

Abstract
The vitamin D endocrine system mediates anti-proliferative and pro-differentiating signaling in multiple epithelial tissues, including mammary gland and breast tumors. The vitamin D metabolite 1α,25(OH)2D3 mediates growth inhibitory signaling via activation of the vitamin D receptor (VDR), a ligand dependent transcription factor. 1α,25(OH)2D3 is synthesized from 25(OH)D3 (the major circulating form of the vitamin) by the mitochondrial enzyme CYP27b1 in renal and other tissues. Human mammary epithelial (HME) cells express VDR and CYP27b1 and undergo growth inhibition when exposed to physiological concentrations of 25(OH)D3, suggesting that autocrine or paracrine vitamin D signaling contributes to maintenance of differentiation and quiescence in the mammary epithelium. In the current studies we tested the hypothesis that cancer cells would exhibit reduced sensitivity to vitamin D mediated negative growth regulation. We used a series of progressively transformed HME cell lines expressing known oncogenic manipulations to study the effects of transformation per se on the vitamin D pathway. We report that mRNA and protein levels of VDR and CYP27b1 were reduced greater than 70% upon stable introduction of known oncogenes (SV40 T antigens and H-rasV12) into HME cells. Oncogenic transformation was also associated with reduced 1α,25(OH)2D3 synthesis, and cellular sensitivity to growth inhibition by 1α,25(OH)2D3 and 25(OH)D3 was decreased approximately 100-fold in transformed cells. These studies provide evidence that disruption of the vitamin D signaling pathway occurs early in the cancer development process.
Introduction
1α,25(OH)2D3 (1,25D), the biologically active form of vitamin D that acts as the ligand for the vitamin D receptor (VDR), is an endogenous regulator of differentiation, proliferation, and apoptosis in mammary cells (Kemmis et al, 2006; Pendas-Franco et al, 2006). VDR expression is dynamically regulated in mammary gland throughout the reproductive cycle and is necessary for proper glandular development during puberty, pregnancy and involution (Zinser et al, 2002; Zinser and Welsh, 2004). VDR agonists, including 1,25D, inhibit ductal proliferation and branching and induce the negative growth regulator TGFβ (Mehta et al, 1997). CYP27b1, the enzyme that synthesizes 1,25D from 25(OH)D3 (25D), is expressed in mammary tissue, as are megalin and cubilin, endocytic proteins which facilitate cellular uptake of circulating 25D bound to its serum vitamin D binding protein (DBP) (Townsend et al, 2005; Rowling et al, 2006). Normal human mammary epithelial (HME) cells express both CYP27b1 and VDR, synthesize 1,25D and undergo growth inhibition when exposed to physiological concentrations of 25D (Kemmis et al, 2006). Collectively, these data support the hypothesis that circulating 25D bound to DBP is internalized by normal mammary cells and converted to 1,25D, which interacts with VDR in an autocrine or paracrine manner to maintain differentiation and quiescence in the mammary epithelium. This hypothesis predicts that disruption of any of the multiple steps in vitamin D transport, metabolism or function could contribute to the development or progression of breast cancer.
Support for the hypothesis that the transformation process is associated with alterations in the vitamin D signaling pathway has been generated in several studies (Bareis et al, 2001; Cross et al, 2001; Anderson et al, 2006). In human colon, VDR expression is abundant in well-differentiated tissue, decreased in cancerous tissue, and almost non-existent in metastases (Cross et al, 2001). Analysis of human breast cancer cell lines has suggested that VDR expression is lower in estrogen independent, less differentiated lines compared to estrogen dependent, well differentiated lines such as MCF-7 (Buras et al, 1994). Furthermore, impaired VDR function has been reported in cells expressing oncoproteins such as the SV40 large T antigen or constitutively active ras (Solomon et al, 1998; Agadir et al, 1999; Stedman et al, 2003; Rozenchan et al, 2004).
There is also evidence that transformation alters the expression of the cytochrome P450 (CYP) metabolizing enzymes that control the steady state levels of the VDR ligand. Comparative analysis of CYP27b1 in normal and malignant human tissues suggested altered expression in a number of cancer types, including expression of splice variants which would generate non-functional proteins (Radermacher et al, 2006; Fischer et al, 2007; Wu et al, 2007). Overexpression of CYP24, the enzyme that catabolizes both 1,25D and 25D, has been observed in lung, colon, ovarian and esophageal cancer (Albertson et al, 2000; Mimori et al, 2004; Anderson et al, 2006). Townsend et al (2005) demonstrated that although both CYP27b1 and CYP24 mRNA were increased in breast tumors compared to normal tissue, CYP24 activity predominated resulting in net 1,25D catabolism.
Although these studies have provided evidence for altered vitamin D signaling in cancer, the data have primarily been derived from clinical specimens or through comparison of heterogeneous, independently-derived cell lines. To study the effects of transformation per se on the vitamin D pathway in mammary cells, we utilized a model of transformation developed by Dr. Robert Weinberg (Elenbaas et al, 2001). All cell lines in this model were derived from primary cultures of human mammary epithelial (HME) cells which were immortalized by introduction of the hTERT component of human telomerase. The resulting HMEtert cells retain the morphology and growth characteristics of normal mammary epithelial cells, and we have demonstrated that these cells express VDR and CYP27b1 and are growth inhibited by both 1,25D and 25D (Kemmis et al, 2006). For the model of transformation, HMEtert cells were stably infected with a retrovirus containing the SV40 early region (which encodes both the large T and small t antigens) to generate the HMELT line, in which p53 function is abrogated. Although transformed, HMELT cells are not fully tumorigenic and do not form tumors when injected into immunocompromised mice. However, stable introduction of the constitutively active H-rasV12 oncogene into HMELT cells resulted in generation of a fully transformed cell line (HMELT+Ras cells) capable of forming tumors in immunocompromised mice (Elenbaas et al, 2001).
In these studies, we used the HMEtert, HMELT, and HMELT+Ras cell culture model to assess whether the transformation process per se alters the integrity of the vitamin D signaling pathway. This model is especially suited to these studies because all cultures were developed and are continuously maintained in defined, serum-free media devoid of vitamin D steroids (which can lead to vitamin D resistance) and binding proteins (which can interfere with cellular uptake). Through comparative analysis of expression and function of VDR and vitamin D metabolizing enzymes in HMEtert , HMELT and HMELT+Ras cell lines, we provide evidence here that disruption of the vitamin D signaling pathway is an early event in the transformation process.
Materials and Methods
Cells and Cell Culture
HMEtert , HMELT and HMELT+Ras cell lines (generously provided by Dr. Robert Weinberg, MIT, Cambridge, MA) were maintained in Medium 171 supplemented with Mammary Epithelial Growth Supplement (MEGS) containing 0.4% bovine pituitary extract, 5mg/L bovine insulin, 0.5mg/L hydrocortisone, and 3 μg/L human epidermal growth factor (Cascade Biologics, Portland, OR). All cell lines were cultured at 37°C and 5%CO2 in a humidified incubator and passaged every 3-4 days. Morphology was assessed on sub-confluent, formalin-fixed cells by phase contrast microscopy.
For CYP27b1 knockdown, HMEtert cells were cultured to 50% confluence in 6-well culture dishes. Four SureSilencing shRNA plasmids designed to target CYP27b1 and a scrambled negative control vector (2μg, SuperArray, Frederick, MD) were individually transfected into the cells via FuGENE6 Transfection Reagent using a 3:2 (v:w) ratio of FuGENE to DNA. Cells expressing the SureSilencing shRNA plasmids were selected for neomycin resistance with 0.2mg/mL geneticin. Media containing geneticin was replenished every 1-2 days for two weeks. After selection was complete, cells were returned to regular media and analyzed for knockdown of CYP27b1 mRNA. Of the four sequences, the sequence 5′- ggcagagcttgaattgcaaat -3′ was found to be the most effective at CYP27b1 knockdown, therefore cells transfected with this sequence, which were termed HME-shCYP27b1, were used for comparison to HMEtert cells transfected with scrambled sequence and/or untransfected HMEtert cells.
Measurement of Cell Density
Cells were seeded in multi-well plates, allowed to attach overnight and then treated with ethanol vehicle or vitamin D metabolites (25D or 1,25D) at the concentrations indicated in the figure legends for up to 96 hours. Under the conditions used, each cell line displayed linear growth over this time period. Adherent cell density was measured after fixing cultures with 1% glutaraldehyde and staining with 0.1% crystal violet. After rinsing excess dye with water, wells were dried overnight and then solubilized with 0.1% Triton X-100. Absorbance at 590nm was measured on a Wallac Victor 2 plate reader as an indicator of cell density.
Quantitative RT-PCR
Cells were grown to subconfluence in 6-well dishes and treated with ethanol vehicle, 100nM 1,25D or 100nM 25D (Leo Pharmaceutical, Ballerup, Denmark) for 24 hours. Total RNA was extracted with RNeasy Mini Kit (Qiagen, Valencia, CA) and cDNA was synthesized with TaqMan Reverse Transcription Reagents (Applied Biosystems, Foster City, CA). Steady state mRNA expression for CYP27b1 and CYP24 was analyzed with real time PCR using TaqMan PCR Core Reagent Kit (Applied Biosystems). Primers and probes specific for VDR (forward:; reverse:, probe: ACTACCGCAAAGAAGGCTACGGGCTG), CYP27b1 (forward: AGTTGCTATTGGCGGGAGTG; reverse: GTGCCGGGAGAGCTCATACA, probe: ACTACCGCAAAGAAGGCTACGGGCTG) and CYP24 (forward: CAAACCGTGGAAGGCCTATC, reverse: AGTCTTCCCCTTCCAGGATCA, probe: ACTACCGCAAAGAAGGCTACGGGCTG) were used. Gene expression was normalized against 18S RNA (forward: AGTCCCTGCCCTTTGTACACA, reverse: GATCCGAGGGCCTCACTAAAC, probe: CGCCCGTCGCTACTACCGATTGG) expression.
Immunoblotting
Cells were seeded (300,000 cells per 100mm dish), allowed to attach overnight and then treated with ethanol vehicle, 100nM 1,25D or 100nM 25D for 48 hours. Monolayers were scraped into Laemmli buffer containing protease and phosphatase inhibitors (10mM benzamidine, 10mM sodium fluoride, 100mM sodium vanadate, 25μg/μl aprotinin, 25μg/μl pepstatin, 25μg/μl leupeptin and 1mM phenylmethylsulfonyl), sonicated and analyzed for total protein concentration with the BCA protein assay (Pierce, Rockford, IL). Routine western blotting was performed as described (1) using primary antibodies against VDR (clone 9A7, NeoMarkers, Fremont, CA), CYP24 (Cytochroma, Markham, Ontario), CYP27b1 (The Binding Site, San Diego, CA) and E-cadherin (BD Biosciences, San Jose, CA). Specific binding was detected with horseradish peroxidase conjugated anti-rat (Santa Cruz Biotechnology, Santa Cruz, CA), anti-mouse (Amsersham Biosciences, Piskataway, NJ) or anti-sheep (Jackson ImmunoResearch, West Grove, PA) antibodies and chemiluminescence (Super Signal West Dura Extended Duration Substrate, Pierce). Equal loading was confirmed by either ponceau staining or immunoblotting with anti-GAPDH antibody (Biogenesis, Poole, England).
Measurement of CYP27b1 activity
Cells were seeded in 6-well culture dishes (500,000 cells/well) and treated the following day with 100nM 25D in a total of 1mL media. Media was collected for analysis of 1α-hydroxylated metabolites with the 1,25D enzyme immunoassay (Immunodiagnostic Systems, Inc, Fountain Hills, AZ). Assays were conducted according to manufacturer directions for analysis of serum 1,25D. Controls included media alone supplemented with 25D to exclude non-cellular modification of 25D and to control for cross-reactivity with 25D in the EIA. Data was normalized by subtracting control values, and 1,25D synthesis was expressed as pg produced/106 cells.
Statistics
Data are expressed as mean ± standard error (SE). Student’s t-test or one-way ANOVA was used to determine statistical significance between treatment means (p<0.5) using GraphPad Prism software (GraphPad Software, SanDiego, CA). Dunnett’s or Tukey post-tests were used where applicable.
Results
In vitro transformation reduces mammary cell sensitivity to 1,25D
To specifically address the effect of transformation on vitamin D mediated growth inhibition, we used an in vitro model consisting of three distinct cell lines generated from normal human mammary epithelial cells (Elenbaas et al, 2001). The HMEtert cell line, which was created by immortalization of primary human mammary epithelial cultures with telomerase, exhibits monolayer growth in a cobblestone pattern characteristic of normal epithelial cells (Figure 1A, left panel). In contrast, HMELT cells (which express both telomerase and SV40 large T antigen) are reduced in size and grow in a less uniform cobblestone pattern, with some cells exhibiting elongated, fibroblast-like morphology (Figure 1A, middle panel). The fully tumorigenic HMELT+Ras cultures (which express telomerase, SV40 large T antigen and constitutively active H-ras) exhibit fibroblastic morphology and grow in a scattered, independent fashion even when confluent (Figure 1A, right panel).
Figure 1. Characteristics of HME cell variants and response to 1,25D.
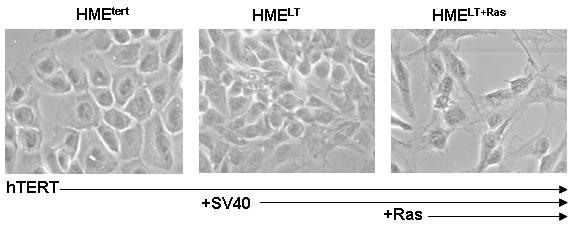
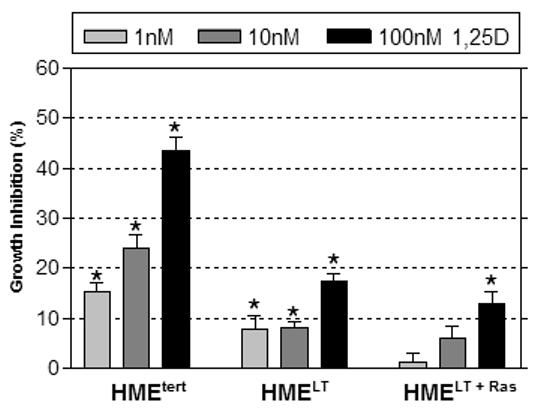
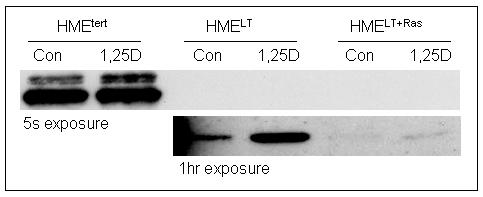
A. Phase contrast morphology of HMEtert, HMELT and HMELT+Ras cultures. hTERT, human telomerase; SV40, simian virus 40; Ras, H-rasV12 oncogene. B. Growth inhibition in response to 1,25D. Adherent cell density was assessed via crystal violet assay in cultures treated with vehicle or 1,25D (1, 10 or 100nM) for 96h. Data are expressed as percent growth inhibition relative to vehicle treated cultures for each cell line. Bars represent mean ± SEM, n=3. *p<0.05, as determined using one-way ANOVA and Dunnett’s multiple comparison test. Data are representative of at least 3 trials. C. Cell lysates from HMEtert, HMELT and HMELT+Ras cultures treated with vehicle or 100nM 1,25D were separated on SDS-PAGE and immunoblotted with antibody directed against E-cadherin. Images of the same blot exposed to film for 5s (top blot) or 1h (bottom blot) are presented.
To assess the integrity of the vitamin D signaling pathway in relation to transformation, we first examined the effect of 1,25D on adherent cell density (which reflects both proliferation and apoptosis) of all three cell lines during the linear growth phase. After attachment, cells in serum free media were treated with increasing concentrations of 1,25D or ethanol vehicle, and cell density was measured after 96h. Data are expressed as percent inhibition relative to ethanol vehicle control treated cultures for each cell line. Density of non-transformed HMEtert cultures was significantly reduced by concentrations of 1,25D as low as 1nM, with 45% inhibition observed in response to 100nM 1,25D (Figure 1B) . HMELT cultures were also inhibited by all concentrations of 1,25D tested, but the maximal inhibition (at 100nM) was less than 20% (comparable to the effect of 1nM 1,25D in HMEtert cultures). HMELT+Ras cultures were unaffected by low (1-10nM) doses of 1,25D, and even the highest dose tested (100nM) produced less than 15% growth inhibition. Time course studies suggested that the inhibitory effect of 1,25D occurred within 24h of treatment in HMEtert cells, but was delayed in HMELT and HMELT+Ras cultures (not shown). The differences in sensitivity to 1,25D between the HME cell variants did not appear to be secondary to altered doubling times, as growth kinetics of the cell lines in basal media were similar for at least 72h after plating. Furthermore, no morphological evidence for apoptosis was detected in any of the cell lines, even upon treatment with the highest concentration (100nM) of 1,25D (data not shown).
In epithelial cells, loss of E-cadherin is associated with cancer progression, and 1,25D has been shown to induce E-cadherin in colon cancer cells (Palmer et al, 2001). We therefore assessed whether basal or 1,25D-inducible E-cadherin expression is altered during mammary cell transformation (Figure 1C). On western blots with 5 second exposure to film, E-cadherin was detected in HMEtert cultures, but not in HMELT or HMELT+Ras cells. By immunofluorescent microscopy, E-cadherin expression was localized at adherens junctions in HMEtert cells but not in HMELT or HMELT+Ras cultures (data not shown). These data indicate that the loss of E-cadherin characteristic of the epithelial-mesenchymal transition is mimicked in this in vitro model of cancer progression. Consistent with findings in colon cancer cells, treatment with 1,25D (100nM, 48h) slightly but consistently increased E-cadherin expression in HMEtert cells. With longer exposure of the same blot to film, E-cadherin could be detected in HMELT cells, and its abundance was also increased in response to 1,25D treatment. In contrast, E-cadherin was only weakly detected HMELT+Ras cells, even upon prolonged exposure of film or after treatment with 1,25D.
Effect of transformation on basal and 1,25D induced CYP24 expression
To assess whether transformation altered genomic signaling through VDR, we examined basal and 1,25D inducible expression of CYP24, a well characterized target gene whose promoter contains multiple vitamin D response elements that are highly induced by the 1,25D-VDR complex (Vaisanen et al, 2005) In a luciferase reporter assay, 100nM 1,25D induced greater than 20-fold induction of the human CYP24 promoter in HMEtert cells whereas induction was only 5-10 fold in HMELT or HMELT+Ras cultures (Figure 2A). A similar profile was observed for expression of the endogenous CYP24 gene as measured by real time PCR (Figure 2B) and the CYP24 protein as measured on western blots (Figure 2C). These data indicate that transformation reduces both the basal and 1,25D inducible expression of a known VDR target gene. These data also suggest that reduced sensitivity to 1,25D mediated growth inhibition in transformed cells is not secondary to increased abundance of CYP24, an enzyme that mediates catabolism of 1,25D (Masuda et al, 2003).
Figure 2. Effect of 1,25D on CYP24 in HME cell variants.
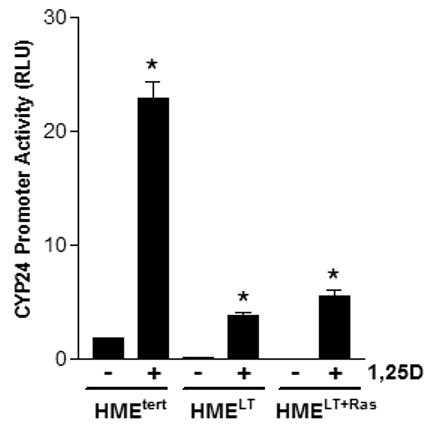
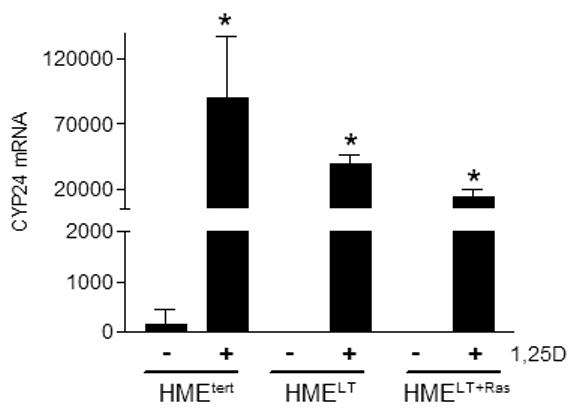
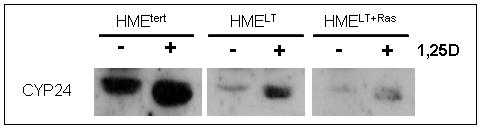
A. Cells were transiently co-transfected with the human CYP24 promoter-driven luciferase reporter gene and the pRL-TK normalization gene and treated with vehicle or 100nM 1,25D. After 24h treatment, activity was measured with the dual luciferase assay kit. Data were corrected for transfection efficiency and expressed as relative luciferase units (RLU). Bars represent mean ± SEM, n=3; *p <0.05, control vs 1,25D treated for each cell line. B. Cultures were treated for 24h with vehicle or 100nM 1,25D. CYP24 mRNA expression was measured by quantitative real time PCR and normalized to 18S. Bars represent mean ± SEM, n= 3. C. Total protein (20μg) from HME, HMELT and HMELT+Ras cultures treated with vehicle or 100nM 1,25D FOR 48h was separated via SDS-PAGE and immunoblotted with anti-CYP24 antibody.
Transformation reduces VDR expression
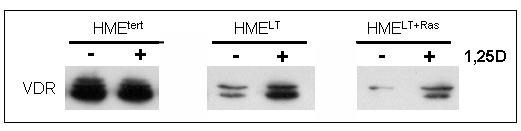
The VDR mediates the genomic effects of 1,25D, and studies in cells derived from VDR knockout mice indicate that the VDR is essential for 1,25D mediated growth inhibition (Zinser et al, 2003). We therefore hypothesized that the reduced sensitivity of transformed cells to 1,25D mediated growth regulation and gene expression could be secondary to reduced VDR expression. This hypothesis was tested by western blotting and real time PCR to compare VDR expression in the three cell lines. As shown in Figure 3A, western blotting of HMEtert cell lysates for VDR revealed a doublet at approximately 50kDa, which likely represents multiple phosphorylated forms of the receptor (Jurukta et al, 2002; Narvaez et al, 2003). In both HMELT and HMELT+Ras cultures, VDR protein expression was dramatically reduced. Treatment with 1,25D (100nM, 48h), which binds VDR and stabilizes it against proteolytic degradation, increased steady state VDR abundance in transformed cells, but the expression remained lower than that detected in the non-transformed HMEtert cells (Figure 3B).
Figure 3. VDR expression in HME cell variants.
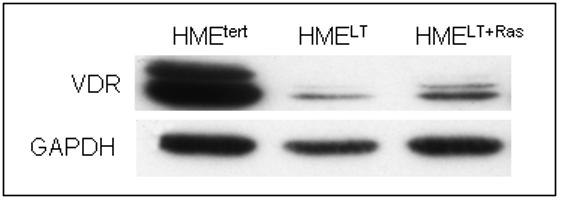

A. Total protein (20μg) from untreated HMEtert, HMELT and HMELT+Ras cultures was separated via SDS-PAGE and immunoblotted with antibodies directed against VDR (top) or GAPDH (bottom).
B. Lysates from cells treated with vehicle or 100nM 1,25D for 48h were separated on SDS-PAGE and immunoblotted with antibody directed against VDR.
C. Relative VDR mRNA expression in untreated cells was measured by quantitative real time PCR and normalized to 18S. Bars represent mean ± SEM, n= 3. a,b,c Bars with different letter superscripts are significantly (*p<0.05) different by one way ANOVA.
Real time PCR was used to assess whether changes in VDR protein expression during transformation were secondary to reduced gene expression. As shown in Figure 3C, steady state levels of VDR mRNA were reduced 80% in HMELT and 60% in HMELT+Ras cells relative to HMEtert cells, thus correlating with the reduction in VDR protein.
Effect of transformation on CYP27b1 expression, 1,25D synthesis and 25D mediated growth inhibition
Previous studies (Kemmis et al, 2006) have demonstrated that both primary and telomerase-immortalized HME cells express CYP27b1, a vitamin D hydroxylase that can synthesize 1,25D from 25D. We therefore tested whether transformation altered the expression or activity of this enzyme. CYP27b1 mRNA, as detected by real time PCR, was markedly reduced in HMELT cells and moderately reduced in HMELT+Ras cultures relative to in HMEtert cells (Figure 4A). CYP27b1 protein levels mirrored the changes in mRNA abundance, and no effects of 1,25D treatment on CYP27b1 protein expression were detected in any of the cell lines (Figure 4B). The CYP27b1 antibody recognized several additional bands in the HMEtert cells that were not present in the HMELT or HMELT+Ras cells. These bands may represent CYP27b1 splice variants (Cordes et al, 2007; Wu et al, 2007), but further studies would be necessary to confirm their identity and determine their functional significance.
Figure 4. Expression and activity of CYP27b1 in HME cell variants.
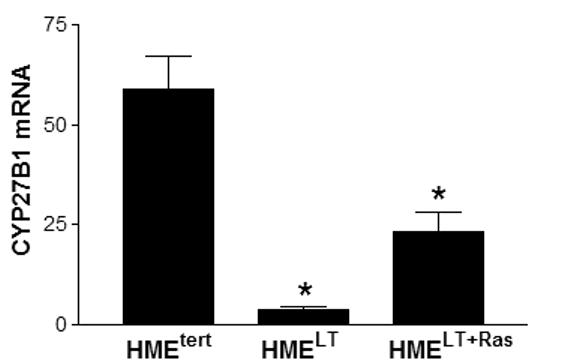
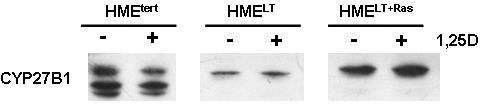
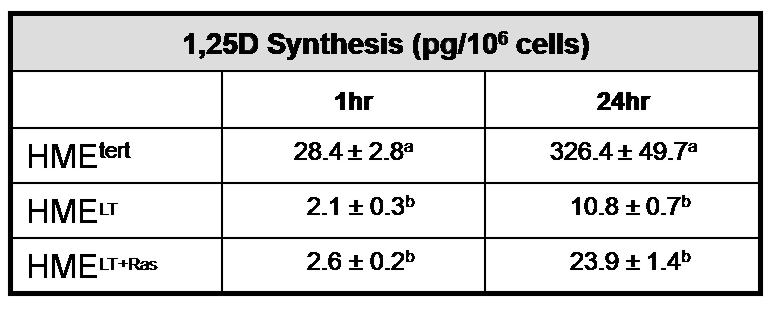
A. Basal CYP27b1 mRNA expression was measured in sub-confluent cultures of HME variant cell lines by quantitative real time PCR. Data was normalized to 18S. Bars represent mean ± SEM, n= 3. a,b,c Bars with different letter superscripts are significantly (*p<0.05) different by one way ANOVA. B. Total protein (20μg) from HMEtert, HMELT and HMELT+Ras cultures treated with vehicle or 100nM 1,25D was separated via SDS-PAGE and immunoblotted with anti-CYP27b1 antibody. C. HME variant cell lines were incubated with 100nM 25D for 1 or 24h as described in Methods. 1,25D was immunoextracted from cultures and quantified with a commercially available 1,25D enzyme-immunoassay. Data are expressed as pg 1,25D synthesized/106 cells (mean ± SEM, n=3). a,b Means with different letter superscripts are significantly (*p<0.05) different by one way ANOVA.
To determine whether the reduction in CYP27b1 expression was of sufficient magnitude to impact on its function, 1,25D synthesis was measured. Preliminary studies indicated that 1,25D synthesis from 25D increased linearly over a 24h period in HMEtert cells (data not shown). We therefore examined 1,25D synthesis in all three cell lines after 1h or 24h incubation with 100nM 25D. As shown in Figure 4C, 1,25D synthesis after 1h was reduced 90% in HMELT and HMELT+Ras cultures, and synthesis remained much lower in both transformed cell lines relative to HMEtert cells after 24h incubation. Interestingly, 1,25D synthesis was two fold higher in HMELT+Ras cells compared to HMELT cells, a finding consistent with the differences in CYP27b1 expression in these cell lines detected by real time PCR and western blotting.
Since CYP27b1 catalyzes the conversion of 25D to 1,25D, the expression of functional CYP27b1 in HMEtert cells confers sensitivity to 25D mediated growth inhibition (Kemmis et al, 2006). To assess whether the reduced synthesis of 1,25D from 25D in HMELT and HMELT+Ras cultures would translate to reduced cellular sensitivity to 25D, we measured adherent cell density of all three cell lines 96h after treatment with increasing concentrations of 25D (Figure 5A). Density of HMEtert cells was dose dependently reduced by 25D beginning at concentrations as low as 1nM, and 40% inhibition was observed at 100nM 25D. In contrast, HMELT or HMELT+Ras cultures were resistant to the inhibitory effects of low (1 or 10nM) doses of 25D, and even at 100nM, the reduction in cell density of transformed cultures by 25D was significantly less than that observed in HMEtert cells.
Figure 5. Growth inhibition of HME cell variants in response to 25D.
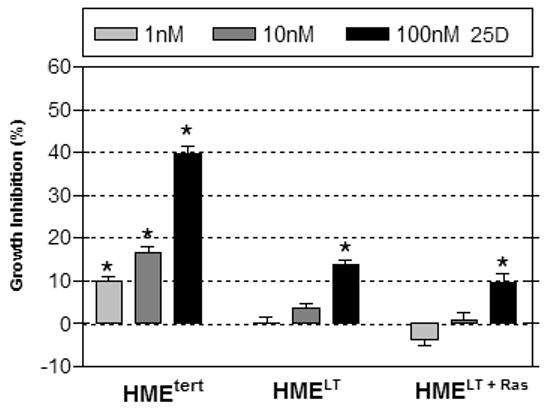
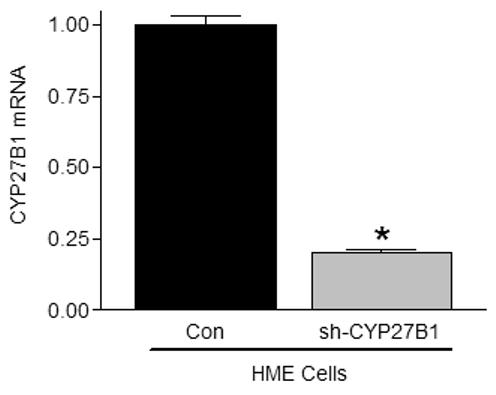
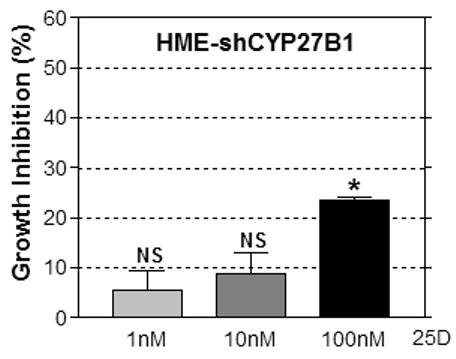
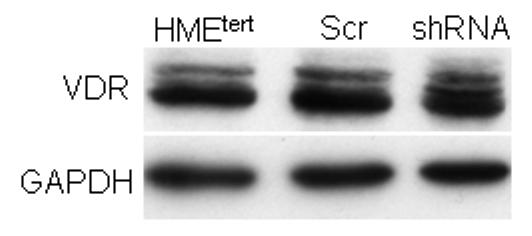
A. Adherent cell density was assessed via crystal violet assay in cultures treated with vehicle or 25D (1, 10 or 100nM) for 96h. Data are expressed as percent growth inhibition relative to vehicle treated cultures. Bars represent mean ± SEM, n=3. Data are representative of at least 3 trials. *p<0.05, treated vs control for each cell line. B. Knockdown of CYP27b1 in HMEtert cultures stably expressing shRNA directed against CYP27b1 was confirmed by real time PCR. Data are expressed as fold change relative to HME values, mean ± SD, n=2. C. Adherent cell density was assessed via crystal violet assay following treatment with ethanol vehicle or 25D for 96 hours. Data are expressed as percent growth inhibition relative to vehicle treated cultures, mean ± SEM (n=3). *p<0.05, control vs. treated. D. Lysates from parental HMEtert cells and HMEtert cultures stably expressing scrambled (scr) shRNA or shRNA directed against CYP27b1 were immunoblotted with an antibodies directed against VDR (top) or GAPDH (bottom).
Because transformed cells exhibited reduced expression of both VDR and CYP27b1, the data presented in Figure 5A do not conclusively demonstrate that loss of CYP27b1 expression in transformed cells contributes to their impaired sensitivity to 25D. We therefore specifically knocked down CYP27b1 expression in HMEtert cells to mimic the loss of this enzyme that occurs in transformed cells. As shown in Figure 5B, CYP27b1 mRNA was reduced 80% in cells stably expressing shRNA designed to target CYP27b1 (designated HMEC-shRNA), a reduction comparable to that observed in the HMELT+Ras cultures. To test whether CYP27b1 knock down altered sensitivity to 25D, cells were treated with increasing concentrations of 25D and cell density was analyzed after 96h (Figure 5B). In contrast to HMEtert cells, which were inhibited by concentrations of 25D as low as 1nM (Figure 5A), density of HMEC-shRNA cultures was inhibited only by 100nM 25D (Figure 5B), and the magnitude of this effect was about half that observed in the parental cell line. Knockdown of CYP27b1 did not alter VDR expression (Figure 5C) or sensitivity to 1,25D mediated growth inhibition (not shown). These data confirm that CYP27b1 mediates the inhibitory effects of 25D via generation of 1,25D, and imply that the 80% reduction in CYP27b1 expression in transformed cells would be of sufficient magnitude to impact on cellular sensitivity to 25D mediated growth inhibition regardless of VDR expression.
Discussion
These studies demonstrate that transformation of human mammary epithelial (HME) cells disrupts the integrity of the vitamin D pathway, which normally transmits anti-proliferative and pro-differentiating signals in these cells (Kemmis et al, 2006). This conclusion is based on relative changes in expression and function of VDR and CYP27b1 upon introduction of SV40 and/or H-rasV12 into HME cells expressing telomerase, an in vitro model of transformation (Elenbaas et al, 2001). More specifically, we report greater than 70% reduction in steady state mRNA levels of both VDR and CYP27b1 in HME cells expressing SV40 or SV40 + H-rasV12. Comparable reductions in protein expression and activity of both VDR and CYP27b1 were also observed in transformed cells. More importantly, oncogenic transformation reduced cellular sensitivity to growth inhibition by 1,25D (the VDR ligand) and 25D (which is converted by 1,25D by CYP27b1) by approximately 100-fold. Thus, these studies provide evidence that loss of function of the vitamin D pathway represents another strategy by which cancer cells evade negative growth regulation, one of the accepted “hallmarks of cancer” (Hanahan and Weinberg, 2000).
We have previously demonstrated that telomerase immortalization of primary HME cultures (which is associated with epigenetic silencing of p16INK4A expression) does not alter expression or function of VDR or CYP27b1 (Kemmis et al, 2006). Furthermore, both primary HME cultures and HMEtert cells are growth inhibited by low nanomolar concentrations of 1,25D and 25D (Kemmis et al, 2006). The current studies were designed to test whether partial or complete transformation of HMEtert cells (mediated by stable introduction of SV40 without or with H-rasV12, respectively) affected vitamin D signaling. Our data indicate that expression of SV40 alone, which abrogates cell cycle control and reduces E-cadherin expression but does not induce anchorage independent growth or tumorigenesis in nude mice (Elenbaas et al, 2001), was sufficient to down-regulate the vitamin D pathway in mammary cells. Thus, although introduction of oncogenic ras to SV40 expressing HMEtert cells induces a more aggressive, fully transformed phenotype, it was not necessary for deregulation of VDR signaling. These observations suggest that impairment in the vitamin D pathway may occur early in the tumorigenesis process.
Consistent with our findings, impaired VDR function and resistance to 1,25D mediated growth inhibition has previously been reported in SV40 immortalized HBL100 breast epithelial cells (Agadir et al, 1999). In HBL100 cells, however, comparative analysis of VDR expression and function in the presence and absence of SV40 was not possible. The SV40 construct utilized in the HME model encodes large T antigen, which simultaneously disables both p53 and Rb tumor suppressor proteins, and small t antigen, which perturbs protein phosphatase 2A (Hahn et al, 2002). Agidir et al (1999) showed that transient transfection of large T antigen disrupted 1,25D mediated VDR transactivation in CV-1 cells, suggesting that large T likely contributes to the reduced VDR function observed in HMELT cells. We hypothesize that one mechanism by which large T antigen down regulates VDR expression is through abrogation of p53 function, since genomic profiling has identified VDR as a gene highly induced by the p53 family (Maruyama et al, 2006; Kommagani et al, 2006; Kommagani et al, 2007). This hypothesis, however, does not exclude the possibility that SV40 mediated deregulation of RB and/or PP2A pathways may also impact on vitamin D signaling, or that SV40-transcribed proteins directly inhibit VDR gene expression.
Because introduction of SV40 alone severely impaired VDR expression in HME cells, the potential contribution of oncogenic ras to VDR deregulation could not be clearly established in our studies. However, oncogenic ras has been shown to destabilize VDR mRNA in mammary cells (Rozenchan et al, 2004) and inhibit VDR transcriptional activity in fibroblasts (Stedman et al, 2003). Furthermore, VDR was identified in a genomic screen for pathways perturbed during ras-mediated transformation (Agudo-Ibanez et al, 2007) and in this study, re-introduction of VDR abrogated ras-induced transformation as judged by foci formation. Collectively, these studies suggest reciprocal cross-talk between the VDR pathway and ras-mediated oncogenic signaling and support the hypothesis that down-regulation of VDR by SV40 in the HME model may facilitate ras-mediated transformation.
Ours are the first studies to examine the effects of specific oncogenic manipulations on the expression of CYP27b1 and CYP24, the cytochrome P450 enzymes that control the cellular concentration of 1,25D, the VDR ligand. Like VDR, expression of CYP27b1 (which generates 1,25D from serum-derived 25D) was markedly reduced in HMELT cells compared to HMEtert cells. This data is consistent with previous reports showing down-regulation of CYP27b1 mRNA in aggressive breast cancer cell lines relative to non-tumorigenic MCF12 cells, but contrary to data from clinical specimens (Townsend et al, 2005). In our study, the down regulation of CYP27b1 mRNA and protein in HMELT cells was of sufficient magnitude to impair the cell’s ability to synthesize 1,25D by more than 30-fold, and to render cells less responsive to the growth inhibitory effects of 25D. Although VDR is also down regulated by SV40, comparison of the effects of 1,25D and 25D on HMELT cell growth suggests that loss of CYP27b1 further reduces cell sensitivity to 25D beyond that caused by loss of VDR alone. This suggestion was supported by our data demonstrating that a comparable reduction in CYP27b1 achieved by shRNA-mediated knockdown in HMEtert cells reduced their sensitivity to 25D mediated growth inhibition in the absence of changes in VDR protein expression.
Surprisingly, the reduced CYP27b1 expression and activity in HMELT cells was partially restored by introduction of oncogenic ras. However, 1,25D synthesis remained more than 10 fold lower, and cellular sensitivity to 25D mediated growth inhibition was not improved, in HMELT+Ras cells compared to HMELT cells. Although multiple factors are known to regulate the human CYP27b1 promoter, including PTH, 1,25D, IFNγ, and various growth factors, there is no evidence as yet for regulation by p53, Rb, protein phosphatase 2A or ras. Thus, dissecting the mechanisms by which SV40 and ras interact to regulate CYP27b1 expression in the HME model system will require additional study.
Basal expression of CYP24, the major enzyme responsible for turnover of 25D and 1,25D, was also reduced in HMELT cells compared to HMEtert cells, and was further reduced in HMELT+Ras cells. Thus, increased CYP24-mediated catabolism of 1,25D and 25D, a frequent cause of vitamin D resistance (Ly et al, 1999), likely does not contribute to the reduced sensitivity of the HMELT and HMELT+Ras cells to these metabolites. This data is in contrast to studies by Townsend et al (2005) that demonstrated up-regulation of CYP24 mRNA in aggressive breast cancer cell lines relative to non-tumorigenic MCF12 cells. These discrepancies may be attributed to the different model systems studied: the HME cell variants share a common genetic background and have been engineered in vitro with known oncogenic manipulations, whereas the established breast cancer cell lines are heterogeneous in origin and have acquired distinct, unknown oncogenic mutations during in vivo tumorigenesis and in vitro propagation. As data from clinical specimens has also been inconsistent (with increased, decreased or no change in CYP24 expression reported in breast cancer compared to normal tissue), additional studies in both the HME model and in established breast cancer cell lines may be necessary to clarify the spectrum of pathways that affect CYP24 expression during tumorigenesis.
In summary, we have demonstrated that expression and function of key components of the vitamin D signaling pathway become deregulated in the course of mammary epithelial cell transformation. These changes enable mammary cells to escape the growth inhibitory signals that are normally triggered through VDR signaling. Further studies are needed to clarify how the balance between proliferation and apoptosis is altered in this model of transformation, and which specific pathways become insensitive to 1,25D. Because the insensitivity to vitamin D mediated signaling occurs early in the course of tumorigenesis, our data supports the concept that deregulation of vitamin D signaling may contribute to or facilitate tumor progression. This concept predicts that strategies to optimize vitamin D signaling would be most effective in preventing cancer during the early, promotional stages.
Acknowledgements
The authors gratefully acknowledge Dr. Robert Weinberg (MIT) for the HME series of cell lines and Cytochroma (Markham, ON) for the CYP24 antibody. This work was supported by NIH grants CA69700 and CA103018 to JW.
References
- Agadir A, Lazzaro G, Zheng Y, Zhang XK, Mehta R. Resistance of HBL100 human breast epithelial cells to vitamin D action. Carcinogenesis. 1999;20:577–582. doi: 10.1093/carcin/20.4.577. [DOI] [PubMed] [Google Scholar]
- Agudo-Ibanez L, Nunez F, Calvo F, Berenjeno IM, Bustelo XR, Crespo P. Transcriptomal profiling of site-specific Ras signals. Cell Signal. 2007;19:2264–2276. doi: 10.1016/j.cellsig.2007.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertson DG, Ylstra B, Segraves R, Collins C, Dairkee SH, Kowbel D, Kuo WL, Gray JW, Pinkel D. Quantitative mapping of amplicon structure by array CGH identifies CYP24 as a candidate oncogene. Nat Genetics. 2000;25:144–146. doi: 10.1038/75985. [DOI] [PubMed] [Google Scholar]
- Anderson MG, Nakane M, Ruan X, Kroeger PE, Wu-Wong JR. Expression of VDR and CYP24A1 mRNA in human tumors. Cancer Chemother Pharmacol. 2006;57:234–240. doi: 10.1007/s00280-005-0059-7. [DOI] [PubMed] [Google Scholar]
- Bareis P, Bises G, Bischof MG, Cross HS, Peterlik M. 25-hydroxy-vitamin D metabolism in human colon cancer cells during tumor progression. Biochem Biophys Res Commun. 2001;285:1012–1017. doi: 10.1006/bbrc.2001.5289. [DOI] [PubMed] [Google Scholar]
- Buras RR, Schumaker LM, Davoodi F, Brenner RV, Shabahang M, Nauta RJ, Evans SR. Vitamin D receptors in breast cancer cells. Breast Cancer Res Treat. 1994;31:191–202. doi: 10.1007/BF00666153. [DOI] [PubMed] [Google Scholar]
- Cordes T, Diesing D, Becker S, Fischer D, Diedrich K, Friedrich M. Expression of splice variants of 1alpha-hydroxylase in MCF-7 breast cancer cells. J Steroid Biochem Mol Biol. 2007;103:326–329. doi: 10.1016/j.jsbmb.2006.12.034. [DOI] [PubMed] [Google Scholar]
- Cross HS, Bareis P, Hofer H, Bischof MG, Bajna E, Kriwanek S, Bonner E, Peterlik M. 25-Hydroxyvitamin D(3)-1alpha-hydroxylase and vitamin D receptor gene expression in human colonic mucosa is elevated during early cancerogenesis. Steroids. 2001;66:287–292. doi: 10.1016/s0039-128x(00)00153-7. [DOI] [PubMed] [Google Scholar]
- Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, Popescu NC, Hahn WC, Weinberg RA. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 2001;15:50–65. doi: 10.1101/gad.828901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer D, Seifert M, Becker S, Ludders D, Cordes T, Reichrath J, Friedrich M. 25-Hydroxyvitamin D3 1alpha-hydroxylase splice variants in breast cell lines MCF-7 and MCF-10. Cancer Genomics Proteomics. 2007;4:295–300. [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hahn WC, Dessain SK, Brooks MW, King JE, Elenbaas B, Sabatini DM, DeCaprio JA, Weinberg RA. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol Cell Biol. 2002;22:2111–2123. doi: 10.1128/MCB.22.7.2111-2123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurutka PW, MacDonald PN, Nakajima S, Hsieh JC, Thompson PD, Whitfield GK, Galligan MA, Haussler CA, Haussler MR. Isolation of baculovirus-expressed human vitamin D receptor: DNA responsive element interactions and phosphorylation of the purified receptor. J Cell Biochem. 2002;85:435–457. doi: 10.1002/jcb.10134. [DOI] [PubMed] [Google Scholar]
- Kemmis CM, Salvador SM, Smith KM, Welsh JE. Human mammary epithelial cells express CYP27b1 and are growth inhibited by 25-hydroxyvitamin D-3, the major circulating form of vitamin D-3. J Nutr. 2006;136:887–892. doi: 10.1093/jn/136.4.887. [DOI] [PubMed] [Google Scholar]
- Kommagani R, Caserta TM, Kadakia MP. Identification of vitamin D receptor as a target of p63. Oncogene. 2006;25:3745–3751. doi: 10.1038/sj.onc.1209412. [DOI] [PubMed] [Google Scholar]
- Kommagani R, Payal V, Kadakia MP. Differential Regulation of Vitamin D Receptor (VDR) by the p53 Family: p73-depdendent induction of VDR upon DNA damage. J Biol Chem. 2007;282:29847–29854. doi: 10.1074/jbc.M703641200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly LH, Zhao XY, Holloway L, Feldman D. Liarozole acts synergistically with 1alpha,25-dihydroxyvitamin D3 to inhibit growth of DU 145 human prostate cancer cells by blocking 24-hydroxylase activity. Endocrinol. 1999;140:2071–2076. doi: 10.1210/endo.140.5.6698. [DOI] [PubMed] [Google Scholar]
- Maruyama R, Aoki F, Toyota M, Sasaki Y, Akashi H, Mita H, Suzuki H, Akino K, Ohe-Toyota M, Maruyama Y, Tatsumi H, Imai K, Shinomura Y, Tokino T. Comparative genome analysis identifies the vitamin D receptor gene as a direct target of p53-mediated transcriptional activation. Cancer Res. 2006;66:4574–4583. doi: 10.1158/0008-5472.CAN-05-2562. [DOI] [PubMed] [Google Scholar]
- Masuda S, Gao M, Zhang A, Kaufmann M, Jones G. Importance of cytochrome P450-mediated metabolism in the mechanism of action of vitamin D analogs. Recent Res Cancer Res. 2003;164:189–202. doi: 10.1007/978-3-642-55580-0_14. [DOI] [PubMed] [Google Scholar]
- Mehta RG, Moriarty RM, Mehta RR, Penmasta R, Lazzaro G, Constantinou A, Guo L. Prevention of preneoplastic mammary lesion development by a novel vitamin D analogue, 1alpha-hydroxyvitamin D5. J Natl Cancer Inst. 1997;89:212–218. doi: 10.1093/jnci/89.3.212. [DOI] [PubMed] [Google Scholar]
- Mimori K, Tanaka Y, Yoshinaga K, Masuda T, Yamashita K, Okamoto M, Inoue H, Mori M. Clinical significance of the overexpression of the candidate oncogene CYP24 in esophageal cancer. Ann Oncol. 2004;15:236–241. doi: 10.1093/annonc/mdh056. [DOI] [PubMed] [Google Scholar]
- Narvaez CJ, Byrne BM, Romu S, Valrance M, Welsh JE. Induction of apoptosis by 1,25-dihydroxyvitamin D3 in MCF-7 vitamin D3-resistant variant can be sensitized by TPA. J Steroid Biochem Mol Biol. 2003;84:199–209. doi: 10.1016/s0960-0760(03)00029-3. [DOI] [PubMed] [Google Scholar]
- Palmer HG, Gonzalez_Sancho JM, Espada J, Berciano MT, Puig I, Baulida J, Quintanilla M, Cano A, de_Herreros AG, Lafarga M, Munoz A. Vitamin D3 promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. J Cell Biol. 2001;154:369–387. doi: 10.1083/jcb.200102028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendas-Franco N, Gonzalez-Sancho JM, Suarez Y, Aguilera O, Steinmeyer A, Gamallo C, Berciano MT, Lafarga M, Munoz A. Vitamin D regulates the phenotype of human breast cancer cells. Differentiation. 2007;75:193–207. doi: 10.1111/j.1432-0436.2006.00131.x. [DOI] [PubMed] [Google Scholar]
- Radermacher J, Diesel B, Seifert M, Tilgen W, Reichrath J, Fischer U, Meese E. Expression analysis of CYP27b1 in tumor biopsies and cell cultures. Anticancer Res. 2006;26:2683–2686. [PubMed] [Google Scholar]
- Rowling MJ, Kemmis CM, Taffany DA, Welsh JE. Megalin-mediated endocytosis of vitamin D binding protein correlates with 25-hydroxycholecalciferol actions in human mammary cells. J Nutr. 2006;136:2754–2759. doi: 10.1093/jn/136.11.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenchan PB, Folgueira MA, Katayama ML, Snitcovsky IM, Brentani MM. Ras activation is associated with vitamin D receptor mRNA instability in HC11 mammary cells. J Steroid Biochem Mol Biol. 2004;92:89–95. doi: 10.1016/j.jsbmb.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Solomon C, Sebag M, White JH, Rhim J, Kremer R. Disruption of vitamin D receptor-retinoid X receptor heterodimer formation following ras transformation of human keratinocytes. J Biol Chem. 1998;273:17573–17578. doi: 10.1074/jbc.273.28.17573. [DOI] [PubMed] [Google Scholar]
- Stedman L, Nickel KP, Castillo SS, Andrade J, Burgess JR, Teegarden D. 1,25-dihydroxyvitamin D inhibits vitamin E succinate-induced apoptosis in C3H10T1/2 cells but not Harvey ras-transfected cells. Nutr Cancer. 2003;45:93–100. doi: 10.1207/S15327914NC4501_11. [DOI] [PubMed] [Google Scholar]
- Townsend K, Banwell CM, Guy M, Colston KW, Mansi JL, Stewart PM, Campbell MJ, Hewison M. Autocrine metabolism of vitamin D in normal and malignant breast tissue. Clin Cancer Res. 2005;11:3579–3586. doi: 10.1158/1078-0432.CCR-04-2359. [DOI] [PubMed] [Google Scholar]
- Vaisanen S, Dunlop TW, Sinkkonen L, Frank C, Carlberg C. Spatio-temporal activation of chromatin on the human CYP24 gene promoter in the presence of 1alpha,25-Dihydroxyvitamin D3. J Mol Biol. 2005;350:65–77. doi: 10.1016/j.jmb.2005.04.057. [DOI] [PubMed] [Google Scholar]
- Wu S, Ren S, Nguyen L, Adams JS, Hewison M. Splice variants of the CYP27b1 gene and the regulation of 1,25-dihydroxyvitamin D3 production. Endocrinol. 2007;148:3410–3418. doi: 10.1210/en.2006-1388. [DOI] [PubMed] [Google Scholar]
- Zinser GM, McEleney K, Welsh JE. Characterization of mammary tumor cell lines from wild type and vitamin D3 receptor knockout mice. Mol Cell Endocrinol. 2003;200:67–80. doi: 10.1016/s0303-7207(02)00416-1. [DOI] [PubMed] [Google Scholar]
- Zinser GM, Packman K, Welsh JE. Vitamin D(3) receptor ablation alters mammary gland morphogenesis. Development. 2002;129:3067–3076. doi: 10.1242/dev.129.13.3067. [DOI] [PubMed] [Google Scholar]
- Zinser GM, Welsh JE. Accelerated mammary gland development during pregnancy and delayed post-lactational involution in vitamin D3 receptor null mice. Mol Endocrinol. 2004;18:2208–2223. doi: 10.1210/me.2003-0469. [DOI] [PubMed] [Google Scholar]