

Abstract
Although low-density lipoprotein (LDL) plays a predominant role in atherogenesis, the low-density lipoproteome has not been fully characterized. Moreover, alterations from a Western diet, diabetes, and physical inactivity on this proteome have yet to be determined. Accordingly, relative quantification was determined in LDL proteins from male Yucatan diabetic dyslipidemic (DD) swine in the early stages of atherosclerosis compared to healthy control (C) and non-diabetic hyperlipidemic (H) swine. Importantly, coronary vascular dysfunction was prevented by aerobic exercise training in these animals (DDX) without altering total LDL concentration. Using 2-DE, Western blot, label-free quantitative MS, and selected reaction monitoring, alterations in the abundance of apolipoproteins A-I, B, C-III, D, E, and J and noncovalently associated proteins were determined in LDL isolated using fast protein liquid chromatography. At least 28 unique proteins, many of which were novel, were identified with high confidence. An apolipoprotein E isoform demonstrated stronger correlation to disease (percent of coronary artery segments with intimal thickening) than some traditional risk factors (total cholesterol, LDL cholesterol, and LDL/HDL cholesterol). Taken together, this work identifies new possible biomarkers, potential therapeutic targets for atherosclerosis, and generates new hypotheses regarding the role of LDL in atherogenesis.
Keywords: Fast protein liquid chromatography, Low density lipoprotein, Two-dimensional gel electrophoresis, Tandem mass spectrometry, Yucatan pig
1 Introduction
Atherosclerosis is the primary cause of coronary heart disease (CHD), the leading cause of mortality in the developed world. Low-density lipoprotein (LDL), the major cholesterol carrier in the blood, has long been associated with CHD. However, surprisingly little is known about the low-density lipoproteome, a term itself only a few years old. It was assumed for some time that LDL contains a single structural protein, apolipoprotein B. However, in 2005 Karlsson et al. [1] reported at least 11 proteins in human LDL isolated by ultracentrifugation, the technique historically used to isolate LDL. Ultracentrifugation is extremely harsh, using several hundred thousand times the force of gravity and high salt concentrations, conditions that could likely remove proteins less tightly associated with the particle. Indeed, Stahlman et al. [2] found that LDL isolated in KBr exhibited lower recovery of total protein and a smaller number of MS protein peaks than LDL isolated in D2O/sucrose. LDL isolated in either medium contained apolipoproteins A-I, B, E, C-I, C-II, C-III, and J and other associated proteins. Preserving protein-protein interactions during LDL isolation is important to achieve a complete and accurate assessment of the functional LDL particle. SEC relies only on resin pore size and mild buffers to separate lipoproteins, dramatically reducing the potential loss of non-covalently associated protein constituents [3]. Hence, fast protein LC (FPLC) was used here to isolate LDL and facilitate a comprehensive characterization of its proteome.
A long-held hypothesis is that atherogenesis begins when excessive LDL accumulates in the subendothelial space, is oxidatively modified, and is taken up selectively by macrophage cells. This leads to foam cell formation and the appearance of “fatty streaks,” the earliest morphological lesion of atherosclerosis [4]. However, the failure of anti-oxidant therapy observed in several studies [5–7] has led some to rethink this “oxidative modification hypothesis” model. We hypothesized that there may be alterations in the proteome of LDL that are associated with disease. Furthermore, effects of important risk factors for disease such as hypercholesterolemia and diabetes have not been determined regarding the LDL proteome. Diabetics are three-to-four times more likely to develop atheroma and exhibit accelerated atherosclerosis [8], yet the underlying reasons remain unknown. It also has been shown that chronic moderate aerobic exercise can reduce the risk for developing CHD [9], yet the underlying mechanisms for this are poorly understood, as well.
The hypotheses tested here were that (i) hyperlipidemia, as a result of an atherogenic diet, is associated with differential protein expression in the low-density lipoproteome; (II) diabetic dyslipidemia results in unique alterations in protein expression; and (iii) exercise attenuates one or more of these diabetic dyslipidemia-induced changes. To test these hypotheses, we chose a porcine model with groups that display hyperlipidemia or diabetic dyslipidemia and that develop coronary atherosclerosis similar to that observed in humans. In vivo measures of early stage atherosclerosis are rare in humans and difficult in smaller animal models. Furthermore, the lipoprotein profile of swine is similar to humans whereas the mouse is not [10–12]. Four conditions were evaluated in this model: (i) healthy control (C) fed a standard mini-pig chow diet; (ii) non-diabetic, hyperlipidemic (H); (iii) diabetic dyslipidemic (DD); and (iv) exercised DD (DDX). Importantly, exercise decreased vascular disease without a change in LDL concentration in these pigs [13].
Relative protein quantification was carried out using 2-DE with identification by LC-MS/MS and by label-free quantitative MS (LFQMS). 2-DE provides reliable and reproducible quantification and has the distinct ability to resolve qualitative alterations in protein PTM as pI micro-heterogeneities in the 2-D gel [14]. Although PTM information is lost in LFQMS, the latter approach is limited less by the dynamic range of protein expression typical of most eukaryotic samples or by the molecular weight or pI of a protein, all of which are disadvantages of 2-DE. LFQMS also has the advantages of simultaneous high-sensitivity and high-confidence protein identification. A third method, Western blotting combined with selected reaction monitoring (SRM), was used to support and confirm alterations in several proteins of interest. The resulting analyses of differential expression of proteins in LDL produced a comprehensive view of how the LDL proteome varied under several metabolic situations that accompany the complex pathophysiological process of atherosclerosis.
2 Materials and methods
2.1 Materials
IPG strips and acrylamide for slab gels were purchased from Bio-Rad (Richmond, CA). Other ultrapure electrophoretic reagents were obtained from Bio-Rad, Sigma-Aldrich Chemical (St. Louis, MO), or BDH (Poole, UK). Sequence-grade trypsin was obtained from Promega (Madison, WI). Ammonium bicarbonate was purchased from Mallinckrodt Chemicals (Paris, KY). Formic acid, iodoethanol, and triethylphosphine were obtained from Sigma-Aldrich. HPLC columns were purchased from Agilent Technologies (Wilmington, DE). ACN and HPLC-grade water were obtained from EMD Chemicals (Gibbstown, NJ). PVDF for Western blotting was obtained from Millipore (Billerica, MA). All other chemicals used were of the highest grade obtainable.
2.2 Porcine model of diabetic dyslipidemia and exercise protocol
Male miniature Yucatan swine (Sinclair Research Center, Columbia, MO) were maintained in facilities accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care. All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) and were in accordance with the guidelines of the IACUC of the National Institute on Drug Abuse and the Guide for the Care and Use of Laboratory Animals of the National Research Council, 1996. Animal handling and experimental design were described in detail previously by Mokelke et al. [13], therefore only information pertinent to the current study will be recapitulated. Briefly, animals were randomly assigned to experimental groups: control, standard mini-pig chow (C); hyperlipidemic, atherogenic mini-pig chow (H); diabetic, atherogenic mini-pig chow (diabetic dyslipidemic, DD); and diabetic, atherogenic mini-pig chow, endurance exercise trained (DDX).
The atherogenic mini-pig chow consisted of standard mini-pig chow supplemented with cholesterol (2%) and coconut oil resulting in a diet that was 46% fat by kilocalorie compared to the control, standard mini-pig chow that had 8% of kilocalories from fat. Animals on the atherogenic diet (H, DD, and DDX) were hypercholesterolemic, and the diabetic pigs (DD and DDX) also became hypertriglyceridemic, hence termed dyslipidemic [13, 15]. Diabetes was induced in the DD and DDX groups using injections of alloxan (125 mg/kg, Sigma), a drug that specifically destroys insulin producing β-cells of the pancreas [16]. The exercise program for the DDX group consisted of running on a motorized treadmill (Good Horsekeeping, Ash Grove, MO) for 30 min/day at their target heart rate, which was 65–75% of maximal heart rate. These 30-min sessions were carried out 4 days/week for 14 weeks and resulted in classical training adaptations, including a decreased resting heart rate, increased work capacity, and increased skeletal muscle oxidative enzymes [13]. Animals in the C, H, and DD groups were individually housed to ensure sedentary behavior. Animals were fed once/day, at the same time of day, and drank water ad libitum for the entire 20-week study. Body weights were monitored weekly, and amount of feed and daily insulin dosage were adjusted as necessary to ensure hyperglycemia and weight gain of 1% of initial body weight per week [17], the normal expected developmental body weight increase for these pigs. At the end of the study, no significant differences in body weight were observed between groups [13]. Blood urea nitrogen, creatinine, and liver enzymes all remained within normal limits in all animals, consistent with previous observations [18]. Early coronary artery disease, i.e. intimal thickening, was quantified by intravascular ultrasound in vivo [11, 13, 16]. Briefly, intravascular ultrasound images of intimal thickening were obtained along about 30–60 mm of each of the left anterior descending and circumflex coronary arteries and quantified as the percentage of 1-mm segments having atheroma (intimal thickening).
2.3 Preparation of LDL
Blood was collected from overnight-fasted pigs and plasma separated by centrifugation. Plasma (1 mL) was chromatographed by fast protein LC (FPLC) on a Superose 6 column (HR 16, Pharmacia) and eluted with w/v 0.9% NaCl, 0.01% Tris, 0.01% EDTA, 0.02% sodium azide, pH 7.6 as previously described [11]. Fractions (2 mL) were collected and assayed for protein and cholesterol. Precise fractions from the LDL peak were then pooled in each animal. Bradford protein assays were then performed on these pooled samples using Bio-Rad reagents. Samples were cryopreserved in 10% sucrose and stored at 80°C.
2.4 2-DE and image analysis
Aliquots of LDL samples from all four groups (n = 5) containing 480 μg of protein (~1–2 mL) were desalted and concentrated by filtration using a Millipore ultrafree-0.5 centrifugal filter device. Each sample was rinsed several times with an IPG solubilizer containing 9 M urea, 4% Igepal CA-630 ([octylphenoxy] polyethoxyethanol), 1% DTT, and 0.2% ampholytes pH 3–10. Filtered protein was resolubilized in 500 μL of the solubilizer and loaded onto IPG strips (24 cm, linear pH 3–10) by overnight, passive rehydration at room temperature. IEF was performed simultaneously on all IPG strips using two Protean IEF Cells (Bio-Rad), using a program of progressively increasing voltage (150 V for 2 h, 300 V for 4 h, 1500 V for 1 h, 5000 V for 5 h, 7000 V for 6 h, and 10 000 V for 3 h) for a total of 100 000 Vh. A computer-controlled gradient casting system was used to prepare the second-dimension SDS gradient slab gels (20×25×0.15 cm), in which the acrylamide concentration varied linearly from 5 to 20% T. First-dimension IPG strips were loaded directly onto the slab gels following equilibration for 10 min in Equilibration Buffer I and 10 mins in Equilibration Buffer II (Equilibration Buffer I: 6 M urea, 2% SDS, 0.375 M Tris-HCl pH 8.8, 20% Glycerol, 130 mM DTT; Equilibration Buffer II: 6 M urea, 2% SDS, 0.375 M Tris-HCl pH 8.8, 20% Glycerol, 135 mM iodoacetamide) for protein alkylation and reduction. HiMark™ Unstained High Molecular Weight Protein Standards (Invitrogen, Carlsbad, CA.) were run on the slab gels’ left margin for MW calibration. Second-dimension slab gels were run in parallel (20-gel capacity) at 8°C for 14 h at 160 V, and stained using a sensitive colloidal CBB procedure [19]. After staining, gels were washed several times with water and scanned at 95.3 μm/pixel resolution using a GS-800 Calibrated Imaging Densitometer (Bio-Rad). The resulting 12-bit images were analyzed using PDQuest™ software (Bio-Rad, v.7.1). Background was subtracted and peaks for the protein spots located and counted. Each gel image was normalized against the total gel density. PDQuest™-resident Student’s t-test was used as a preliminary statistical analysis to determine differential expression candidates. This dataset was then exported to Sigma Stat 3.0 (Systat Software, San Jose, CA) for one-way ANOVA. Linear regression analyses were performed against percent atheroma (neointimal formation) [11, 13, 16] and correlation coefficients calculated using Spotfire® (Spotfire, Cambridge, MA) and Sigma Stat 3.0. Proteins determined to be differentially expressed or with a significant correlation to disease were selected for MS identification based on these calculations.
2.5 MS identification of 2-D gel protein spots
2.5.1 In-gel tryptic digestion
Each pre-selected detectable protein spot was cut manually from each of four separate gels using a 1.5-mm gel cutting tool on a light box. The four replicate gel plugs were placed in one well of a 96-well plate and processed using the Multi-Probe II (Perkin-Elmer, Boston, MA). In this automated system, the excised protein spots were first destained with 50 mM ammonium bicarbonate-50% ACN followed by 100% ACN. Protein reduction with 10 mM DTT and alkylation with 55 mM iodoacetamide were carried out, followed by overnight tryptic digestion using modified trypsin at 6 ng/μL and 37°C with shaking. The resulting peptides were extracted from gel plugs with vigorous shaking via Jitterbug (Boekel Scientific) for 20 min in two rounds of the following three phases: (I) 15 μL 0.2% formic acid and 15 μL of ACN solution (50% aqueous); (ii) 21 μL 0.2% formic acid and 9 μL of ACN solution (70% aqueous), and (iii) with 30 μL of ACN solution (0% aqueous). After each phase, the extraction solution was placed in a separate 96-well plate and dried via speedvac without heat. The dehydrated peptides were then reconstituted in 20 μL of 0.1% TFA with continuous shaking for 5 min before being concentrated and desalted using μ-C-18 Millipore ZipTip® pipette tips (Billerica, MA) according to the manufacturer’s protocol. Purified peptides were eluted in a new 96-well plate, dried via speedvac, and then reconstituted in 5% ACN and 0.1% formic acid.
2.5.2 LC-nESI-MS/MS
Peptide samples (40 μL) were injected into a Thermo Scientific LTQ linear IT mass spectrometer using a Michrom Paradigm AS1 auto-sampler coupled to a Paradigm MS4 HPLC (Michrom BioResources, Auburn, CA). The peptide solution was automatically loaded at a flow rate of 0.5 μL/min across a Paradigm Platinum Peptide Nanotrap (Michrom BioResources) and onto a 150 mm× 0.099 mm capillary column (Polymicro Technologies, L.L.C., Phoenix, AZ) packed in house using a 5 μm, 100-Å pore size Magic C18 AQ stationary phase (Michrom BioResources). The mobile phases A, B, and C were 2% ACN in 0.1% formic acid, 98% ACN in 0.1% formic acid, and 5% ACN in 0.1% formic acid, respectively, all in HPLC-grade water. Buffer C was used to load the sample, and the gradient elution profile was as follows: 5% B (95% A) for 10 min; 5–55% B (95–45% A) for 30 min; 55–80% B (45–20% A) for 5 min; and 80–5% B (20–95% A) for 10 min. The data were collected in a “Triple-Play” (MS scan, Zoom scan, and MS/MS scan) mode using nanospray ionization (nESI) with a normalized collision energy of 35%.
2.6 Bioinformatic analysis of identified proteins
The acquired MS data were searched against a FASTA format pig (Sus scrofa) database assembled in-house using gene annotations publicly available from PIR (Protein Information Resource, http://pir.georgetown.edu/) using the SEQUEST (v. 28 rev. 12) program in Bioworks (v. 3.3). General parameters were set as follows: peptide tolerance 2.0 amu, fragment ion tolerance 1.0 amu, enzyme limits set as “fully enzymatic – cleaves at both ends” and missed cleavage sites set at 2. The searched peptides and proteins were subjected to the validation processes PeptideProphet [20] and ProteinProphet [21] in the Trans-Proteomic Pipeline (TPP, v. 3.3.0) (http://tools.proteomecenter.org/software.php), and only those proteins with greater than 90% confidence (containing multiple peptides with greater than 90% confidence) were considered positive identifications.
2.7 Label-free quantitative MS
The following was carried by Monarch LifeSciences (Indianapolis, IN). LDL proteins (20 μg) were denatured with an 8 M urea, 10 mM DTT solution and spiked with chicken lysozyme. Samples were reduced and alkylated by triethylphosphine and iodoethanol and tryptically digested as previously described [22]. Peptides were prepared and subjected to LC-MS/MS analysis as previously described [23]. All samples were analyzed in duplicate from all four groups (n = 4) for 32 HPLC injections. Peptides were loaded and separated prior to MS analysis on a C18 microbore column (Zorbax 300SB-C18, 1 mm × 5 cm) in the Surveyor HPLC system and eluted off with linear gradient from 5 to 45% ACN over 120 min (50 μL/min). An electrospray interface was used with the LTQ mass spectrometer in the Triple Play (MS scan, zoom scan, and MS/MS scan) mode to collect data, which were then analyzed by a proprietary algorithm developed by Higgs et al. [23]. Pig and human databases were downloaded from the NCBI (National Center for Biotechnology Information) and IPI (International Protein Index) public domains and concatenated, and redundant entries were removed. Because pig databases are incomplete, a human database was incorporated to provide coverage of the genome not yet sequenced in the pig. The success of this depends on the similarity of gene/protein sequences, which is yet to be determined. Nonetheless, searches against this pig/human database were executed using both the X!Tandem and SEQUEST programs. Protein quantification was completed using a proprietary protein quantification algorithm licensed from Eli Lilly & Company [23]. Briefly, all extracted ion chromatograms (XIC) from raw data files were aligned by retention time. Criteria for quantification included, matched parent ion, charge state, daughter ions (MS/MS data), and retention time (within a 1-min window) in aligned peaks. The area-under-the-curve (AUC) could then be calculated for each aligned peak from each sample, quantile-normalized, and relative abundance compared across groups. All peak intensities were transformed to a log2 scale before quantile normalization [20]. Quantile normalization was included to produce peptide intensity histograms of the same scale, location and shape. This helped to remove trends introduced by sample handling and preparation, differences in total protein, and variation in instrument sensitivity over multiple sample runs. Quantile normalized log2 intensities peptides with the same protein identification were averaged to obtain log2 protein intensities. The log2 protein intensity was then analyzed by an ANOVA statistical model for each protein:
Group effect refers to the effect caused by the experimental conditions or treatments being evaluated. Sample effect represents the random effects from individual biological samples. It also includes random effects from sample preparation. The replicate effect refers to the random effects from replicate injections of the same sample. All of the injections were randomized and the instrument was operated by the same operator for this study. The inverse log2 of each sample mean was calculated to determine the fold change between samples.
2.8 1-DE and Western blotting
Aliquots of LDL samples from all four groups (n = 3) containing 20 μg (~50–100 μL) of protein were concentrated by filtration using a Millipore ultrafree-0.5 centrifugal filter device then washed with several times with a standard 1-DE Laemmli sample buffer [21] (100 μL) followed by resuspension in NuPAGE® LDS Sample Buffer and NuPAGE® Reducing Agent per the manufacturer’s instructions (Invitrogen, Carlsbad, CA.). Samples were heated for 10 min at 70°C and loaded onto a NuPAGE® Novex® 3–8% Tris-Acetate Midi Gel (1.0 mm×12+2 well), for apolipoprotein B, (Invitrogen) in a Bio-Rad Criterion™ Cell using Midi Gel Adapters (Invitrogen) with HiMark™ Prestained HMW Protein Standard (Invitrogen) in the outer wells. Gels were run using a Power Pac HC (Bio-Rad) set at 200 V on ice until the dye front ran off the bottom of the gel (~80 min) in NuPAGE® Tris-Acetate SDS Running Buffer along with NuPAGE® Antioxidant (Invitrogen) in the upper buffer chamber. For adiponectin, NuPAGE® Novex® 4–20% Tris-Glycine Midi Gels were used with NuPAGE® Tris-Glycine SDS Running buffer.
Upon completion of 1-DE, the gel was equilibrated in transfer buffer (49 mM TRIS, 39 mM Glycine, 0.04% SDS, 20% MeOH, pH 9.2) for 30 min at room temperature. Proteins were transferred to a PVDF membrane in a Transblot® SD Semi-Dry Transfer Cell (Bio-Rad) at 25 V for 90 min. The PVDF membrane was blocked with 0.3% PBST for 2 h using 3–5 washes then incubated overnight with a primary sheep polyclonal anti-apolipoprotein B antibody (1:20 000) in 0.1% PBST (Abcam Cambridge, MA). The unbound antibody was rinsed off with 5–6 changes of 0.1% PBST for 30 min. The secondary antibody (AP-conjugated polyclonal rabbit anti-sheep IgG) (Abcam), was diluted 1:20 000 in 0.1% PBST and incubated for 2 h. The unbound antibody was rinsed off with 5–6 changes of 0.1% PBST for 30 min. Membranes were developed in NBT/BCIP (Pierce, Rockford, IL) for ~5 min at room temperature then air-dried and scanned at 95.3 μm/pixel resolution using a GS-800 Calibrated Densitometer in the reflective mode. Integrated density of the stained bands was measured using the Scion Image program (version ALPHA 4.0.3.2, Scion Corporation, Frederick, MD.) and comparisons calculated using one-way ANOVA. For adiponectin, the PVDF membrane was blocked with 5% NFDM and 0.1% PBST for 1 h and then incubated overnight at 4°C with a primary polyclonal rabbit anti-pig adiponectin antibody, (1:1000). The secondary antibody (HRP-conjugated goat anti-rabbit IgG, Upstate/Millipore), diluted 1:5000 in 3% NFDM and 0.1% PBST was incubated for 2 h. Membranes were incubated with Pierce ECL Western Blotting Substrate (Pierce) for ~5 min, exposed for 3 min to Classic Blue Autoradiography Film BX (Molecular Technologies, St. Louis, MO), and developed using Hope micromax film processor. Because LDL lacks an internal standard that could be utilized for normalization, loading was controlled for initially by precise determination of protein concentration.
2.9 Selected reaction monitoring
LDL proteins (100 μg) were reduced and alkylated by triethylphosphine and iodoethanol as described by Hale et al. [24]. Briefly, each sample was spiked with 0.5 μg chicken lysozyme as an internal standard. The reduction/alkylation cocktail was added to the protein solution and incubated at 37°C for 90 min, then dried by Speedvac and reconstituted with 100 μL of 100 mM NH4HCO3 at pH 8.0. A 150-μL aliquot of a 20 μg/mL trypsin solution was added to the sample and incubated at 37°C for 3 h after which another 150 μL of trypsin was added and the solution incubated at 37°C overnight.
Specific tryptic peptides were quantified on a relative basis by LTQ linear IT MS using the SRM mode coupled to a Surveyor auto-sampler and MS HPLC system (Thermo-Fisher Scientific). Tryptic peptides were injected onto the C18 microbore reversed-phase column (Zorbax SB-C18, 1.0 mm × 150 mm) at a flow rate of 50 μL/min. The mobile phases A, B, and C were 0.1% formic acid, 50% ACN in 0.1% formic acid, and 80% ACN in 0.1% formic acid, respectively, in HPLC-grade water. The gradient elution profile was as follows: 10% B (90% A) for 5 min; 10–95% B (90–5% A) for 120 min; 100% C for 5 min; and 10% B (90% A) for 12 min. The data were collected with the electrospray interface using a normalized collision energy of 35%. Samples from all four groups (n = 4) were injected in triplicate for 48 HPLC injections.
Known peptides of chicken lysozyme (Swiss-Prot no. P00698), pig apolipoprotein B (Swiss-Prot no. Q29021), apolipoprotein A1 (Swiss-Prot no. P18648), apolipoprotein C-III (Swiss-Prot no. P27917), and pig apolipoprotein E (Swiss-Prot no. P18650) were targeted and quantified. The SRM setting used to identify the selected peptides was as follows: chicken lysozyme (peptide: 2_NTDGSTDY-GILQINSR; precursor ion = 877.78; product ions = 1063.59, 1178.62 and 1279.67), apolipoprotein C-III (peptide 1: 2_TAQDALTSVKESEVAQQAR; precursor ion = 1016.90; product ions = 1244.66, 1331.69 and 1432.74; peptide 2: 2_GWVTDSISSLKDYWSTFK; precursor ion = 1060.83; product ions = 1361.67, 1474.76 and 1561.79), apolipoprotein A1 (peptide: 2_LLDNWDSLGSTFTK; precursor ion = 799.38; product ions = 1141.55, 1255.60 and 1370.62), apolipoprotein B (peptide: 2_LLAQEDQGDFQ-GLR; precursor ion = 795.75; product ions = 920.46, 1035.49 and 1164.53), and apolipoprotein E (peptide: 2_VTQELTELIEESMKEVK; precursor ion = 1004.01; product ions = 1435.74).
3 Results
3.1 2-DE
LDL proteins (480 μg) were separated simultaneously, in parallel, using high-resolution, large-format 2-DE. An average of 585 protein spots were detected in each gel, with an average 73.7% of each gels’ spots being matched to the master gel pattern. As a preliminary statistical analysis to determine differential expression candidates, Student’s t-test was performed via PDQuest™ (p <0.05). The union of these six comparisons totaled 171 unique protein spots that were then subjected to one-way ANOVA. Those that were statistically significant after one-way ANOVA and had sufficient staining intensity (>200 ppm), were cut, tryptically digested, and peptides analyzed by MS/MS. High-confidence identification required multiple peptides with ≥90% confidence after validation by PeptideProphet and ProteinProphet via the Transproteomic Pipeline (TPP), and these are listed in Table 1 and illustrated in Fig. 1, with their corresponding coordinate positions in the gel pattern. If more than two proteins in a charge train were identified with high confidence, it was assumed the pI microheterogeneities were PTM of the same protein. Relative quantification by 2-DE revealed a significant increase in apolipoproteins E, A-I, and C-III with exercise (Figs. 2A, 3A, and 4A, respectively). A significant increase was observed in all three isoforms (alpha, beta, and gamma) of fibrinogen in all three treatment groups, with the exception of beta fibrinogen in H vs. control (Fig. 5). An atherogenic diet was the common variable in these three treatment groups (H, DD, and DDX). In this regard, the 2-DE data are consistent with the LFQMS results also illustrated in Fig. 5.
Table 1.
Differentially expressed protein spots detected by 2-DE and identified by LC-MS/MS
| Swiss-Prot entry | Protein name | TPP confidence | Number of peptides | Number of spots | Theoretical MW (kDa) | Theoretical pI | Percent coverage |
|---|---|---|---|---|---|---|---|
| P18648 | Apolipoprotein A-I precursor [Sus scrofa] | 1.0000 | 24 | 3 | 30 | 5.5 | 46.4 |
| P27917 | Apolipoprotein C-III precursor [Sus scrofa] | 1.0000 | 3 | 1 | 10 | 4.8 | 19.8 |
| P18650 | Apolipoprotein E precursor [Sus scrofa] | 1.0000 | 16 | 4 | 37 | 5.6 | 21.5 |
| Q28936 | Fibrinogen A-alpha-chain (Fragment) [Sus scrofa] | 1.0000 | 22 | 13 | 47 | 6.6 | 30.1 |
| P14477 | Fibrinogen beta chain (Fragment) [Sus scrofa] | 0.9999 | 2 | 2 | 2.2 | 4.3 | 68.4 |
| Q6R6M8 | Fibrinogen gamma polypeptide (Fragment) [Sus scrofa] | 1.0000 | 7 | 9 | 10 | 4.4 | 45.7 |
| P01846 | Ig lambda chain C region | 1.0000 | 7 | 5 | 11 | 6.8 | 59 |
Significant differences were determined by one-way ANOVA, proteins identified by SEQUEST and validated using the transproteomic pipeline (TPP) via PeptideProphet and ProteinProphet; listed from the left are the Swiss-Prot primary accession number, the protein name, percent confidence, the highest number of peptides sequenced of all the protein spots identified as that particular protein, the number of spots identified as that particular protein, the theoretical MW of that Swiss-Prot protein, the theoretical pI of that Swiss-Prot protein, and the percent of the protein’s sequence that was covered when the number of peptides were as indicated.
Figure 1.

Proteins identified with high confidence via LC-NSI-MS/MS. A representative large format 2-DE gel of Yucatan pig LDL stained with colloidal CBB with groups of protein spots highlighted with their corresponding protein identity as listed in Table 1. This particular gel contains DDX pig LDL. Representative images from all four groups can be found in the Supporting Information. Axes were calibrated based on a high MW protein standard (HiMark™, Invitrogen, Carlsbad, CA.) and using known pI values given as percent distance from the acidic end by the IPG strip manufacturer (Bio-Rad, Richmond, CA).
Figure 2.

Apolipoprotein E in LDL. (Left) The 2-DE analysis (n = 5) revealed a significant increase in apolipoprotein E in LDL with exercise treatment compared to control and diabetic dyslipidemic groups (C and DD). The F-ratio was highest in the 2-DE experiment. (Middle) LFQMS analysis (n = 4) confirmed the significant increase in apolipoprotein E in exercised pigs compared to C and DD groups. (Right) SRM (n = 4) revealed no significant alterations in apolipoprotein E abundance with any treatment groups, although the expression profile resembled that in 2-DE and LFQMS. * Denotes a significant difference compared to control animals; †denotes a significant difference compared to diabetic dyslipidemic animals (p ≤0.05).
Figure 3.

Apolipoprotein A-I in LDL. (Left) The 2-DE analysis (n = 5) revealed a significant increase in apolipoprotein A-I in LDL with exercise treatment compared to all other treatment groups (C, H, and DD). The F-ratio was highest in the 2-DE experiment. (Middle) LFQMS analysis (n = 4) confirmed the significant increase in apolipoprotein A-I in exercised pigs compared to C and DD groups, but not compared to H. (Right) SRM (n = 4) confirmed the significant increase in apolipoprotein A-I in exercised pigs compared to all treatment groups (C, H, and DD). The expression profiles were similar with all three methods used. * Denotes a significant difference compared to control (C) animals; # denotes a significant difference from hyperlipidemic (H) animals; †denotes a significant difference compared to diabetic dyslipidemic (DD) animals (p ≤0.01).
Figure 4.

Apolipoprotein C-III in LDL. (Left) The 2-DE analysis (n = 5) revealed a significant increase in apolipoprotein C-III in LDL with exercise treatment compared to all other treatment groups (C, H, and DD). The F-ratio was highest in the 2-DE experiment. (Middle) LFQMS analysis (n = 4) and SRM (Right) revealed no significant increases in apolipoprotein C-III with any treatment group. * Denotes a significant difference compared to control (C) animals; # denotes a significant difference from hyperlipidemic (H) animals; †denotes a significant difference compared to diabetic dyslipidemic (DD) animals (p ≤0.01).
Figure 5.
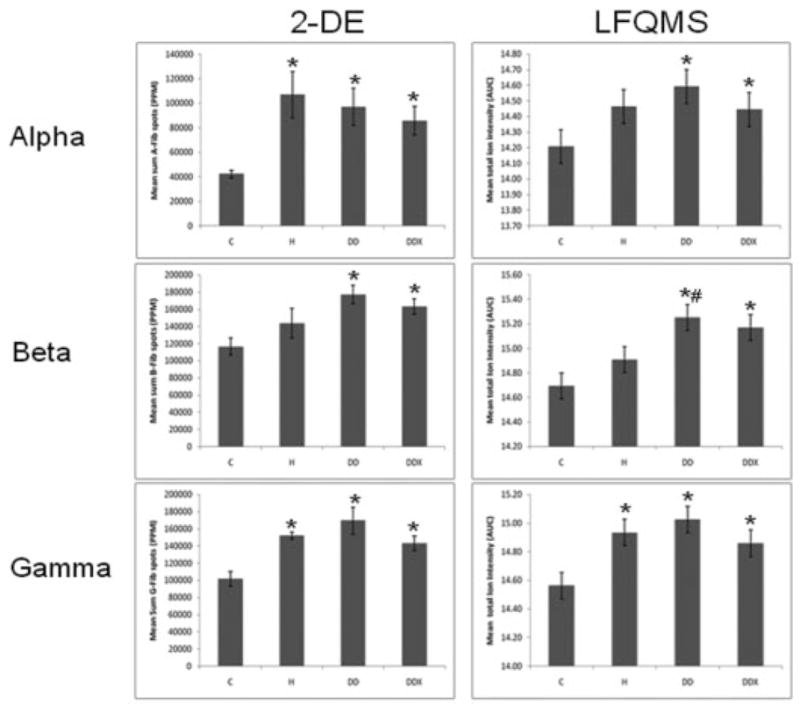
Fibrinogen in LDL. (Left) Results of 2-DE analysis (n = 5) revealed a significant increase in all three iso-forms of LDL Fibrinogen with all three treatment groups where the common variable is an atherogenic diet compared to control (C). (Right) Results of LFQMS analysis (n = 4) revealed similar trends in all three Fibrinogen isoforms as in the 2-DE analysis. * Denotes a significant difference compared to control (C) animals; # denotes a significant difference from hyperlipidemic (H) animals (p ≤0.05).
Linear regression analyses were performed using Spot-fire® to determine which protein spots, if any, were significantly correlated to atherosclerotic disease (percent atheroma, or coronary artery segments with intimal thickening). There were 79 protein spots associated with percent segments atheroma (p <0.01), and 25 of these were differentially expressed across groups. All 79 had an F-ratio of at least 7.9 and a correlation coefficient (R2) of at least 0.377, with the highest being p <0.00000001, F-ratio = 74.5, and R2 = 0.85, although it was not identified successfully. Nine of these protein spots were identified as the following three proteins: apolipoprotein E, alpha fibrinogen, and immunoglobulin lambda chain. To compare these proteins with traditional risk factors in terms of prediction of disease, linear regression analyses were performed for total cholesterol, total triglycerides, LDL cholesterol, and LDL/HDL cholesterol ratio versus percent segments atheroma (Fig. 6). Only total cholesterol and LDL-C were significantly correlated with atheroma (p <0.05). An apolipoprotein E charge train is shown in the inset of Fig. 6E. It is important to note that the two spots with the most acidic pI (most leftward) were strongly correlated with disease (p = 0.0001), whereas the presumably unmodified apolipoprotein E (far right) did not correlate with coronary artery intimal thickening.
Figure 6.

Linear regression analyses of traditional risk factors for atherosclerosis and atheroma. Traditional risk factors have lower F-ratios and correlation coefficients than many proteins associated with LDL, including acidic forms of apolipoprotein E. Therefore, acidic apolipoprotein E in LDL may be a superior prognostic indicator of atherosclerotic disease.
3.2 Label-free quantitative MS
LDL proteins (20 μg) were digested with trypsin and the resultant peptides analyzed serially via LC-MS/MS. Proteins were identified using SEQUEST and X!Tandem, while protein quantification was carried out based on total ion chromatograms using the proprietary technology of Monarch LifeSciences. In total, 434 proteins were identified and 56 qualified as priority 1, those identified with multiple high confidence peptides (Table 2). The 56 ID corresponded to 31 unique proteins, 18 of which were altered significantly (p <0.05) and are listed in Table 3.
Table 2.
Label-free quantitative MS protein identification results
| Protein priority | Peptide ID confidence | Multiple sequences | Median number of sequences | Number of proteins |
|---|---|---|---|---|
| 1 | HIGH | YES | 7 | 56 |
| 2 | HIGH | NO | 1 | 149 |
| 3 | MODERATE | YES | 2 | 11 |
| 4 | MODERATE | NO | 1 | 218 |
| Overall | 1 | 434 |
Proteins identified by LC-MS/MS were assigned to one of four categories based on the confidence in their identity, with priority 1 the highest confidence and priority 4 the lowest. To be categorized as priority 1, peptide ID confidence must be high (>90%) and multiple unique peptide sequences (median number per group listed) must be determined. Priority 4 requires a single peptide with ID confidence (>70%). Altogether, 434 proteins were identified. (Statistical summary provided by INCAPS, K. Bemis).
Table 3.
High-confidence protein identifications determined by MS/MS during LFQMS
| Protein ID | Annotation | Best sequence | Max FC | p min | Number of sequences |
|---|---|---|---|---|---|
| IPI00020019 | Adiponectin [Sus scrofa] | AVLFTYDQYQDK | 2.0 | 0.001 | 3 |
| 51235682 | Albumin [Sus scrofa] | LSCAEDYLSLVLNR | 1.2 | NS | 15 |
| IPI00431656 | Alpha-1-antichymotrypsin 1 [Sus scrofa] | MAAVEAKLLPETLR | 1.6 | 0.002 | 2 |
| 9968807 | Alpha-1-antichymotrypsin 3 [Sus scrofa] | VFTHEADLSGVTGDNK | 1.2 | NS | 2 |
| IPI00478003.1 | Alpha-2-macroglobulin precursor | QFSFPLSSEPFQGSYK | 1.6 | 0.02 | 33 |
| IPI00015180.1 | Apical-like protein | ELIESISR | 1.2 | NS | 2 |
| IPI00021841 | Apolipoprotein A-I [Sus scrofa] | FEALKEGGGSLAEYQAK | 2.1 | 0.007 | 14 |
| 2695742 | Apolipoprotein A-IV [Sus scrofa] | LGEVNTYTEDLQK | 1.2 | NS | 10 |
| IPI00022229.1 | Apolipoprotein B-100 precursor | NLTDFAEQYSIQDWAK | 1.2 | NS | 24 |
| 50657386 | Apolipoprotein C-III [Sus scrofa] | FTDFWDYTPKPEPS | 1.7 | NS | 17 |
| IPI00006662.1 | Apolipoprotein D precursor | CPNPPVQENFDVNK | 1.7 | 0.01 | 2 |
| 2388609 | Apolipoprotein-E [Sus scrofa] | WVQSLSDQVQEELLSTK | 1.8 | 0.02 | 12 |
| 47522716 | Apolipoprotein R [Sus scrofa] | CDEGYTLVGEDR | 1.3 | NS | 1 |
| 51095266 | Apolipoprotein N [Sus scrofa] | LPSTELVR | 1.4 | NS | 1 |
| IPI00291262 | Clusterin [Sus scrofa] (Apolipoprotein J) | LFDSYPITLIIPQEVSDPK | 1.5 | 0.004 | 4 |
| 21464594 | Complement regulator factor H [Sus scrofa] | EGFYPEIQGNVAR | 1.5 | 0.03 | 5 |
| IPI00643948 | Complement C1qB [Sus scrofa] | FDQVITNANENYESR | 1.5 | 0.002 | 2 |
| 1236646 | Ch4 and secrete domains of swine IgM [Sus scrofa] | VVSGVSTGPVETLQSSPVTYR | 1.3 | NS | 43 |
| 1304179 | Fibrinogen A-alpha-chain [Sus scrofa] | SGPGDTTPSNPDWGTFK | 1.3 | 0.03 | 34 |
| IPI00298497 | Fibrinogen Beta | YCGLPGEYWLGNDK | 1.5 | 0.003 | 11 |
| IPI00021891 | Fibrinogen Gamma [Sus scrofa] | AIQISYNPEDLSKPDR | 1.4 | 0.004 | 8 |
| IPI00022418 | Fibronectin [Sus scrofa] | IYLYTLNDNAR | 1.4 | 0.002 | 22 |
| 47523126 | Ficolin [Sus scrofa] | YLGGSHGSFANGVNWSSGK | 1.5 | 0.05 | 3 |
| IPI00010463.2 | GTPBP1 protein | SMAEQIEADVILLRER | 1.5 | 0.07 | 2 |
| 47522826 | Haptoglobin alpha 1S [Sus scrofa] | TAEYGVYVR | 1.3 | NS | 2 |
| IPI00472345 | IgG heavy chain [Sus scrofa] | DNSQNTAYLQMNSLR | 1.3 | 0.003 | 4 |
| 55167422 | Immunoglobulin alpha heavy chain [Sus scrofa] | YLVWESLPEPGQAIPTYAVTSVLR | 1.7 | 0.04 | 10 |
| 33150403 | Immunoglobulin delta heavy chain [Sus scrofa] | VLLPSVSIPQDPEAFLVCEVQHPSGTK | 1.3 | NS | 8 |
| 41323535 | Immunoglobulin kappa variable region [Sus scrofa] | AIVLTQTPLSLSVSPGEPASISCR | 1.4 | NS | 5 |
| 253878 | Immunoglobulin mu heavy chain [Sus scrofa] | GEASVCVEDWESGDR | 1.3 | NS | 35 |
| 1915956 | Inter-alpha-inhibitor heavy-chain 1 [Sus scrofa] | IYEDHDAAQQLQGFYDQVANPLLK | 1.6 | 0.02 | 2 |
| IPI00179357.1 | Titin | LKPPPPKVPEEPK | 1.2 | NS | 2 |
| 833800 | Transferrin [Sus scrofa] | SAGWIIPMGLLYDQLPEPR | 1.1 | NS | 7 |
All proteins listed are priority 1 with the exception of apolipoproteins R and N, which were priority 2. Identification results are indicated by the accession number (Protein ID), protein name or annotation, amino acid sequence of the peptide with highest ID confidence (Best Sequence), and the number of distinct amino acid sequences for this protein. Relative quantification results are indicated by the maximum absolute fold change observed between all two-group comparisons and the minimum p-value among all two-group comparisons when p≤0.05. NS indicates not significant the p-value was higher than 0.05.
Several differential expression patterns detected by LFQMS are shown in Figs. 2B, 3B, 4B, 5B, 7B, 8B, and 9 for apolipoproteins E, A-I, C-III, fibrinogen, apolipoprotein B, and adiponectin, alpha-2-macroblobulin, complement C1q, ficolin, and apolipoprotein J, respectively. LFQMS revealed a significant increase in apolipoproteins E and A-I in exercised animals compared to control and diabetic dyslipidemic animals. The expression profiles of apolipoproteins E, A-I, C-III, and B were also determined using SRM. The relative changes in expression of apolipoprotein B were confirmed by Western blot. A similar pattern was observed for apolipoprotein E by 2DE, LFQMS, and SRM methods. Apolipoprotein E was increased in hyperlipidemic pigs but not in diabetic dyslipidemic pigs. Exercise increased apolipoprotein E in diabetic dyslipidemic pigs. There were significant increases in all three isoforms (alpha, beta, and gamma) of fibrinogen with diabetic dyslipidemia and exercise treatment groups, as well as H in beta-fibrinogen, compared to control, where an atherogenic diet was the common variable in three treatment groups (H, DD, and DDX). Importantly, the expression profiles are consistent with the 2-DE results. There was a significant decrease in adiponectin in all atherogenic-diet fed groups compared to control; the decrease was consistent with Western blot analysis, where the differences showed trends, but were not statistically significant.
Figure 7.

Apolipoprotein B in LDL. (Left) Western blot analysis (n = 3) revealed no significant changes in apolipoprotein B in LDL diseased pigs, although there was a wide variation in apolipoprotein B content in exercised pigs. The 500-kDa bands reacting with the anti-apolipoprotein B antibody are shown at the top of the bar graph. (Middle) LFQMS analysis (n = 4) revealed no significant changes in apolipoprotein B with any treatment groups. (Right) SRM (n = 4) revealed an increase in apolipoprotein B with exercise (DDX) compared to control and diabetic dyslipidemic groups (C and DD). However, there was substantial variation in apolipoprotein B level in exercised pigs using both Western blot and SRM analysis methods. The F-ratio was highest in the SRM experiment. Apolipoprotein B is too large to enter 2-D gels. * Denotes a significant difference compared to control (C) animals; †denotes a significant difference compared to diabetic dyslipidemic (DD) animals (p ≤0.01).
Figure 8.

Adiponectin in LDL. (Left) Results of a Western blot analysis (n = 3) revealed no significant decreases of adiponectin in LDL with any treatment groups. The anti-adiponectin antibody reacted with two bands as shown at the top of the bar graph. (Right) Results of LFQMS analysis (n = 4) revealed significant decreases in adiponectin with in all groups on an atherogenic diet (H, DD, DDX) compared to control. * Denotes a significant difference compared to control (C) animals (p ≤0.05).
Figure 9.

Effects of diabetic dyslipidemia and exercise. (A) Effects of diabetic dyslipidemia: relative quantification of complement C1q in LDL (LFQMS, n = 4) revealed a significant increase in complement C1q with DD and DDX compared to control and hyperlipidemic animals. Relative quantification of alpha-2-macroglobulin in LDL (LFQMS, n = 4) revealed a significant decrease in alpha-2-macroglobulin with DD and DDX compared to control animals. (B) Effects of exercise: relative quantification of ficolin in LDL (LFQMS, n = 4) revealed a significant decrease in ficolin with exercise, DDX vs. DD. Relative quantification of apolipoproteins J in LDL (LFQMS, n = 4) revealed a significant increase in apolipoprotein J with exercise, DDX vs. C and H animals. * Denotes a significant difference compared to control (C) animals; # denotes a significant difference from hyperlipidemic (H) animals; †denotes a significant difference compared to diabetic dyslipidemic (DD) animals (p ≤0.05).
4 Discussion
A primary objective of this study was to characterize proteins associated with the LDL particle, i.e. the low-density lipoproteome. This was accomplished via 2-DE/LC-MS/MS and LFQMS analyses, the results of which are listed in Tables 1 and 3, respectively. We sought to detect the alterations in this proteome that occur in an atherogenic state. To this end, we tested the effects of two important risk factors for CHD, hypercholesterolemia and diabetic dyslipidemia, and sought to determine the effects of exercise on this proteome. First, it is important to note that LDL was isolated from pigs after 20 weeks, still in the early stages of atherosclerosis. Therefore, alterations in LDL-associated proteins observed in this study likely preceded the development of severe disease and may be involved in the initiating stages of disease.
Recent lipoprotein research has revealed the presence of more proteins than previously known in both LDL and HDL fractions, even when using harsh techniques such as ultracentrifugation. In 2005, Karlsson et al. [1] reported finding 11 proteins in an ultracentrifuged LDL sample. Stahlman et al. [25] compared the protein profile of lipoproteins isolated using ultracentrifugation in either KBr or D2O/sucrose buffers and determined that more total protein and more MS protein peaks were observed in LDL isolated with D2O/sucrose. Although individual proteins were not fully identified and quantified, their 2-DE gel patterns for LDL were similar to our 2-DE gel patterns. Our results from 2-DE analysis of FPLC-purified LDL revealed almost 600 protein spots per gel, corresponding to a much smaller number of actual proteins. Many spots are actually micro-heterogeneities, isoforms, and fragments of far fewer constituent proteins. In fact, only 7 differentially expressed proteins were identified with high confidence from 2-DE as shown in Fig. 2 and listed in Table 1, corresponding to approximately 100 protein spots. If this ratio represents the entire sample, then 600 protein spots correspond to only 42 unique proteins. Nevertheless, there are clearly more than 7 proteins in this sample, as verified by LFQMS, but the exact number requires further experimentation. It is also important to note that the pig database is incomplete, and this explains why several protein spots were not identified successfully. These proteins had sufficient staining intensity and were cut, tryptically digested, and analyzed by LC-MS/MS multiple times yielding high-quality spectra, yet no high-confidence identifications resulted. This suggests that those proteins are products of genes not yet sequenced or added to the publicly available Sus scrofa databases (PIR, NCBInr, etc.). When searched against the human database, no significant matches were made, emphasizing a lack of sequence homology for these proteins between Sus scrofa and Homo sapiens. In the LFQMS experiment, 434 proteins were identified; however, only 56 met priority 1 criteria. Indeed, many experts would view any protein identification outside of priority 1 (>90% confidence with multiple unique peptides) as questionable [26]. Of the 56 priority 1 proteins identified, many of these were the same protein with different gene annotations, leaving only 31 unique, high-confidence protein ID (Table 3), which is similar to the number estimated by 2-DE (42 unique proteins).
Most of the proteins identified in our samples have not been associated traditionally with LDL, but several of them have been shown either directly or indirectly to be non-covalently associated with the LDL particle. Karlsson et al. [1] were the first to characterize the low-density lipoproteome using ultracentrifugation to isolate LDL. Using SEC, they also identified several proteins observed here, including alpha, beta, and gamma fibrinogen, alpha-2-macroglobulin, fibronectin, and albumin. Geiss et al. [27] used membrane differential filtration to remove lipoproteins from plasma on the basis of particle size. In so doing, they depleted the plasma of LDL as well as most of the plasma’s fibrinogen and IgM and a significant amount of IgG and alpha-2-macro-globulin, which we identified to be in LDL, and thus implying an association of these proteins with LDL. Another study identified the soluble proteins IgA, IgG, IgM, alpha-2-macroglobulin, fibrinogen, albumin, and transferrin as well as LDL in human aortic atherosclerotic intimal tissue [28], all of which were identified in our study to be associated with LDL (Table 3).
4.1 Effects of an atherogenic diet
Adiponectin is a protein hormone that is secreted directly into the blood exclusively by adipocytes and modulates a number of processes including glucose regulation and fatty acid metabolism [29]. It has well-known direct anti-diabetic, anti-inflammatory, and anti-atherogenic properties, including suppressing cellular superoxide generation, inhibiting monocyte adhesion to endothelial cells by preventing adhesion molecule expression, and decreasing macrophage scavenger receptor expression [30]. Marso et al. [31] concluded that low adiponectin levels are associated with atherogenic lipoproteins, increased plaque volume, and intimal thickening in humans, suggesting it may play an anti-atherogenic role early in lesion development. We observed a decrease in adiponectin in H, DD, and DDX animals compared to C animals in the LFQMS experiment (Fig. 9), but no significant differences between H, DD, and DDX animals, suggesting the alteration was the result of an atherogenic diet, the common variable among all three treatment groups. Adiponectin was not identified via 2-DE, emphasizing the importance of using multiple, complementary approaches. To our knowledge, it has not been identified previously in LDL, although one report suggested the association after finding adiponectin in an apolipoprotein B immunoprecipitate [32]. Having less adiponectin associated with LDL would increase the propensity to develop atherosclerosis, especially considering that LDL is known to be retained in subendothelial spaces where adiponectin could exert its anti-atherogenic effects. Thus, we hypothesize that decreased adiponectin in LDL increases the atherogenic potential of the particle.
All three isoforms of fibrinogen had similar expression profiles as determined by both 2-DE and LFQMS. There was an increase in alpha, beta, and gamma fibrinogen mean abundance in the H, DD, and DDX pigs compared to C, a significant change apparently elicited by an atherogenic diet. Fibrinogen has been identified directly in LDL previously using size exclusion chromatography [1] and implicated to be associated with LDL in other reports [27], but to our knowledge this is the first report that fibrinogen in LDL increases with diabetic dyslipidemia. We also observed a significant correlation in one alpha fibrinogen spot’s abundance with percent atheroma (p <0.0005, R2 = 0.61). This particular protein spot had the most alkaline pI, presumably the unmodified form. Fibrinogen is a well-accepted independent risk factor for CVD [33], and its levels are positively correlated with LDL cholesterol in type 1 diabetics [34]. It is also known to contribute to blood viscosity and therefore shear stress, which is known to initiate sites of lesion formation. Further, it is interesting to speculate that increased fibrinogen in LDL may contribute to increased platelet aggregation in diabetic dyslipidemic swine [35].
4.2 Effects of diabetic dyslipidemia
We observed significant increases in the mean abundance of complement C1qB in LDL in DD and DDX compared to C and H animals (p <0.05, LFQMS). Complement C1q combines with C1s and C1r to form C1, the first component in the serum complement cascade of the innate immune system. Interestingly, A. Festa et al. [36] discovered an oxLDL immune complex in insulin-dependent diabetes mellitus (IDDM) patients that could interact with C1q and activate the complement cascade, eventually leading to phagocytosis by macrophage cells. In the case of LDL, this could contribute to foam cell formation. It is now well known that the complement component expression is up-regulated and the cascade is extensively activated in atherosclerotic plaques [37]. Therefore, we hypothesize that an increase in complement C1q in LDL of diabetic patients could potentially contribute to the increased incidence of atherosclerotic disease.
There was a decrease in alpha-2-macroglobulin (A2M) in the DD and DDX animals (Fig. 10) compared to control. A2M has been identified at least once before in an LDL sample isolated by size exclusion chromatography [1]. Interestingly, A2M is increased in the serum of diabetic patients [38–40] where it is thought to cause an increase in serum viscosity and shear stress, and is associated with dyslipoproteinemias and atherosclerosis [41]. A2M has also been identified in human atherosclerotic tissue [28] and is significantly associated with coronary atherosclerosis [42]. Geiss et al. [27] observed almost a 50% decrease in A2M when LDL was depleted by apheresis, indicating a substantial portion of plasma A2M may be associated with LDL. Thus, it is plausible that the increase in plasma A2M observed in diabetics could result from a decreased association of A2M with LDL, as observed here. In addition, A2M has been shown to interact directly and specifically with adiponectin [43] and apolipoprotein E, suggesting possible protein-protein interactions leading to its association with LDL.
Figure 10.
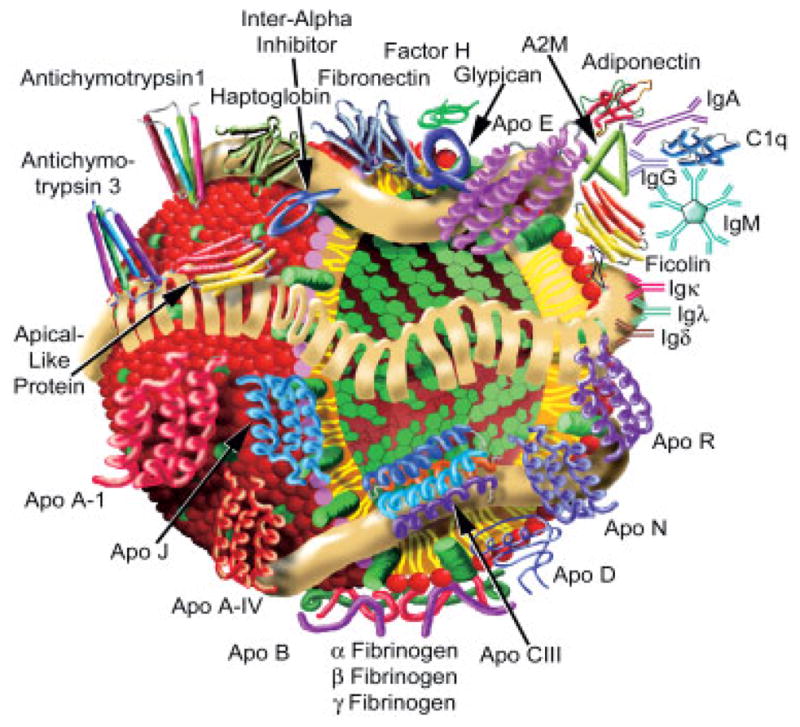
A hypothetical new model of LDL. Our current model of LDL isolated by gel filtration chromatography is a lipoprotein particle with a more complex and extensive proteome than previous models based on LDL isolated using high salt and high centrifugal force. Apolipoprotein B, with its amphipathic domains, forms a tight belt-like structure around the phospholipid surface and predominantly cholesteryl ester core. A constellation of other plasma proteins (Table 3) may interact with LDL transiently or for longer periods. Several of the interactions shown between peripheral proteins have been noted previously in other studies. This model of LDL serves to stimulate new experiments concerning the atherogenicity of LDL. Surface phospholipids: red spheres with yellow tails; surface free cholesterol: green barrels; core cholesteryl ester: green multi-rings.
4.3 Effects of exercise
LDL-associated apolipoproteins B, E, A-I, C-III, D, and J all increased with exercise. Apolipoprotein B and E facilitate the hepatic removal of cholesterol from the plasma, thus an up-regulation of these may facilitate this process and add to the cardioprotective effects of exercise. Apolipoprotein A-I’s function in LDL is unclear, but it is known to facilitate reverse cholesterol transport in HDL. Therefore, it may play some beneficial role in LDL as well. Apolipoprotein D may facilitate lipoprotein metabolism thus enhancing clearance of plasma LDL. The only protein in this expression profile that is unexpected is the up-regulation of apolipoprotein C-III with exercise because it is positively correlated with cardiovascular disease. However, the increase in apolipoprotein C-III may be responsible for the lack of an improvement in plasma lipids in the exercised pigs.
Also known as clusterin, apolipoprotein J has been associated previously with VLDL, the precursor particle of LDL, as well as LDL isolated by ultracentrifugation [1] and has an affinity for hydrophobic proteins and membranes. Apolipoprotein J has anti-inflammatory properties as indicated in studies of apolipoprotein J-deficient animals [44]. Atherosclerosis is an inflammatory disease as lesions often occur at sites of inflammation, and inflammatory cells are involved in lipid uptake. Exercise is known to be cardioprotective, reducing the incidence of CHD, possibly through its anti-inflammatory effects. Studies have demonstrated acute and chronic anti-inflammatory effects of exercise in animals and humans, including a reduced accumulation of macrophage cells involved in foam cell formation [45]. In addition to its anti-inflammatory properties, apolipoprotein J can induce cholesterol export from macrophage-foam cells [46], making it an unmistakable anti-atherogenic protein. Therefore, we hypothesize that the exercise induced up-regulation of apolipoprotein J is at least a partial mediator of the cardioprotective effects of exercise.
In addition to the strong effects of exercise on apolipoproteins B, E, A-I, C-III, J, and D, there was a significant exercise-induced alteration in ficolin. Ficolin is a serum protein that decreased significantly with exercise (DDX) compared to its control counterpart, DD, and there was no significant difference between C and DDX. A recent report by Faro et al. [47] showed that ficolin reacts with N-acetylglucosamine (GlcNAc) and acetylated LDL. It is also known that apolipoprotein B has defined points of GlcNAc modification [48]. Therefore, this finding of ficolin associated with LDL is reasonable. Ficolin is a known stimulator of the lectin pathway of complement activation, which has the same endpoint as complement C1q activation: macrophage uptake of the marked “pathogen.” Ficolin’s association with LDL could play a heretofore unknown role in atherogenesis. To our knowledge this is the first report of an exercise effect on ficolin expression and, certainly, in LDL-associated ficolin. We hypothesize that exercise partly exerts its anti-inflammatory, cardioprotective effects by reducing expression of ficolin in LDL.
4.4 Final remarks
Using multiple discovery-oriented proteomic techniques provides a powerful platform by which to perform exploratory semi-quantitative analyses. The work presented in this study corroborates and extends previous findings regarding the constituents of the low-density lipoproteome. Many of the proteins identified with high confidence here have support in the literature regarding their potential association with LDL. However, this appears to be the first report demonstrating their direct association with LDL for the vast majority identified. Thus, one of the most important conclusions from this work is that the low-density lipoproteome is more extensive than previously thought. This implies that the LDL particle is also more complex than previously thought, which is consistent with the latest findings regarding its function in physiology. A new hypothetical model of LDL based on our findings is illustrated in Fig. 10. The proteins presented can be found in Table 3 with the exception of Glypican, a priority 2 category protein identification. Glypican is a heparin-sulfate proteoglycan, which are known to bind apolipoprotein B [49] and has been shown to be able to bind lipoprotein particles in other model organisms [50]. The protein-protein interactions posed in this model have not been proven in LDL but are supported by the aforementioned references.
The alterations observed in the LDL proteome with an atherogenic diet and diabetic dyslipidemia appear to be novel, several of which may be involved in atherogenesis. Approximately half of those LDL-associated proteins identified with high confidence have known roles in either lipoprotein metabolism or inflammation, two processes important in atherogenesis. Their up- or down-regulation is consistent with facilitating disease rather than compensating for it, and exercise appears capable of attenuating several of these changes. However, the exact mechanisms by which diabetic dyslipidemia and exercise alter protein association with LDL are not yet understood. Future work is needed to investigate this and the many hypotheses generated by this study. The identification of additional proteins associated with LDL and changes in their LDL content with different risk factors will facilitate our understanding of the physiological function of LDL and will potentially lead to the identification of more specific biomarkers and potential therapeutic targets for atherosclerosis.
Acknowledgments
The authors would like to thank Dr. Michael E. Spurlock for providing the adiponectin antibody and assistance in optimizing the Western blot. The authors would also like to thank James Scherschel for his assistance with the Spotfire regression analysis. Finally, we are extremely grateful for James Byrd’s effort and artistry in constructing the LDL image in Fig. 10. This study was supported in part by PHS grants RR013223 and HL062552 to M. S., HL47586 to J. L. D., and AA015698 to F. A. W.
Abbreviations
- CHD
coronary heart disease
- FPLC
fast protein liquid chromatography
- LDL
low-density lipoprotein
- LFQMS
label-free quantitative MS
- SRM
selected reaction monitoring
Footnotes
The authors have declared no conflict of interest.
References
- 1.Karlsson H, Leanderson P, Tagesson C, Lindahl M. Lipoproteomics I: mapping of proteins in low-density lipoprotein using two-dimensional gel electrophoresis and mass spectrometry. Proteomics. 2005;5:551–565. doi: 10.1002/pmic.200300938. [DOI] [PubMed] [Google Scholar]
- 2.Stahlman M, Davidsson P, Kanmert I, Rosengren B, et al. Proteomics and lipids of lipoproteins isolated at low salt concentrations in D2O/sucrose or in KBr. J Lipid Res. 2008;49:481–490. doi: 10.1194/jlr.D700025-JLR200. [DOI] [PubMed] [Google Scholar]
- 3.Ordovas JM, Osgood D. Preparative isolation of plasma lipoproteins using fast protein liquid chromatography (FPLC) Methods Mol Biol. 1998;110:105–111. doi: 10.1385/1-59259-582-0:105. [DOI] [PubMed] [Google Scholar]
- 4.Stocker R, Keaney JF., Jr Role of oxidative modifications in atherosclerosis. Physiol Rev. 2004;84:1381–1478. doi: 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]
- 5.Hennekens CH, Buring JE, Manson JE, Stampfer M, et al. Lack of effect of long-term supplementation with beta carotene on the incidence of malignant neoplasms and cardiovascular disease. N Engl J Med. 1996;334:1145–1149. doi: 10.1056/NEJM199605023341801. [DOI] [PubMed] [Google Scholar]
- 6.Omenn GS, Goodman GE, Thornquist MD, Balmes J, et al. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. N Engl J Med. 1996;334:1150–1155. doi: 10.1056/NEJM199605023341802. [DOI] [PubMed] [Google Scholar]
- 7.Yusuf S, Dagenais G, Pogue J, Bosch J, Sleight P. Vitamin E supplementation and cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:154–160. doi: 10.1056/NEJM200001203420302. [DOI] [PubMed] [Google Scholar]
- 8.Bucala R. What is the effect of hyperglycemia on atherogenesis and can it be reversed by aminoguanidine? Diabetes Res Clin Pract. 1996;30(Suppl):123–130. doi: 10.1016/s0168-8227(96)80048-9. [DOI] [PubMed] [Google Scholar]
- 9.Blair SN, Kampert JB, Kohl HW, 3rd, Barlow CE, et al. Influences of cardiorespiratory fitness and other precursors on cardiovascular disease and all-cause mortality in men and women. JAMA. 1996;276:205–210. [PubMed] [Google Scholar]
- 10.Cohn JS, Rodriguez C, Jacques H, Tremblay M, Davignon J. Storage of human plasma samples leads to alterations in the lipoprotein distribution of apoC-III and apoE. J Lipid Res. 2004;45:1572–1579. doi: 10.1194/jlr.D300041-JLR200. [DOI] [PubMed] [Google Scholar]
- 11.Dixon JL, Shen S, Vuchetich JP, Wysocka E, et al. Increased atherosclerosis in diabetic dyslipidemic swine: protection by atorvastatin involves decreased VLDL triglycerides but minimal effects on the lipoprotein profile. J Lipid Res. 2002;43:1618–1629. doi: 10.1194/jlr.m200134-jlr200. [DOI] [PubMed] [Google Scholar]
- 12.Weng S, Zemany L, Standley KN, Novack DV, et al. Beta3 integrin deficiency promotes atherosclerosis and pulmonary inflammation in high-fat-fed, hyperlipidemic mice. Proc Natl Acad Sci USA. 2003;100:6730–6735. doi: 10.1073/pnas.1137612100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mokelke EA, Hu Q, Song M, Toro L, et al. Altered functional coupling of coronary K+ channels in diabetic dyslipidemic pigs is prevented by exercise. J Appl Physiol. 2003;95:1179–1193. doi: 10.1152/japplphysiol.00972.2002. [DOI] [PubMed] [Google Scholar]
- 14.Witzmann FA, Richardson MR. Two-dimensional gels for toxicological drug discovery applications. Expert Opin Drug Metab Toxicol. 2006;2:103–111. doi: 10.1517/17425255.2.1.103. [DOI] [PubMed] [Google Scholar]
- 15.Steiner G. The dyslipoproteinemias of diabetes. Atherosclerosis. 1994;110(Suppl):S27–33. doi: 10.1016/0021-9150(94)05373-q. [DOI] [PubMed] [Google Scholar]
- 16.Wamhoff BR, Dixon JL, Sturek M. Atorvastatin treatment prevents alterations in coronary smooth muscle nuclear Ca+ signaling in diabetic dyslipidemia. J Vasc Res. 2002;39:208–220. doi: 10.1159/000063686. [DOI] [PubMed] [Google Scholar]
- 17.Boullion RD, Mokelke EA, Wamhoff BR, Otis CR, et al. Porcine model of diabetic dyslipidemia: insulin and feed algorithms for mimicking diabetes mellitus in humans. Comp Med. 2003;53:42–52. [PubMed] [Google Scholar]
- 18.Dixon JL, Stoops JD, Parker JL, Laughlin MH, et al. Dyslipidemia and vascular dysfunction in diabetic pigs fed an atherogenic diet. Arterioscler Thromb Vasc Biol. 1999;19:2981–2992. doi: 10.1161/01.atv.19.12.2981. [DOI] [PubMed] [Google Scholar]
- 19.Neuhoff V, Arold N, Taube D, Ehrhardt W. Improved staining of proteins in polyacrylamide gels including iso-electric focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis. 1988;9:255–262. doi: 10.1002/elps.1150090603. [DOI] [PubMed] [Google Scholar]
- 20.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 21.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 22.Reed E. Platinum-DNA adduct, nucleotide excision repair and platinum based anti-cancer chemotherapy. Cancer Treat Rev. 1998;24:331–344. doi: 10.1016/s0305-7372(98)90056-1. [DOI] [PubMed] [Google Scholar]
- 23.Higgs RE, Knierman MD, Gelfanova V, Butler JP, Hale JE. Comprehensive label-free method for the relative quantification of proteins from biological samples. J Proteome Res. 2005;4:1442–1450. doi: 10.1021/pr050109b. [DOI] [PubMed] [Google Scholar]
- 24.Hale JE, Butler JP, Gelfanova V, You JS, Knierman MD. A simplified procedure for the reduction and alkylation of cysteine residues in proteins prior to proteolytic digestion and mass spectral analysis. Anal Biochem. 2004;333:174–181. doi: 10.1016/j.ab.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 25.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 26.Carr S, Aebersold R, Baldwin M, Burlingame A, et al. The need for guidelines in publication of peptide and protein identification data: Working Group on Publication Guidelines for Peptide and Protein Identification Data. Mol Cell Proteomics. 2004;3:531–533. doi: 10.1074/mcp.T400006-MCP200. [DOI] [PubMed] [Google Scholar]
- 27.Geiss HC, Parhofer KG, Donner MG, Schwandt P. Low density lipoprotein apheresis by membrane differential filtration (cascade filtration) Ther Apher. 1999;3:199–202. doi: 10.1046/j.1526-0968.1999.00157.x. [DOI] [PubMed] [Google Scholar]
- 28.Hollander W, Colombo MA, Kirkpatrick B, Paddock J. Soluble proteins in the human atherosclerotic plaque. With spectral reference to immunoglobulins, C3-complement component, alpha 1-antitrypsin and alpha 2-macroglobulin. Atherosclerosis. 1979;34:391–405. doi: 10.1016/0021-9150(79)90064-9. [DOI] [PubMed] [Google Scholar]
- 29.Karbowska J, Kochan Z. Role of adiponectin in the regulation of carbohydrate and lipid metabolism. J Physiol Pharmacol. 2006;57(Suppl 6):103–113. [PubMed] [Google Scholar]
- 30.Matsuzawa Y. Adiponectin: Identification, physiology and clinical relevance in metabolic and vascular disease. Atherosclerosis Suppl. 2005;6:7–14. doi: 10.1016/j.atherosclerosissup.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 31.Marso SP, Mehta SK, Frutkin A, House JA, et al. Low adiponectin levels are associated with atherogenic dyslipidemia and lipid-rich plaque in nondiabetic coronary arteries. Diabetes Care. 2008;31:989–994. doi: 10.2337/dc07-2024. [DOI] [PubMed] [Google Scholar]
- 32.Kobayashi K, Inoguchi T, Sonoda N, Sekiguchi N, Nawata H. Adiponectin inhibits the binding of low-density lipoprotein to biglycan, a vascular proteoglycan. Biochem Biophys Res Commun. 2005;335:66–70. doi: 10.1016/j.bbrc.2005.07.048. [DOI] [PubMed] [Google Scholar]
- 33.Kannel WB. Overview of hemostatic factors involved in atherosclerotic cardiovascular disease. Lipids. 2005;40:1215–1220. doi: 10.1007/s11745-005-1488-8. [DOI] [PubMed] [Google Scholar]
- 34.Klein RL, Hunter SJ, Jenkins AJ, Zheng D, et al. Fibrinogen is a marker for nephropathy and peripheral vascular disease in type 1 diabetes: studies of plasma fibrinogen and fibrinogen gene polymorphism in the DCCT/EDIC cohort. Diabetes Care. 2003;26:1439–1448. doi: 10.2337/diacare.26.5.1439. [DOI] [PubMed] [Google Scholar]
- 35.Shukla SD, Kansra SV, Reddy MA, Shukla SM, et al. Diabetic pig platelets exhibit hypersensitivity to thrombin. Comp Med. 2008;58:481–484. [PMC free article] [PubMed] [Google Scholar]
- 36.Festa A, Kopp HP, Schernthaner G, Menzel EJ. Auto-antibodies to oxidised low density lipoproteins in IDDM are inversely related to metabolic control and microvascular complications. Diabetologia. 1998;41:350–356. doi: 10.1007/s001250050914. [DOI] [PubMed] [Google Scholar]
- 37.Kostner KM. Activation of the complement system: a crucial link between inflammation and atherosclerosis? Eur J Clin Invest. 2004;34:800–802. doi: 10.1111/j.1365-2362.2004.01431.x. [DOI] [PubMed] [Google Scholar]
- 38.Ceriello A, Giugliano D, Quatraro A, Stante A, et al. Increased alpha 2-macroglobulin in diabetes: a hyperglycemia related phenomenon associated with reduced antithrombin III activity. Acta Diabetol Lat. 1989;26:147–154. doi: 10.1007/BF02581366. [DOI] [PubMed] [Google Scholar]
- 39.James K, Merriman J, Gray RS, Duncan LJ, Herd R. Serum alpha 2-macroglobulin levels in diabetes. J Clin Pathol. 1980;33:163–166. doi: 10.1136/jcp.33.2.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Inoue W. Immunopathological analysis of acute phase reactant (APR) proteins in glomeruli from patients with diabetic nephropathy. Nippon Jinzo Gakkai Shi. 1989;31:211–219. [PubMed] [Google Scholar]
- 41.Otto C, Ritter MM, Richter WO, Minkenberg R, Schwandt P. Hemorrheologic abnormalities in defined primary dyslipoproteinemias with both high and low atherosclerotic risks. Metabolism. 2001;50:166–170. doi: 10.1053/meta.2001.20192. [DOI] [PubMed] [Google Scholar]
- 42.Mori T, Sasaki J, Kawaguchi H, Handa K, et al. Serum glycoproteins and severity of coronary atherosclerosis. Am Heart J. 1995;129:234–238. doi: 10.1016/0002-8703(95)90003-9. [DOI] [PubMed] [Google Scholar]
- 43.Wang Y, Xu LY, Lam KS, Lu G, et al. Proteomic characterization of human serum proteins associated with the fat-derived hormone adiponectin. Proteomics. 2006;6:3862–3870. doi: 10.1002/pmic.200500840. [DOI] [PubMed] [Google Scholar]
- 44.Savkovic V, Gantzer H, Reiser U, Selig L, et al. Clusterin is protective in pancreatitis through anti-apoptotic and anti-inflammatory properties. Biochem Biophys Res Commun. 2007;356:431–437. doi: 10.1016/j.bbrc.2007.02.148. [DOI] [PubMed] [Google Scholar]
- 45.Woods JA, Vieira VJ, Keylock KT. Exercise, inflammation, and innate immunity. Neurol Clin. 2006;24:585–599. doi: 10.1016/j.ncl.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 46.Gelissen IC, Hochgrebe T, Wilson MR, Easterbrook-Smith SB, et al. Apolipoprotein J (clusterin) induces cholesterol export from macrophage-foam cells: a potential anti-atherogenic function? Biochem J. 1998;331(Pt 1):231–237. doi: 10.1042/bj3310231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Faro J, Chen Y, Jhaveri P, Oza P, et al. L-ficolin binding and lectin pathway activation by acetylated low-density lipoprotein. Clin Exp Immunol. 2008;151:275–283. doi: 10.1111/j.1365-2249.2007.03538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harazono A, Kawasaki N, Kawanishi T, Hayakawa T. Site-specific glycosylation analysis of human apolipoprotein B100 using LC/ESI MS/MS. Glycobiology. 2005;15:447–462. doi: 10.1093/glycob/cwi033. [DOI] [PubMed] [Google Scholar]
- 49.Paananen K, Saarinen J, Annila A, Kovanen PT. Proteolysis and fusion of low density lipoprotein particles strengthen their binding to human aortic proteoglycans. J Biol Chem. 1995;270:12257–12262. doi: 10.1074/jbc.270.20.12257. [DOI] [PubMed] [Google Scholar]
- 50.Eugster C, Panakova D, Mahmoud A, Eaton S. Lipoprotein-heparan sulfate interactions in the Hh pathway. Dev Cell. 2007;13:57–71. doi: 10.1016/j.devcel.2007.04.019. [DOI] [PubMed] [Google Scholar]