

Abstract
TEA domain transcription factor-1 (TEAD-1) is essential for proper heart development and is implicated in cardiac specific gene expression and the hypertrophic response of primary cardiomyocytes to hormonal and mechanical stimuli, and its activity increases in the pressure-overloaded hypertrophied rat heart. To investigate whether TEAD-1 is an in vivo modulator of cardiac specific gene expression and hypertrophy, we developed transgenic mice expressing hemagglutinin-tagged TEAD-1 under the control of the muscle creatine kinase promoter. We show that a sustained increase in TEAD-1 protein leads to an age-dependent dysfunction. Magnetic resonance imaging revealed decreases in cardiac output, stroke volume, ejection fraction, and fractional shortening. Isolated TEAD-1 hearts revealed decreased left ventricular power output that correlated with increased βMyHC protein. Histological analysis showed altered alignment of cardiomyocytes, septal wall thickening, and fibrosis, although electrocardiography displayed a left axis shift of mean electrical axis. Transcripts representing most members of the fetal heart gene program remained elevated from fetal to adult life. Western blot analyses revealed decreases in p-phospholamban, SERCA2a, p-CX43, p-GSK-3α/β, nuclear β-catenin, GATA4, NFATc3/c4, and increased NCX1, nuclear DYKR1A, and Purα/β protein. TEAD-1 mice did not display cardiac hypertrophy. TEAD-1 mice do not tolerate stress as they die over a 4-day period after surgical induction of pressure overload. These data provide the first in vivo evidence that increased TEAD-1 can induce characteristics of cardiac remodeling associated with cardiomyopathy and heart failure.
Keywords: Diseases, DNA/Transcription, Exchange/Sodium/Calcium, Gene/Transcription, Phosphorylation/Kinases/Serine-Threonine, Signal Transduction/Protein Kinases/Serine/Threonine, Beta-Catenin, GSK-3, TEAD-1, Cardiac Muscle
Introduction
Heart disease develops over a long period of time, with patients relatively unaware of their disease status until the functional properties of the heart have been significantly compromised leading to heart failure. Approximately 5 million people are living with heart failure in the United States, making it one of the leading causes of morbidity and mortality. The heart adapts to an increased workload as a result of physiological or pathological stimuli, such as exercise or hypertension, respectively, by undergoing a complex remodeling process characterized by significant changes in cardiac chamber size, shape, wall thickness (or hypertrophy, defined as an increase in cell size or length), and contractile function (1–4). These functional changes are driven, at the molecular level, by significant alterations in signal transduction, transcription of cardiac specific genes, and protein expression and modification (2, 5–9). This adaptive phase enables the heart to maintain a certain level of function and appears to contribute to the slow progression of disease. Unfortunately, the initial beneficial adaptations to increased stress eventually transition to a pathological phase, which is distinguished by altered alignment of cardiomyocytes, fibrosis, arrhythmias, and eventually heart failure as defined as an insufficiency of the heart to adequately pump blood to meet systemic demands (3, 4). Because the heart is a post-mitotic organ with very low levels of cellular replacement, a better understanding of the mechanistic basis underlying cardiac remodeling in response to pathological stress is essential (10).
Remodeling of the adult heart in response to disease states is accompanied by the re-expression of a set of genes primarily expressed in the fetal heart (fetal gene program: αMyHC, βMyHC, skeletal muscle α-actin, atrial natriuretic peptide, and brain natriuretic peptide (BNP)3). In association with the induction of the fetal gene program is the activation of a set of transcription factors, such as GATA4, NFAT, and MEF2 (myocyte enhancer factor-2), which have previously been demonstrated to be necessary for proper heart development, thereby raising the possibility that they are responsible for fetal gene re-expression in the remodeling heart (5, 7–9). In this regard, it is possible that TEAD-1 may participate in adult-stage disease-induced cardiac remodeling and fetal gene re-expression as TEAD-1 gene inactivation results in embryonic lethality (day E11.5) due to defects in heart morphology, thereby revealing its critical role in fetal heart development (11, 12).
The vertebrate TEAD genes encode a family of transcription factors that include TEAD-1 (NTEF-1/TEF-1), TEAD-2 (ETEF-1/TEF-4), TEAD-3 (DTEF-1/TEF-5), and TEAD-4 (RTEF-1/TEF-3). All TEAD family members contain an evolutionarily conserved 72-amino acid DNA binding domain (TEAD) that is found in plant (ABAA), fly (Scalloped), and yeast (TEC1) transcription factors. TEAD proteins have been shown to serve a regulatory role by binding to canonical MCAT elements (5′-CATTCC(T/A)-3′), located in the promoter/enhancer region of numerous striated and smooth muscle genes, and by combinatorial interactions with adjacently bound transcriptional regulators (MEF2, SRF, poly(ADP-ribose)polymerase, and MAX)), ubiquitous and/or tissue-specific coactivators (TAZ (transcriptional coactivator with PDZ-binding motif), YAP65 (Yes-associated protein 65 kDa), P160 steroid hormone receptor family of coactivators (Src1, TIF2, and RAC3), TONDU/Vgl-1 (Vestigial-like protein-1), and Vgl2–4, and modification by protein kinases (13–15). Although most mammalian tissues express more than one TEAD protein during embryonic and adult life, several recent studies involving single or double TEAD gene inactivation (TEAD-1, TEAD-2, and TEAD-4) have shown that these transcription factors serve both unique as well as overlapping functional roles during embryonic development (11, 12, 16–18).
Although TEAD-1 has been shown to play a vital role in fetal heart development, a definitive role for TEAD-1 in the postnatal heart has not been established. Nevertheless, evidence gathered from numerous investigations support a role for TEAD-1 in postnatal cardiac specific gene expression and fetal gene re-expression in the heart and primary cardiomyocytes in response to hormonal and mechanical stimuli. Studies using primary rat cardiomyocytes have shown a dependence on intact MCAT elements for the basal expression of α-sk-actin, αMyHC, βMyHC, and BNP reporter genes, TEAD-1 transactivation, and responsiveness to stimulation by α1-adrenergic signaling (19–25). An increase in TEAD-1 binding at αMyHC and βMyHC canonical MCAT elements was shown to occur in the pressure-overloaded rat heart (26). In addition, Hasegawa et al. (27) have shown that the inducible expression of βMyHC reporter genes directly injected into pressure-overloaded rat hearts was conferred by an A/T-rich element that we have recently shown avidly binds TEAD proteins (28). It is also worth noting that we have demonstrated that the TEAD proteins are capable of binding to a subset of muscle gene A/T-rich and MEF2 elements and of transcriptional regulation of a MEF2-dependent reporter gene (28). Taking into account the latter finding, it is notable that TEAD-1 has been shown to physically interact with MEF2 and SRF to collaboratively activate cardiac gene expression (29, 30). Furthermore, because transcription factors important for fetal heart development, such as MEF2 and SRF, are re-employed in adult heart remodeling in response to disease-induced work overload, it is significant that TEAD-1 is required for proper fetal heart development and that it displays an expression pattern similar to MEF2 and SRF in the developing embryo. It is also of relevance that the cardiac specific transgenic overexpression of TEAD-4 was shown to result in cardiac conduction defects; however, whether increased TEAD-4 induced remodeling of the heart and/or altered contractile function was not assessed (31).
To investigate the in vivo role of TEAD-1 in the postnatal heart, we developed transgenic mice expressing HA-tagged TEAD-1 under the control of the muscle creatine kinase (MCK) promoter (32). Our current results provide the first in vivo evidence that TEAD-1 overexpression in the mouse heart can induce an age-dependent dysfunction characterized by decreased cardiac output, stroke volume, ejection fraction, fractional shortening, left ventricular power output, and increased fibrosis. Furthermore, we provided evidence that a persistent increase in TEAD-1 protein directly and/or indirectly activated the fetal gene program and leads to increased nuclear DYRK1A (dual specificity tyrosine phosphorylation-regulated kinase 1A) protein and GSK-3α/β activation resulting in decreased nuclear β-catenin, GATA4, and NFATc3/c4 protein. Our data suggest that TEAD-1 activated an anti-hypertrophy program because GSK-3β and DYRK1A have been shown to act as negative regulators of cardiac hypertrophy. When considered collectively, these data indicate that TEAD-1-overexpressing mice may provide a valuable in vivo model to study the time course progression of cardiac remodeling and dysfunction.
EXPERIMENTAL PROCEDURES
Generation of Transgenic Mice
TEAD-1 transgenic lines were generated by pronuclear injection, and three independent lines were chosen for analysis. The transgene was designed as described previously (32). In short, the mouse TEAD-1 cDNA containing an in-frame HA tag at its 5′-end was subcloned at the 3′-end of the MCK regulatory sequences, which consist of a 3.3-kb promoter, untranslated exon 1, intron 1 containing a muscle-specific enhancer, and exon 2 that terminated prior to the MCK start site. The TEAD-1 cDNA was followed by the SV40 polyadenylation signal. The incorporation of a HA tag allowed us to easily make a distinction between endogenous TEAD-1 protein and the transgene-encoded HA-TEAD-1 protein by Western blot analysis. Transgenic founders and offspring were identified and genotyped by PCR using the following primers: TEAD1 primer, 5′-ATCCATGCTTGTTACCTTCAG-3′, and Tag primer, 5′-ACTACAAGGACGATGACAAG-3′. Transgenic mice were bred with nontransgenic (C57/Bl6 × DBA/2) F1 mice and analyzed at 2, 5, and 10 months of age. All mice were housed in an Association for the Assessment and Accreditation of Laboratory Animal Care International accredited institution according to animal care and use guidelines.
Protein Isolation for Western Blots
For total protein extracts, hearts were homogenized in 1.0 ml of buffer (50 mm Tris, pH 7.5, 10 mm EGTA, 5 mm EDTA, phenylmethylsulfonyl fluoride, phosphatase inhibitor mixture 1 and 2, and proteinase inhibitor mixture (Sigma)). For isolation of nuclear protein, the homogenate was centrifuged at 14,000 × g for 10 min at 4 °C; the cytoplasmic fraction (supernatant) was removed, and the pellet was resuspended in buffer. Sonication of homogenates was followed by addition of 1% Triton X-100 and incubation on ice for 30 min. Membrane fractions were isolated by homogenizing tissues in 1.5 ml of buffer (50 mm Tris-HCl, pH 7.4, 50 mm mannitol, 2 mm EDTA), centrifugation at 500 × g for 10 min at 4 °C, mixing the supernatant with 3.2 ml of buffer (50 mm Tris-HCl, pH 7.4, 300 mm mannitol, 2 mm EDTA), and centrifugation at 40,000 rpm for 45 min. Protein concentrations were determined using a protein assay kit (Bio-Rad), and extracts were stored at −80 °C. Protein separation and analysis were performed as described previously (28). Experimental and standard bands were scanned and quantified using the MultiGauge software, and the experimental data were normalized by dividing by the signal of the standard. The antibodies used in this study are as follows: TEAD-1 (BD Transduction Laboratories); HA (Cell Signaling); Akt, p-Akt, Akt1, and Akt2 (Cell Signaling); GSK-3α/β (Santa Cruz Biotechnology); p-Ser-GSK-3α and p-Ser-GSK-3β (Cell Signaling); glyceraldehyde-3-phosphate dehydrogenase and β-catenin (Cell Signaling); NFATc3 and NFATc4 (Santa Cruz Biotechnology); histone H1 (Santa Cruz Biotechnology); IP90 anti-peptide antibody (Abcam); and horseradish peroxidase-linked anti-rabbit IgG and anti-mouse IgG (Cell Signaling).
Protein Extraction for High Resolution Gel Electrophoresis
Protein extraction and high resolution gel electrophoresis for MyHC isoform separation was performed as described previously (33). Briefly, 50 mg of heart muscle tissue of adult wild type and TEAD-1 Tg mice was processed, and 0.75 μg of total protein was separated on an 8% acrylamide gel by 104 V for 24 h at 4 °C. The gel was then stained with SilverSNAP stain (Thermo Scientific) according to the manufacturer's recommendations.
In Vivo Cardiac Magnetic Resonance Imaging (MRI) for Mice
In vivo MRI was performed on a 7T Varian Unity INOVA horizontal bore MRI system (Varian Inc., Palo Alto, CA) equipped with a gradient insert (400 milliteslas/m, 115 mm inner diameter). A quadrature-driven birdcage radiofrequency coil (38 mm inner diameter) was employed for the imaging of mice. Animals were anesthetized with 1–2.5% isoflurane in oxygen via a nose cone and placed into the coil in prone position. Electrodes were attached to right front paw and left leg for ECG gating. A respiratory sensor was placed on abdomen for respiratory gating and monitoring of vital signs. Body temperature was maintained at 37 °C with warm air circulating in the magnet bore. Physiological monitoring and gating were performed using a Physiological Monitoring System (SA Instruments, Inc., Stony Brook, NY).
ECG-triggered cine images in both long and short axis views were acquired at 12 equally spaced time points corresponding to different cardiac phases throughout the entire cardiac cycle. The resulting temporal resolution is in a range of 10–13 ms per frame. A long axis coronal slice was acquired to view left and right ventricles. Short axis view was chosen perpendicular to the long axis of the left ventricle (from root of aorta to apex). A short axis midventricular slice was imaged at 50% of the distance between the atrioventricular valve plane and the apex. For quantitative measurements of left ventricle volumes, 5 or 6 consecutive short axis slices were imaged from basal to apical levels for coverage of the entire left ventricle. Cine images were acquired using gated gradient echo sequence with the following parameters: flip angle, 30°; echo time, 1.2 ms; slice thickness, 1.0 mm; image matrix, 128 × 192; and a field of view of 30 × 40 mm2 for long axis view and 30 × 30 mm2 for short axis view. Repetition time was adjusted according to the R-R interval of the heart. The acquisition time for each cine imaging was ∼4 min with two signal averages. Data were processed and zero-filled into a 512 × 512 data matrix using VnmrJ (Varian, Inc.; Palo Alto, CA). Septal wall thickness was determined using a short axis midventricular slice at 0-ms delay after the R wave and averaging five measurements per heart. Left ventricle (LV) ejection fraction was calculated by using the formula (EDV − ESV/EDV) × 100, where EDV is the end diastolic volume; ESV is the end systolic volume. LV volumes at different cardiac phase cycles were calculated by summing the endocardial areas of the short axis slices covering basal to apical levels and then multiplying by the slice thickness. The lowest volume obtained was considered as the end systolic volume, and the largest volume obtained was the end diastolic volume. The septal wall thickness, LV volume, and ejection fraction were measured using VnmrJ (Varian Inc.) and Segment (34). All values are expressed as means ± S.D. Statistical analyses were performed using paired or unpaired Student's t tests. Significance was accepted as p < 0.05.
Power Output Measurements on Isolated Working Hearts
For isolated whole heart experiments, mice were anesthetized with sodium pentobarbital (30 mg/kg body weight intraperitoneally). After treatment with heparin (100 units), hearts were quickly removed and the aorta cannulated followed by 10 min of Langendorff perfusion with oxygenated perfusion buffer; the pulmonary vein was then cannulated, and hearts were switched to a working heart system (35). Heart rate, blood pressure, aortic flow, and coronary flow were constantly monitored at a preload of 20 cm H2O at 32 °C. Left ventricular power was calculated using Equation 1,
where MAP is mean arterial pressure; LAP is left atrial pressure; and CO is cardiac output (36).
The relative expression of each MyHC isoform was determined for each whole heart following power output measurements. Once perfusion was completed, hearts were removed from cannulas, and the ventricles were isolated, weighed, freeze-clamped, and then stored at −80 °C. Frozen ventricular samples then were used for SDS-PAGE analysis of relative MyHC isoform expression. The gel electrophoresis procedure was similar to one described previously (37, 38). The gels were prepared with 3.5% acrylamide in the stacking gel and 12% acrylamide in the resolving gel. Myocyte proteins were separated at constant current (12 mA) for 8.5 h. The separated proteins were fixed in an acid/alcohol solution followed by glutaraldehyde. MyHC isoforms were visualized by ultrasensitive silver staining, and then gels were dried and stored between Mylar sheets. The relative expression of each MyHC isoform was determined by using QuantiScan (Biosoft) software and an Epson scanner to measure the relative intensity and area of each MyHC band.
Electrocardiographic Methods
Mice were positioned in sternal recumbency. The ECG leads were placed according to the hexaxial lead system using 27-gauge platinum subdermal needle electrodes. The needles were inserted subcutaneously, as distal on the limbs as possible. Measurements of intervals and amplitudes were performed using a paper recording speed of 200 mm/s and a calibration of 2 cm/mV. Evaluations of heart rate and rhythm disturbances were made from recordings obtained at 50 mm/s. The mean electrical axis was calculated from the net deflections of leads I and III using previously described methods (39). The results between control and transgenic mice were analyzed using one-way analysis of variance. When indicated, post hoc analysis was performed using the Tukey Kramer multiple comparison test to determine which values were significant.
RNA Samples and Isolation
Hearts of adult wild type and transgenic (RTN12) mice were surgically removed and transferred to RNA-later (Ambion) according to the manufacturer's instructions. Total RNA was isolated from ∼20 to 50 mg of tissue from each animal using RNA STAT-60 according to the manufacturer's instructions (TelTest Inc.). Equal amounts of total RNA from each of four replicate tissue samples were pooled for cDNA synthesis after RNA quality control tests.
RNA Quality Control
Immediately prior to cDNA synthesis, the purity and concentration of RNA samples were determined from A260/280 readings using a dual beam UV spectrophotometer, and RNA integrity was determined by capillary electrophoresis using the RNA 6000 Nano Lab-on-a-Chip kit and the Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA) as per the manufacturer's instructions.
Reverse Transcription, PCR, and Real Time Quantitative PCR Analysis
Single strand cDNA was synthesized from 0.02 to 2.0 μg for each total RNA sample using the High Capacity cDNA synthesis kit (Applied Biosystems), according to the manufacturer's instructions. Approximately 20 ng of each cDNA (9 μl) was mixed with 10 μl of TaqMan® Gene Expression 2× PCR Master Mix (Applied Biosystems, Roche Applied Science) and 1 μl of each indicated TaqMan® Gene Expression Assay (Applied Biosystems) in 384-well plates and analyzed on the 7900HT Fast Real Time PCR system according to the manufacturer's instructions (Applied Biosystems). Primary analysis of the acquired signal data was performed in SDS 2.3 and RQ Manager 1.2 (Applied Biosystems). Outlier reactions were removed after Grubb's test identification, and differential expression was calculated using the ΔΔCT method.
Oligonucleotide Array Hybridization and Analysis
Labeled cRNA was resuspended in RNase-free H2O, and 15.0 μg was fragmented by ion-mediated hydrolysis at 95 °C for 35 min in 200 mm Tris acetate, pH 8.1, 500 mm potassium acetate, 150 mm magnesium acetate. The fragmented cRNA was hybridized for 16 h at 45 °C to Affymetrix Mouse Genome 430 2.0 short oligomer arrays, which detect ∼44,000 mouse transcripts representing over 34,000 well characterized mouse genes (Affymetrix, Santa Clara, CA).
Arrays were washed and stained using a Fluidics Station 450 (Affymetrix) according to the manufacturer's recommended procedures. The arrays were stained with phycoerythrin-conjugated streptavidin (Invitrogen), and the fluorescence intensities were determined using a GCS 3000 7G high resolution confocal laser scanner (Affymetrix). The scanned images were analyzed using programs resident in GeneChip Operating System version 1.4 (Affymetrix). Quality control metrics for cRNA integrity, sample loading, and variations in staining were determined after background correction and signal summarization by MAS 5.0 statistical algorithms resident in GeneChip Operating System version 1.4 and standardization of each array by global scaling the average of the fluorescent intensities of all genes on an array to a constant target intensity (TGT) of 250.
Data Analysis
Expression data were analyzed following background correction, probe set signal summarization, and normalization by MAS5.0 with global scaling (TGT = 250) (40–43). Probe sets exhibiting significant differential expressions were selected using MAS 5.0-generated Signal log ratios ≥0.6, MAS 5.0-generated detection p values ≤0.05, and MAS 5.0-generated change calls = decrease (D) or increase (I).
Gene annotation, gene ontology information, and biochemical pathway information were obtained from the National Center for Biotechnology Information (www.ncbi.nlm.nih.gov), NetAffx, the Gene Ontology Consortium, the Kyoto Encyclopedia of Genes and Genomes, and WebGestalt. Significant enrichment of specific GO categories or KEGG pathways in each comparison is estimated by χ2, z-score, or hypergeometric tests. Literature-based pathway analysis was performed by Pathway Architect (Stratagene).
RESULTS
Increased HA-TEAD-1 Protein in the Mouse Heart Is Associated with Fetal Gene Induction but Not Hypertrophy or Regulation of Other TEAD Family Members
In our previous work, we developed three independent lines of transgenic mice expressing a HA-tagged TEAD-1 transgene under the control of the MCK promoter, and we demonstrated that all three lines expressed HA-TEAD-1 protein exclusively in skeletal muscle and the heart, as expression in nonstriated muscle tissues was not detected (32). We also showed that a persistent increase in TEAD-1 protein resulted in a slower skeletal muscle contractile phenotype in postnatal mice (32). Based on the above-mentioned results, and the findings that α1-adrenergic stimulation of primary rat neonatal cardiomyocytes is associated with TEAD protein induction of several fetal genes and hypertrophic growth, this study was undertaken with the goal of determining the functional consequences of solely increasing TEAD-1 protein chronically in the postnatal mouse heart. In this study, we have chosen to primarily present our analysis of HA-TEAD-1 Tg line 12 because our previous work demonstrated a similar magnitude of TEAD-1 expression (3.3-fold) in the heart for all three independent MCK-HA-TEAD-1 transgenic lines (32). A comparison of body weight, heart weight, and normalized heart weight (heart weight/tibia length) between age-matched WT and TEAD-1 Tg male mice did not reveal a difference (Table 1). These data demonstrate that an increase in total TEAD-1 protein (endogenous and HA-TEAD-1) neither altered the growth rate of the TEAD-1 Tg mice nor was it sufficient to directly and/or indirectly induce cardiac hypertrophy (see under “Discussion”).
TABLE 1.
Mouse heart weight
Heart weights of TEAD-1 Tg mice (line 12) at 2, 5, and 10 months of age show no significant difference when compared with age-matched WT mice. HW/TL is heart weight/tibia length.
| WT, n = 10 | Tg, L12, n = 10 | p value | |
|---|---|---|---|
| 2 months | |||
| Heart weight | 142.0 ± 2.64 mg | 138.7 ± 5.96 mg | 0.64 |
| normalized (HW/TL) | 7.18 ± 0.18 mg/mm | 7.03 ± 0.34 mg/mm | 0.72 |
| 5 months | |||
| Heart weight | 160.9 ± 3.79 mg | 167.7 ± 6.03 mg | 0.33 |
| Normalized (HW/TL) | 7.85 ± 0.22 mg/mm | 8.11 ± 0.27 mg/mm | 0.45 |
| 10 months | |||
| Heart weight | 190.8 ± 7.76 mg | 221.4 ± 14.08 mg | 0.10 |
| Normalized (HW/TL) | 8.86 ± 0.38 mg/mm | 10.01 ± 0.59 mg/mm | 0.14 |
In the mouse, endogenous MCK gene expression is restricted to striated muscle (skeletal muscle and heart), is first detected in the heart at approximately embryonic day 13.5, and is expressed at moderate levels in the adult heart (44). The mouse MCK promoter has been well studied in cultured cardiomyocytes and transgenic mouse hearts (45–52). In transgenic mice, a 3300-nucleotide region of the MCK 5′-flanking region was found to be sufficient to drive expression of a reporter gene at high levels in skeletal muscle, at moderate levels in the heart, and at barely detectable levels in non-muscle tissue, which is consistent with the pattern and magnitude of endogenous MCK gene expression. To determine the expression pattern of the MCK-driven HA-TEAD-1 transgene, we performed a Western blot analysis using anti-HA antibody and total protein extracts isolated from embryonic day 17, neonatal day 14, and 5- and 10-month-old hearts of WT and TEAD-1 Tg line 12 mice. Consistent with the expression pattern of the endogenous MCK gene, only a single band representing HA-TEAD-1 protein was detected in total heart protein extracts of embryonic day 17, neonatal day 14, and 5- and 10-month-old TEAD-1 Tg line 12 mice, whereas HA-TEAD-1 protein was not detected in total heart protein extracts obtained from WT mice (Fig. 1). By using an anti-TEAD-1-specific antibody, endogenous TEAD-1 protein was detectable in total protein extracts isolated from both WT and TEAD-1 transgenic mouse hearts; however, barely detectable levels were found in fetal day 17 total heart extracts for both WT and TEAD-1 transgenic mice (Fig. 1). Multiple bands were detected when using anti-TEAD-1 antibody because this antibody recognizes a common epitope (amino acid 86–199) present in both HA-TEAD-1 (top band only) and endogenous TEAD-1 protein (bottom band of doublet, Fig. 1). Because adult heart muscle has been shown to express multiple TEAD protein isoforms, we employed quantitative reverse transcription-PCR to assess whether the mRNA levels encoding other TEAD isoproteins in the hearts of TEAD-1 Tg line 12 mice would be altered due to a persistent increase in TEAD-1 protein. With the exception of the combined endogenous TEAD-1 and HA-TEAD-1 mRNA levels, mRNAs encoding TEAD-2, TEAD-3, and TEAD-4 did not differ significantly from the levels detected in WT mouse heart muscle (Table 2). Collectively, these data demonstrate that the MCK-driven HA-TEAD-1 transgene mimicked the expression pattern of the endogenous MCK gene and that the sustained increase in total TEAD-1 protein did not directly or indirectly alter the basal gene expression level of other members of the TEAD gene family.
FIGURE 1.

Expression pattern of transgenic and endogenous TEAD-1 at different ages. Western blot analysis was performed using 100 μg of total protein extract isolated from hearts of fetal, neonatal, and 5- and 10-month-old WT and TEAD-1 transgenic mice (line 12). Analysis using either anti-HA or anti-TEAD-1 antibody revealed the expression of the TEAD-1 transgene (HA-tagged TEAD-1 protein) at all ages.
TABLE 2.
TEAD gene expression analysis
Quantitative reverse transcription-PCR analysis of TEAD1–4 mRNA expression in adult TEAD-1 Tg heart (line 12; n = 3) shows an increase in HA-tagged TEAD1 mRNA without a compensatory alteration in TEAD-2, TEAD-3, or TEAD-4 mRNA abundance. qRT-PCR, quantitative reverse transcription-PCR.
| Gene | qRT-PCR | Regulation | p value |
|---|---|---|---|
| TEAD-1 + HA-TEAD-1 | 9.3-Folda | Increased | 0.011 |
| TEAD-1 | 1.5-Fold | No change | 0.112 |
| TEAD-2 | 1.3-Fold | No change | 0.349 |
| TEAD-3 | 1.1-Fold | No change | 0.284 |
| TEAD-4 | 1.1-Fold | No change | 0.759 |
a p < 0.05.
TEAD-1 has been shown to participate in the transcriptional regulation of α-sk-actin, αMyHC, βMyHC, and BNP in primary rat neonatal cardiomyocytes in response to α1-adrenergic signaling (19–21, 23–25, 53). To determine whether TEAD-1 overexpression in the mouse heart resulted in the transcription of genes encoding proteins representative of the fetal gene program, we used quantitative reverse transcription-PCR to obtain an assessment of transcripts encoding the αMyHC, βMyHC, α-sk-actin, ANP, and BNP at fetal day 17, neonatal day 2, and in the adult (5 and 10 months) (Table 3). At all time points, transcripts representing the βMyHC, α-sk-actin, and total TEAD-1 were increased, whereas transcripts encoding the α-MyHC were decreased in the hearts of TEAD-1 Tg line 12 mice when compared with WT hearts. Transcripts representing ANP and BNP were not increased in the TEAD-1 Tg line 12 hearts until neonatal day 2 as compared with the WT mouse heart. These data provide in vivo evidence suggesting that TEAD-1 can participate directly and/or indirectly in the transcriptional regulation of all members of the fetal gene program.
TABLE 3.
Time course of fetal gene expression
Quantitative reverse transcription-PCR analysis of fetal gene mRNA expression in fetal, neonatal, and adult TEAD-1 Tg hearts (line 12) shows the re-expression of gene programs in neonatal and adult mice (fold change).
| WT | 14-Day fetal, n = 12 | 2-day neonatal, n = 7 | 5 months, n = 5 | 10 months, n = 3 | |
|---|---|---|---|---|---|
| ANP | 1 | 1.1 | 1.4 | 3.0 | 49.7 |
| BNP | 1 | 1.0 | 1.6 | 2.1 | 24.4 |
| α-MHC | 1 | 0.3 | 0.8 | 0.8 | 0.2 |
| β-MHC | 1 | 1.5 | 2.2 | 8.7 | 7.4 |
| α-sk-actin | 1 | 2.1 | 8.7 | 12.7 | 18.0 |
| TEAD-1 | 1 | 5.4 | 3.4 | 12.1 | 9.3 |
HA-TEAD-1 Transgenic Mouse Hearts Display Altered MyHC Expression and a Decrease in Contractile Function
The adult mouse heart primarily expresses the αMyHC isoform; however, βMyHC gene expression can be induced under conditions of increased workload. Although in vitro data support a role for TEAD-1 proteins in stress-related regulation of MyHC gene expression, in vivo evidence is lacking (19–21, 23–26). Herein, our quantitative reverse transcription-PCR data demonstrate differential transcription of the αMyHC and βMyHC genes in response to a sustained increase in TEAD-1 protein at all developmental stages (Table 3). These data are important because it has been well established that the functional properties of the heart are strongly correlated to MyHC protein content. To determine whether the measured change in αMyHC and βMyHC transcripts was reflected by a corresponding change in αMyHC and βMyHC myofibrillar protein content, we performed a comparative high resolution gel electrophoresis analysis of heart myofibrillar protein extract isolated from WT mice and from three independent TEAD-1 Tg mouse lines (L4, L12, and L14) at 5 months of age. Myofibrillar protein extract isolated from the slow-twitch mouse soleus muscle, which expresses high levels of βMyHC, served as a standard for identifying βMyHC protein. As expected, myofibrillar protein extract isolated from 5-month-old WT hearts displayed only a single intense silver-stained band representing αMyHC protein. In contrast, analysis of heart myofibrillar protein extract isolated from three distinct 5-month-old TEAD-1 Tg mouse lines revealed two silver-stained bands of similar intensity reflecting a significant decrease in αMyHC protein content and a significant increase in βMyHC protein content. Likewise, a comparative analysis of myofibrillar protein extract obtained from right and left ventricles and the septum of WT and TEAD-1 Tg line 12 mice at 10 months of age revealed a significant transition to a greater proportion of βMyHC protein with a resultant decrease in αMyHC protein (Fig. 2C).
FIGURE 2.

TEAD-1 induced changes in cardiac myosin heavy chain protein and LV power output of isolated working hearts. High resolution glycerol gel electrophoresis of protein extract (0.75 μg) isolated from the left ventricle of 5-month-old WT and Tg lines 4, 12, and 14 revealed that a sustained increase in TEAD-1 protein leads to an αMyHC to βMyHC transition in TEAD-1 transgenic mice (A). LV power output of isolated TEAD-1 transgenic hearts of 5-month-old mice confirm a contractile dysfunction (B). High resolution glycerol gel electrophoresis of protein extract (0.75 μg) isolated from LV, right ventricle (RV), and septum of 10-month-old WT and TEAD-1 Tg line 12 mice (C) display a significant induction of βMyHC protein (n = 3). *, p < 0.05.
To determine whether the TEAD-1-induced increase in myofibrillar βMyHC protein content (≅50%) of the TEAD-1 Tg mouse heart altered function, we measured LV power output using an isolated working heart preparation. As can be seen in Fig. 2B, all three 5-month-old TEAD-1 transgenic lines demonstrated significantly reduced LV power output, confirming a decrease in contractile function that occurs in conjunction with an αMyHC to βMyHC transition.
To further assess whole heart function, we performed in vivo cardiac MRI on 10-month-old male TEAD-1 Tg mice and age-matched WT mice. At 5 months, no significant differences between WT and Tg line 12 hearts could be detected. However, at 10 months of age, male Tg line 12 hearts displayed significantly decreased myocardial contractile function as compared with age-matched WT hearts (Fig. 3, A–D, and Table 4). As illustrated in (Fig. 3A), short axis MRI images show significantly increased end systolic volume and decreased LV ejection fraction (Fig. 3, C and D). Long axis images show changes in the morphology of the TEAD-1 Tg line 12 mouse hearts as revealed by a significantly thickened LV septal wall (1.45 ± 0.14 mm) when compared with WT hearts (1.26 ± 0.10 mm) (Fig. 3B and Table 4). LV end systolic volume was significantly increased in the TEAD-1 Tg line 12 hearts (57.38 ± 21.46 μl) as compared with the WT hearts (28.68 ± 1.38 μl) (Fig. 3D and Table 4). Cardiac output and stroke volume were also significantly decreased in the TEAD-1 Tg line 12 mice when compared with WT mice (Table 4). TEAD-1 Tg line 12 hearts displayed decreased LV ejection fraction (35.40 ± 15.06%) as compared with WT hearts (66.95 ± 3.55%), and their LV short axis fractional shortening (16.72 ± 10.25%) was significantly decreased when compared with WT values (39.59 ± 6.30%) (Table 4). Collectively, these data provide in vivo evidence that a sustained increase in TEAD-1 protein has a notable effect on adult heart contractile properties.
FIGURE 3.

MRI. Short axis MRI images show a systolic dysfunction represented by a significant increase in end systolic (ES) volume and significantly decreased LV ejection fraction due to decreased % fractional shortening in the TEAD-1 transgenic heart (A). Long axis MRI images reveal a change in the morphology of 10-month-old TEAD-1 Tg line 12 hearts (B). Significantly decreased LV ejection fraction (EF) (C) and significantly increased end systolic (ES) volume (D) are shown. (n = 5). *, p < 0.05; **, p < 0.01. ED, end diastolic labeling.
TABLE 4.
Morphological and functional heart measurements
In vivo MRI heart functional measurements of 5- and 10-month-old TEAD-1 Tg (line 12) mice versus their wild-type littermates (n = 5) are shown. NS means not significant.
| Morphological and functional remodeling of heart | |||
|---|---|---|---|
| Wild type | TEAD-1 Tg | p value | |
| 5 months of age | |||
| End diastolic septal wall thickness | 1.16 ± 0.11 mm | 1.23 ± 0.22 mm | NS |
| Ejection fraction | 66.20 ± 6.13% | 71.80 ± 2.53% | NS |
| Stroke volume | 39.28 ± 3.54 μl | 42.32 ± 3.85 μl | NS |
| Cardiac output | 13.77 ± 1.08 ml/min | 12.69 ± 1.82 ml/min | NS |
| End systolic volume | 20.25 ± 4.65 μl | 16.60 ± 1.71 μl | NS |
| End diastolic volume | 59.53 ± 5.27 μl | 58.92 ± 4.46 μl | NS |
| Short axis fractional shortening | 43.51 ± 4.10% | 45.45 ± 3.88% | NS |
| Long axis fractional shortening | 18.62 ± 5.52% | 17.93 ± 6.50% | NS |
| 10 months of age | |||
| End diastolic septal wall thickness | 1.26 ± 0.10 mm | 1.45 ± 0.14 mma | 0.045 |
| Ejection fraction | 66.95 ± 3.55% | 35.40 ± 15.06%b | 0.0025 |
| Stroke volume | 58.89 ± 10.04 μl | 33.07 ± 11.62 μlb | 0.005 |
| Cardiac output | 25.56 ± 3.45 ml/min | 11.56 ± 5.22 ml/minc | 0.0002 |
| End systolic volume | 28.68 ± 1.38 μl | 57.38 ± 21.46 μla | 0.019 |
| End diastolic volume | 87.57 ± 10.16 μl | 87.70 ± 21.50 μl | NS |
| Short axis fractional shortening | 39.59 ± 6.30% | 16.72 ± 10.25%b | 0.004 |
| Long axis fractional shortening | 14.04 ± 3.95 | 8.40 ± 4.06 | 0.075 |
a p < 0.05 versus WT.
b p < 0.005.
c p < 0.0005.
Decreased contractility of the heart can occur when calcium cycling at the sarcoplasmic reticulum (SR) is impaired. The SR Ca-ATPase (SERCA2a) mediates the uptake of cytoplasmic calcium into the SR following contraction, and its activity is regulated by the phosphorylation status of phospholamban. In this regard, previous work has shown that decreases in phospholamban levels or phosphorylation status (activity) or decreases in SR Ca-ATPase protein and/or activity impair calcium cycling at the SR and thus contractility of the heart (54–58). To determine whether a sustained increase in TEAD-1 protein altered SERCA2a or phospholamban gene transcription, we assessed global gene expression by microarray analysis. As compared with WT hearts, microarray analysis revealed that SERCA2a and phospholamban transcripts were each decreased by 70% in the TEAD-1 Tg line 12 hearts at 10 months (Table 5). To determine whether TEAD-1 overexpression resulted in altered SERCA2a protein levels and/or the phosphorylation status of phospholamban, we performed a Western blot analysis using protein extract (membrane fraction) isolated from WT and TEAD-1 Tg line 12 hearts (Fig. 4). Western blot analysis using anti-SERCA2a or anti-phospho-phospholamban-specific antibodies revealed a decrease in both SERCA2a protein and phosphorylated phospholamban levels in the TEAD-1 Tg line 12 hearts when compared with WT hearts (Fig. 4 and Table 7). In addition, Western blot analysis using anti-sodium-calcium exchanger antibody revealed an increase in sodium-calcium exchanger protein in the Tg line 12 hearts when compared with WT hearts, which is consistent with previous studies showing a compensatory increase in the sodium-calcium exchanger protein in response to decreased SERCA2a protein (Fig. 4 and Table 7). Additional changes in calcium-handling proteins, such as the sarcomeric cardiac/slow skeletal troponin C and the SR-ryanodine receptor are listed in Table 5. Taken together, these results provide strong in vivo evidence indicating that TEAD-1 serves a role in the regulation of proteins involved in calcium handling at the SR and sarcomeric level.
TABLE 5.
Expression profile of wild type and transgenic hearts
Microarray analysis of TEAD-1 Tg (line 12, n = 3) versus WT 10-month-old hearts showed significant changes in the mRNA expression of collagen, extracellular matrix, and calcium handling genes.
| Cardiac remodeling | |||
|---|---|---|---|
| Gene title | WT | Fold | p value |
| Calcium handling | |||
| Calcium channel, voltage-dependent, α 2/δ subunit 3 | 1 | 3.0 | 0.037 |
| Phospholamban | 1 | 0.7 | 0.003 |
| Ryanodine receptor 2, cardiac (Ryr2), mRNA | 1 | 2.1 | 0.000 |
| RyR1 mRNA for skeletal muscle ryanodine receptor | 1 | 12.1 | 0.030 |
| Sarco/endoplasmic reticulum Ca2+-ATPase, Serca2a | 1 | 0.7 | 0.020 |
| Sarco/endoplasmic reticulum Ca2+-ATPase; Serca2b | 1 | 0.5 | 0.000 |
| Triadin | 1 | 0.5 | 0.022 |
| Troponin C, cardiac/slow skeletal | 1 | 0.7 | 0.014 |
| Collagen and extracellular matrix | |||
| A disintegrin and metallopeptidase domain 23 | 1 | 0.5 | 0.009 |
| Annexin A2 | 1 | 1.7 | 0.020 |
| Biglycan | 1 | 5.7 | 0.002 |
| Coagulation factor II (thrombin) receptor | 1 | 2.0 | 0.022 |
| Collagen, type I, α 1 | 1 | 2.4 | 0.004 |
| Collagen, type I, α 2 | 1 | 3.3 | 0.000 |
| Collagen, type III, α 1 | 1 | 3.3 | 0.007 |
| Collagen, type V, α 2 | 1 | 3.0 | 0.005 |
| Collagen, type VIII, α 1 | 1 | 4.3 | 0.016 |
| Collagen, type VIII, α 2 | 1 | 3.1 | 0.002 |
| Collagen, type XIV, α 1 | 1 | 1.7 | 0.037 |
| Connective tissue growth factor | 1 | 4.1 | 0.048 |
| Fibrillin 1 | 1 | 3.2 | 0.003 |
| Fibronectin type III domain containing 3B | 1 | 2.1 | 0.035 |
| Hydroxysteroid (17-β ) dehydrogenase 12 | 1 | 1.7 | 0.031 |
| Integrin β 1 (fibronectin receptor β) | 1 | 2.8 | 0.007 |
| Integrin β 1-binding protein 1 | 1 | 1.5 | 0.007 |
| Matrix metallopeptidase 2 | 1 | 2.1 | 0.006 |
| Microtubule-associated serine/threonine kinase 2 | 1 | 0.5 | 0.029 |
| Microtubule-associated protein 1 light chain 3α | 1 | 0.6 | 0.006 |
| Microtubule-associated protein 1B | 1 | 2.6 | 0.039 |
| Microtubule-associated protein 4 | 1 | 1.7 | 0.026 |
| Microtubule-associated protein, RP/EB family, member 1 | 1 | 1.8 | 0.003 |
| Procollagen C-endopeptidase enhancer protein | 1 | 2.6 | 0.019 |
| Procollagen-proline, 2-oxoglutarate 4-dioxygenase, α 1 | 1 | 2.1 | 0.013 |
| Procollagen-proline, 2-oxoglutarate 4-dioxygenase, α II | 1 | 1.6 | 0.030 |
| TGFβ 2 | 1 | 4.0 | 0.017 |
| TGFβ 3 | 1 | 2.7 | 0.012 |
| Thrombospondin 1 | 1 | 4.3 | 0.003 |
| Thrombospondin 2 | 1 | 0.4 | 0.011 |
| Thrombospondin 4 | 1 | 4.7 | 0.025 |
| Tissue inhibitor of metalloproteinase 1 (TIMP1) | 1 | 2.5 | 0.002 |
| Tissue inhibitor of metalloproteinase 2 (TIMP2) | 1 | 2.4 | 0.002 |
FIGURE 4.
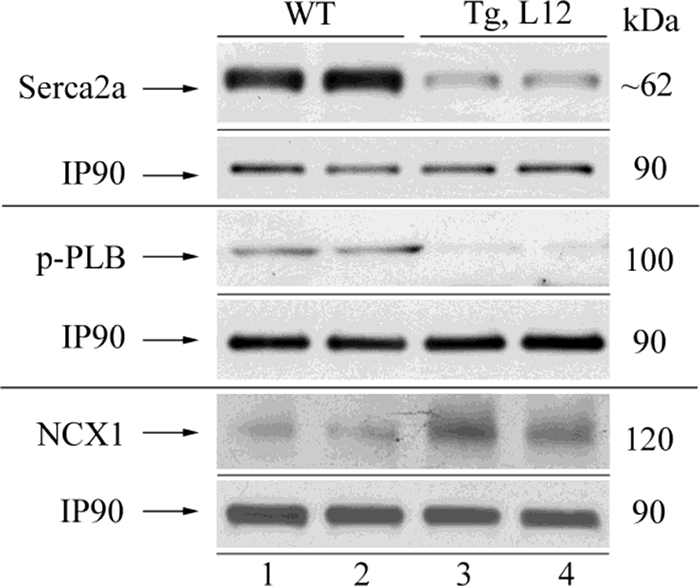
Western blot analysis of total Serca2a, phosphorylated phospholamban (p-PLB), and NCX1 in the heart. Membrane protein extracts (100 μg) obtained from 10-month-old wild type and TEAD-1 Tg L12 hearts revealed a significant decrease in the total level of Serca2a protein, the phosphorylation of phospholamban, and increased NCX1 protein (n = 4).
TABLE 7.
Densitometry quantification of Western blots of 10-month-old TEAD-1 transgenic line 12 versus wild-type heart muscle tissue
| WT | Tg, line 12 | p value | n | Fig. | |
|---|---|---|---|---|---|
| Serca2a (membrane) | 1 | 0.174 ± 0.055 | 0.019 | 4 | 4 |
| p-Phospholamban (membrane) | 1 | 0.483 ± 0.056 | 0.048 | 4 | 4 |
| Sodium-calcium exchanger 1 (membrane) | 1 | 2.931 ± 0.063 | 0.036 | 4 | 4 |
| Connexin43 (membrane) | 1 | NSa | 0.213 | 3 | 6 |
| p-Connexin43 (membrane) | 1 | 0.148 ± 0.041 | 0.00008 | 3 | 6 |
| GSK-3α (total) | 1 | NS | 0.70 | 8 | 7 |
| GSK-3β (total) | 1 | NS | 0.79 | 8 | 7 |
| p-GSK-3α (total) | 1 | 0.217 ± 0.006 | 0.005 | 8 | 7 |
| p-GSK-3β (total) | 1 | 0.22 ± 0.002 | 0.0004 | 8 | 7 |
| DYRK1A (total) | 1 | 2.01 ± 0.243 | 0.021 | 5 | 7 |
| DYRK1A (nuclear) | 1 | 1.86 ± 0.077 | 0.025 | 5 | 7 |
| β-Catenin (nuclear) | 1 | 0.33 ± 0.057 | 0.02 | 6 | 7 |
| GATA-4 (nuclear) | 1 | 0.62 ± 0.057 | 0.02 | 7 | 7 |
| NFATc3 (nuclear) | 1 | 0.43 ± 0.009 | 0.09 | 8 | 7 |
| NFATc4 (nuclear) | 1 | 0.44 ± 0.089 | 0.04 | 8 | 7 |
| Akt (total) | 1 | NS | 0.44 | 8 | 9 |
| p-Akt (total) | 1 | NS | 0.63 | 8 | 9 |
| Akt 1 (total) | 1 | NS | 0.70 | 8 | 9 |
| Akt 2 (total) | 1 | NS | 0.93 | 8 | 9 |
| Purα (nuclear) | 1 | 1.7 ± 0.08 | 0.002 | 4 | 10 |
| Purβ (nuclear) | 1 | 1.9 ± 0.14 | 0.005 | 4 | 10 |
| Purα (total) | 1 | 3.1 ± 0.578 | 0.042 | 4 | 10 |
| Purβ (total) | 1 | 6.0 ± 0.070 | 0.00002 | 4 | 10 |
a NS means not significant.
Histological Analysis Reveals Interstitial Fibrosis and Myocyte Disarray in TEAD-1 Transgenic Hearts
Fibrosis of WT and TEAD-1 Tg line 12 hearts was assessed by picrosirius red staining (collagen staining). Longitudinal sections through 10-month-old WT and TEAD-1 transgenic hearts were stained with picrosirius red to assess cardiac fibrosis (n = 3 per group). As shown in Fig. 5, A and B, moderate fibrosis was found in all areas of the transgenic hearts. Apart from a higher level of fibrosis in the septum, there were no regional preferences. In terms of transmural distribution, fibrotic areas were mainly found mid-wall and occasionally closer to the subepicardium, with none in the subendocardium. The increase in TEAD-1 heart collagen content as assessed by picrosirius red staining was supported by microarray analysis, which revealed significant increases in the gene expression level of collagens and components of the extracellular matrix, both of which are associated with cardiac remodeling and LV dysfunction (Table 5). In addition to increased fibrosis, hematoxylin and eosin revealed myocyte disarray (Fig. 5B).
FIGURE 5.

Picrosirius red staining of heart sections. PSR staining of heart sections obtained from 10-month-old TEAD-1 transgenic mice (line 12, n = 3) revealed increased levels of fibrosis (collagen stained pink) in the septum, left and right ventricle free wall, and apex at ×20 magnification (A). Hematoxylin and eosin (H&E) staining of these heart sections showed misalignment of cardiomyocytes at ×40 magnification (B).
Surface ECG Analysis Reveals a Conduction Disturbance
There was no significant difference in heart rate between 10-month-old WT and TEAD-1 Tg line 12 mice (Table 6). Although the TEAD-1 Tg line 12 mice displayed an increase in P wave amplitude, the increase was not significantly different when compared with WT mice. However, when compared with WT mice, the TEAD-1 Tg line 12 mice did display a significant increase in P wave duration. Taken together, these changes are consistent with left atrial and potentially right atrial enlargement. There was a marked left axis shift of the mean electrical axis in the TEAD-1 Tg line 12 mice as compared with the WT mice (Fig. 6A). This indicates a conduction disturbance that is most consistent with a left anterior fascicular block. The R wave amplitude was significantly decreased in the TEAD-1 Tg line 12 hearts, which was merely a reflection of the axis shift. Left anterior fascicular block results in a delay in activation of the anterior and lateral walls of the left ventricle. Ventricular conduction delays have been shown to occur when the phosphorylation levels of connexin43, a major gap junction channel protein in the heart, are decreased (59). To determine whether this conduction disturbance was related to remodeling of ventricular gap junctions, we performed a Western blot analysis to determine the levels of total and phosphorylated connexin43 using anti-connexin43- and phospho-anti-connexin43-specific antibodies and membrane protein extract isolated from 10-month-old WT and TEAD-1 Tg line 12 hearts. A significant decrease in phospho-connexin43 was revealed in heart membrane protein extract of TEAD-1 Tg line 12 hearts when compared with those obtained from WT hearts (Fig. 6B and Table 7).
TABLE 6.
ECG measurements
ECG measurements on the hearts of 10-month-old TEAD-1 Tg (line 12) mice versus their wild-type littermates are shown. bpm is beats/min. NS means not significant.
| 10 months of age | Wild type | TEAD-1 Tg | p value |
|---|---|---|---|
| Heart rate | 447 ± 82 bpm | 435 ± 101 bpm | NS |
| P-wave duration | 0.013 ± 0.002 s | 0.017 ± 0.003 sa | 0.005 |
| P-wave amplitude | 0.031 ± 0.015 mV | 0.044 ± 0.014 mV | NS |
| P-R duration | 0.042 ± 0.006 s | 0.043 ± 0.008 s | NS |
| Q-R-S duration | 0.014 ± 0.002 s | 0.015 ± 0.003 s | NS |
| R-wave amplitude | 0.300 ± 0.134 mV | 0.132 ± 0.098 mVb | 0.0012 |
| Q-T duration | 0.034 ± 0.004 s | 0.036 ± 0.005 s | NS |
| Mean electrical axis | 61.7 ± 29.269 | −36.4 ± 66.3b | 0.0013 |
a p < 0.006 versus WT, n = 10.
b p < 0.002.
FIGURE 6.
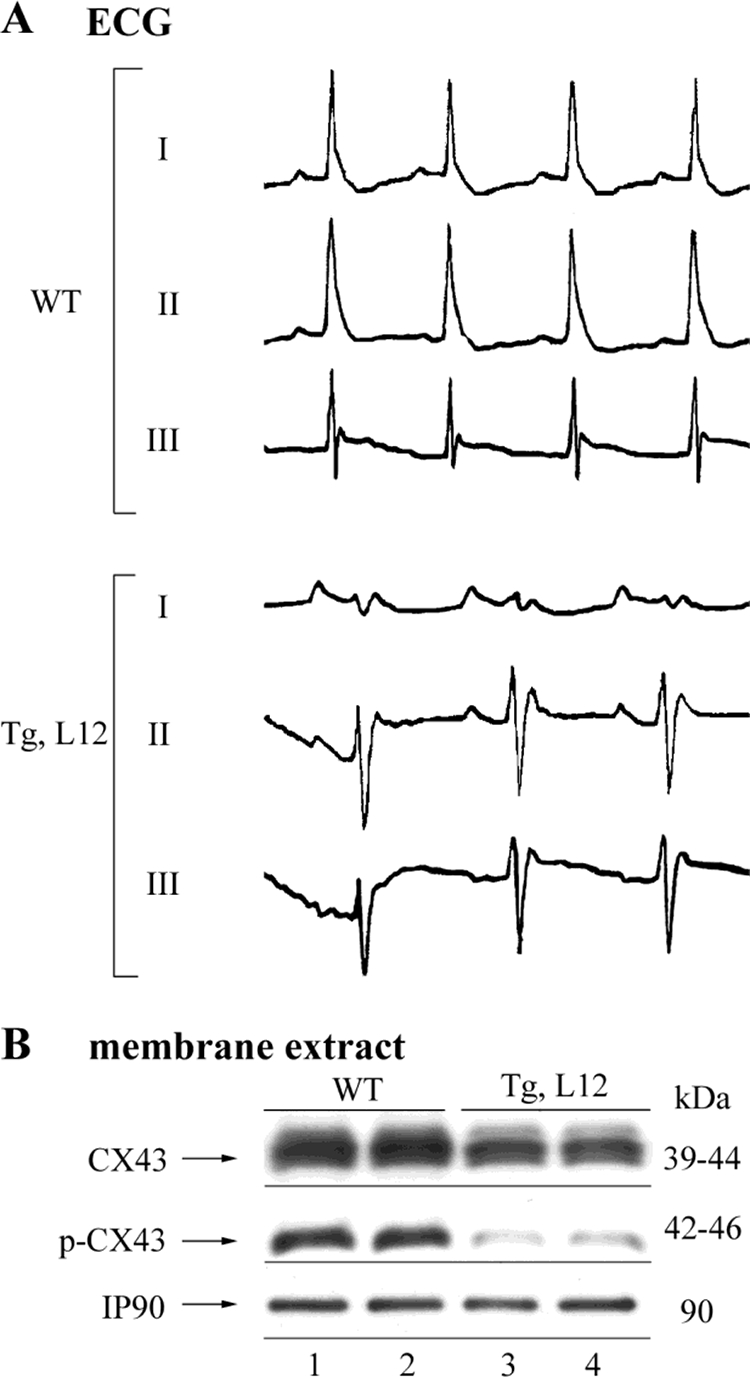
ECG and Western blot analysis of total and phosphorylated connexin43 in the heart. ECG tracing of leads I, II, and III obtained from 10-month-old WT and TEAD-1 Tg (line 12, n = 10) mice show a mean electrical axis shift (A). Membrane protein extracts (100 μg) obtained from 10-month-old WT and TEAD-1 Tg (line 12, n = 3) hearts revealed a significant decrease in the phosphorylation of connexin43 (B).
TEAD-1 Overexpression Increases Glycogen Synthase Kinase-3 Activity
In our previous work, we have shown that overexpression of TEAD-1 in skeletal muscle resulted in the unexpected activation of GSK-3α/β (dephosphorylation) and decreases in nuclear NFATc1/c3 and β-catenin, which are downstream targets of GSK-3 (32). To determine whether TEAD-1 overexpression also leads to altered GSK-3 activity in the heart, we performed a Western blot analysis using anti-GSK-3α- or anti-GSK-3β-specific antibody to assess GSK-3α/β content and activity. No significant difference in total GSK-3α or GSK-3β was detected in total protein extract obtained from day 17 fetal, day 14 neonatal, and 5- and 10-month-old TEAD-1 Tg line 12 hearts when compared with those obtained from WT hearts (Fig. 7 and Table 7). The activity of GSK-3 is tightly regulated whereby phosphorylation of Ser-21 of GSK-3α and Ser-9 of GSK-3β decreases kinase activity, whereas dephosphorylation of these serine residues activates kinase activity (60, 61). Western blot analysis using either phospho-GSK-3α (Ser-21)- or phospho-GSK-3β (Ser-9)-specific antibodies revealed a significant decrease in GSK-3α and GSK-3β phosphorylation (activation) in the protein extracts obtained from the hearts of 10-month-old TEAD-1 Tg mice when compared with those obtained from WT hearts (Fig. 7A and Table 7). Most importantly, decreased levels of phospho-GSK-3β were detected in the hearts of day 17 fetal TEAD-1 Tg mice, which corresponded to the early expression of the MCK-driven TEAD-1 transgene (Figs. 1 and 8A and Table 7).
FIGURE 7.
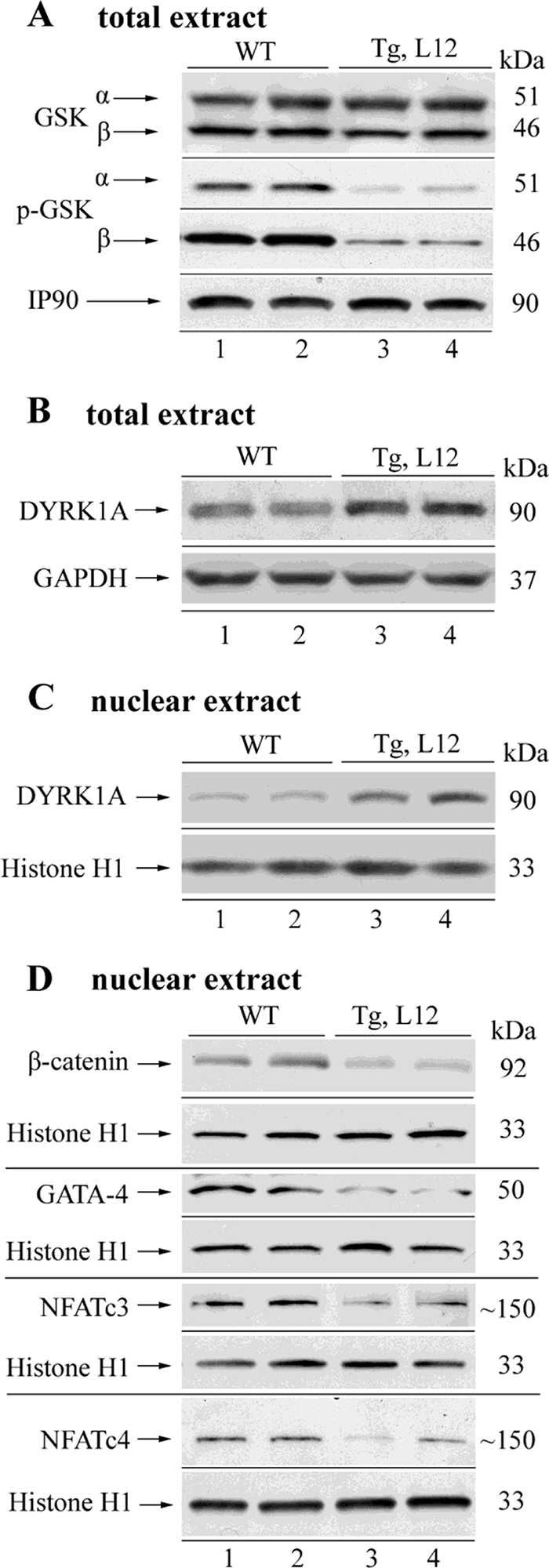
Western blot analysis of total and phosphorylated GSK-3α/β, total dual specificity tyrosine-phosphorylated and regulated kinase 1A (DYRK1A), β-catenin, GATA-4, NFATc3, and NFATc4 in WT and TEAD-1 Tg hearts. Total protein extracts (100 μg) obtained from 10-month-old WT and TEAD-1 Tg (line 12) hearts revealed no change in total GSK-3α/β and a significant decrease in phospho-GSK-3α/β (A). TEAD-1 Tg hearts displayed a significant increase in DYRK1A protein in total and nuclear extract (B and C) and decreases in nuclear β-catenin, GATA-4, NFATc3, and NFATc4 (D) (n = 5–8). GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
FIGURE 8.

Western blot analysis of total and phosphorylated GSK-3β, β-catenin, GATA-4, NFATc3, and NFATc4 in WT and TEAD-1 Tg hearts. Total protein extracts (100 μg) obtained from fetal, neonatal, and 5- and 10-month-old WT and TEAD-1 Tg (line 12) hearts revealed a significant decrease in phospho-GSK-3β at all ages and no change in total GSK-3β (A). TEAD-1 Tg neonatal (day 14) hearts displayed a significant decrease in nuclear β-catenin, GATA-4, NFATc3- and NFATc4 (B) (n = 4).
Signaling by Akt family members (Akt1, Akt2, and Akt3) has been shown to inhibit GSK-3α/β activity via phosphorylation of Ser-21 of GSK-3α and Ser-9 of GSK-3β (60, 62). To evaluate whether TEAD-1 overexpression altered Akt signaling activity, we performed a Western blot analysis using an anti-Akt antibody that recognizes all three Akt isoforms (Akt1, Akt2, and Akt3), as well as antibodies that specifically recognize either Akt1 or Akt2 protein. No difference in total Akt (1–3), Akt1, or Akt2 protein was detected between the cellular extracts isolated from 10-month-old hearts of WT and TEAD-1 Tg line 12 mice (Fig. 9 and Table 7). Similarly, phospho-Ser-73 Akt antibody did not detect significant differences in Akt phosphorylation between WT and TEAD-1 Tg heart muscle extracts (Fig. 9 and Table 7). These data show that activation of GSK-3α and GSK-3β in the hearts of TEAD-1 transgenic mice was not due to decreases in total Akt or Akt activity (phospho-Akt).
FIGURE 9.
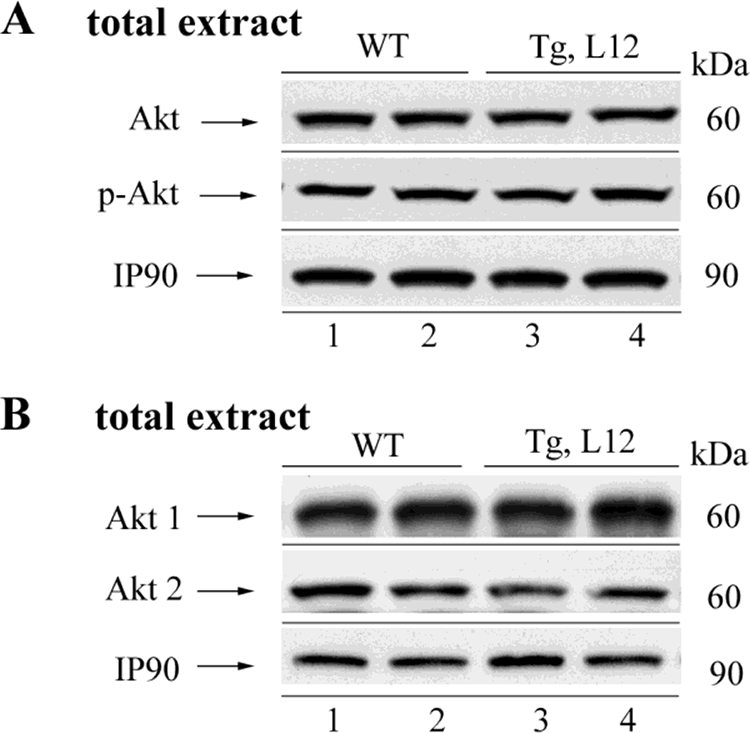
Western blot analysis of phosphorylated and total Akt protein in WT and TEAD-1 Tg hearts. Total protein extracts (100 μg) from the hearts of 10-month-old WT and TEAD-1 Tg (line 12) mice revealed no significant changes in total Akt and phospho-Akt (n = 8).
Nuclear NFAT Levels Are Decreased in TEAD-1-overexpressing Hearts
Activation of the calcium-dependent phosphatase calcineurin results in the nuclear translocation of members of the NFAT transcription factor family where they interact with other transcriptional activators (e.g. GATA4) to mediate cardiac hypertrophy (63). Because activated GSK-3 works in opposition to calcineurin by phosphorylation of NFAT transcription factors, thereby excluding them from the nucleus, it was of importance to determine the nuclear levels of NFAT protein. Western blot analysis using both anti-NFATc3- and NFATc4-specific antibodies revealed a significant decrease in NFATc3 and NFATc4 protein in nuclear extracts obtained from the hearts of day 14 neonatal and 10-month-old TEAD-1 line 12 Tg mice when compared with nuclear extracts isolated from the hearts of WT mice (Figs. 7D and 8B and Table 7). In contrast, we did not detect a difference in NFATc3 and NFATc4 protein by Western blot when using total cellular extracts (data not shown).
Expression of DYRK1A Is Increased in TEAD-1 Tg Hearts
The dual specificity tyrosine-phosphorylated and regulated kinase 1A (DYRK1A) is predominantly localized to the nucleus and has recently been shown to negatively regulate cardiomyocyte hypertrophy presumably by phosphorylation of NFAT transcription factors, thereby eliminating them from the nucleus (64). To evaluate whether TEAD-1 overexpression altered DYRK1A expression, we performed a Western blot analysis using total and nuclear extract isolated from WT and TEAD-1 Tg line 12 hearts and an anti-DYRK1A-specific antibody. A significant increase in DYRK1A protein was revealed in the total and nuclear extracts of 10-month-old TEAD-1 Tg line 12 hearts when compared with those obtained from WT hearts (Fig. 7, B and C, and Table 7).
Increased GSK-3 Activity Alters the Nuclear Levels of Its Downstream Targets GATA4 and β-Catenin
GATA4 has been shown to regulate cardiac specific gene expression and to mediate cardiac hypertrophy as transgenic overexpression of GATA4 induced hypertrophic growth of the adult heart, whereas conditional gene targeting of GATA4 diminished hypertrophy (9, 65). As with the NFAT transcription factors, the nuclear level of GATA4 is regulated by activated GSK-3β (66). To determine the nuclear level of GATA4, we performed a Western blot analysis using nuclear extract isolated from WT and TEAD-1 Tg line 12 hearts and a GATA4-specific antibody. A significant decrease in GATA4 protein was revealed in the nuclear extracts of day 14 neonatal and 10-month-old TEAD-1 Tg line 12 hearts when compared with those obtained from WT hearts (Figs. 7D and 8B and Table 7).
Like NFAT and GATA4, the nuclear level of β-catenin is regulated by GSK-3β. GSK-3β-mediated phosphorylation of β-catenin results in proteosome-mediated degradation and altered transcriptional regulation of β-catenin target genes (60, 61). Immunoblot analysis using anti-β-catenin-specific antibody detected reduced levels of β-catenin in the nuclear extracts obtained from the hearts of day 14 neonatal and 10-month-old TEAD-1 Tg line 12 mice as compared with those obtained from WT hearts (Figs. 7D and 8B and Table 7). We did not detect a difference in β-catenin protein by Western blot when using total cellular extract, which is likely due to the large amount of total cellular β-catenin (data not shown).
TEAD-1-overexpressing Hearts Display Increased Nuclear Purα/β
Purα and Purβ are single-stranded DNA- and RNA-binding proteins that are located in the nucleus and cytoplasmic domains. Recently, Purα and Purβ were shown to negatively regulate αMyHC gene expression at both the transcriptional and translational levels (67). Notably, the levels of Purα and Purβ were found to be elevated in the failing rabbit and human heart (67). Thus, it was of interest to determine whether the nuclear and/or cytoplasmic levels of Purα and Purβ were increased in the TEAD-1 Tg mouse line 12 heart. Western blot analysis using anti-Purα- or anti-Purβ-specific antibodies detected significantly enriched levels of both Purα and Purβ protein in heart cytoplasmic extract, whereas primarily Purβ was increased in heart nuclear extract obtained from the hearts of 10-month-old TEAD-1 Tg line 12 mice as compared with those obtained from WT hearts (Fig. 10, A and B, and Table 7).
FIGURE 10.

Western blot analysis of Purα and Purβ in the heart. Total protein extracts (A) and nuclear protein extract (B) obtained from 10-month-old WT and TEAD-1 Tg (line 12) hearts revealed a significant increase in Purα and Purβ (n = 4).
DISCUSSION
Our study provides the first in vivo evidence that in the postnatal mouse heart a persistent increase in TEAD-1 can induce characteristics of cardiac remodeling that occur over time and are associated with pathological hypertrophy and the failing heart. We also present evidence suggesting that a sustained increase in TEAD-1 protein had an anti-hypertrophic effect due, at least in part, to GSK-3α/β activation and increased nuclear DYRK1A protein; both demonstrated suppressors of cardiac hypertrophy that collectively reduced the nuclear levels of the pro-hypertrophic transcription factors β-catenin, NFATc3/c4, and GATA4.
Evidence gathered from TEAD-1 gene inactivation and retroviral gene trapping in embryonic stem cells has revealed an essential role for TEAD-1 in proper heart development (11, 12). However, the in vivo role of TEAD-1 in the postnatal mouse heart has not been determined. Here, we show that a sustained increase in TEAD-1 protein (by 3.3-fold) in the postnatal mouse heart leads to the differential regulation of transcripts encoding members of the fetal gene program as has been shown to occur in most animal models that induce characteristics of pathological cardiac hypertrophy (19–21, 23–26, 68–70). This was evidenced in quantitative reverse transcription-PCR analysis wherein the abundance of mRNAs encoding βMyHC, α-sk-actin, ANP, and BNP were significantly increased in neonatal and adult hearts of TEAD-1 transgenic mice. In contrast, the abundance of mRNAs encoding α-MyHC, which is normally highly expressed in the adult mouse heart, was significantly decreased in neonatal and adult hearts of TEAD-1 Tg mice. Although this analysis does not demonstrate direct transcriptional regulation of the above-mentioned genes by TEAD-1, it is noteworthy that the 5′-regulatory region of these genes contains multiple canonical TEAD-binding elements (MCAT elements), which is suggestive of a potential role for TEAD-1 in the direct regulation of these genes in response to stimuli that induce pathological hypertrophy. This notion is supported by experiments using in vitro systems that model in vivo pathological hypertrophy in which TEAD-1 was shown to participate in the regulated transcription of the α-sk-actin, αMyHC, βMyHC, and BNP reporter genes (19–21, 23–25). Furthermore, an increase in nuclear TEAD-1 binding activity and TEAD-1-dependent induction of βMyHC reporter genes was shown to occur in response to pressure overload, a demonstrated inducer of pathological hypertrophy (26, 27). It is also reasonable to suggest that the sustained increase in TEAD-1 protein indirectly regulated αMyHC gene expression via up-regulation of the Pur proteins. This is supported by the notable increase in the levels of cytoplasmic and nuclear Purα and Purβ in the TEAD-1 transgenic hearts because previous work has shown that these proteins negatively regulate αMyHC gene expression at the level of transcription and translation (67). It is also interesting that these proteins are both up-regulated in the failing human heart (67).
Most importantly, we have shown that the increase in βMyHC and decrease in αMyHC mRNA abundance were reflected by corresponding changes at the protein level in all three TEAD-1 transgenic lines examined at 5 and 10 months of age. Because the adult mouse heart primarily expresses the αMyHC (high ATPase activity and rapid Vmax), the observed transition from αMyHC to βMyHC (low ATPase and slow Vmax) in the TEAD-1 Tg hearts would be expected to be of functional importance. Indeed, the physiological significance of the αMyHC to βMyHC shift was underscored by a corresponding decrease in LV power output (Vmax × tension) as the proportion of βMyHC content increased in the TEAD-1 transgenic hearts. Our results are consistent with those of Korte et al. (71), who demonstrated that the unloaded shortening velocity of isolated rat myocytes and the LV power output (Vmax × tension) of isolated working rat heart preparations decreased in a linear fashion as the proportion of βMyHC content increased. Likewise, transgenic mice with cardiac restricted overexpression of βMyHC have shown that substitution of αMyHC with βMyHC (40–73% replacement) leads to decreases in Vmax and LV power output in isolated working heart preparations, and similar results were obtained when βMyHC content was increased as a result of lower thyroid hormone levels (hypothyroid) (72). Similarly, the acute replacement of αMyHC with βMyHC in isolated adult rat cardiomyocytes using a recombinant adenoviral vector was shown to reduce contractility in proportion to the increase in βMyHC content (73). In related experiments, a small amount of αMyHC content was shown to significantly increase power output of cardiac myocytes in the predominantly βMyHC-expressing hypothyroid rat heart (38).
A further assessment of cardiac function by MRI analysis (intact mouse) revealed that at 10 months of age the hearts of TEAD-1 Tg mice displayed a significant reduction in cardiac output, stroke volume, ejection fraction, and the percent fractional shortening with an increase in end systolic volume, which is consistent with impaired systolic function of the left ventricle. A contributing factor underlying the impaired systolic function of the TEAD-1 Tg hearts could have been reduced calcium handling at the SR. In support of this, the levels of SERCA2a mRNA and protein were significantly decreased in the hearts of 10-month-old TEAD-1 Tg mice. It is also noteworthy that the levels of p-phospholamban were significantly decreased because previous work has shown that the activity of SERCA2a is regulated by the phosphorylation status of phospholamban whereby decreased levels of p-phospholamban inhibit SERCA2a function, and thus cardiac contractility (74). Even though we did not determine diastolic calcium levels, it is conceivable that because the primary function of the SR Ca-ATPase (SERCA2a)-phospholamban interaction was to mediate re-uptake of cytoplasmic calcium into the SR following contraction (diastole), that the SR calcium stores in the TEAD-1 Tg hearts were decreased thereby resulting in LV contractile dysfunction. Evidence that SERCA2a protein serves as an important determinant of cardiac contractility comes from several studies employing either adenoviral gene transfer or transgenic overexpression wherein increased levels of SERCA2a protein were shown to augment cardiac contractility, although a decrease in SERCA2a protein by inactivation of one SERCA2a allele in mice depressed cardiac contractility (54, 55, 58, 75–77). Consistent with these observations, decreased levels of SERCA2a protein have been reported to occur in various animal models of cardiomyopathy and in the failing human heart (54, 78). Additionally, the reduced levels of SERCA2a and p-phospholamban in the TEAD-1 Tg mice may underlie their inability to tolerate stress as they die over a 4-day period after surgical induction of pressure overload (Fig. 11). In this regard, mice heterozygous for SERCA2a were shown to transition to heart failure more rapidly than wild type mice in response to pressure overload (54, 55). Phospholamban has also been shown to be an important regulator of cardiac contractility. Gene inactivation of phospholamban, which decreases phospholamban inhibition of SERCA2a calcium affinity, has been shown to enhance basal cardiac contractility and to restore cardiac contractility in several models of mouse heart failure and to failing human ventricular cardiomyocytes (56, 57, 79, 80). In addition to decreases in SERCA2a protein and p-phospholamban, we detected an increase in sarcolemma sodium-calcium exchanger 1 protein (NCX1). Several studies have reported a compensatory increase in the sodium-calcium exchanger in response to decreased SERCA2a protein, and because its function is to enhance calcium efflux across the sarcolemma, this response is thought to compensate for impaired SERCA2a diastolic calcium re-uptake into the SR in the failing heart (58, 81–84). Thus, it is possible that the combined effects of increased NCX1 and reduced SERCA2a protein and activity (phospholamban inhibition) may have contributed to reduced SR calcium stores leading to LV contractile dysfunction in the TEAD-1 Tg hearts.
FIGURE 11.

Kaplan-Meier plot of survival. This plot illustrates a striking reduction in survival of TEAD-1 Tg mice as a function of time after surgical induction of pressure overload as compared with WT mice. Fifteen TEAD-1 Tg and 15 WT mice were used in this study.
It is also plausible that the presence of cardiomyocyte disarray and an increased level of fibrosis throughout the TEAD-1 Tg hearts contributed to their impaired LV systolic function (3). Collectively, alterations in these two parameters would be expected to result in disruption of normal cell-cell contacts, increased stiffness of the heart, and conduction disturbance as has been documented to occur in several cardiac disease states such as hypertension-induced pressure overload hypertrophy and dilated and familial hypertrophic cardiomyopathy (3, 85–88). In agreement with this notion, we detected a marked left axis shift of the mean electrical axis in the TEAD-1 Tg line 12 mice, which indicates a conduction disturbance that is most consistent with a left anterior fascicular block, which is a delay in activation of the anterior and lateral walls of the left ventricle. Interestingly, we also detected a decrease in phosphorylated connexin43, a major heart gap junction channel protein that when dephosphorylated has been shown to alter gap junction function leading to ventricular conduction delay (59). Similarly, cardiac specific overexpression of TEAD-4 resulted in conduction defects that were related to altered connexin43 phosphorylation and correlated with increased expression of protein phosphatase 1β (31). Conversely, we did not detect significant changes in protein phosphatase 1α, 1β, or 1γ mRNA or protein, which may point toward a difference in target gene specificity between TEAD-1 and TEAD-4.
Another important finding in this study was that a sustained increase in TEAD-1 protein leads to a reduction in the phosphorylation status of both isoforms of glycogen synthase kinase-3 (GSK-3α and GSK-3β; dephosphorylation activates GSK-3 kinase activity) in the hearts of TEAD-1 Tg mice. The observed decrease in the phosphorylation status of GSK-3α/β could not be attributed to an alteration in signaling from Akt, a primary upstream regulator of GSK phosphorylation, because we did not detect changes in total or phosphorylated levels of Akt (60, 62). In association with the decrease in phospho-GSK-3α/β, we detected a significant decrease in the nuclear levels of GATA4, NFATc3/4, and β-catenin. This finding is in line with previous work showing that activated GSK-3, whether in primary cardiomyocytes or transgenic mice, can function as a regulator of transcription factor activity and/or subcellular location, which may in part account for its antihypertrophic action (66, 89–98). GSK-3β-mediated phosphorylation excludes GATA4 and NFAT from the nucleus, whereas phosphorylation of β-catenin targets it for proteosome-mediated degradation thereby effectively altering transcription of their target genes (60, 99). Although GATA4, NFATc3/4, and β-catenin have collectively been shown to be important regulators of fetal heart development and cardiomyocyte maturation, we did not detect overt differences in heart growth rate or normalized weight (heart weight/tibia length) between wild type and TEAD-1 Tg mice despite the significant increase in TEAD-1 protein and activated GSK-3 at embryonic day 17, indicating that these factors are required at an earlier time during cardiogenesis or that the overall magnitude of their nuclear decrease in our experiment was not sufficient to impede heart growth. In contrast to our findings, the transgenic overexpression of WT GSK-3α, WT GSK-3β, or constitutively active GSK-3β (GSK-3βS9A) was shown to impair maturational growth of the heart (89, 93, 95). There are several possible explanations that could account for the observed disparity in maturational heart growth reported between the current and previously mentioned studies. In our study, increased TEAD-1 expression did not alter the total amount of GSK-3α or GSK-3β, only their phosphorylation status, whereas the transgenic overexpression of WT GSK-3α, WT GSK-3β, or constitutively active GSK-3β (GSK-3βS9A) leads to a 4-fold (GSK-3α) or to a 9-fold (GSK-3β) increase in total GSK-3α/β (endogenous and transgenic), which is likely to have significantly increased the proportion of active GSK-3α/β. It is also possible that the dissimilarity in promoters used to drive transgene expression could account for the observed difference. We used the MCK promoter to drive TEAD-1 transgene expression (MCK-HA-TEAD-1), and MCK gene expression is first detected at approximately embryonic day 13.5 in the mouse heart. However, the αMyHC promoter was used to drive cardiac specific expression of WT GSK-3α, WT GSK-3β, or constitutively active GSK-3β (GSK-3βS9A), and αMyHC gene expression is detected at approximately embryonic day 8.5, which likely represents a more critical time point during cardiogenesis.
Although overexpression of TEAD-1 in the mouse heart induces many characteristics of cardiac remodeling that are associated with pathological hypertrophy and the failing heart, it did not promote cardiac hypertrophy. However, this observation is not surprising given that TEAD-1 overexpression leads to increased GSK-3α and GSK-3β activity, which is antihypertrophic. In primary cultures of cardiomyocytes and transgenic mice, cardiac specific overexpression of wild type or constitutively active forms of either GSK-3α (GSK-3S21A) or GSK-3β (GSK-3S9A) reduced cardiac hypertrophic growth induced by pressure overload (transverse aortic constriction), isoprenaline infusion, or overexpression of constitutively activated calcineurin (89, 93, 98). However, inhibition of GSK-3β activity by cardiac specific transgenic overexpression of a dominant negative (kinase-inactive) GSK-3β (GSK-3βK85M/K86I) resulted in compensated hypertrophy at base line, up-regulation of αMyHC gene expression, and improved cardiac contractility (91). In addition to increased GSK-3α/β activity, we also found increased nuclear expression of DYRK1A, a dual specificity tyrosine phosphorylation-regulated kinase recently shown to negatively regulate the hypertrophic response of primary cardiomyocytes to phenylephrine and constitutively activated calcineurin presumably by inhibition of nuclear NFAT factors (64). The absence of cardiac hypertrophy in this study may, at least in part, be due to GSK-3s phosphorylation of transcription factors (GATA4, NFAT, and β-catenin) previously shown to mediate hypertrophic gene expression (9, 94). Given the antihypertrophic effects of GSK-3α/β and DYRK1A, a definitive evaluation as to whether TEAD-1 is solely sufficient to induce hypertrophic growth was precluded in this study.
In summary, this study provides the first in vivo evidence that TEAD-1 can induce many characteristics of pathological cardiac remodeling and reveals a novel correlative link between increased TEAD-1 expression and activation of GSK-3α/β, which, in turn, decreased nuclear GATA4, NFATc3/4, and β-catenin protein. Despite the considerable effort that will be required, it will be important to decipher how increased TEAD-1 leads to activation of GSK-3α/β (↑TEAD-1→???→↑GSK-3α/β activity). As most models of cardiomyopathy rapidly progress to heart failure, the observed slow time course of cardiac remodeling herein suggests that the TEAD-1-overexpressing mice may serve as a model system with which to study the progression of cardiac remodeling and dysfunction.
Acknowledgments
We thank Dr. Mark Hannink for critical review of the manuscript and Katie Capkovic for production of Fig. 11 graph. In vivo MRI was performed at Veterans Affairs Biomolecular Imaging Center (supported by the Harry S. Truman Veterans Affairs Hospital and the University of Missouri, Columbia). The transgenic mice were produced by the University of Missouri-Columbia Transgenic Facility.
This work was supported, in whole or in part, by National Institutes of Health Grants AR41464 and AR47197 (to R. W. T.) from USPHS, NIAMS.
- BNP
- brain natriuretic peptide
- MRI
- magnetic resonance imaging
- TEAD
- TEA domain
- ECG
- electrocardiogram
- MCK
- muscle creatine kinase
- NFAT
- nuclear factor of T-cells
- SRF
- serum-response factor
- HA
- hemagglutinin
- Tg
- transgenic
- WT
- wild type
- SR
- sarcoplasmic reticulum
- MCAT
- muscle CAT element
- LV
- left ventricle
- α-sk-actin
- α-skeletal actin
- ANP
- atrial natriuretic peptide.
REFERENCES
- 1.Dorn G. W., 2nd, Robbins J., Sugden P. H. (2003) Circ. Res. 92, 1171–1175 [DOI] [PubMed] [Google Scholar]
- 2.Heineke J., Molkentin J. D. (2006) Nat. Rev. 7, 589–600 [DOI] [PubMed] [Google Scholar]
- 3.Swynghedauw B. (2006) J. Exp. Biol. 209, 2320–2327 [DOI] [PubMed] [Google Scholar]
- 4.Opie L. H., Commerford P. J., Gersh B. J., Pfeffer M. A. (2006) Lancet 367, 356–367 [DOI] [PubMed] [Google Scholar]
- 5.Akazawa H., Komuro I. (2003) Circ. Res. 92, 1079–1088 [DOI] [PubMed] [Google Scholar]
- 6.Dorn G. W., 2nd (2007) Hypertension 49, 962–970 [DOI] [PubMed] [Google Scholar]
- 7.Clerk A., Cullingford T. E., Fuller S. J., Giraldo A., Markou T., Pikkarainen S., Sugden P. H. (2007) J. Cell. Physiol. 212, 311–322 [DOI] [PubMed] [Google Scholar]
- 8.Barry S. P., Davidson S. M., Townsend P. A. (2008) Int. J. Biochem. Cell Biol. 40, 2023–2039 [DOI] [PubMed] [Google Scholar]
- 9.Oka T., Xu J., Molkentin J. D. (2007) Semin. Cell Dev. Biol. 18, 117–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bergmann O., Bhardwaj R. D., Bernard S., Zdunek S., Barnabé-Heider F., Walsh S., Zupicich J., Alkass K., Buchholz B. A., Druid H., Jovinge S., Frisén J. (2009) Science 324, 98–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Z., Friedrich G. A., Soriano P. (1994) Genes Dev. 8, 2293–2301 [DOI] [PubMed] [Google Scholar]
- 12.Sawada A., Kiyonari H., Ukita K., Nishioka N., Imuta Y., Sasaki H. (2008) Mol. Cell. Biol. 28, 3177–3189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacquemin P., Davidson I. (1997) Trends Cardiovasc. Med. 7, 192–197 [DOI] [PubMed] [Google Scholar]
- 14.Larkin S. B., Ordahl C. P. (1999) in Heart Development (Horvey R. P., Rosenthal N. eds) pp. 307–329, Academic Press, San Diego, CA [Google Scholar]
- 15.Yoshida T. (2008) Arterioscler. Thromb. Vasc. Biol. 28, 8–17 [DOI] [PubMed] [Google Scholar]
- 16.Kaneko K. J., Kohn M. J., Liu C., DePamphilis M. L. (2007) Genesis 45, 577–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishioka N., Yamamoto S., Kiyonari H., Sato H., Sawada A., Ota M., Nakao K., Sasaki H. (2008) Mech. Dev 125, 270–283 [DOI] [PubMed] [Google Scholar]
- 18.Yagi R., Kohn M. J., Karavanova I., Kaneko K. J., Vullhorst D., DePamphilis M. L., Buonanno A. (2007) Development 134, 3827–3836 [DOI] [PubMed] [Google Scholar]
- 19.McLean B. G., Lee K. S., Simpson P. C., Farrance I. K. (2003) J. Mol. Cell. Cardiol. 35, 461–471 [DOI] [PubMed] [Google Scholar]
- 20.Maeda T., Mazzulli J. R., Farrance I. K., Stewart A. F. (2002) J. Biol. Chem. 277, 24346–24352 [DOI] [PubMed] [Google Scholar]
- 21.Karns L. R., Kariya K., Simpson P. C. (1995) J. Biol. Chem. 270, 410–417 [DOI] [PubMed] [Google Scholar]
- 22.Kariya K., Farrance I. K., Simpson P. C. (1993) J. Biol. Chem. 268, 26658–26662 [PubMed] [Google Scholar]
- 23.Kariya K., Karns L. R., Simpson P. C. (1994) J. Biol. Chem. 269, 3775–3782 [PubMed] [Google Scholar]
- 24.Stewart A. F., Suzow J., Kubota T., Ueyama T., Chen H. H. (1998) Circ. Res. 83, 43–49 [DOI] [PubMed] [Google Scholar]
- 25.Ueyama T., Zhu C., Valenzuela Y. M., Suzow J. G., Stewart A. F. (2000) J. Biol. Chem. 275, 17476–17480 [DOI] [PubMed] [Google Scholar]
- 26.Molkentin J. D., Markham B. E. (1994) Mol. Cell. Biol. 14, 5056–5065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hasegawa K., Lee S. J., Jobe S. M., Markham B. E., Kitsis R. N. (1997) Circulation 96, 3943–3953 [DOI] [PubMed] [Google Scholar]
- 28.Karasseva N., Tsika G., Ji J., Zhang A., Mao X., Tsika R. (2003) Mol. Cell. Biol. 23, 5143–5164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maeda T., Gupta M. P., Stewart A. F. (2002) Biochem. Biophys. Res. Commun. 294, 791–797 [DOI] [PubMed] [Google Scholar]
- 30.Gupta M., Kogut P., Davis F. J., Belaguli N. S., Schwartz R. J., Gupta M. P. (2001) J. Biol. Chem. 276, 10413–10422 [DOI] [PubMed] [Google Scholar]
- 31.Chen H. H., Baty C. J., Maeda T., Brooks S., Baker L. C., Ueyama T., Gursoy E., Saba S., Salama G., London B., Stewart A. F. (2004) Circulation 110, 2980–2987 [DOI] [PubMed] [Google Scholar]
- 32.Tsika R. W., Schramm C., Simmer G., Fitzsimons D. P., Moss R. L., Ji J. (2008) J. Biol. Chem. 283, 36154–36167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Talmadge R. J., Roy R. R. (1993) J. Appl. Physiol. 75, 2337–2340 [DOI] [PubMed] [Google Scholar]
- 34.Heiberg E., Wigström L., Carlsson M., Bolger A. F., Karlsson M. (2005) Proc. IEEE Comput. Cardiol. 32, 599–602 [Google Scholar]
- 35.Neely J. R., Rovetto M. J. (1975) Methods Enzymol. 39, 43–60 [DOI] [PubMed] [Google Scholar]
- 36.Rovetto M. J. (1980) Proc. Soc. Exp. Biol. Med. 164, 13–17 [DOI] [PubMed] [Google Scholar]
- 37.Herron T. J., Korte F. S., McDonald K. S. (2001) Am. J. Physiol. Heart Circ. Physiol. 281, H1217–H1222 [DOI] [PubMed] [Google Scholar]
- 38.Herron T. J., McDonald K. S. (2002) Circ. Res. 90, 1150–1152 [DOI] [PubMed] [Google Scholar]
- 39.Fine D. (2006) Vet. Med. 101, 28–36 [Google Scholar]
- 40.Lockhart D. J., Dong H., Byrne M. C., Follettie M. T., Gallo M. V., Chee M. S., Mittmann M., Wang C., Kobayashi M., Horton H., Brown E. L. (1996) Nat. Biotechnol. 14, 1675–1680 [DOI] [PubMed] [Google Scholar]
- 41.Liu W. M., Mei R., Di X., Ryder T. B., Hubbell E., Dee S., Webster T. A., Harrington C. A., Ho M. H., Baid J., Smeekens S. P. (2002) Bioinformatics 18, 1593–1599 [DOI] [PubMed] [Google Scholar]
- 42.Hubbell E., Liu W. M., Mei R. (2002) Bioinformatics 18, 1585–1592 [DOI] [PubMed] [Google Scholar]
- 43.Irizarry R. A., Wu Z., Jaffee H. A. (2006) Bioinformatics 22, 789–794 [DOI] [PubMed] [Google Scholar]
- 44.Lyons G. E. (1994) Trends Cardiovasc. Med. 4, 70–77 [DOI] [PubMed] [Google Scholar]
- 45.Amacher S. L., Buskin J. N., Hauschka S. D. (1993) Mol. Cell. Biol. 13, 2753–2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Donoviel D. B., Shield M. A., Buskin J. N., Haugen H. S., Clegg C. H., Hauschka S. D. (1996) Mol. Cell. Biol. 16, 1649–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnson J. E., Wold B. J., Hauschka S. D. (1989) Mol. Cell. Biol. 9, 3393–3399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nguyen Q. G., Buskin J. N., Himeda C. L., Fabre-Suver C., Hauschka S. D. (2003) Transgenic Res. 12, 337–349 [DOI] [PubMed] [Google Scholar]
- 49.Nguyen Q. G., Buskin J. N., Himeda C. L., Shield M. A., Hauschka S. D. (2003) J. Biol. Chem. 278, 46494–46505 [DOI] [PubMed] [Google Scholar]
- 50.Fabre-Suver C., Hauschka S. D. (1996) J. Biol. Chem. 271, 4646–4652 [DOI] [PubMed] [Google Scholar]
- 51.Himeda C. L., Ranish J. A., Hauschka S. D. (2008) Mol. Cell. Biol. 28, 6521–6535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Himeda C. L., Ranish J. A., Angello J. C., Maire P., Aebersold R., Hauschka S. D. (2004) Mol. Cell. Biol. 24, 2132–2143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thuerauf D. J., Glembotski C. C. (1997) J. Biol. Chem. 272, 7464–7472 [DOI] [PubMed] [Google Scholar]
- 54.Periasamy M., Huke S. (2001) J. Mol. Cell. Cardiol. 33, 1053–1063 [DOI] [PubMed] [Google Scholar]
- 55.Periasamy M., Reed T. D., Liu L. H., Ji Y., Loukianov E., Paul R. J., Nieman M. L., Riddle T., Duffy J. J., Doetschman T., Lorenz J. N., Shull G. E. (1999) J. Biol. Chem. 274, 2556–2562 [DOI] [PubMed] [Google Scholar]
- 56.Koss K. L., Kranias E. G. (1996) Circ. Res. 79, 1059–1063 [DOI] [PubMed] [Google Scholar]
- 57.Luo W., Grupp I. L., Harrer J., Ponniah S., Grupp G., Duffy J. J., Doetschman T., Kranias E. G. (1994) Circ. Res. 75, 401–409 [DOI] [PubMed] [Google Scholar]
- 58.Ji Y., Lalli M. J., Babu G. J., Xu Y., Kirkpatrick D. L., Liu L. H., Chiamvimonvat N., Walsh R. A., Shull G. E., Periasamy M. (2000) J. Biol. Chem. 275, 38073–38080 [DOI] [PubMed] [Google Scholar]
- 59.Solan J. L., Lampe P. D. (2009) Biochem. J. 419, 261–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Doble B. W., Woodgett J. R. (2003) J. Cell Sci. 116, 1175–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Forde J. E., Dale T. C. (2007) Cell. Mol. Life Sci. 64, 1930–1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Manning B. D., Cantley L. C. (2007) Cell 129, 1261–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wilkins B. J., Molkentin J. D. (2004) Biochem. Biophys. Res. Commun. 322, 1178–1191 [DOI] [PubMed] [Google Scholar]
- 64.Kuhn C., Frank D., Will R., Jaschinski C., Frauen R., Katus H. A., Frey N. (2009) J. Biol. Chem. 284, 17320–17327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liang Q., De Windt L. J., Witt S. A., Kimball T. R., Markham B. E., Molkentin J. D. (2001) J. Biol. Chem. 276, 30245–30253 [DOI] [PubMed] [Google Scholar]
- 66.Morisco C., Seta K., Hardt S. E., Lee Y., Vatner S. F., Sadoshima J. (2001) J. Biol. Chem. 276, 28586–28597 [DOI] [PubMed] [Google Scholar]
- 67.Gupta M., Sueblinvong V., Raman J., Jeevanandam V., Gupta M. P. (2003) J. Biol. Chem. 278, 44935–44948 [DOI] [PubMed] [Google Scholar]
- 68.Komuro I., Kaida T., Shibazaki Y., Kurabayashi M., Katoh Y., Hoh E., Takaku F., Yazaki Y. (1990) J. Biol. Chem. 265, 3595–3598 [PubMed] [Google Scholar]
- 69.Komuro I., Yazaki Y. (1993) Annu. Rev. Physiol. 55, 55–75 [DOI] [PubMed] [Google Scholar]
- 70.Sadoshima J., Jahn L., Takahashi T., Kulik T. J., Izumo S. (1992) J. Biol. Chem. 267, 10551–10560 [PubMed] [Google Scholar]
- 71.Korte F. S., Herron T. J., Rovetto M. J., McDonald K. S. (2005) Am. J. Physiol. Heart Circ. Physiol. 289, H801–H812 [DOI] [PubMed] [Google Scholar]
- 72.Krenz M., Sanbe A., Bouyer-Dalloz F., Gulick J., Klevitsky R., Hewett T. E., Osinska H. E., Lorenz J. N., Brosseau C., Federico A., Alpert N. R., Warshaw D. M., Perryman M. B., Helmke S. M., Robbins J. (2003) J. Biol. Chem. 278, 17466–17474 [DOI] [PubMed] [Google Scholar]
- 73.Herron T. J., Vandenboom R., Fomicheva E., Mundada L., Edwards T., Metzger J. M. (2007) Circ. Res. 100, 1182–1190 [DOI] [PubMed] [Google Scholar]
- 74.Simmerman H. K., Jones L. R. (1998) Physiol. Rev. 78, 921–947 [DOI] [PubMed] [Google Scholar]
- 75.del Monte F., Harding S. E., Dec G. W., Gwathmey J. K., Hajjar R. J. (2002) Circulation 105, 904–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Giordano F. J., He H., McDonough P., Meyer M., Sayen M. R., Dillmann W. H. (1997) Circulation 96, 400–403 [DOI] [PubMed] [Google Scholar]
- 77.He H., Giordano F. J., Hilal-Dandan R., Choi D. J., Rockman H. A., McDonough P. M., Bluhm W. F., Meyer M., Sayen M. R., Swanson E., Dillmann W. H. (1997) J. Clin. Invest. 100, 380–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kawase Y., Hajjar R. J. (2008) Nat. Clin. Pract. 5, 554–565 [DOI] [PubMed] [Google Scholar]
- 79.Minamisawa S., Hoshijima M., Chu G., Ward C. A., Frank K., Gu Y., Martone M. E., Wang Y., Ross J., Jr., Kranias E. G., Giles W. R., Chien K. R. (1999) Cell 99, 313–322 [DOI] [PubMed] [Google Scholar]
- 80.Hajjar R. J., MacRae C. A. (2002) N. Engl. J. Med. 347, 1196–1199 [DOI] [PubMed] [Google Scholar]
- 81.Reed T. D., Babu G. J., Ji Y., Zilberman A., Ver Heyen M., Wuytack F., Periasamy M. (2000) J. Mol. Cell. Cardiol. 32, 453–464 [DOI] [PubMed] [Google Scholar]
- 82.Vetter R., Studer R., Reinecke H., Kolár F., Ostádalová I., Drexler H. (1995) J. Mol. Cell. Cardiol. 27, 1689–1701 [DOI] [PubMed] [Google Scholar]
- 83.Cernohorský J., Kolár F., Pelouch V., Korecky B., Vetter R. (1998) Am. J. Physiol. 275, H264–H273 [DOI] [PubMed] [Google Scholar]
- 84.Studer R., Reinecke H., Bilger J., Eschenhagen T., Böhm M., Hasenfuss G., Just H., Holtz J., Drexler H. (1994) Circ. Res. 75, 443–453 [DOI] [PubMed] [Google Scholar]
- 85.Gürtl B., Kratky D., Guelly C., Zhang L., Gorkiewicz G., Das S. K., Tamilarasan K. P., Hoefler G. (2009) Int. J. Exp. Pathol. 90, 338–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Luk A., Ahn E., Soor G. S., Butany J. (2009) J. Clin. Pathol. 62, 219–225 [DOI] [PubMed] [Google Scholar]
- 87.Rajamannan N. M. (2006) Drug Discov. Today 3, 291–295 [Google Scholar]
- 88.Soor G. S., Luk A., Ahn E., Abraham J. R., Woo A., Ralph-Edwards A., Butany J. (2009) J. Clin. Pathol. 62, 226–235 [DOI] [PubMed] [Google Scholar]
- 89.Antos C. L., McKinsey T. A., Frey N., Kutschke W., McAnally J., Shelton J. M., Richardson J. A., Hill J. A., Olson E. N. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 907–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Haq S., Choukroun G., Kang Z. B., Ranu H., Matsui T., Rosenzweig A., Molkentin J. D., Alessandrini A., Woodgett J., Hajjar R., Michael A., Force T. (2000) J. Cell Biol. 151, 117–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hirotani S., Zhai P., Tomita H., Galeotti J., Marquez J. P., Gao S., Hong C., Yatani A., Avila J., Sadoshima J. (2007) Circ. Res. 101, 1164–1174 [DOI] [PubMed] [Google Scholar]
- 92.Kerkelä R., Woulfe K., Force T. (2007) Trends Cardiovasc. Med. 17, 91–96 [DOI] [PubMed] [Google Scholar]
- 93.Michael A., Haq S., Chen X., Hsich E., Cui L., Walters B., Shao Z., Bhattacharya K., Kilter H., Huggins G., Andreucci M., Periasamy M., Solomon R. N., Liao R., Patten R., Molkentin J. D., Force T. (2004) J. Biol. Chem. 279, 21383–21393 [DOI] [PubMed] [Google Scholar]
- 94.Sugden P. H., Fuller S. J., Weiss S. C., Clerk A. (2008) Br. J. Pharmacol. 153, S137–S153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhai P., Gao S., Holle E., Yu X., Yatani A., Wagner T., Sadoshima J. (2007) J. Biol. Chem. 282, 33181–33191 [DOI] [PubMed] [Google Scholar]
- 96.Liao W., Wang S., Han C., Zhang Y. (2005) FEBS J. 272, 1845–1854 [DOI] [PubMed] [Google Scholar]
- 97.Philips A. S., Kwok J. C., Chong B. H. (2007) J. Biol. Chem. 282, 24915–24927 [DOI] [PubMed] [Google Scholar]
- 98.Sanbe A., Gulick J., Hanks M. C., Liang Q., Osinska H., Robbins J. (2003) Circ. Res. 92, 609–616 [DOI] [PubMed] [Google Scholar]
- 99.Eastman Q., Grosschedl R. (1999) Curr. Opin. Cell Biol. 11, 233–240 [DOI] [PubMed] [Google Scholar]