scholar]
- [4].Cane DE. Acc. Chem. Res. 1985;18:220–226. [Google Scholar]
- [5].Mercke P, Crock J, Croteau R, Brodelius PE. Arch. Biochem. Biophys. 1999;369:213–222. doi: 10.1006/abbi.1999.1358. [DOI] [PubMed] [Google Scholar]
- [6].Steele CL, Crock J, Bohlmann J, Croteau R. J. Biol. Chem. 1998;273:2078–2089. doi: 10.1074/jbc.273.4.2078. [DOI] [PubMed] [Google Scholar]
- [7].Colby SM, Crock J, Dowdle-Rizzo B, Lemaux PG, Croteau R. Proc. Natl. Acad. Sci. USA. 1998;95:2216–2221. doi: 10.1073/pnas.95.5.2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Whitehead IM, Threlfall DR, Ewing DF. Phytochemistry. 1989;28:775. [Google Scholar]
- [9].Whitehead IM, Ewing DF, Threlfall DR. Phytochemistry. 1988;27:1365. [Google Scholar]
- [10].Whitehead IM, Prabhakaran PC, Ewing DF, Cane DE, Threlfall DR. Phytochemistry. 1990;29:479. [Google Scholar]
- [11].Vogeli U, Chappell J. Plant Physiol. 1988;88:1291. doi: 10.1104/pp.88.4.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Facchini PJ. J. Chappell, Proc. Natl. Acad. Sci. USA. 1992;89:11088–11092. doi: 10.1073/pnas.89.22.11088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mau CJ, West CA. Proc. Natl. Acad. Sci. USA. 1994;91:8497–8501. doi: 10.1073/pnas.91.18.8497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Colby SM, Alonso WR, Katahira EJ, McGarvey DJ, Croteau R. J. Biol. Chem. 1993;268:23016–23024. [PubMed] [Google Scholar]
- [15].Back K, Chappell J. J. Biol. Chem. 1995;270:7375–7381. doi: 10.1074/jbc.270.13.7375. [DOI] [PubMed] [Google Scholar]
- [16].Back K, Yin S, Chappell J. Arch. Biochem. Biophys. 1994;315:527–532. doi: 10.1006/abbi.1994.1533. [DOI] [PubMed] [Google Scholar]
- [17].Mathis JR, Back K, Starks C, Noel J, Poulter CD, Chappell J. Biochemistry. 1997;36:8340–8348. doi: 10.1021/bi963019g. [DOI] [PubMed] [Google Scholar]
- [18].Starks CM, Back K, Chappell J, Noel JP. Science. 1997;277:1815–1820. doi: 10.1126/science.277.5333.1815. [DOI] [PubMed] [Google Scholar]
- [19].Bouwmeester HJ, Kodde J, Verstappen FW, Altug IG, de Kraker JW, Wallaart TE. Plant Physiol. 2002;129:134–144. doi: 10.1104/pp.001024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Benedict CR, Lu JL, Pettigrew DW, Liu J, Stipanovic RD, Williams HJ. Plant Physiol. 2001;125:1754–1765. doi: 10.1104/pp.125.4.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kollner TG, Schnee C, Gershenzon J, Degenhardt J. Plant Cell. 2004;16:1115–1131. doi: 10.1105/tpc.019877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Schenk DJ. Dissertation Thesis. Department of Chemistry University of Illinois; Urbana, IL: 2000. [Google Scholar]
- [23].Zhao Y, Schenk DJ, Takahashi S, Chappell J, Coates RM. J. Org. Chem. 2004;69:7428–7435. doi: 10.1021/jo049058c. [DOI] [PubMed] [Google Scholar]
- [24].Cane DE, Ha HJ. J. Am. Chem. Soc. 1988;110:6865–6870. [Google Scholar]
- [25].O’Maille PE, Chappell J, Noel JP. Anal. Biochem. 2004;335:210–217. doi: 10.1016/j.ab.2004.09.011. [DOI] [PubMed] [Google Scholar]
- [26].Konig WA, Joulain D, Hochmuth DH. Mass Finder. version 3 University of Hamburg; 2004. [Google Scholar]
- [27].de Kraker JW, Franssen MC, de Groot A, Konig WA, Bou-wmeester HJ. Plant Physiol. 1998;117:1381–1392. doi: 10.1104/pp.117.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rising KA, Starks CM, Noel J, Chappell J. J. Am. Chem. Soc. 2000 [Google Scholar]
- [29].Greenhagen BT. Dissertation Thesis. Department of Agriculture, The University of Kentucky; 2003. [Google Scholar]
- [30].O’Maille PE, Greenhagen BT, Chappell J, Zhao Y, Coates RM, Noel JP. Terpnet. Kentucky; Lexington: 2003. 2003. [Google Scholar]
- [31].Eggers MD, Sinnwell V, Stahl-Biskup E. Phytochemistry. 1999;51:987–990. [Google Scholar]
- [32].Vettel PR, Coates RM. J. Org. Chem. 1980;45:5430–5432. [Google Scholar]
- [33].Calvert MJ, Ashton PR, Allemann RK. J. Am. Chem. Soc. 2002;124:11636–11641. doi: 10.1021/ja020762p. [DOI] [PubMed] [Google Scholar]
- [34].Rynkiewicz MJ, Cane DE, Christianson DW. Proc. Natl. Acad. Sci. USA. 2001;98:13543–13548. doi: 10.1073/pnas.231313098. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.