

Abstract
The TAK1-NLK cascade is a mitogen-activated protein kinase-related pathway that plays an inhibitory role in canonical Wnt/β-catenin signaling through regulating the LEF1/TCF family transcriptional factors. TAB2 (TAK1-binding protein 2) is a putative TAK1 interacting protein that is involved in the regulation of TAK1. Here, we found that TAB2 could directly interact with NLK and function as a scaffold protein to facilitate the interaction between TAK1 and NLK. Knocking down TAB2 using small interfering RNA abolished the interaction of TAK1 with NLK in mammalian cells. The intermediate region (residues 292–417) of TAB2 was mapped for its binding to NLK. TAB2-ΔM, a TAB2 mutant lacking this region, showed a lower affinity for NLK and became defective in its scaffolding function. In addition, TAB2, but not TAB2-ΔM, mediated TAK1-dependent activation of NLK and LEF1 polyubiquitylation, resulting in the inhibition of canonical Wnt signaling. Moreover, Wnt3a stimulation led to an increase in the interaction of TAB2 with NLK and the formation of a TAK1·TAB2·NLK complex, suggesting that this TAK1-TAB2-NLK pathway may constitute a negative feedback mechanism for canonical Wnt signaling.
Keywords: Signal Transduction/Protein Kinases/MAP, MAP Kinases (MAPKs), Protein Phosphorylation, Protein-Protein Interactions, Wnt Pathway, NLK, TAB2, TAK1, Scaffold Proteins
Introduction
The TAK1-NLK cascade is a mitogen-activated protein kinase (MAPK)2-related pathway that regulates a number of biological processes (1–7). TAK1 (transforming growth factor-β-activated kinase 1) is a MAPK kinase kinase that was identified by complementation assays to substitute for the MAPKKK ste11 in yeast (8). NLK (Nemo-like kinase) is an atypical MAPK that adopts some critical variations in its putative MAPK domain that differs from classic MAPK (9, 10). The TAK1-NLK cascade was first found in Caenorhabditis elegans to modulate Wnt signaling in endoderm induction during embryogenesis, and this cascade was later confirmed in mammalian cells and other organisms (2, 3, 11, 12). In this cascade TAK1 is the upstream activator of the kinase activity of NLK, which in turn phosphorylates and regulates several transcriptional factors (4, 6, 13), including lymphoid enhancer factor 1/T-cell factor (LEF1/TCF) family proteins (2, 14).
The Wnt/β-catenin pathway, i.e. canonical Wnt pathway, functions in various physiological and pathophysiological processes through stabilizing cytoplasmic β-catenin. Accumulated β-catenin stimulated by Wnt signals enters the nucleus to form a complex with the high mobility group box class of DNA binding transcription factors, including LEF1 and TCFs, and initiates the transcription of Wnt target genes (15–17). In addition, many activators or inactivators have been found to converge to regulate the transcriptional complex of β-catenin and LEF1/TCFs (1, 18–20). The TAK1-NLK pathway plays a negative role in regulating the canonical Wnt pathway. NLK can directly phosphorylate LEF1/TCFs to prevent the β-catenin-LEF1/TCFs complex from binding to DNA (2, 14). The phosphorylation also facilitates the ubiquitylation of LEF1/TCFs, which leads to their degradation (21) and, hence, the inhibition of β-catenin-dependent transcription.
Scaffold proteins play a determinative role in modulating the signaling strength and fidelity by assembling cognate components, in particular signal transductions, especially in the MAPK pathway. TAK1 is regulated by several distinct extracellular stimuli as previously reported (8, 22–24). However, only some of them can stimulate the TAK1-NLK cascade (5). This notion suggests that some specific regulation might be required for the signal transduction from TAK1 to NLK. Specific interaction or particular scaffold proteins are likely to be required to ensure the fidelity and efficiency of signal flow through kinase cascades (25–27). However, TAK1 as the activator cannot directly interact with its effector, NLK (4, 5). Thus, it is a great possibility that a scaffold protein is involved in the signal transduction from TAK1 to NLK. TAB2 (TAK1-binding protein 2) is one well characterized TAK1-binding protein that is required in several TAK1 functions (28, 29). TAB2 cannot directly regulate TAK1 kinase activity (30) as TAB1 (TAK1-binding protein 1) does (22, 30), whereas it acts as an adaptor or scaffold protein to modulate TAK1 in different signal transductions by linking it to other molecules (28, 31–36). In this work we report that TAB2 functions as a scaffold protein for TAK1 and NLK to bridge their interaction in repressing canonical Wnt signaling.
EXPERIMENTAL PROCEDURES
Plasmids, siRNAs, and Antibodies
Full-length mouse NLK was identified from the pPC86-based mouse embryonic E10.5 cDNA library as a positive clone, which showed the interaction with TAB2, and subcloned into pCMV-HA, pCMV-EE (containing a Glu-Glu epitope tag), pCMV-FLAG, pGEX-4T2, and pET28C plasmids. Mouse TAK1 and TAB2 plasmids were cloned from mouse cDNA and subcloned into pCMV plasmids to express in mammalian cells. The plasmid of IκB super suppressor was offered from the Chen Wang laboratory (Institute of Biochemistry and Cell Biology). The plasmid of ubiquitin-HA was from the Bing Sun laboratory (Institute of Biochemistry and Cell Biology). Other plasmids have been used in our previous work (37).
Two siRNAs targeting NLK were designed and synthesized according to the sequences as siNLK-1 (5′-GGATGTTGGTCTTTGATCCATCCAA-3′) and siNLK-2 (5′-GCTGCTACAGTTAAGGCGCACCATC-3′). TAB2 RNAi was synthesized according to the sequence as 5′-CCTCCAGCACTTCCTCTTCAGTCAA-3′ (siTAB2-1) and 5′-GGTTTTACATGAGGTGCGACAAAAA-3′ (siTAB2-2). Control siRNA was designed with the sequence targeting renilla luciferase (5′-AATGAACGTGCTGGACTCCTTCATC-3′).
TAB2, TAK1, and NLK antibodies were purchased from Santa Cruz Biotechnology, Inc. Anti-HA, Myc, and EE (Covance), anti-FLAG (Sigma), anti-β-catenin (BD Biosciences), and c-Jun N-terminal kinase (JNK) and phospho-JNK (Cell Signaling Technology) were used in this work.
Yeast Two-hybrid Screen
Yeast two-hybrid screens system and pPC86-based mouse embryonic E10.5 cDNA library were purchased from Invitrogen. Full-length mouse TAB2 was subcloned into pDB plasmid, which was used as bait. A yeast two-hybrid assay was performed according to the manufacturer's manual.
Cell Culture, Transfection, and Reporter Gene Assay
Cells of HEK293T or HEK293 stably transfected NLK (NLK-293) were maintained in Dulbecco's modified Eagle's medium plus 10% fetal bovine serum (Invitrogen). Plasmids were transfected using Lipofectamine/Plus reagent (Invitrogen) according to the manufacturer's instructions. For reporter gene assays, HEK293T cells were used and seeded in 24-well plates. Each well was transfected with 250 ng of plasmids in total, including 10 ng of TOPFLASH, 10 ng of EGFP-C1, and other plasmids indicated in each figure. At 24 h after transfection, cells were lysed, and experiments were done as previously described (37). Relative luciferase activity was calculated as the -fold luciferase activity over the basal activity from cells transfected with blank control plasmid (LacZ).
Co-immunoprecipitation and Western Blot
HEK293T cells were seeded in 35-mm dishes and cultured for 24 h before transfection. At 24 h post transfection, cells were lysed in 500 μl of lysis buffer (20 mm Tris-HCl, pH 8.0, 1% Nonidet P-40, 10% glycerol, 138 mm NaCl) plus 10 mm NaF, 2 mm Na3VO4, 1 mm pyrophosphoric acid, and CompleteTM protease inhibitors (Roche Applied Science). The lysates were centrifuged for 15 min at 14,000 rpm at 4 °C. The supernatants were mixed with antibodies and Protein A/G plus-agarose (Santa Cruz Technology) for 3 h at 4 °C. Then immunoprecipitates were washed with lysis buffer three times, denatured in SDS sample buffer for 10 min at 95 °C, and resolved by SDS-PAGE. For experiments using the NLK-293 cell line, cells were lysed, and co-IP was carried out by incubating relevant antibodies or FLAG affinity gel (Sigma) with cell lysates overnight at 4 °C. Then cell lysates with antibodies were added with protein A/G plus-agarose and mixed for another 2 h at 4 °C. For immunoblotting, proteins were transferred to nitrocellulose (Schleicher & Schüll) or polyvinylidene difluoride (Millipore) membranes. After being blocked with 5% nonfat milk, membranes were incubated with the primary antibodies for 1 h at room temperature or overnight at 4 °C and then IRDye 800 (Rockland) or horseradish peroxidase-conjugated secondary antibodies (Thermo Scientific) for 0.5–1 h at room temperature. Results were visualized by the Odyssey Infrared Imaging System 9120 (LI-COR) or FujiFilm Las 4000 (FujiFilm).
Free β-Catenin Assay
For the free β-catenin assay, a 35-mm dish of HEK293T cells was used. Cells were washed once with cold phosphate-buffered saline (PBS) and scraped into 1 ml of PBS. Then cells were collected by spinning at 3000 rpm for 10 min and resuspended in buffer A (10 mm Hepes-KOH, pH 7.9, 10 mm KCl). After 10 min on ice, cells were homogenized and centrifuged at 3000 rpm for 5 min at 4 °C, and the deposited parts were preserved for nuclear extraction. The supernatant was collected and centrifuged at 100,000 × g for 1 h at 4 °C. After centrifugation, the supernatant was collected as the cytoplasmic fraction and subjected to a Western blot to detect free β-catenin. For nuclear extraction, the pellet was resuspended in buffer C (20 mm Hepes-KOH, pH 7.9, 25% glycerol, 500 mm NaCl). After cooling on ice for 30 min, the nuclear extract was centrifuged at 14,000 rpm for 15 min at 4 °C, and the supernatant was obtained as the nuclear fraction.
Reverse Transcription-PCR and Quantitative Real-time PCR
Total RNAs were extracted from siRNA-transfected HEK293T cells with TRIzol, and reverse transcription of purified RNA was performed using oligo(dT) priming and superscript III reverse transcription according to the manufacturer's instructions (Invitrogen). The quantification of AXIN2 and GAPDH transcripts was done by quantitative PCR using the SYBR Premix Ex TaqTM (Perfect Real Time) PCR kit (TaKaRa) and a Rotor-Gene RG-3000A apparatus (Corbett Research). The primer pairs used for human Axin2 gene were 5′-AGTGTGAGGTCCACGGAAAC-3′ and 5′-CTTCACACTGCGATGCATTT-3′. The primer pairs used for the human GAPDH gene were 5′-GCACCACCAACTGCTTA-3′ and 5′-AGTAGAGGCAGGGATGAT-3′. The relative expression of Axin2 was quantitated by normalization over the expression of GAPDH.
NLK Kinase Assay
HEK293T cells seeded in 35-mm dishes were transfected with indicated plasmids. At 24 h post-transfection, cells were lysed, and NLK or LEF1 was immunoprecipitated. The beads were washed twice with lysis buffer and twice with kinase assay buffer (30 mm Hepes, pH 7.4, 3.7 mm MgCl2, 4.6 mm MnCl2). Then, the immunoprecipitates were incubated in kinase buffer containing 5 μCi of [γ-32P]ATP at 25 °C for 20 min as reported (38). For NLK kinase assays with NLK-293 cells, stably transfected NLK was pulled down with FLAG affinity gel (Sigma). After washing, the immunoprecipitates were eluted off from the beads with FLAG peptide. Kinase assays were performed at 25 °C for 5 min in kinase assay buffer (25 mm Hepes, pH 7.4, 10 mm MgCl2) including 5 μCi of [γ-32P]ATP. Reactions were terminated by the addition of SDS loading buffer, and proteins were separated on SDS-PAGE. Phosphorylated proteins were visualized by autoradiography.
RNA Interference
Efficiencies of each siRNA were validated with Western blot. RNA were resolved in diethyl pyrocarbonate water and stored at the concentration of 20 μm. For transfection of siRNA, 0.5–1 μl was used for each well of the 24-well plate. Transfections of siRNA were performed with Lipofectamine 2000 reagent (Invitrogen). Transfected cells were subjected to different assays 48 h after transfection.
RESULTS AND DISCUSSION
NLK Is Identified as a Novel TAB2-binding Protein
Previous studies have shown that TAB2 interacted with TAK1 in a number of TAK1-mediated signaling events (28, 29). In the search of additional TAB2-binding proteins, we screened a mouse embryonic cDNA library using the approach of yeast two-hybrid with TAB2 as bait and identified the full-length NLK. We first confirmed the interaction using co-IP. As shown in Fig. 1A, overexpressed NLK and TAB2 can be co-immunoprecipitated in HEK293T cells. In an in vitro pulldown assay, recombinant GST-TAB2 protein efficiently pulled down His6-NLK (Fig. 1B), suggesting that TAB2 can directly interact with NLK. Besides NLK, TAK1 as MAPKKK (MAPK kinase kinase) can also activate JNK and p38 MAPK (23, 31). However, TAB2 only interacted with NLK, but not JNK and p38 in co-IP assays (Fig. 1C), suggesting that the interaction of TAB2 and NLK is specific. NLK binding with TAB2 not only requires its kinase domain, which is similar with that of JNK and p38 (10), but also its C-terminal region (Fig. 1D). This result may explain why TAB2 specifically binds NLK. Because we could not find a NLK-specific antibody that can efficiently pull down endogenous NLK, we stably expressed FLAG-tagged NLK in HEK293 cells (NLK293) to detect the interaction of NLK and TAB2 in vivo. The expression of NLK in NLK-293 cells was similar to the endogenous level of NLK in HEK293T as detected by Western blot (supplemental Fig. S1A). In NLK-293 cells, FLAG-tagged NLK can be efficiently immunoprecipitated from cell lysates, and endogenous TAB2 was detected in the immunoprecipitates pulled down by NLK (Fig. 1E). These results together indicate that NLK is a novel TAB2-binding protein.
FIGURE 1.

NLK is identified as a novel TAB2 interacting protein. A, NLK interacts with TAB2. HEK293T cells transfected with indicated plasmids were used for the co-IP assay. Interactions were determined by Western blot. TAK1-KD (kinase dead mutant), K63W, was used as a positive control of TAB2-interacting protein. B, TAB2 physically interacts with NLK. Purified His6-NLK mixed with GST-TAB2 or GST bound GSH-Sepharose beads for 1 h, then beads were washed with purification buffer 3 times. The samples from the GST pulldown assay were subjected to SDS-PAGE and Western blot. C, TAB2 specifically interacts with NLK, but not JNK and p38 MAPK. D, interactions of various NLK mutants with TAB2 are shown. A schematic map of various NLK mutants shows amino acid numbers and their interactions with TAB2. The solid rectangle stands for the identified kinase domain of NLK. E, TAB2, as well as TAK1, can be co-immunoprecipitated with NLK in NLK-293 cells. NLK-293 cells were used to perform the co-IP assay as described under “Experimental Procedures.” A Western blot was carried out with the indicated antibodies. F, the interactions of TAB2 mutants with TAK1 and NLK are shown. A schematic map of various TAB2 mutants shown with amino acid numbers and their interactions with TAK1 and NLK were shown as in D. The solid rectangle stands for the identified TAK1 interacting region of TAB2.
To understand how TAB2 interacts with NLK, we mapped the NLK binding region in TAB2 with co-immunoprecipitation assays. We generated four N-terminal truncation mutants of TAB2. They are C1 (amino acids (aa) 178–693), C2 (aa 292–693), C3 (aa 417–693), and C4 (aa 477–693) (Fig. 1F). Because all of these mutants contain the TAK1 binding region (amino acids 574–653) (29), they efficiently interacted with TAK1. However, these mutants exhibited different affinities for NLK. TAB2-C3 and C4 lost the ability to interact with NLK, indicating that NLK binding region of TAB2 is near the N terminus, which does not overlap with its interacting domain for TAK1. Therefore, it provides the possibility that TAK1 and NLK bind to TAB2 at the same time by interacting with different region.
TAB2 Scaffolds the Interaction of TAK1 and NLK
In the light of above findings, we speculated that TAB2 might play a role in the interaction of TAK1 and NLK. TAK1 activates NLK during signaling, but they cannot bind to each other as previously reported (4, 5) (Fig. 2A, left lane). However, when TAB2 was present, TAK1 and NLK were recruited to form a complex (Fig. 2A, right lane), indicating that TAB2 could act as a scaffold protein for TAK1 and NLK to bridge their interaction. Consistent with this idea, not only TAB2 was pulled down by NLK in NLK-293 cells, but also TAK1 appeared in the co-IP fractions (Fig. 1E), suggesting TAK1, TAB2, and NLK form a protein complex in vivo. Moreover, overexpressing TAB2 can also increase the affinity of NLK for TAK1 in NLK-293 cells (Fig. 2B). Most importantly, knocking down endogenous TAB2 with a specific siRNA (siTAB2-1) disrupted the interaction of TAK1 and NLK (Fig. 2C). These results support the conclusion that TAB2 acts as the scaffold protein for TAK1 and NLK.
FIGURE 2.

TAB2 bridges the interaction of TAK1 and NLK as a scaffold protein. A, NLK interacts with TAK1 in the presence of TAB2. Indicated plasmids were transfected into HEK293T cells, and a co-IP assay was done as described in Fig. 1A. B, ectopically expressed TAB2 can increase the interaction of TAK1 and NLK in vivo. NLK-293 cells transfected with TAB2 or LacZ plasmid were used to evaluate endogenous TAK1-NLK interaction. C, RNAi of TAB2 (siTAB2-1) blocks the interaction of TAK1 and NLK in vivo. NLK-293 cells were used for a co-IP assay 48 h after transfection. Co-IP experiments in B and C were done as described in Fig. 1D. Ctrl, control. D, TAB2-ΔM (ΔM) shows a lower affinity for NLK, but not TAK1, than wild type (WT) TAB2. Upper, the schematic map shows the sequences of the wild type TAB2 and TAB2-ΔM. The deletion region of amino acids 292–417 is represented with a line. Lower, the interaction of TAB2 with TAK1 and NLK was evaluated by co-IP assay. E, TAB2-ΔM is defective in bridging the interaction of TAK1 and NLK compared with wild type TAB2. HEK293T cells were used, and the co-IP experiment was done as described above.
Because TAB2-C1 and C2, not C3 and C4, bind to NLK (Fig. 1F), the region of amino acids 292–417 in TAB2 may be critical for its binding to NLK. Thus, we generated the deletion mutant of TAB2, TAB2-ΔM, which lacks the region of 292–417 (Fig. 2D, upper panel). TAB2-ΔM showed a weaker affinity for NLK than wild type TAB2 but retained its intact ability to interact with TAK1 (Fig. 2D, lower panel). Consistently, TAB2-ΔM was less capable of scaffolding TAK1 and NLK together (Fig. 2E). This conclusion was also confirmed by the results from TAB2-C1 and C2 (supplemental Fig. S2). Thus, TAB2 scaffolding ability depends on its interaction with NLK.
TAB2 Inhibits Canonical Wnt Signaling through Scaffolding TAK1 and NLK
The TAK1-NLK cascade plays a negative role in the canonical Wnt pathway (1–3, 38). The aforementioned findings suggest that TAB2 may function together with TAK1 and NLK in the negative regulation of canonical Wnt signaling. Indeed, TAB2 could effectively block the TOPFLASH activity induced by both Wnt1 and ΔN-β-catenin, a constitutive active form of β-catenin, without affecting the accumulation and nuclear translocation of free β-catenin induced by Wnt3a conditioned medium (Wnt3a CM) (Fig. 3, A and B). These results suggest that TAB2 inhibits Wnt signaling by targeting the transcriptional complex. In addition, TAB2 siRNAs (supplemental Fig. S1B) effectively increase the activation of Wnt3a CM induced TOPFLASH activity (Fig. 3C) as well as expression of Axin2 (Fig. 3D), an authentic Wnt target gene (39–41), which suggests that TAB2 could act as an inhibitor to canonical Wnt signaling in physiological conditions.
FIGURE 3.

TAB2 inhibits canonical Wnt signaling without affecting the stability of β-catenin. A, TAB2 blocks the TOPFLASH activities induced by Wnt1 or ΔN-β-catenin. Plasmids of Wnt1 or ΔN-β-catenin were used to activate TOPFLASH reporter gene in HEK293T cells. RLA, relative luciferase activity. B, TAB2 down-regulates Wnt signaling without affecting the accumulation and subcellular translocation of β-catenin. Transfected cells were treated with control or Wnt3a CM for 8 h and then for free β-catenin assays. Expression (Exp.) of transfected TAB2 and GSK3β is shown. C and D, TAB2 siRNAs efficiently increase the activation of TOPFLASH (C) and expression of Axin2 (D). siRNAs were transfected into HEK293T cells with or without TOPFLASH plasmids. 48 h later cells were treated with control or Wnt3a CM for 8 h and then lysed for a TOPFLASH assay (C) or the expression of Axin2 by quantitative real-time PCR (D). Data are representative results from at least two independent experiments. Student's t test was used to determine statistical significance based on at least three independent experiments. *, p < 0.05; **, p < 0.01. Ctrl, control.
TAB2 can assist TAK1 to activate NFκB (28). On the other hand, RelA, a component of NFκB, was reported to suppress β-catenin/TCF-dependent transcription (42). To check whether RelA is involved in the inhibition of TAB2 to Wnt signaling, we blocked the activation of RelA with IκB super repressor (IκBα-S32A/S36A) or IKKβ-KD (kinase dead). Neither of them could rescue the inhibition of TAB2 (Fig. 4, A and B), suggesting RelA might not be the effector of TAB2 in repressing Wnt signaling. Instead, TAB2 regulates the canonical Wnt pathway via NLK because NLK-KD (K155M) (Fig. 4C) blocked the effect of TAB2 on Wnt signaling. Moreover, two NLK-specific siRNAs prevented TAB2 from the suppressing Wnt pathway (Fig. 4D and supplemental Fig. S1C), suggesting that NLK is the downstream effector of TAB2.
FIGURE 4.

TAB2 inhibits canonical Wnt signaling through scaffolding TAK1 and NLK. A and B, TAB2 does not inhibit Wnt1 induced TOPFLASH activity through NFκB pathway. The IκB super repressor (ISR) (A) or the kinase dead mutant of IκB kinase (IKK-KD) (B) can block the activity of NFκB in the NFκB luciferase reporter gene assay (right panel) but could not rescue the inhibition of TAB2 to Wnt1 or ΔN-β-catenin-induced signaling. HEK293T cells were transfected with the indicated plasmids and reporter genes. C, TAK1-KD and NLK-KD rescues the inhibition of TAB2 to Wnt signaling. The activation (%) was calculated as the percentage of -fold activation induced by Wnt1 under TAB2 expression compared with control plasmid (LacZ) in the cells co-transfected with LacZ, TAK1-KD, or NLK-KD. D, NLK RNAi rescues the inhibition of TAB2 to Wnt signaling. Control or NLK siRNAs were co-transfected into HEK293T cells with the indicated plasmids. Luciferase assays were carried out 48 h after transfection. E, TAB2 and its N-terminal truncated mutants show different inhibition to Wnt signaling. F, TAB2-ΔM shows weaker inhibition to Wnt signaling compared with wild type TAB2. Plasmids of TAB2 and TAB2-ΔM were transfected in two different doses to evaluate their inhibitory effects. Right panel, TAB2 and TAB2-ΔM have the same efficiency in activating NFκB signaling. TOPFLASH assays were done with HEK293T cells transfected with the indicated plasmids. In all TOPFLASH assays, IκB super repressor were co-transfected to avoid the interference caused by NF-κB, and GFP was co-transfected to normalize the transfection efficiency. Data are representative results from at least two independent experiments. Bars depict the relative luciferase activity (RLA) to the basal activity from cells transfected with LacZ, and error bars were calculated from duplicate samples. Student's t test was used to determine statistical significance based on at least three independent experiments. **, p < 0.01.
In contrast to wild type TAB2, those N-terminal-truncated mutants of TAB2 exhibited inhibitory effects corresponding to their scaffolding abilities (Fig. 4E). Moreover, TAB2-ΔM, which is defective in scaffolding TAK1 and NLK, showed reduced inhibition of Wnt signaling compared with the wild type TAB2 (Fig. 4F, left panel), whereas TAB2 and TAB2-ΔM have no difference in helping TAK1 to activate NFκB (Fig. 4F, right panel) and JNK (supplemental Fig. S3). Thus, the inhibition of TAB2 to Wnt signaling relies on its ability to scaffold the complex of TAK1 and NLK.
TAB2 Scaffolds TAK1 and NLK to Regulate Functions of NLK
As previous reports, NLK kinase activity could be examined by an in vitro kinase assay according to the level of its autophosphorylation (2, 9, 38) or phosphorylation of LEF1/TCFs in vitro (38). To further clarify the role of TAB2 in the TAK1-NLK cascade, an in vitro kinase assay of NLK activity was performed. TAK1 alone cannot activate NLK (Fig. 5A), as previously reported (2). However, NLK kinase activity was markedly increased in the presence of TAK1 with TAB2 but not TAB2-ΔM (Fig. 5A). However, JNK activation by TAB2 or TAB2-ΔM via TAK1 was equal (supplemental Fig. S3), indicating that TAB2 scaffolds TAK1 and NLK to specifically potentiate NLK kinase activity. NLK was shown to regulate Wnt signaling by phosphorylating LEF1/TCFs. Activation of NLK by upstream activators could enhance the phosphorylation of LEF1/TCFs (38). We confirmed the finding and found that TAB2, not TAB2-ΔM, could increase the phosphorylation of LEF1 (Fig. 5B). Moreover, the phosphorylation mediated by NLK can augment polyubiquitylation of LEF1/TCFs by NARF, an E3 ubiquitin ligase (21). We examined the effect of TAB2 in ubiquitylation of LEF1. In the presence of ubiquitin, only the combination of TAK1, TAB2, and NLK markedly increased the polyubiquitylation of LEF1, whereas NLK-KD did not work as NLK in the same experiment (Fig. 5C), suggesting TAK1 and TAB2 cooperated to potentiate the ubiquitylation of LEF1 through activating NLK kinase activity. Furthermore, neither TAB2-ΔM nor -C3 worked as well as the full-length TAB2 in stimulating LEF1 ubiquitylation (Fig. 5D). Thus, we conclude that the interaction of TAB2 and NLK is indispensable for phosphorylating or polyubiquitylating LEF1 induced by the TAK1-NLK cascade.
FIGURE 5.

TAB2 scaffolds TAK1 and NLK to promote functions of NLK. A, TAB2, but not TAB2-ΔM, cooperates with TAK1 in activating kinase activity of NLK. B, overexpression of TAB2, but not TAB2-ΔM (ΔM), leads to an increase in phosphorylation of LEF1. WT, wild type. FLAG-tagged NLK or LEF1 was co-transfected with other plasmids and immunoprecipitated for in vitro kinase assay (IVK) of NLK autophosphorylation (A) or LEF1 phosphorylation (B). The results of in vitro kinase assay were visualized with phosphorimaging. C and D, shown are the effects of TAB2, TAB2-ΔM, and TAB2-C3 on the TAK1-NLK cascade-mediated LEF1 polyubiquitylation (Ub). To check the polyubiquitylated LEF1, the indicated plasmids were co-transfected into HEK293T cells, and LEF1-FLAG was immunoprecipitated with FLAG antibody and resolved to Western blot. Polyubiquitylated LEF1 (immunoblotted against HA) was shown at 100–250 kDa.
Wnt3a-conditioned Medium Enhances the Formation of TAK1·TAB2·NLK Complex
Both canonical and non-canonical Wnt proteins have been reported to regulate the TAK1-NLK pathway (4, 7, 38, 43). In our study we found that Wnt3a CM could enhance TAB2 binding with NLK (Fig. 6A). Consistently, Wnt3a CM treatment also increased the interaction of TAK1 and NLK (Fig. 6B) and led to the activation of NLK kinase activity (Fig. 6C), indicating that Wnt signals could promote the formation of TAK1·TAB2·NLK complex. As knockdown of TAB2 could disrupt the complex of TAK1 and NLK (Fig. 2C), siRNA of TAB2 (siTAB2-1) also inhibited the effects of Wnt on NLK (Fig. 6C), suggesting that TAB2 is required to mediate the TAK1-NLK pathway in this process. Thus, Wnt proteins may promote the formation of TAK1·TAB2·NLK complex to activate the TAK1-NLK cascade as a negative feedback mechanism to down-regulate canonical Wnt signaling (38).
FIGURE 6.

Formation of TAK1·TAB2·NLK complex is induced by Wnt3a CM. A and B, Wnt3a stimulation increases the interaction of NLK with TAB2 (A) and TAK1 (B), respectively. NLK-293 cells were treated with Wnt3a CM for the indicated times, then cells were lysed and immunoprecipitated with TAB2 specific antibody (A) or FLAG affinity gel (B). C, NLK kinase activity is potentiated by Wnt3a CM stimulation. NLK-293 cells were transfected with control or TAB2 siRNA (siTAB2-1). 48 h later, cells were treated with control or Wnt3a CM for 6 h, then cells were lysed and immunoprecipitated with FLAG affinity gel. Immunoprecipitates were used for the NLK kinase assay. Ctrl, control.
TAB2 has been reported as an adaptor protein to link TAK1 into several signaling complexes (28, 32, 33) in which TAK1 can be activated by the polyubiquitin chain (31, 44). On the other hand, a recent study showed that TAB2 could also bridge TAK1 with its substrate, such as RCAN1 (36). In the TAK1-NLK cascade, TAK1 activates NLK, but these two proteins do not directly interact with each other (4, 5). The finding that TAB2 can scaffold TAK1 and NLK fills the gap and declares the role of TAB2 in directing TAK1 signaling to its specific downstream effector. In a recent study TAB2 was also used to specifically potentiate signaling between TAK1 and NLK. However, it did not touch the mechanism (7). Nevertheless, the results from Takada et al. (7) are consistent with ours that TAB2 can function in the TAK1-NLK cascade. TAB2-ΔM with defects in scaffolding NLK exhibits functional loss in TAK1-NLK signaling, but it works almost the same as wild type TAB2 in TAK1 stimulated NFκB and JNK activity (Fig. 4F and supplemental Fig. S3). These facts indicate that the interaction of NLK and TAB2 endows TAB2 with the ability to modulate the TAK1-NLK pathway, which is independent of its other functions with TAK1.
In addition, TAB3 was identified as a TAB2 -like molecule, which is highly homologous to TAB2 (33, 45). Subsequently, several works proved that TAB3 has some redundant function with TAB2 both in vitro and in vivo (29, 33, 34, 46, 47). In our work we also studied the function of TAB3 in the TAK1-NLK cascade. As expected, TAB3, with a similar structure of TAB2, can also interact with NLK, bridge the interaction of TAK1 and NLK, and inhibit β-catenin-activated TOPFLASH activity as TAB2 (data not shown), which indicates that TAB3 may work as TAB2 in mediating TAK1-NLK signaling. However, more evidence is required to further examine this function of TAB3 in vivo.
Scaffold proteins are usually regulated by particular extracellular stimulus (25–27). STAT3 was reported to enhance TAK1-NLK signaling only in response to interleukin-6 (5). With our findings, TAB2 is involved in the signaling of TAK1-NLK induced by canonical Wnt (Fig. 6). Wnt3a stimulation can strengthen TAK1·TAB2·NLK complex formation via enhancing the interaction of TAB2 and NLK (Fig. 6, A and B). The function of NLK is highly related with Wnt signaling, and a number of reports show that Wnt regulates NLK (1–4, 43, 48, 49). Based on our and others' findings, as depicted in Fig. 7, Wnt proteins can activate LEF1/TCF-dependent transcription of a series of target genes (15–17, 50) and, on the other hand, activate different intracellular signals (51, 52), among which the TAK1-NLK pathway functions as a negative modulator through regulating LEF1/TCFs (38). In this model, we describe that Wnt can induce TAK1·TAB2·NLK complex formation, which in turn leads to NLK activation and inhibition on LEF1/TCF family transcriptional factors. We believe that canonical Wnt might use this mechanism to balance the signaling itself. The canonical Wnt pathway works in several developmental processes and tumorigenesis, and TAB2-mediated activation of the TAK1-NLK pathway may play some role in particular biological events.
FIGURE 7.
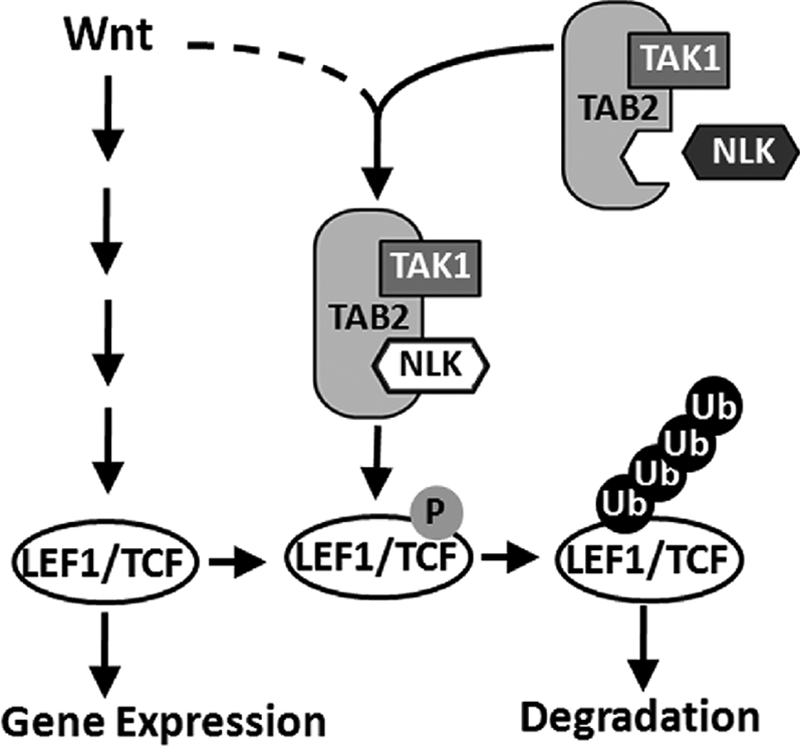
Model of TAK1·TAB2·NLK complex negatively regulating canonical Wnt pathway.
Supplementary Material
Acknowledgments
We greatly appreciated the gift of IκB super repressor and ubiquitin-HA plasmid from Dr. C. Wang and B. Sun (Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Shanghai), respectively. We are grateful to D. Wu for critical reading and commenting on this paper.
This work was supported by Ministry of Science and Technology of China Grants 2010CB912100 and 2007CB914500, by Natural Science Foundation of China Grants 30821065, 30930052, and 90813024 (to L. L.) and 30600305 (to J. W.), and by Science and Technology Commission of Shanghai Municipality grants.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
- MAPK
- MAP kinase
- siRNA
- small interfering RNA
- TAK1
- transforming growth factor-β-activated kinase 1
- NLK
- Nemo-like kinase
- TCF
- T-cell factors
- LEF1
- lymphoid enhancer factor 1
- TAB2
- TAK1-binding protein 2
- TAB1
- TAK1-binding protein 1
- CM
- conditioned medium
- HA
- hemagglutinin
- JNK
- c-Jun N-terminal kinase
- IP
- immunoprecipitation
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- KD
- kinase dead
- GST
- glutathione S-transferase.
REFERENCES
- 1.Behrens J. (2000) J. Cell Sci. 113, 911–919 [DOI] [PubMed] [Google Scholar]
- 2.Ishitani T., Ninomiya-Tsuji J., Nagai S., Nishita M., Meneghini M., Barker N., Waterman M., Bowerman B., Clevers H., Shibuya H., Matsumoto K. (1999) Nature 399, 798–802 [DOI] [PubMed] [Google Scholar]
- 3.Meneghini M. D., Ishitani T., Carter J. C., Hisamoto N., Ninomiya-Tsuji J., Thorpe C. J., Hamill D. R., Matsumoto K., Bowerman B. (1999) Nature 399, 793–797 [DOI] [PubMed] [Google Scholar]
- 4.Kanei-Ishii C., Ninomiya-Tsuji J., Tanikawa J., Nomura T., Ishitani T., Kishida S., Kokura K., Kurahashi T., Ichikawa-Iwata E., Kim Y., Matsumoto K., Ishii S. (2004) Genes Dev. 18, 816–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kojima H., Sasaki T., Ishitani T., Iemura S., Zhao H., Kaneko S., Kunimoto H., Natsume T., Matsumoto K., Nakajima K. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 4524–4529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohkawara B., Shirakabe K., Hyodo-Miura J., Matsuo R., Ueno N., Matsumoto K., Shibuya H. (2004) Genes Dev. 18, 381–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takada I., Mihara M., Suzawa M., Ohtake F., Kobayashi S., Igarashi M., Youn M. Y., Takeyama K., Nakamura T., Mezaki Y., Takezawa S., Yogiashi Y., Kitagawa H., Yamada G., Takada S., Minami Y., Shibuya H., Matsumoto K., Kato S. (2007) Nat. Cell Biol. 9, 1273–1285 [DOI] [PubMed] [Google Scholar]
- 8.Yamaguchi K., Shirakabe K., Shibuya H., Irie K., Oishi I., Ueno N., Taniguchi T., Nishida E., Matsumoto K. (1995) Science 270, 2008–2011 [DOI] [PubMed] [Google Scholar]
- 9.Brott B. K., Pinsky B. A., Erikson R. L. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 963–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coulombe P., Meloche S. (2007) Biochim. Biophys. Acta 1773, 1376–1387 [DOI] [PubMed] [Google Scholar]
- 11.Shin T. H., Yasuda J., Rocheleau C. E., Lin R., Soto M., Bei Y., Davis R. J., Mello C. C. (1999) Mol. Cell 4, 275–280 [DOI] [PubMed] [Google Scholar]
- 12.Röttinger E., Croce J., Lhomond G., Besnardeau L., Gache C., Lepage T. (2006) Development 133, 4341–4353 [DOI] [PubMed] [Google Scholar]
- 13.Hyodo-Miura J., Urushiyama S., Nagai S., Nishita M., Ueno N., Shibuya H. (2002) Genes Cells 7, 487–496 [DOI] [PubMed] [Google Scholar]
- 14.Ishitani T., Ninomiya-Tsuji J., Matsumoto K. (2003) Mol. Cell. Biol. 23, 1379–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Logan C. Y., Nusse R. (2004) Annu. Rev. Cell Dev. Biol. 20, 781–810 [DOI] [PubMed] [Google Scholar]
- 16.Moon R. T., Kohn A. D., De Ferrari G. V., Kaykas A. (2004) Nat. Rev. Genet 5, 691–701 [DOI] [PubMed] [Google Scholar]
- 17.Reya T., Clevers H. (2005) Nature 434, 843–850 [DOI] [PubMed] [Google Scholar]
- 18.Städeli R., Hoffmans R., Basler K. (2006) Curr. Biol. 16, R378–R385 [DOI] [PubMed] [Google Scholar]
- 19.Shitashige M., Hirohashi S., Yamada T. (2008) Cancer Sci. 99, 631–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gan X. Q., Wang J. Y., Xi Y., Wu Z. L., Li Y. P., Li L. (2008) J. Cell Biol. 180, 1087–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamada M., Ohnishi J., Ohkawara B., Iemura S., Satoh K., Hyodo-Miura J., Kawachi K., Natsume T., Shibuya H. (2006) J. Biol. Chem. 281, 20749–20760 [DOI] [PubMed] [Google Scholar]
- 22.Shibuya H., Yamaguchi K., Shirakabe K., Tonegawa A., Gotoh Y., Ueno N., Irie K., Nishida E., Matsumoto K. (1996) Science 272, 1179–1182 [DOI] [PubMed] [Google Scholar]
- 23.Ninomiya-Tsuji J., Kishimoto K., Hiyama A., Inoue J., Cao Z., Matsumoto K. (1999) Nature 398, 252–256 [DOI] [PubMed] [Google Scholar]
- 24.Irie T., Muta T., Takeshige K. (2000) FEBS Lett. 467, 160–164 [DOI] [PubMed] [Google Scholar]
- 25.Dhanasekaran D. N., Kashef K., Lee C. M., Xu H., Reddy E. P. (2007) Oncogene 26, 3185–3202 [DOI] [PubMed] [Google Scholar]
- 26.Dard N., Peter M. (2006) BioEssays 28, 146–156 [DOI] [PubMed] [Google Scholar]
- 27.Morrison D. K., Davis R. J. (2003) Annu. Rev. Cell Dev. Biol. 19, 91–118 [DOI] [PubMed] [Google Scholar]
- 28.Takaesu G., Kishida S., Hiyama A., Yamaguchi K., Shibuya H., Irie K., Ninomiya-Tsuji J., Matsumoto K. (2000) Mol. Cell 5, 649–658 [DOI] [PubMed] [Google Scholar]
- 29.Besse A., Lamothe B., Campos A. D., Webster W. K., Maddineni U., Lin S. C., Wu H., Darnay B. G. (2007) J. Biol. Chem. 282, 3918–3928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ono K., Ohtomo T., Sato S., Sugamata Y., Suzuki M., Hisamoto N., Ninomiya-Tsuji J., Tsuchiya M., Matsumoto K. (2001) J. Biol. Chem. 276, 24396–24400 [DOI] [PubMed] [Google Scholar]
- 31.Wang C., Deng L., Hong M., Akkaraju G. R., Inoue J., Chen Z. J. (2001) Nature 412, 346–351 [DOI] [PubMed] [Google Scholar]
- 32.Takaesu G., Ninomiya-Tsuji J., Kishida S., Li X., Stark G. R., Matsumoto K. (2001) Mol. Cell. Biol. 21, 2475–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ishitani T., Takaesu G., Ninomiya-Tsuji J., Shibuya H., Gaynor R. B., Matsumoto K. (2003) EMBO J. 22, 6277–6288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kanayama A., Seth R. B., Sun L., Ea C. K., Hong M., Shaito A., Chiu Y. H., Deng L., Chen Z. J. (2004) Mol. Cell 15, 535–548 [DOI] [PubMed] [Google Scholar]
- 35.Ea C. K., Deng L., Xia Z. P., Pineda G., Chen Z. J. (2006) Mol. Cell 22, 245–257 [DOI] [PubMed] [Google Scholar]
- 36.Liu Q., Busby J. C., Molkentin J. D. (2009) Nat. Cell Biol. 11, 154–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen T., Li M., Ding Y., Zhang L. S., Xi Y., Pan W. J., Tao D. L., Wang J. Y., Li L. (2009) J. Biol. Chem. 284, 6683–6689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smit L., Baas A., Kuipers J., Korswagen H., van de Wetering M., Clevers H. (2004) J. Biol. Chem. 279, 17232–17240 [DOI] [PubMed] [Google Scholar]
- 39.Jho E. H., Zhang T., Domon C., Joo C. K., Freund J. N., Costantini F. (2002) Mol. Cell. Biol. 22, 1172–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yan D., Wiesmann M., Rohan M., Chan V., Jefferson A. B., Guo L., Sakamoto D., Caothien R. H., Fuller J. H., Reinhard C., Garcia P. D., Randazzo F. M., Escobedo J., Fantl W. J., Williams L. T. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 14973–14978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu W., Dong X., Mai M., Seelan R. S., Taniguchi K., Krishnadath K. K., Halling K. C., Cunningham J. M., Boardman L. A., Qian C., Christensen E., Schmidt S. S., Roche P. C., Smith D. I., Thibodeau S. N. (2000) Nat. Genet. 26, 146–147 [DOI] [PubMed] [Google Scholar]
- 42.Masui O., Ueda Y., Tsumura A., Koyanagi M., Hijikata M., Shimotohno K. (2002) Int. J. Mol. Med. 9, 489–493 [PubMed] [Google Scholar]
- 43.Ishitani T., Kishida S., Hyodo-Miura J., Ueno N., Yasuda J., Waterman M., Shibuya H., Moon R. T., Ninomiya-Tsuji J., Matsumoto K. (2003) Mol. Cell. Biol. 23, 131–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xia Z. P., Sun L., Chen X., Pineda G., Jiang X., Adhikari A., Zeng W., Chen Z. J. (2009) Nature 461, 114–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muñoz-Sanjuán I., Bell E., Altmann C. R., Vonica A., Brivanlou A. H. (2002) Development 129, 5529–5540 [DOI] [PubMed] [Google Scholar]
- 46.Cheung P. C., Nebreda A. R., Cohen P. (2004) Biochem. J. 378, 27–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jin G., Klika A., Callahan M., Faga B., Danzig J., Jiang Z., Li X., Stark G. R., Harrington J., Sherf B. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 2028–2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thorpe C. J., Moon R. T. (2004) Development 131, 2899–2909 [DOI] [PubMed] [Google Scholar]
- 49.Zeng Y. A., Verheyen E. M. (2004) Development 131, 2911–2920 [DOI] [PubMed] [Google Scholar]
- 50.Clevers H. (2006) Cell 127, 469–480 [DOI] [PubMed] [Google Scholar]
- 51.Gordon M. D., Nusse R. (2006) J. Biol. Chem. 281, 22429–22433 [DOI] [PubMed] [Google Scholar]
- 52.Veeman M. T., Axelrod J. D., Moon R. T. (2003) Dev. Cell 5, 367–377 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.