

Abstract
SNAP25 occurs on chromosome 20p12.2, which has been linked to schizophrenia in some samples, and recently linked to latent classes of psychotic illness in our sample. SNAP25 is crucial to synaptic functioning, may be involved in axonal growth and dendritic sprouting, and its expression may be decreased in schizophrenia. We genotyped 18 haplotype-tagging SNPs in SNAP25 in a sample of 270 Irish high-density families. Single marker and haplotype analyses were performed in FBAT and PDT. We adjusted for multiple testing by computing q values. Association was followed up in an independent sample of 657 cases and 411 controls. We tested for allelic effects on the clinical phenotype by using the method of sequential addition and 5 factor-derived scores of the OPCRIT. Nine of 18 SNPs had P values <0.05 in either FBAT or PDT for one or more definitions of illness. Several two-marker haplotypes were also associated. Subjects inheriting the risk alleles of the most significantly associated two-marker haplotype were likely to have higher levels of hallucinations and delusions. The most significantly associated marker, rs6039820, was observed to perturb 12 transcription-factor binding sites in in silico analyses. An attempt to replicate association findings in the case–control sample resulted in no SNPs being significantly associated. We observed robust association in both single marker and haplotype-based analyses between SNAP25 and schizophrenia in an Irish family sample. Although we failed to replicate this in an independent sample, this gene should be further tested in other samples.
Keywords: schizophrenia, SNAP25, polymorphism, association, clinical features
Introduction
Schizophrenia is a debilitating neuropsychiatric condition, candidate susceptibility genes for which have only been identified by positional cloning in the last 5 years [Harrison and Weinberger, 2005; Riley and Kendler, 2006]. Since its first descriptions, writers have noted that it is clinically heterogeneous [Berrios, 1995]. Affected persons can have myriad combinations of positive symptoms such as hallucinations and delusions, as well as negative and affective symptoms.
Of the current list of replicated susceptibility genes for schizophrenia, it has been suggested that a common role is direct or indirect impact on synaptic functioning [Harrison and Weinberger, 2005]. In recent years, close attention has been paid to expression levels of presynaptic proteins. These have often been used as an index of synaptic density, which has been demonstrated to be altered in schizophrenia [Knable et al., 2004; Sweet et al., 2007]. One of the most commonly studied proteins is Synaptosomal-Associated Protein of 25 kDa (SNAP25). It is a component of the soluble N-ethylmaleimide-sensitive factor attachment receptor (SNARE) complex, along with syntaxin-1A and vesicle-associated membrane protein (VAMP). One of the most important roles of this complex is to mediate exocytosis in neurons, neuroendocrine cells, and pancreatic β-cells [Graham et al., 2002]. Specifically, the components of the SNARE complex form a four-helix bundle that allows vesicles to dock and fuse with the presynaptic cell membrane, which is crucial for synaptic transmission [Graham et al., 2002]. It is also involved in axonal growth [Osen-Sand et al., 1993] and neurite sprouting [Kimura et al., 2003]. Dysfunction or underexpression of a component of this mechanism would be consistent with the decreased release of both dopamine [Hetey et al., 1991] and glutamate [Sherman et al., 1991] from presynaptic terminals as has been described in schizophrenic brain. It could also contribute to neurodevelopmental abnormalities thought to be involved in the etiology of schizophrenia [Harrison and Weinberger, 2005]. Studies have reported reduced SNAP25 immunoreactivity in hippocampus [Thompson et al., 1998, 2003a; Young et al., 1998; Fatemi et al., 2001], prefrontal cortex [Thompson et al., 1998; Karson et al., 1999], temporal cortex [Thompson et al., 1998], and cingulate cortex [Gabriel et al., 1997], as well as increased levels of CSF SNAP25 [Thompson et al., 1998, 1999, 2003b] in schizophrenia.
The SNAP25 gene is located at chromosome 20p12.2. While this region has not been implicated in many previous genome scans, 20p12.3-p11 was ranked #11 in the Genome Scan Meta-Analysis of Schizophrenia [Lewis et al., 2003]. In a scan of several European and one Taiwanese samples, this was the most highly linked region in the genome [Moises et al., 1995]. A recent study in a Japanese sib-pair sample using a high-density SNP map reported linkage to 20p11.2 [Arinami et al., 2005]. Although there was little evidence of linkage to this chromosome in a previous genome scan of the family sample we report here [Straub et al., 2002], we recently reported evidence of linkage to 20p using latent classes of psychotic illness as the phenotypes of interest [Fanous et al., 2008]. One previous single-marker association study of SNAP25 in schizophrenia has been negative [Tachikawa et al., 2001]. However, an association between SNAP25 and decreased prepulse inhibition, a schizophrenia-related sensory gating defect, has recently been reported in mice [Jeans et al., 2007]. Furthermore, overexpression of the schizophrenia susceptibility gene DTNBP1 [Harrison and Weinberger, 2005; Riley and Kendler, 2006] increases expression of SNAP25 in cultured primary cortical neurons [Numakawa et al., 2004]. Because of the convergence of evidence from gene expression and animal model studies suggesting involvement of SNAP25 in the etiology of schizophrenia, as well as its presence in a region of linkage in our sample and others, we tested this gene for association using 18 haplotype-tagging SNPs genotyped in two independent schizophrenia samples.
Materials and Methods
Subjects
In this study, we use a family-based sample to test for association initially, then attempted to confirm results in an independent case–control sample. The Irish Study of High-Density Schizophrenia Families (ISHDSF) was jointly sponsored by the Medical College of Virginia, the Health Research Board, Dublin and the Queen's University, Belfast. The field work was completed between April 1987 and November 1992. Families were ascertained through public psychiatric services in Ireland and Northern Ireland that together provided over 90% of all in-patient psychiatric care in the island of Ireland. The main diagnostic instruments utilized in the field work were the structured clinical interview for DSM-III-R [Spitzer et al., 1987].
We used eight diagnostic categories reflecting the probable genetic relationship of these syndromes to classic schizophrenia, as influenced by previous twin, family and adoption studies of schizophrenia, and especially by the results of the Roscommon Family Study (RFS) [Kendler et al., 1993]. This hierarchy contains three definitions of affection: (i) Narrow—categories D1 and D2, or “core schizophrenia,” including schizophrenia, poor-outcome schizoaffective disorder (PO-SAD) and simple schizophrenia; (ii) Intermediate—categories D1 through D5, or a narrow definition of the schizophrenia spectrum, adding to the narrow definition schizotypal personality disorder, schizophreniform disorder, delusional disorder, atypical psychosis, and good-outcome SAD; (iii) Broad—categories D1-D8, including all disorders which significantly aggregated in relatives of schizophrenic probands in the RFS and adding to the intermediate definition mood incongruent and mood congruent psychotic affective illness, and paranoid, avoidant and schizoid personality disorder.
Final diagnoses sometimes disagreed with field diagnoses, always in the more conservative direction. Final inclusion criteria for pedigrees in the ISHDSF sample was, therefore, based on final diagnoses and required two or more first, second or third degree relatives with a diagnosis of D1–D5, one or more of whom had a D1–D2 diagnosis. This criterion excluded two pedigrees from the sample. Inclusion criteria for families for the linkage subsample of the ISHDSF required that DNA samples be available on two or more first, second, or third degree relatives with a diagnosis of D1–D5, one or more of whom had a D1–D2 diagnosis.
The ISHDSF sample contains 270 pedigrees and 1,425 individuals with adequate clinical information and DNA. The number of individuals defined as “affected” under our three definitions were: narrow—577, intermediate—700, and broad—754. Of the 270 pedigrees in the sample, using the narrow, intermediate, and broad definitions of illness, 68 (25.7%), 113 (42.6%), 133 (50.2%), respectively, contained three or more affected individuals. Using the narrow, intermediate, and broad definitions of illness, the ISHDSF sample contains 389, 627, and 777 affected relative pairs of whom 285, 420, and 505, respectively, are affected sibling pairs. Subjects with a history of probable psychotic illness were rated using the Operational Criteria Checklist for Psychotic Illness (OPCRIT) [McGuffin et al., 1991] by KSK.
The Irish Case–Control Study of Schizophrenia (ICCSS) sample was collected in the same geographic regions as that of the ISHDSF sample. In this study, we used 657 affected subjects and 411 controls. The affected subjects were selected from in-patient and out-patient psychiatric facilities in the Republic of Ireland and Northern Ireland. Subjects were eligible for inclusion if they had a diagnosis of schizophrenia or PO-SAD by DSM-III-R criteria. Controls, selected from several sources, including blood donation centers, were included if they denied a lifetime history of schizophrenia. Both cases and controls were included only if they reported all four grandparents as being born in Ireland or the United Kingdom.
SNP Selection and Genotyping
In order to minimize genotyping load while maximizing association information, implementation of haplotypes-tagging SNPs (tSNPs) selection is now common. For this study, tSNPs were identified using HAPLOVIEW 3.2 [Barrett et al., 2005] using the method of Gabriel et al. [2002] SNPs were obtained from Hapmap CEU data, phase I. Multiple methods for SNP genotyping are in current use in our lab. Some were genotyped by template-directed dye-terminator incorporation with fluorescence polarization detection (FP-TDI) using the relevant AcycloPrime FP SNP detection kit (PerkinElmer Life Sciences, Boston, MA) according to the manufacturer's instructions, and an automated allele scoring platform [Van Den Oord et al., 2003]. Multiplex genotyping of additional SNPs was conducted on the GenomeLab SNPstream (Beckman Coulter, Fullerton, CA) following manufacturer's protocols, in panels SNPs matched for their extension type. SNP sequences were screened for repeats and homology with other genomic sequences prior to using the proprietary Beckman Coulter primer design program, Autoprimer, for constructing the multiplex panels. In cases where SNP sequences were repeat-rich or matching extension types were necessary for successful SNPstream multiplex paneling, proxy SNPs with matching r2 > 0.8 and and MAF > 0.1 were substituted. tSNPs failing on the SNPstream platform and those which could not be paneled were genotyped as monoplex reactions using Taqman Assays-on-Demand (Applied Biosystems, Foster City, CA).
Statistical Analysis
Prior to analysis, SNP data in the family sample were tested for Mendelian inconsistencies using the program PEDCHECK [O'Connell and Weeks, 1998], and for unlikely genotypes using Merlin [Abecasis et al., 2002]. A genotyping error rate of 0.1% was observed, and all erroneous genotypes were removed. We used two family-based methods to test for association between SNAP25 SNPs and schizophrenia: the family-based association test, operationalized in FBAT [Laird et al., 2000], and the pedigree disequilibrium test (PDT) [Martin et al., 2000], operationalized in PDT. Individual marker genotypes were tested for Hardy–Weinberg equilibrium, and analyzed for linkage disequilibrium relationships, in Haploview [Barrett et al., 2005]. We tested for association using two programs as a means of confirming results, in an attempt to reduce the probability of false positives.
We first tested for excessive transmission of alleles at each marker separately, and then for excessive transmission of multiple-marker haplotypes. Haplotypes were reconstructed using the HBAT command in FBAT for FBAT analyses, which utilizes the EM algorithm. In these analyses, we used the empirical variance estimator option (−e) as our primary hypothesis was of association in the presence of linkage. Haplotypes were constructed for PDT analyses using PDTPHASE, part of the UNPHASED [Dudbridge, 2003] software suite v.2.404, also using the EM algorithm. For all PDT analyses, we used the “sum PDT” statistic, which assigns weights to pedigrees on the basis of size. We tested for association in the ISTS using the CoCaPhase module of UNPHASED package v. 2.4.04. The P values obtained were χ2 distributions.
To control the risk of false discoveries, we calculated for each P-value a so-called q-value [Storey, 2003; Storey and Tibshirani, 2003]. A q-value is an estimate of the proportion of false discoveries among all significant markers (i.e., q values are false discovery rates or FDRs) when the corresponding P-value is used as the threshold for declaring significance. As argued previously [Van den Oord and Sullivan, 2003b] we preferred this FDR-based approach because it (a) provides a good balance between the competing goals of finding true effects versus controlling false discoveries, (b) allows the use of more similar standards in terms of the proportion of false discoveries produced across studies because it is much less affected by number of tests, which is an arbitrary factor, (c) is relatively robust against having correlated tests [Benjamini and Hochberg, 1995; Brown and Russell, 1997; Sabatti et al., 2003; Storey, 2003; Tsai et al., 2003; Van den Oord and Sullivan, 2003a; Fernando et al., 2004; Korn et al., 2004; Van den Oord, 2005], and (d) rather than an all-or-nothing conclusion about whether a study produces significant results, gives a more subtle picture of the possible role of the tested markers.
To test for possible heterogeneity between the family-based and case–control samples, we generated combined odds ratios using the information included in the primary analysis approaches and standard meta-analytic techniques. The PDT statistic compares the number of times a given parental allele (“risk” allele) is transmitted versus non-transmitted and examines allele sharing between affected and unaffected sibling pairs while standard case–control approaches examine allele frequencies in cases versus controls. The parental transmission odds ratio is constructed as (a/c)/(b/d), where a = transmissions of the high-risk allele, c = non -transmissions of the high-risk allele, b = transmissions of all other alleles, and d = non-transmissions of all other alleles. In the sibling pair sample and the population-based samples, which compare case to control allele frequencies, we construct an odds ratio as (a/c)/(b/d) where a is the number of major alleles present in cases, c is the number of minor alleles present in cases, b is the number of major alleles in controls, and d is the number of minor alleles present in controls. We used formal meta-analytic techniques to combine odds ratios across study types. We performed a fixed-effects (Mantel–Haenszel) approach to meta-analysis, and calculated a P value for heterogeneity.
We tested an associated haplotoype for effects on the clinical phenotype using UNPHASED. To determine whether a subset of the subjects was driving the observed association between SNAP25 and schizophrenia, all cases were rank ordered according to each of five symptom factors previously extracted from the OPCRIT in this sample: negative symptoms, depressive symptoms, manic symptoms, delusions, and hallucinations [Fanous et al., 2005]. The sequential addition method [Macgregor et al., 2006] was used, as follows: first, a Chi-square test was performed for all cases and controls. Next, for each factor, cases with the highest scale scores were compared with all control subjects using a Chi-square test. Cases with incrementally lower scores were progressively included until the Chi-square value met or exceeded that of the full case sample. To assess the probability of a factor–haplotype association under the null hypothesis, permutations were carried out in which the same proportion of cases achieving a significant association was randomly sampled from the total case population using SAS 9.1.3 [SAS Institute, 1999] and compared to all controls using UNPHASED. Empirical P values were determined from these permutations by the formula P = d + 1/n + 1, where d is the number of Chi-square statistics observed in n = 5000 permutations that exceeded the Chi-square observed in the full sample.
SNPs demonstrating significant association with multiple diagnostic categories and/or in both association packages, were explored for putative functionality via bioinformatic approaches to detect allele-specific sequence motifs. Surrounding sequences were examined for evidence of evolutionary conservation. In order to identify gain or loss of physical transcription-factor binding sites generated by the nucleotide exchange corresponding to alternative SNP alleles, 50 bp of sequence surrounding each SNP was analyzed using MatInspector v 7.6.2 and SNPinspector v 2.2 [Cartharius et al., 2005] (http://www.genomatrix.de). MatInspector identifies physical transcription-factor binding sites whilst SNPInspector pinpoints those binding sites that are affected by SNPs, which hence have putative regulatory activity. In both cases the vertebrate transcription factor matrices and optimized matrix threshold were used to reduce the incidence of false positives [Cartharius et al., 2005].
VISTA was used to define conserved regions in the genomic sequence of SNAP25 surrounding specific SNPs [Couronne et al., 2003] and visualized as added tracks on the University of California Santa Cruz genome browser (http://genome.ucsc.edu/). Forty basepairs of DNA sequence including each SNP allele was analyzed for putative splice sites using the predictor from the Berkeley Drosophila genome project (http://www.fruitfly.org/seq_tools/splice.html. Database similarity searches, and protein open-reading frame identification were performed using BLAST and ORF-finder via the National Centre for Biotechnology Information (http://www.ncbi.nlm.nih.gov). Hairpin structures indicative of miRNA precursors were sought using the software ProMiR-II [Nam et al., 2006] and MiRPred [Brameier and Wiuf, 2007].
Results
Eighteen single nucleotide polymorphisms in the SNAP25 gene were genotyped. There was significant linkage disequilibrium between them, as exhibited in Figure 1. These formed five haplotype blocks. Haplotype block structure was very similar across samples.
FIG. 1.
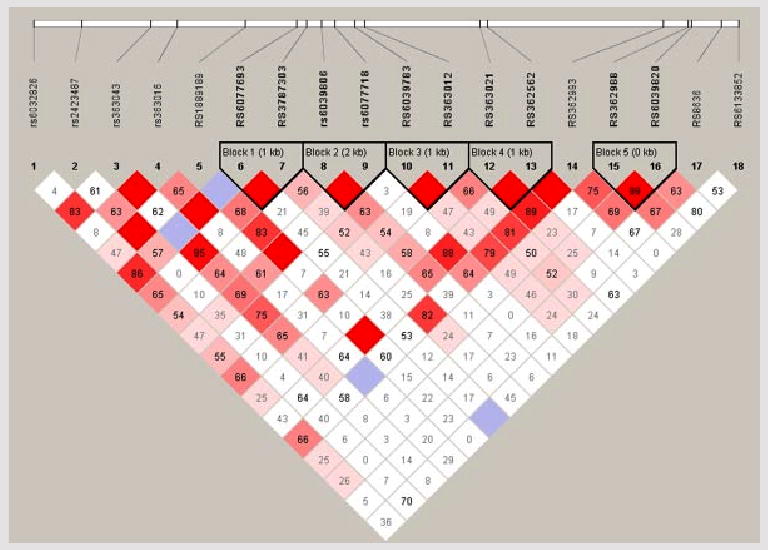
Intermarker linkage disequilibrium and haplotype blocks in SNAP25. Marker to marker LD, as well as haplotype blocks, in the SNAP25 gene as determined using haplotypes-tagging SNPs obtained from HapMap. Bright red represents D' = 1 and LOD ≥ 2, blue represents D' = 1 and LOD < 2, pink represents D' < 1 (figure displayed), and LOD ≥ 2, and white represents D' < 1 and LOD < 2.
In the family sample, nine of these SNPs were significantly associated at the 0.05 significance level, for at least one of the four diagnostic definitions used, as displayed in Table I. We calculated q values that can be viewed as estimates of the proportion of false discoveries among all significant markers (i.e., q values are false discovery rates or FDRs) when the corresponding P-value is used as the threshold for declaring significance. Q values were calculated conservatively assuming that the proportion of markers without effect, P0, equals one. Because SNAP25 is a positional candidate; that is, a gene with considerable a priori biological evidence of association in a linked region, we also calculated q values using the lowest slope estimator [Benjamini and Hochberg, 2000] that resulted P0 = 0.68. We have previously suggested that a q-value of 0.1 could be a good threshold for declaring significance in genetic studies [Van den Oord and Sullivan, 2003b]. This threshold corresponded with a P-value of 0.0185, resulting in q values of 0.122 with P0 = 1 and 0.084 with P0 = 0.68, in the present study. SNPs with P < 0.0185 included SNPs 2, 3, 8, 15, 16, and 18. Haplotype block 5, comprising SNPs 15 and 16, was significant in all diagnostic definitions, and in both FBAT and PDTPHASE. Using the intermediate definition, haplotype blocks 2 (SNPs 8 and 9), 3 (SNPs 9 and 10), and 4 (SNPs 11 and 12) were significant, but were not more significant than the trend level using the other diagnostic definitions. Results of haplotype analyses are presented in Table II.
TABLE I. Marker Characteristics and Single-SNP Association Results in the Family Sample.
| SNP # | Name | Position | Polymorphism (minor allele) | MAF | Risk allele | FBAT | PDT | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Narrow | Intermediate | Broad | Very broad | Narrow | Intermediate | Broad | Very broad | ||||||
| 1 | rs1889189 | 10192086 | C/T (T) | 0.365 | T | 0.767 | 0.637 | 0.778 | 0.412 | 0.807 | 0.248 | 0.364 | 0.881 |
| 2 | rs6032826 | 10198817 | A/G (G) | 0.189 | G | 0.570 | 0.210 | 0.108 | 0.009 | 0.556 | 0.884 | 0.734 | 0.073 |
| 3 | rs6077693 | 10201917 | A/G (A) | 0.164 | G | 0.028 | 0.006 | 0.005 | 0.010 | 0.152 | 0.069 | 0.074 | 0.172 |
| 4 | rs3787303 | 10203748 | C/T (C) | 0.145 | C | 0.496 | 0.887 | 0.380 | 0.068 | 0.330 | 0.105 | 0.448 | 0.770 |
| 5 | rs2423487 | 10208095 | C/T (C) | 0.092 | C | 0.854 | 0.588 | 0.498 | 0.445 | 0.647 | 0.896 | 0.900 | 0.260 |
| 6 | rs6039783 | 10213034 | C/T (C) | 0.056 | C | 0.797 | 0.634 | 0.823 | 0.621 | 0.463 | 0.264 | 0.417 | 0.620 |
| 7 | rs363012 | 10214799 | A/G (G) | 0.351 | G | 0.734 | 0.147 | 0.191 | 0.610 | 0.597 | 0.129 | 0.210 | 0.322 |
| 8 | rs363043 | 10221146 | C/T (T) | 0.275 | C | 0.093 | 0.013 | 0.017 | 0.056 | 0.054 | 0.010 | 0.010 | 0.031 |
| 9 | rs363016 | 10226174 | C/T (T) | 0.426 | T | 0.759 | 0.408 | 0.315 | 0.112 | 0.630 | 0.882 | 0.776 | 0.284 |
| 10 | rs363021 | 10236811 | C/T (T) | 0.464 | T | 0.141 | 0.127 | 0.201 | 0.030 | 0.357 | 0.236 | 0.352 | 0.131 |
| 11 | rs362562 | 10238186 | A/G (A) | 0.414 | G | 0.100 | 0.029 | 0.069 | 0.019 | 0.325 | 0.110 | 0.199 | 0.164 |
| 12 | rs6039806 | 10253654 | A/C (A) | 0.494 | C | 0.230 | 0.049 | 0.185 | 0.128 | 0.185 | 0.038 | 0.123 | 0.170 |
| 13 | rs6077718 | 10256142 | G/T (G) | 0.126 | T | 0.282 | 0.144 | 0.223 | 0.140 | 0.426 | 0.504 | 0.587 | 0.631 |
| 14 | rs362993 | 10271716 | C/T (T) | 0.118 | C | 0.064 | 0.105 | 0.224 | 0.139 | 0.132 | 0.273 | 0.494 | 0.434 |
| 15 | rs362988 | 10276370 | A/G (A) | 0.456 | G | 0.013 | 0.012 | 0.014 | 0.004 | 0.159 | 0.092 | 0.074 | 0.030 |
| 16 | rs6039820 | 10277027 | C/T (T) | 0.481 | C | 0.005 | 0.004 | 0.002 | 0.002 | 0.007 | 0.002 | 0.001 | 0.004 |
| 17 | rs8636 | 10282742 | C/T (T) | 0.303 | C | 0.206 | 0.181 | 0.124 | 0.525 | 0.289 | 0.334 | 0.259 | 0.437 |
| 18 | rs6133852 | 10285960 | A/G (A) | 0.032 | G | 0.176 | 0.067 | 0.036 | 0.006 | 1.000 | 0.387 | 0.223 | 0.024 |
Bold type denotes P < 0.05.
TABLE II. Haplotype-Based Association Results in the Family Sample.
| Block | SNPs | Allele | FBAT | PDT | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Freq | Z | P | S | E (S) | Global, P | Freq | Z | P | Trio-T | Trio-NT | Aff Sib | Unaff Sib | Global, P | |||
| Narrow | ||||||||||||||||
| 1 | 6–7 | 1-1 | 0.68 | 0.40 | 0.69 | 294.97 | 292.44 | 0.395 | 0.65 | −0.38 | 0.70 | 68 | 58 | 635 | 656 | 0.606 |
| 1-2 | 0.27 | 0.37 | 0.71 | 170.03 | 167.99 | 0.33 | 0.66 | 0.51 | 24 | 28 | 292 | 270 | ||||
| 2-2 | 0.045 | −1.33 | 0.18 | 34.97 | 39.53 | 0.02 | −0.96 | 0.34 | 4 | 10 | 17 | 18 | ||||
| 2 | 8–9 | 1-1 | 0.32 | 1.05 | 0.29 | 191 | 184.23 | 0.366 | 0.38 | 0.00 | 1.00 | 35 | 37 | 259 | 257 | 0.728 |
| 1-2 | 0.42 | 0.21 | 0.83 | 243 | 241.53 | 0.39 | 0.51 | 0.61 | 44 | 46 | 266 | 249 | ||||
| 2-1 | 0.26 | −1.30 | 0.19 | 156 | 164.25 | 0.22 | −0.83 | 0.40 | 25 | 21 | 157 | 176 | ||||
| 3 | 10–11 | 1-1 | 0.11 | 1.43 | 0.15 | 82.03 | 74.87 | 0.114 | 0.12 | 1.28 | 0.20 | 13 | 11 | 134 | 110 | 0.237 |
| 1-2 | 0.42 | −2.01 | 0.04 | 198.97 | 213.35 | 0.41 | −1.51 | 0.13 | 36 | 46 | 371 | 402 | ||||
| 2-1 | 0.46 | 1.19 | 0.23 | 250.97 | 242.72 | 0.47 | 0.64 | 0.52 | 53 | 44 | 503 | 496 | ||||
| 4 | 12–13 | 1-1 | 0.56 | 1.40 | 0.16 | 292.89 | 282.30 | 0.267 | 0.53 | 0.97 | 0.33 | 72.98 | 62.97 | 651.4 | 631.4 | 0.459 |
| 2-1 | 0.34 | −0.64 | 0.53 | 207.11 | 211.50 | 0.35 | −1.00 | 0.32 | 43.02 | 49.03 | 420.6 | 443.6 | ||||
| 2-2 | 0.10 | −1.35 | 0.18 | 74.89 | 81.08 | 0.12 | −0.64 | 0.53 | 9.982 | 15.97 | 162.4 | 169.4 | ||||
| 5 | 15–16 | 1-1 | 0.51 | 2.82 | 0.0048 | 295.97 | 275.66 | 0.045 | 0.51 | 2.48 | 0.01 | 51 | 35 | 555 | 513 | 0.019 |
| 2-2 | 0.46 | −2.58 | 0.0098 | 231.97 | 251.26 | 0.46 | −2.18 | 0.03 | 46 | 59 | 414 | 457 | ||||
| Intermediate | ||||||||||||||||
| 1 | 6–7 | 1-1 | 0.68 | −0.76 | 0.45 | 351.97 | 356.682 | 0.280 | 0.65 | −1.11 | 0.27 | 81 | 71 | 681 | 722 | 0.26 |
| 1-2 | 0.27 | 1.43 | 0.15 | 209.04 | 201.24 | 0.33 | 1.41 | 0.16 | 29 | 33 | 334 | 292 | ||||
| 2-2 | 0.05 | −0.85 | 0.39 | 44.97 | 48.032 | 0.02 | −0.91 | 0.36 | 4 | 10 | 19 | 20 | ||||
| 2 | 8–9 | 1-1 | 0.32 | 2.12 | 0.0344 | 232.00 | 218.43 | 0.0496 | 0.38 | 1.13 | 0.26 | 49 | 49 | 310 | 286 | 0.38 |
| 1-2 | 0.42 | −0.16 | 0.87 | 288.00 | 289.11 | 0.39 | 0.04 | 0.97 | 54 | 54 | 271 | 270 | ||||
| 2-1 | 0.26 | −1.98 | 0.0475 | 190.00 | 202.46 | 0.22 | −1.27 | 0.20 | 29 | 29 | 173 | 198 | ||||
| 3 | 10–11 | 1-1 | 0.11 | 1.54 | 0.12 | 101.09 | 93.16 | 0.0437 | 0.12 | 1.62 | 0.11 | 17 | 15 | 150 | 121 | 0.06308 |
| 1-2 | 0.42 | −2.40 | 0.0163 | 254.91 | 272.69 | 0.41 | −2.12 | 0.0337 | 41 | 54 | 385 | 432 | ||||
| 2-1 | 0.46 | 1.51 | 0.13 | 297.91 | 286.78 | 0.47 | 1.08 | 0.28 | 60 | 48 | 565 | 547 | ||||
| 4 | 12–13 | 1-1 | 0.56 | 2.12 | 0.0342 | 353.89 | 337.40 | 0.0504 | 0.53 | 1.65 | 0.0982 | 92.96 | 82.97 | 721.3 | 678.4 | 0.13 |
| 2-1 | 0.34 | −1.06 | 0.29 | 247.11 | 254.93 | 0.35 | −1.73 | 0.0834 | 48.04 | 58.03 | 431.7 | 474.6 | ||||
| 2-2 | 0.10 | −1.92 | 0.0544 | 91.89 | 100.54 | 0.12 | −0.69 | 0.49 | 16.96 | 18.97 | 169.3 | 179.4 | ||||
| 5 | 15–16 | 1-1 | 0.51 | 2.81 | 0.0050 | 344.94 | 324.10 | 0.0433 | 0.51 | 2.91 | 0.0036 | 58 | 39 | 608 | 553.9 | 0.00613 |
| 2-2 | 0.46 | −2.60 | 0.0093 | 290.94 | 310.74 | 0.46 | −0.37 | 0.71 | 1 | 0.0000 | 8.047 | 4.052 | ||||
| Broad | ||||||||||||||||
| 1 | 6–7 | 1-1 | 0.68 | −0.61 | 0.54 | 379.94 | 384.143 | 0.178 | 0.65 | −0.90 | 0.37 | 83 | 74 | 671 | 709 | 0.30 |
| 1-2 | 0.27 | 1.54 | 0.12 | 225.06 | 216.171 | 0.33 | 1.28 | 0.20 | 30 | 34 | 330 | 287 | ||||
| 2-2 | 0.05 | −1.14 | 0.26 | 46.94 | 51.626 | 0.02 | −1.09 | 0.28 | 5 | 10 | 19 | 24 | ||||
| 2 | 8–9 | 1-1 | 0.32 | 2.09 | 0.0366 | 254 | 239.25 | 0.067 | 0.38 | 0.91 | 0.36 | 52 | 52 | 285 | 264 | 0.58 |
| 1-2 | 0.42 | −0.32 | 0.75 | 308 | 310.24 | 0.39 | −0.10 | 0.92 | 55 | 55 | 258 | 261 | ||||
| 2-1 | 0.26 | −1.87 | 0.0615 | 202 | 214.51 | 0.22 | −0.90 | 0.37 | 29 | 29 | 167 | 185 | ||||
| 3 | 10–11 | 1-1 | 0.11 | 1.11 | 0.27 | 105.15 | 99.66 | 0.122 | 0.12 | 1.00 | 0.32 | 18 | 16 | 141 | 123 | 0.23 |
| 1-2 | 0.42 | −1.90 | 0.0572 | 274.85 | 290.33 | 0.41 | −1.59 | 0.11 | 44 | 54 | 376 | 415 | ||||
| 2-1 | 0.46 | 1.33 | 0.18 | 317.85 | 306.62 | 0.47 | 0.93 | 0.35 | 60 | 51 | 577 | 556 | ||||
| 4 | 12–13 | 1-1 | 0.56 | 1.37 | 0.17 | 380.86 | 369.54 | 0.190 | 0.53 | 1.07 | 0.28 | 95.96 | 87.97 | 701.3 | 673.4 | 0.38 |
| 2-1 | 0.34 | −0.50 | 0.61 | 272.14 | 276.03 | 0.35 | −1.16 | 0.25 | 50.04 | 59.03 | 427.7 | 455.6 | ||||
| 2-2 | 0.10 | −1.64 | 0.10 | 99.86 | 107.28 | 0.12 | −0.67 | 0.51 | 17.96 | 18.97 | 165.3 | 175.4 | ||||
| 5 | 15–16 | 1-1 | 0.51 | 2.88 | 0.0040 | 369.93 | 346.61 | 0.0231 | 0.51 | 3.14 | 0.0017 | 59 | 42 | 613 | 543.9 | 0.00359 |
| 2-2 | 0.46 | −2.55 | 0.0109 | 308.93 | 329.99 | 0.46 | −2.67 | 0.0076 | 55 | 69 | 418 | 481.9 | ||||
| Very broad | ||||||||||||||||
| 1 | 6–7 | 1-1 | 0.68 | −0.12 | 0.91 | 496.94 | 497.69 | 0.228 | 0.65 | −0.45 | 0.65 | 110 | 101 | 552 | 572 | 0.19 |
| 1-2 | 0.27 | 1.28 | 0.20 | 281.06 | 273.40 | 0.33 | 1.23 | 0.22 | 41 | 42 | 261 | 231 | ||||
| 2-2 | 0.05 | −1.47 | 0.14 | 55.94 | 62.84 | 0.02 | −1.80 | 0.0719 | 5 | 13 | 13 | 23 | ||||
| 2 | 8–9 | 1-1 | 0.32 | 2.03 | 0.0421 | 320 | 306.11 | 0.103 | 0.38 | 1.38 | 0.17 | 70 | 66 | 248 | 224 | 0.38 |
| 1-2 | 0.42 | −0.65 | 0.51 | 398 | 402.86 | 0.39 | −0.79 | 0.43 | 77 | 79 | 225 | 240 | ||||
| 2-1 | 0.26 | −1.38 | 0.17 | 268 | 277.03 | 0.22 | −0.62 | 0.53 | 41 | 43 | 145 | 154 | ||||
| 3 | 10–11 | 1-1 | 0.11 | 0.83 | 0.41 | 131.25 | 126.99 | 0.0624 | 0.12 | 0.52 | 0.60 | 22 | 23 | 101 | 93.01 | 0.19 |
| 1-2 | 0.42 | −2.25 | 0.0243 | 342.75 | 360.86 | 0.41 | −1.66 | 0.0974 | 57 | 73 | 325 | 352 | ||||
| 2-1 | 0.46 | 1.82 | 0.0687 | 409.75 | 394.28 | 0.47 | 1.42 | 0.15 | 79 | 61 | 440 | 421 | ||||
| 4 | 12–13 | 1-1 | 0.56 | 1.53 | 0.13 | 495.86 | 482.79 | 0.0932 | 0.53 | 0.89 | 0.37 | 125.9 | 115 | 570.5 | 556.5 | 0.47 |
| 2-1 | 0.34 | −0.31 | 0.75 | 364.14 | 366.69 | 0.35 | −0.72 | 0.47 | 73.05 | 81.04 | 378.5 | 389.5 | ||||
| 2-2 | 0.10 | −2.06 | 0.0390 | 134.86 | 144.88 | 0.12 | −0.97 | 0.33 | 28.95 | 33.96 | 137.5 | 147.5 | ||||
| 5 | 15–16 | 1-1 | 0.51 | 2.79 | 0.0053 | 463.87 | 439.45 | 0.0392 | 0.51 | 2.69 | 0.0071 | 75 | 51 | 486 | 443 | 0.00916 |
| 2-2 | 0.46 | −2.66 | 0.0079 | 389.87 | 413.27 | 0.46 | −2.52 | 0.0119 | 71 | 94 | 366 | 410 | ||||
Analysis of clinical covariates using sequential addition yielded non-significant results for the negative, manic, and depressive factors of the OPCRIT. However, for the delusions factor, inclusion of 58% of the cases with the highest symptom levels resulted in a Chi-square value exceeding that of the full sample (7.40 compared to 5.46, empirical P = 0.01). Including 59% of the cases with the highest hallucination scores resulted in a Chi-square value of 8.21 (compared to 5.46 in the full sample, empirical P = 0.006). All analyses of clinical covariates tested only one haplotype block, block 5, as it was the only one that was significant across diagnostic groups and in both analysis packages.
Based on the low q values of several SNPs, as well as the significance of block 5 across diagnostic categories and statistical packages, we attempted a gene-based replication of SNAP25 in the case–control sample. We performed meta-analyses of these SNPs across the two samples to test for heterogeneity (results presented in Table III). Only one SNP had P < 0.05 for heterogeneity, which could very likely arise due to chance. None of the haplotype blocks in the case–control sample had significant haplotype frequency differences between cases and controls (results available on request).
TABLE III. Odds Ratios and 95% Confidence Intervals for Trios, Discordant Sibpairs, and Case–Control Analyses Using Narrow Diagnostic Criteria.
| Marker | Trios | Sibs | Case–control | Heterogeneity, P | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| OR | Lower 95% CI | Upper 95% CI | OR | Lower 95% CI | Upper 95% CI | OR | Lower 95% CI | Upper 95% CI | ||
| rs1889189 | 1.34 | 0.76 | 2.35 | 0.94 | 0.79 | 1.12 | 1.06 | 0.91 | 1.23 | 0.2443 |
| rs6032826 | 1 | 0.52 | 1.94 | 0.92 | 0.74 | 1.15 | 0.9 | 0.76 | 1.07 | 0.8229 |
| rs6077693 | 1.86 | 0.89 | 3.9 | 1.24 | 1 | 1.54 | 0.87 | 0.71 | 1.06 | 0.3029 |
| rs3787303 | 1.23 | 0.59 | 2.55 | 0.84 | 0.66 | 1.08 | 1.07 | 0.88 | 1.3 | 0.3316 |
| rs2423487 | 0.81 | 0.33 | 2 | 0.95 | 0.71 | 1.26 | 0.82 | 0.64 | 1.06 | 0.744 |
| rs6039783 | 2.33 | 0.78 | 6.93 | 0.66 | 0.46 | 0.96 | 1.01 | 0.74 | 1.38 | 0.0327 |
| rs363012 | 1.29 | 0.73 | 2.29 | 0.91 | 0.76 | 1.09 | 1.01 | 0.87 | 1.18 | 0.2515 |
| rs363043 | 1.04 | 0.59 | 1.85 | 1.27 | 1.06 | 1.51 | 1.17 | 0.99 | 1.37 | 0.5239 |
| rs363016 | 1 | 0.6 | 1.67 | 0.94 | 0.8 | 1.11 | 1.07 | 0.92 | 1.24 | 0.8352 |
| rs363021 | 0.73 | 0.43 | 1.27 | 0.94 | 0.8 | 1.12 | 1.02 | 0.88 | 1.18 | 0.3895 |
| rs362562 | 1.36 | 0.79 | 2.34 | 1.08 | 0.91 | 1.29 | 0.9 | 0.78 | 1.05 | 0.4372 |
| rs6039806 | 1.48 | 0.91 | 2.39 | 1.1 | 0.94 | 1.28 | 1.07 | 0.92 | 1.24 | 0.2534 |
| rs6077718 | 1.54 | 0.77 | 3.09 | 1.07 | 0.86 | 1.33 | 1.15 | 0.92 | 1.44 | 0.3272 |
| rs362993 | 1.58 | 0.68 | 3.69 | 1.23 | 0.95 | 1.61 | 0.95 | 0.76 | 1.2 | 0.585 |
| rs362988 | 1.59 | 0.92 | 2.75 | 1.12 | 0.94 | 1.33 | 0.99 | 0.85 | 1.15 | 0.2275 |
| rs6039820 | 1.63 | 0.95 | 2.81 | 1.24 | 1.04 | 1.47 | 1.01 | 0.88 | 1.17 | 0.3366 |
| rs8636 | 1.14 | 0.64 | 2.05 | 1.15 | 0.96 | 1.38 | 0.93 | 0.8 | 1.08 | 0.9849 |
| rs6133852 | 3.55 | 0.95 | 13.22 | 0.85 | 0.55 | 1.3 | 1.05 | 0.73 | 1.52 | 0.042 |
MatInspector and SNPinspector analysis showed that rs6077693, rs362562, rs362988, and rs6039820 (SNPs 3, 11, 15, and 16) all perturbed transcription-factor binding sites (Table IV). Of note is the large number of sites influenced by rs6039820 (SNP 16), the most significantly associated SNP in the family sample (Table I). Although functionality for these binding sites is currently putative, it is possible that these SNPs could influence the regulation of SNAP25 gene expression. There was no evidence of cryptic splice site creation for any of the associated SNPs.
TABLE IV. List of Putative Transcription-Factor Binding Sites Modified by SNAP25 Schizophrenia-Associated SNPs in the Family Sample.
| SNP | Position (bp)a | Allele change | Gain/loss | Vertebrate matrix | Position (bp) | Strand | Name | Matrix simb | REc |
|---|---|---|---|---|---|---|---|---|---|
| SNP 3 (rs6077693) | 10154917 | A→G | Loss | V$FAST/FAST1.02 | 10154910–10154926 | − | Forkhead box H1 (Foxh1) | 0.950 | 0.03 |
| SNP 11 (rs362562) | 10191186 | A→G | Loss | VSGFI1/GFI1.02 | 10191177–10191191 | − | Growth factor independence 1 | 0.963 | 1.29 |
| Loss | V$HOXC/PBX_HOXA9.01 | 10191180–10191196 | + | PBX-HOXA9 binding site | 0.804 | 0.01 | |||
| Gain | V$CAAT/CAAT.01 | 10191176–10191190 | − | Cellular and viral CCAAT box | 0.900 | 2.35 | |||
| Gain | V$HAML/AML3.01 | 10191179–10191193 | + | Runt-related transcription factor 2/CBFA1 (core-binding factor, runt domain, alpha subunit 1) | 0.945 | 0.11 | |||
| SNP 15 (rs362988) | 10229370 | A→G | Loss | V$CLOX/CDPCR3HD.01 | 10229355–10229377 | − | Cut-like homeodomain protein (cut repeat III/homeodomain) | 0.961 | 3.06 |
| Loss | V$CLOX/CLOX.01 | 10229360–10229382 | + | Cut-like homeodomain protein | 0.888 | 0.14 | |||
| SNP 16 (rs362988) | 10230027 | C→T | Loss | V$XBBF/MIF1.01 | 10230027–10230055 | − | MIBP1/RFX1 complex | 0.762 | 0.01 |
| Gain | V$HOMF/S8.01 | 10230015–10230031 | + | Binding site for S8 type homeodomains | 0.997 | 1.90 | |||
| Gain | V$LHXF/LMX1B.01 | 10230016–10230032 | + | LIM-homeodomain transcription factor | 0.918 | 0.09 | |||
| Gain | V$BRNF/BRN3.02 | 10230017–10230035 | + | Brn-3, POU-IV protein class | 0.921 | 1.62 | |||
| Gain | V$HOXF/GSH2.01 | 10230017–10230033 | − | Homeodomain transcription factor Gsh-2 | 0.952 | 1.84 | |||
| Gain | V$OCTP/OCT1P.01 | 10230017–10230029 | − | Octamer-binding factor-1, POU specific domain | 0.875 | 0.61 | |||
| Gain | V$BRNF/BRN3.01 | 10230018–10230036 | − | Brn-3, POU-IV protein class | 0.792 | 0.00 | |||
| Gain | V$HOXF/GSH1.01 | 10230020–10230036 | − | Homeobox transcription factor Gsh-1 | 0.899 | 0.14 | |||
| Gain | V$HOXF/GSH2.01 | 10230020–10230036 | + | Homeodomain transcription factor Gsh-2 | 0.955 | 1.84 | |||
| Gain | V$LHXF/LMX1B.01 | 10230021–10230037 | − | LIM-homeodomain transcription factor | 0.932 | 0.09 | |||
| Gain | V$ATBF/ATBF1.01 | 10230022–10230038 | − | AT-binding transcription factor 1 | 0.830 | 0.07 | |||
| Gain | V$HOMF/8.01 | 10230022–10230038 | − | Binding site for S8 type homeodomain | 0.992 | 1.90 |
Human genome build 36.1.
Matrix similarity: A perfect match to the matrix gets a score of 1.00—a “good” match to the matrix usually has a similarity ≥ 0.80.
Random expectation (matches per 1,000 bp).
BLASTN sequence similarity search against the database of expressed sequence tags (dbEST) revealed that rs6039820 (SNP 16) also lies within a 99% full-length match to a human intronless and same strand EST representing the 5′ sequence of amygdala cDNA clone DKFZp761D1514 (Genbank accession AL133826). No overlapping ESTs were detected by further database sequence searching, but incorporation of selected VISTA tracks onto the UCSC genome browser showed this genomic region to be conserved in mammals, for example, mouse and dog (Fig. 2). Although no encoded open-reading frame was strongly suggestive of a functional protein, and we detected no features currently ascribed to miRNA precursors, we cannot rule out the possibility that rs6039820 is a biologically meaningful SNP.
FIG. 2.
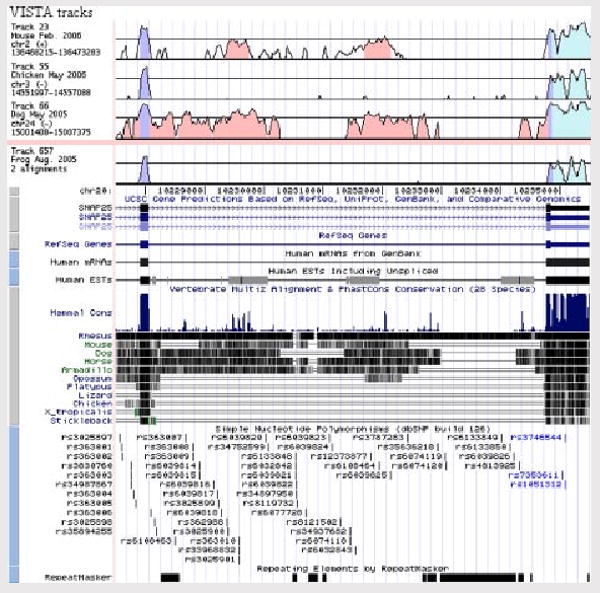
SNAP25 in the UCSC human genome browser. Schematic showing UCSC genome browser window of chr20:10,227,500-10,235,500 spanning exon 7 to the 3′-UTR of the SNAP25 gene. VISTA tracks are at the top, showing evolutionarily conserved regions of intron 7 (pink), coding sequence (purple) plus 3′-UTR (blue). EST AL133826 is depicted by the first gray box on the human EST track with evidence shown for mammalian conservation and directly above SNP rs6039820.
Discussion
In this study, we tested the positional candidate gene SNAP25 for association with schizophrenia in an Irish sample of 270 high-density families. This was followed up with an attempt at replication using an independent sample of 657 cases and 411 controls, also from Ireland. As we hypothesized, our family-based association tests yielded considerable evidence for association in several markers that were not in significant LD with each other. This was supported by the fact that several SNPs had q values lower than 0.1 (i.e., a threshold for declaring significance leading, on average, to less than 10% false positive findings) as well as analysis of two marker haplotypes.
Although there have been no positive reports of association between SNAP25 and schizophrenia, several association studies of ADHD have been positive [Brophy et al., 2002; Mill et al., 2004; Feng et al., 2005; Mill et al., 2005; Kim et al., 2007; Renner et al., 2008]. There is modest evidence of a familial relationship between schizophrenia and symptoms of ADHD in relatives [Keshavan et al., 2003, 2008]. It is nevertheless intriguing to speculate about such potential shared factors. Some evidence lending modest face validity to such a supposition includes the presence of inattention as a cardinal feature of ADHD and as a negative symptom of schizophrenia, as well as the response of both ADHD and negative symptoms to psychostimulants [Angrist et al., 1980]. Both negative symptoms [Wolkin et al., 1992; Goto et al., 2007] and ADHD are thought to be etiologically related to abnormalities in dopaminergic tone [Ludolph et al., 2008]. Although speculative now, it will be possible to begin to address the question of genetic overlap between schizophrenia and ADHD in the near future in the Psychiatric GWAS Consortium (PGC) cross-disorder analyses (AJP, submitted).
We found that the most significantly associated haplotoype of SNAP25 was preferentially overtransmitted to subjects in the upper ∼60% of the hallucinations and delusions factors. These factors, as previously described, are correlated in this sample, as would be expected due to their frequent co-occurrence as “positive symptoms.” If the marker-disease associations observed in this sample are true, this would suggest that SNAP25 is what we have termed a susceptibility-modifier gene. This class of genes is one that predisposes to more or less specific clinical subtypes of illness, and may affect the distribution of clinical features in a population [Fanous and Kendler, 2005, 2008]. This genotype–phenotype correlation will of course need to be confirmed in an independent sample, especially given the inflation of the false positive rate due to our testing of multiple, albeit somewhat correlated, symptom factors. Again, this is likely to be possible in the PGC analyses. We did attempt to relate these results to the linkage results of the latent class phenotypes we previously published [Fanous et al., 2008]. Although the manic, schizomanic, and deficit syndrome classes were suggestively linked to chromosome 20p, no latent class was associated with SNAP25 SNPs (results available on request). However, we would consider such an approach underpowered for association, since the latent classes each comprised only 18–25% of the sample.
We attempted to replicate the marker-disease association we observed in the family sample in an independent sample of cases and controls from the same country. We observed no significant markers or haplotypes in this replication sample. Therefore, we are currently unable to confirm association between SNAP25 and schizophrenia. It has been noted, however, that none of the most replicated susceptibility genes for schizophrenia, such as DTNBP1 and NRG1, have been uniformly supported in all studies [Harrison and Weinberger, 2005; Riley and Kendler, 2006]. This would suggest that SNAP25 should be studied in several additional independent samples before a judgment is made. Our formal tests for heterogeneity indicated that the non-replication was not likely to be due to heterogeneity, and that it may still be possible to replicate the association observed in our family sample in a larger case–control sample. It is possible that genetic heterogeneity may account for the non-replication, and that a clinical subtype of illness may be associated with SNAP25 in this sample. However, we do not have OPCRIT ratings in this sample currently and would not be able to relate the OPCRIT-based analyses in the family sample with comparable ones in this sample.
Despite the current lack of replication, functional studies of associated variants observed in this study may be informative, given the weight of previous expression studies (please see Introduction Section for review). It is therefore of interest that the most significant SNP in the family sample was observed to occur in 12 transcription-factor binding sites in our in silico analyses. In vitro studies of this SNP may further elucidate its effect on gene expression, and possibly, on its role in schizophrenia susceptibility.
Acknowledgments
This work was supported by a Department of Veterans Affairs Merit Review Program grant to AF, as well as NIH grants MH-41953, MH-52537, and MH-45390. Data collection was conducted under the supervision of S. Humphries, M. Healy, and A. Finnerty. Additional interviews were conducted by J. Burke, B. Murphy, F. Duke, R. Shinkwin, M. Ni Nuallain, F. McMahon, J. Downing, T. Hebron, B. Hanratty, E. Crowe, M. Doherty, J. Bray, and L. Lowry. This project would not have been possible without the cooperation of the families themselves and the staffs of the many psychiatric hospitals and units in Ireland and Northern Ireland and their efforts are greatly acknowledged.
Footnotes
Financial Disclosures: Drs. Fanous, Zhao, van den Oord, Thiselton, Amdur, O'Neill, Walsh, Kendler, Riley, Maher, Mr. Wormley, Mr. Bigdeli, and Ms. Bergen have no financial conflicts of interest to declare.
References
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- Angrist B, Rotrosen J, Gershon S. Differential effects of amphetamine and neuroleptics on negative vs. positive symptoms in schizophrenia. Psychopharmacology (Berl) 1980;72:17–19. doi: 10.1007/BF00433802. [DOI] [PubMed] [Google Scholar]
- Arinami T, Ohtsuki T, Ishiguro H, Ujike H, Tanaka Y, Morita Y, Mineta M, Takeichi M, Yamada S, Imamura A, Ohara K, Shibuya H, Ohara K, Suzuki Y, Muratake T, Kaneko N, Someya T, Inada T, Yoshikawa T, Toyota T, Yamada K, Kojima T, Takahashi S, Osamu O, Shinkai T, Nakamura M, Fukuzako H, Hashiguchi T, Niwa SI, Ueno T, Tachikawa H, Hori T, Asada T, Nanko S, Kunugi H, Hashimoto R, Ozaki N, Iwata N, Harano M, Arai H, Ohnuma T, Kusumi I, Koyama T, Yoneda H, Fukumaki Y, Shibata H, Kaneko S, Higuchi H, Yasui-Furukori N, Numachi Y, Itokawa M, Okazaki Y. Genomewide high-density SNP linkage analysis of 236 Japanese families supports the existence of schizophrenia susceptibility loci on chromosomes 1p, 14q, and 20p. Am J Hum Genet. 2005;77:937–944. doi: 10.1086/498122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- Benjamini Y, Hochberg Y. On the adaptive control of the false discovery rate in multiple testing with independent statistics. J Educ Behav Stat. 2000;25:60–83. [Google Scholar]
- Berrios GE. Unitary psychosis concept. In: Berrios GE, Porter R, editors. A history of clinical psychiatry. London: The Athlone Press; 1995. pp. 313–335. [Google Scholar]
- Brameier M, Wiuf C. Ab initio identification of human microRNAs based on structure motifs. BMC Bioinformatics. 2007;8:478. doi: 10.1186/1471-2105-8-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brophy K, Hawi Z, Kirley A, Fitzgerald M, Gill M. Synaptosomal-associated protein 25 (SNAP-25) and attention deficit hyperactivity disorder (ADHD): Evidence of linkage and association in the Irish population. Mol Psychiatry. 2002;7:913–917. doi: 10.1038/sj.mp.4001092. [DOI] [PubMed] [Google Scholar]
- Brown BW, Russell K. Methods of correcting for multiple testing: Operating characteristics. Stat Med. 1997;16:2511–2528. doi: 10.1002/(sici)1097-0258(19971130)16:22<2511::aid-sim693>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T. MatInspector and beyond: Promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21:2933–2942. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]
- Couronne O, Poliakov A, Bray N, Ishkhanov T, Ryaboy D, Rubin E, Pachter L, Dubchak I. Strategies and tools for whole-genome alignments. Genome Res. 2003;13:73–80. doi: 10.1101/gr.762503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudbridge F. Pedigree disequilibrium tests for multilocus haplotypes. Genet Epidemiol. 2003;25:115–121. doi: 10.1002/gepi.10252. [DOI] [PubMed] [Google Scholar]
- Fanous AH, Kendler KS. Genetic heterogeneity, modifier genes, and quantitative phenotypes in psychiatric illness: Searching for a framework. Mol Psychiatry. 2005;10:6–13. doi: 10.1038/sj.mp.4001571. [DOI] [PubMed] [Google Scholar]
- Fanous AH, Kendler KS. Genetics of clinical features and subtypes of schizophrenia: A review of the recent literature. Curr Psychiatry Rep. 2008;10:164–170. doi: 10.1007/s11920-008-0028-z. [DOI] [PubMed] [Google Scholar]
- Fanous AH, Van Den Oord EJ, Riley BP, Aggen SH, Neale MC, O'Neill FA, Walsh D, Kendler KS. Relationship between a high-risk haplotype in the DTNBP1 (dysbindin) gene and clinical features of schizophrenia. Am J Psychiatry. 2005;162:1824–1832. doi: 10.1176/appi.ajp.162.10.1824. [DOI] [PubMed] [Google Scholar]
- Fanous AH, Neale MC, Webb BT, Straub RE, O'Neill FA, Walsh D, Riley BP, Kendler KS. Novel linkage to chromosome 20p using latent classes of psychotic illness in 270 Irish high-density families. Biol Psychiatry. 2008;64:121–127. doi: 10.1016/j.biopsych.2007.11.023. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Earle JA, Stary JM, Lee S, Sedgewick J. Altered levels of the synaptosomal associated protein SNAP-25 in hippocampus of subjects with mood disorders and schizophrenia. Neuroreport. 2001;12:3257–3262. doi: 10.1097/00001756-200110290-00023. [DOI] [PubMed] [Google Scholar]
- Feng Y, Crosbie J, Wigg K, Pathare T, Ickowicz A, Schachar R, Tannock R, Roberts W, Malone M, Swanson J, Kennedy JL, Barr CL. The SNAP25 gene as a susceptibility gene contributing to attention-deficit hyperactivity disorder. Mol Psychiatry. 2005;10:998–1005. 973. doi: 10.1038/sj.mp.4001722. [DOI] [PubMed] [Google Scholar]
- Fernando RL, Nettleton D, Southey BR, Dekkers JC, Rothschild MF, Soller M. Controlling the proportion of false positives in multiple dependent tests. Genetics. 2004;166:611–619. doi: 10.1534/genetics.166.1.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel SM, Haroutunian V, Powchik P, Honer WG, Davidson M, Davies P, Davis KL. Increased concentrations of presynaptic proteins in the cingulate cortex of subjects with schizophrenia. Arch Gen Psychiatry. 1997;54:559–566. doi: 10.1001/archpsyc.1997.01830180077010. [DOI] [PubMed] [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- Goto Y, Otani S, Grace AA. The Yin and Yang of dopamine release: A new perspective. Neuropharmacology. 2007;53:583–587. doi: 10.1016/j.neuropharm.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham ME, Washbourne P, Wilson MC, Burgoyne RD. Molecular analysis of SNAP-25 function in exocytosis. Ann NY Acad Sci. 2002;971:210–221. doi: 10.1111/j.1749-6632.2002.tb04465.x. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: On the matter of their convergence. Mol Psychiatry. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. [DOI] [PubMed] [Google Scholar]
- Hetey L, Schwitzkowsky R, Ott T, Barz H. Diminished synaptosomal dopamine (DA) release and DA autoreceptor supersensitivity in schizophrenia. J Neural Transm Gen Sect. 1991;83:25–35. doi: 10.1007/BF01244449. [DOI] [PubMed] [Google Scholar]
- Jeans AF, Oliver PL, Johnson R, Capogna M, Vikman J, Molnar Z, Babbs A, Partridge CJ, Salehi A, Bengtsson M, Eliasson L, Rorsman P, Davies KE. A dominant mutation in Snap25 causes impaired vesicle trafficking, sensorimotor gating, and ataxia in the blind-drunk mouse. Proc Natl Acad Sci USA. 2007;104:2431–2436. doi: 10.1073/pnas.0610222104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karson CN, Mrak RE, Schluterman KO, Sturner WQ, Sheng JG, Griffin WS. Alterations in synaptic proteins and their encoding mRNAs in prefrontal cortex in schizophrenia: A possible neurochemical basis for ‘hypofrontality’. Mol Psychiatry. 1999;4:39–45. doi: 10.1038/sj.mp.4000459. [DOI] [PubMed] [Google Scholar]
- Kendler KS, McGuire M, Gruenberg AM, O'Hare A, Spellman M, Walsh D. The Roscommon Family Study. I. Methods, diagnosis of probands, and risk of schizophrenia in relatives. Arch Gen Psychiatry. 1993;50:527–540. doi: 10.1001/archpsyc.1993.01820190029004. [DOI] [PubMed] [Google Scholar]
- Keshavan MS, Sujata M, Mehra A, Montrose DM, Sweeney JA. Psychosis proneness and ADHD in young relatives of schizophrenia patients. Schizophr Res. 2003;59:85–92. doi: 10.1016/s0920-9964(01)00400-5. [DOI] [PubMed] [Google Scholar]
- Keshavan M, Montrose DM, Rajarethinam R, Diwadkar V, Prasad K, Sweeney JA. Psychopathology among offspring of parents with schizophrenia: Relationship to premorbid impairments. Schizophr Res. 2008;103:114–120. doi: 10.1016/j.schres.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Biederman J, Arbeitman L, Fagerness J, Doyle AE, Petty C, Perlis RH, Purcell S, Smoller JW, Faraone SV, Sklar P. Investigation of variation in SNAP-25 and ADHD and relationship to co-morbid major depressive disorder. Am J Med Genet Part B. 2007;144B:781–790. doi: 10.1002/ajmg.b.30522. [DOI] [PubMed] [Google Scholar]
- Kimura K, Mizoguchi A, Ide C. Regulation of growth cone extension by SNARE proteins. J Histochem Cytochem. 2003;51:429–433. doi: 10.1177/002215540305100404. [DOI] [PubMed] [Google Scholar]
- Knable MB, Barci BM, Webster MJ, Meador-Woodruff J, Torrey EF. Molecular abnormalities of the hippocampus in severe psychiatric illness: Postmortem findings from the Stanley Neuropathology Consortium. Mol Psychiatry. 2004;9:609–620. 544. doi: 10.1038/sj.mp.4001471. [DOI] [PubMed] [Google Scholar]
- Korn EL, Troendle J, McShane L, Simon R. Controlling the number of false discoveries: Application to high-dimensional genomic data. J Stat Plann Inference. 2004;124:379–398. [Google Scholar]
- Laird NM, Horvath S, Xu X. Implementing a unified approach to family-based tests of association. Genet Epidemiol. 2000;19(Suppl1):S36–S42. doi: 10.1002/1098-2272(2000)19:1+<::AID-GEPI6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Lewis CM, Levinson DF, Wise LH, DeLisi LE, Straub RE, Hovatta I, Williams NM, Schwab SG, Pulver AE, Faraone SV, Brzustowicz LM, Kaufmann CA, Garver DL, Gurling HM, Lindholm E, Coon H, Moises HW, Byerley W, Shaw SH, Mesen A, Sherrington R, O'Neill FA, Walsh D, Kendler KS, Ekelund J, Paunio T, Lonnqvist J, Peltonen L, O'Donovan MC, Owen MJ, Wildenauer DB, Maier W, Nestadt G, Blouin JL, Antonarakis SE, Mowry BJ, Silverman JM, Crowe RR, Cloninger CR, Tsuang MT, Malaspina D, Harkavy-Friedman JM, Svrakic DM, Bassett AS, Holcomb J, Kalsi G, McQuillin A, Brynjolfson J, Sigmundsson T, Petursson H, Jazin E, Zoega T, Helgason T. Genome scan meta-analysis of schizophrenia and bipolar disorder, part II: Schizophrenia. Am J Hum Genet. 2003;73:34–48. doi: 10.1086/376549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludolph AG, Kassubek J, Schmeck K, Glaser C, Wunderlich A, Buck AK, Reske SN, Fegert JM, Mottaghy FM. Dopaminergic dysfunction in attention deficit hyperactivity disorder (ADHD), differences between pharmacologically treated and never treated young adults: A 3,4-dihdroxy-6-[(18)F]fluorophenyl-l-alanine PET study. Neuroimage. 2008;41:718–727. doi: 10.1016/j.neuroimage.2008.02.025. [DOI] [PubMed] [Google Scholar]
- Macgregor S, Craddock N, Holmans PA. Use of phenotypic covariates in association analysis by sequential addition of cases. Eur J Hum Genet. 2006;14:529–534. doi: 10.1038/sj.ejhg.5201604. [DOI] [PubMed] [Google Scholar]
- Martin ER, Monks SA, Warren LL, Kaplan NL. A test for linkage and association in general pedigrees: The pedigree disequilibrium test. Am J Hum Genet. 2000;67:146–154. doi: 10.1086/302957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffin P, Farmer A, Harvey I. A polydiagnostic application of operational criteria in studies of psychotic illness. Development and reliability of the OPCRIT system. Arch Gen Psychiatry. 1991;48:764–770. doi: 10.1001/archpsyc.1991.01810320088015. [DOI] [PubMed] [Google Scholar]
- Mill J, Richards S, Knight J, Curran S, Taylor E, Asherson P. Haplotype analysis of SNAP-25 suggests a role in the aetiology of ADHD. Mol Psychiatry. 2004;9:801–810. doi: 10.1038/sj.mp.4001482. [DOI] [PubMed] [Google Scholar]
- Mill J, Xu X, Ronald A, Curran S, Price T, Knight J, Craig I, Sham P, Plomin R, Asherson P. Quantitative trait locus analysis of candidate gene alleles associated with attention deficit hyperactivity disorder (ADHD) in five genes: DRD4, DAT1, DRD5, SNAP-25, and 5HT1B. Am J Med Genet Part B. 2005;133B:68–73. doi: 10.1002/ajmg.b.30107. [DOI] [PubMed] [Google Scholar]
- Moises HW, Yang L, Kristbjarnarson H, Wiese C, Byerley W, Macciardi F, Arolt V, Blackwood D, Liu X, Sjogren B. An international two-stage genome-wide search for schizophrenia susceptibility genes. Nat Genet. 1995;11:321–324. doi: 10.1038/ng1195-321. [DOI] [PubMed] [Google Scholar]
- Nam JW, Kim J, Kim SK, Zhang BT. ProMiR II: A web server for the probabilistic prediction of clustered, nonclustered, conserved and nonconserved microRNAs. Nucleic Acids Res. 2006;34:W455–W458. doi: 10.1093/nar/gkl321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numakawa T, Yagasaki Y, Ishimoto T, Okada T, Suzuki T, Iwata N, Ozaki N, Taguchi T, Tatsumi M, Kamijima K, Straub RE, Weinberger DR, Kunugi H, Hashimoto R. Evidence of novel neuronal functions of dysbindin, a susceptibility gene for schizophrenia. Hum Mol Genet. 2004;13:2699–2708. doi: 10.1093/hmg/ddh280. [DOI] [PubMed] [Google Scholar]
- O'Connell JR, Weeks DE. PedCheck: A program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osen-Sand A, Catsicas M, Staple JK, Jones KA, Ayala G, Knowles J, Grenningloh G, Catsicas S. Inhibition of axonal growth by SNAP-25 antisense oligonucleotides in vitro and in vivo. Nature. 1993;364:445–448. doi: 10.1038/364445a0. [DOI] [PubMed] [Google Scholar]
- Renner TJ, Walitza S, Dempfle A, Eckert L, Romanos M, Gerlach M, Schafer H, Warnke A, Lesch KP, Jacob C. Allelic variants of SNAP25 in a family-based sample of ADHD. J Neural Transm. 2008;115:317–321. doi: 10.1007/s00702-007-0840-3. [DOI] [PubMed] [Google Scholar]
- Riley B, Kendler KS. Molecular genetic studies of schizophrenia. Eur J Hum Genet. 2006;14:669–680. doi: 10.1038/sj.ejhg.5201571. [DOI] [PubMed] [Google Scholar]
- Sabatti C, Service S, Freimer N. False discovery rate in linkage and association genome screens for complex disorders. Genetics. 2003;164:829–833. doi: 10.1093/genetics/164.2.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAS Institute. SAS/STAT Software, version 8. Cary, N.C.: SAS Institute; 1999. [Google Scholar]
- Sherman AD, Hegwood TS, Baruah S, Waziri R. Deficient NMDA-mediated glutamate release from synaptosomes of schizophrenics. Biol Psychiatry. 1991;30:1191–1198. doi: 10.1016/0006-3223(91)90155-f. [DOI] [PubMed] [Google Scholar]
- Spitzer RL, Williams JB, Gibbon J. Structured clinical interview for DSM-III-R patient version. New York: New York Psychiatric Institute; 1987. [Google Scholar]
- Storey J. The positive false discovery rate: A Bayesian interpretation and the q-value. Ann Stat. 2003;31:2013–2035. [Google Scholar]
- Storey J, Tibshirani R. Statistical significance for genome-wide studies. Proc Natl Acad Sci. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub RE, MacLean CJ, Ma Y, Webb BT, Myakishev MV, Harris-Kerr C, Wormley B, Sadek H, Kadambi B, O'Neill FA, Walsh D, Kendler KS. Genome-wide scans of three independent sets of 90 Irish multiplex schizophrenia families and follow-up of selected regions in all families provides evidence for multiple susceptibility genes. Mol Psychiatry. 2002;7:542–559. doi: 10.1038/sj.mp.4001051. [DOI] [PubMed] [Google Scholar]
- Sweet RA, Bergen SE, Sun Z, Marcsisin MJ, Sampson AR, Lewis DA. Anatomical evidence of impaired feedforward auditory processing in schizophrenia. Biol Psychiatry. 2007;61:854–864. doi: 10.1016/j.biopsych.2006.07.033. [DOI] [PubMed] [Google Scholar]
- Tachikawa H, Harada S, Kawanishi Y, Okubo T, Suzuki T. Polymorphism of the 5′-upstream region of the human SNAP-25 gene: An association analysis with schizophrenia. Neuropsychobiology. 2001;43:131–133. doi: 10.1159/000054880. [DOI] [PubMed] [Google Scholar]
- Thompson PM, Sower AC, Perrone-Bizzozero NI. Altered levels of the synaptosomal associated protein SNAP-25 in schizophrenia. Biol Psychiatry. 1998;43:239–243. doi: 10.1016/S0006-3223(97)00204-7. [DOI] [PubMed] [Google Scholar]
- Thompson PM, Rosenberger C, Qualls C. CSF SNAP-25 in schizophrenia and bipolar illness. A pilot study. Neuropsychopharmacology. 1999;21:717–722. doi: 10.1016/S0893-133X(99)00068-8. [DOI] [PubMed] [Google Scholar]
- Thompson PM, Egbufoama S, Vawter MP. SNAP-25 reduction in the hippocampus of patients with schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2003a;27:411–417. doi: 10.1016/S0278-5846(03)00027-7. [DOI] [PubMed] [Google Scholar]
- Thompson PM, Kelley M, Yao J, Tsai G, van Kammen DP. Elevated cerebrospinal fluid SNAP-25 in schizophrenia. Biol Psychiatry. 2003b;53:1132–1137. doi: 10.1016/s0006-3223(02)01599-8. [DOI] [PubMed] [Google Scholar]
- Tsai CA, Hsueh HM, Chen JJ. Estimation of false discovery rates in multiple testing: Application to gene microarray data. Biometrics. 2003;59:1071–1081. doi: 10.1111/j.0006-341x.2003.00123.x. [DOI] [PubMed] [Google Scholar]
- Van den Oord EJCG, Sullivan PF. A framework for controlling false discovery rates and minimizing the amount of genotyping in the search for disease mutations. Hum Hered. 2003a;56:188–199. doi: 10.1159/000076393. [DOI] [PubMed] [Google Scholar]
- Van den Oord EJCG, Sullivan PF. False discoveries and models for gene discovery. Trends Genet. 2003b;19:537–542. doi: 10.1016/j.tig.2003.08.003. [DOI] [PubMed] [Google Scholar]
- Van Den Oord EJ, Jiang Y, Riley BP, Kendler KS, Chen X. FP-TDI SNP scoring by manual and statistical procedures: A study of error rates and types. Biotechniques. 2003;34:610–620. 622. doi: 10.2144/03343dd04. [DOI] [PubMed] [Google Scholar]
- Van den Oord EJCG. Controlling false discoveries in candidate gene studies. Mol Psychiatry. 2005;10:230–231. doi: 10.1038/sj.mp.4001581. [DOI] [PubMed] [Google Scholar]
- Wolkin A, Sanfilipo M, Wolf AP, Angrist B, Brodie JD, Rotrosen J. Negative symptoms and hypofrontality in chronic schizophrenia. Arch Gen Psychiatry. 1992;49:959–965. doi: 10.1001/archpsyc.1992.01820120047007. [DOI] [PubMed] [Google Scholar]
- Young CE, Arima K, Xie J, Hu L, Beach TG, Falkai P, Honer WG. SNAP-25 deficit and hippocampal connectivity in schizophrenia. Cereb Cortex. 1998;8:261–268. doi: 10.1093/cercor/8.3.261. [DOI] [PubMed] [Google Scholar]