

Abstract
Curcumin is the yellow pigment of turmeric that interacts irreversibly forming an adduct with thioredoxin reductase (TrxR), an enzyme responsible for redox control of cell and defence against oxidative stress. Docking at both the active sites of TrxR was performed to compare the potency of three naturally occurring curcuminoids, namely curcumin, demethoxy curcumin and bis-demethoxy curcumin. Results show that active sites of TrxR occur at the junction of E and F chains. Volume and area of both cavities is predicted. It has been concluded by distance mapping of the most active conformations that Se atom of catalytic residue SeCYS498, is at a distance of 3.56 from C13 of demethoxy curcumin at the E chain active site, whereas C13 carbon atom forms adduct with Se atom of SeCys 498. We report that at least one methoxy group in curcuminoids is necessary for interation with catalytic residues of thioredoxin. Pharmacophore of both active sites of the TrxR receptor for curcumin and demethoxy curcumin molecules has been drawn and proposed for design and synthesis of most probable potent antiproliferative synthetic drugs.
Keywords: , , , , ,
Background
Turmeric, a culinary food additive (spice) is powdered rhizome of the plant Curcuma longa. It has been used for centuries in indigenous medicine in India for the treatment of a variety of diseases. [1] The yellow pigment (curcuminoids), forming 3-4% by weight of turmeric is a mixture of curcumin (1, 94%) demethoxy curcumin (2, 6%) and bis-demethoxy curcumin (3, 0.3%) (Figure 1). Aggarwal et al [2] have shown that curcumin is potent inhibitor of tumor initiation in vivo and in vitro studies. Many workers have shown that curcumin induces apoptosis and is a potent chemopreventive agent in several cancers. e.g breast, colon , skin, oral, intestinal carcinogenesis. Fang et al [3] characterized an irreversible inhibition mechanism of thioredoxin reductase (TrxR) by curcumin. Curcumin binds at the active site of thioredoxin reductase, and inhibits the process of electron transfer from NADPH to ribonucleotide TrxR is present in all living cells; but is overexpressed in tumor cells. However, tumor growth seems to be crucially dependent on an active thioredoxin redox system, making it a potential target for anticancer drugs.
Figure 1.

Curcuminoids: (a) Curcumin; (b) Bis-demethoxy curcumin; (c) Demethoxy curcumin
Thioredoxin is a key enzyme for DNA synthesis by directly serving as an electron donor to ribonucleotide reductase. TrxR is an important enzyme for DNA metabolism which is directly related to cell proliferation. Thioredoxin reductase (TrxR) catalyzes NADPHdependent reduction of the redox-active disulfide (S-S) in thioredoxin (Trx), which serves a wide range of functions in cellular proliferation, defense against oxidative stress, apoptosis and redox control. [4] The inhibition by curcumin and demethoxy derivatives was caused by covalent modification of active site CYS496 and SEC497 residues resulting in the destruction of the Trx reduction activity. In addition and more importantly, the curcumin-modified enzyme was converted into an NADPH oxidase with production of ROS to which cancer cells appear more sensitive. The threedimensional structure of the SeCYS498 mutant of rat TrxR in complex with NADP (+) has been determined to 3.0 resolution by X-ray crystallography. The overall structure is similar to that of glutathione reductase (GR), including conserved amino acid residues binding the cofactors FAD and NADPH. [5] TrxR can serve as a potential target for anticancer drugs and curcumin has been reported to be a potent anticancer drug. The combination of these two factors motivated us for in silico study of three naturally occurring curcuminoids in terms of binding and docking energies which is likely to help in designing a new synthetic analog of curcumin as a potent anticancer drug.
Methodology
The docking study was performed with three naturally occuring curcuminoids at the active site (CYS497-SEC498) of thioredoxin reductase. The PDB file that we considered for docking has S atom so we replaced it by Se atom with same coordinates because naturally occurring TrxR has SeCys498 amino acids. However, AutoDock does not recognize Se atom, which might be due to there is no parameter for Se atom in AutoDock. The grid maps were calculated with Auto Grid and grid's size was decided accordingly and the volume calculated by CAST-p program. The dimensions of the grids for E chain cavity were thus 50 • 50 • 50 Å, with a spacing of 0.375 Å between the grid points and the center (30.074, 0.267 and -1.792) close to the (S/Se) atom of the E chain of the catalytic residue CYS498. Similarly for F chain active site of TrxR, the center of catalytic residue CYS498 (S/Se) was used as the center (28.922, 7.243 and 42.311) of grid, with the grid points of 50 • 50 • 50 Å and the spacing of 0.375 Å. AutoDock 3 with a genetic search algorithm was chosen for all dockings. The maximum number of energy evaluations, the maximum number of generation in the genetic algorithm and the number of GA per run were 2,500,000, 27, 00000 and 50 respectively. All other run parameters were maintained at their default settings. As far as the search methods are concerned, we used genetic algorithm only.
Curcumin was obtained from Cambridge structure database (CSD), whereas demethoxy curcumin and bis demethoxy curcumin were generated by drawing the 2-D on ChemSketch and 3-D structures from CORINA server and optimized with Insight II program (Accelrys Inc.). Atomic charges were added using calibration given in the AutoDock empirical free energy function and the numbers of flexible torsions were defined as seven for all three curcumin molecules. To find out hydrogen bonds and hydrophobic interactions we used Ligplot [7] and HBPLUS programes [8] that automatically generated 2-D diagrams of protein-ligand interactions from the 3D coordinates in a PDB file. Ligbuilder [9] program was used to design new potent curcumin derivatives and for lead optimization i.e.to improve bioactivity by precalculated key site of interaction and pharmacophore of the receptor.
Discussion
TrxR is inhibited by curcumin irreversibly forming an adduct. Volume and area of the E and F chains of curcumin binding site were simulated by the CASTp program. [10] Volume of E and Fchain cavity were 2451 Å3 and 4996.9 Å3 respectively, while area of both active sites was1437.5 Å2 and 3320.9Å2. In E chain active site 59 amino acid residues and at F chain 135 residues were accessible to the solvent. However, twenty-two amino acids are common at both the active sites. All the three naturally occurring curcuminoids were docked at the active site of E chain whereas the docking energies were -9.17 kcal,-9.48kcal,-8.48 kcal, and binding energies were -6.75 kcal,-7.17 kcal, -7.0 kcal respectively [Table 1 in supplementary material]. 2D plot of hydrogen bonds and hydrophobic interactions of curcumin at the E-chain active site were simulated. Four amino acids namely, HIS108(F), ARG351(F), LYS29(F) and LEU112(F) of F chain interact with curcumin molecule. Amongst these four F chain residues three are basic amino acids. HIS108(F) forms hydrogen bond where ARG351(F), LYS29(F) and LEU112(F) are in hydrophobic contacts. The continuous stretch of carboxyl terminal residues of E chain from SER495 to GLY499 are important in either hydrophobic or in hydrogen bond formation. SER 483 and TRP467 of E chain are also important at the active site. Se atom SeCYS498 residue forms hydrophobic contact with C11 atom of curcumin at a distance of 3.23 Å. Demethoxy curcumin forms two hydrogen bonds with docking energy of -9.48 and binding energy -7.17. Binding of demethoxy curcumin at E chain active site is very precise in comparison to curcumin itself [Figure 2(a)]. 2D plot of hydrogen bond and hydrophobic interaction of demethoxy curcumin of the Echain active site has shown that F-chain does not interact with demethoxy curcumin molecule [Figure 2(b)]. The residues ILE478, THR481, VAL484, ILE492, GLN494, CYS498 and GLY499 are in hydrophobic interactions and the residues SER404, PHE405 SER483 and GLY496 are involved in hydrogen bond interactions. The carboxyl terminal residues of E chain ILE492, GLN494, GLY496, SEC498, GLY499 are important in either hydrophobic or in hydrogen bond formation. The Se atom of SeCYS498 is at the distance of 3.56Å, 3.55 Å and 3.25Å from C13, C12 and C11 respectively.
Figure 2.
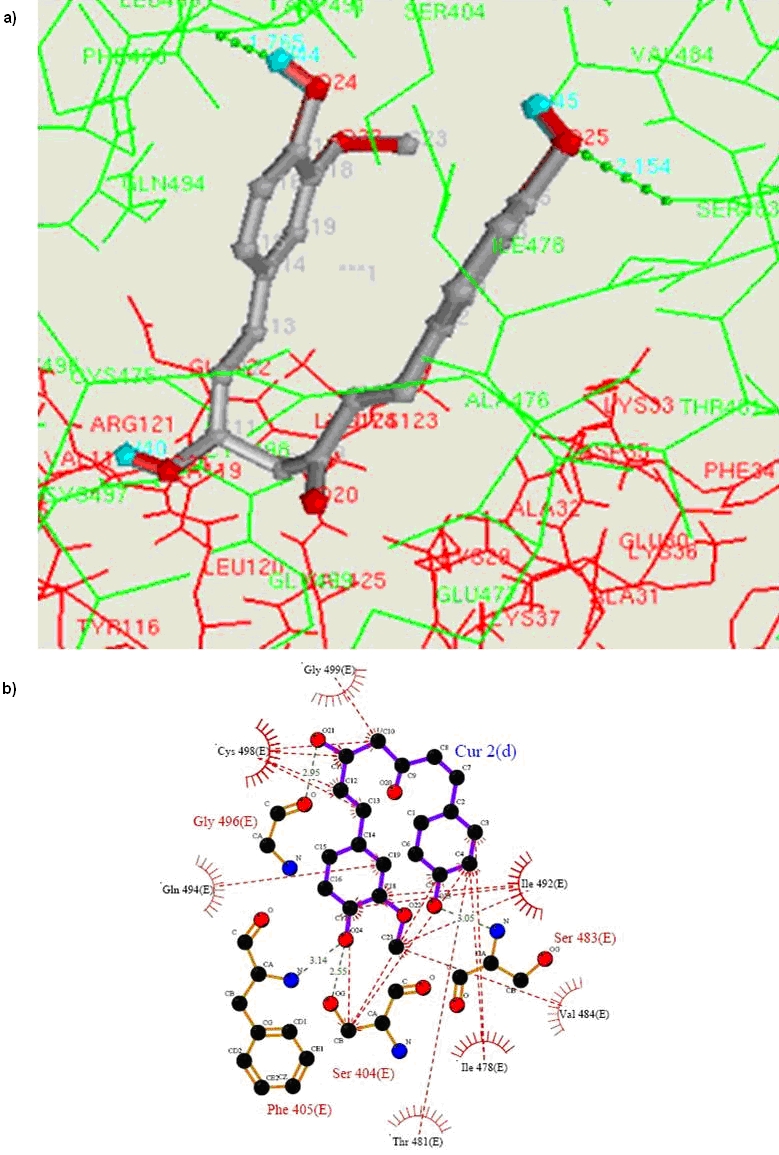
(a) Bound conformation of demethoxy curcumin visualized by ADT in the active site of E-chain of TrxR. Curcumin and TrxR both molecules are in line model with all interacting residues. (b) 2D representation of 3D structure of [Figure 2 (a)] created with LigPlot. [7] The hydrogen bonds were identified with HBPLUS [8] as were the hydrophobic contacts formed between demethoxy curcumin and the TrxR residues.
Bis demethoxy curcumin has only one hydrogen bond with the docking energy of -8.84 kcal and binding energy -7.0. Bis demethoxycurcumin has shown very poor binding in comparison to Curcumin and demthoxy curcumin at same (E chain) active site. The 2D plot of hydrogen bond and hydrophobic interaction of bisdemethoxy curcumin at the E-chain active site has shown that the residue of F-chain does not interact with demthoxy curcumin molecule at E-chain active site. The aromatic residues PHE405 PHE406 TRP407 and SER495 involved in hydrophobic contacts and THR481 SER483 GLN494 ILE492 forming hydrogen bond with bisdemethoxycurcumin molecule. Bis demethoxy curcumin is not as close as curcumin and demethoxy curcumin were to the catalytic residues Cys497 and SeCys498 so it can be concluded methoxy group is crucial for interaction at E chain active site. All the three naturally occurring curcuminoids were also docked at the active site of F chain with docking energies -9.75 kcal,-8.13 kcal,- 10.28 kcal, and -7.29 kcal,-9.09 kcal, -8.25 kcal binding energy were respectively, Table 2 (see supplementary material). Docked conformations of F chain active site and hydrogen bond interaction given in the Table 2 (see supplementary material). The 2D plot of Hydrogen bond and hydrophobic interaction of Curcumin-1 at the F-chain active site is as shown that the residue of E-chain does not interact with curcumin-1 molecule at F-chain active site. Three aromatic amino acids, PHE405 PHE406 TRP407, two acidic amino acids, GLU477 and ASP491 and two nonpolar amino acid residues i.e.VAL484 and ILE492 are involved in hydrophobic interactions. GLN494 is crucial for interaction and stabilizes the docked conformation by hydrogen bonds. The catalytic residues Cys497 and SeCys498 do not approach close to the main chain carbon atoms of curcumin molecule. Demethoxy curcumin also forms two hydrogen bonds with docking energy of -8.13 and binding energy of -9.09. The 2D plot of hydrogen bond and hydrophobic interactions of demethoxy curcumin at the F-chain active site has been shown that TYR106 residue of E-chain forms hydrogen bond with demethoxy curcumin molecule at F-chain active site. Three amino acids PRO473, HIS472 and GLU477 are involved in hydrophobic contacts. Five amino acids viz. LEU409 (F), TYR116(E), CYS498(F), GLY499(F) and TRP407(F), form hydrogen bonds with demethoxy curcumin and also both hydrophobic interactions (at the distance of 3.60Å from C12 and 3.36Å from C11 atom of demethoxy curcumin) and forms hydrogen bond with NH of SeCYS498 and O21 of demethoxy curcumin. Bis-demethoxy curcumin forms three hydrogen bonds with the docking energy of - 10.28 kcal and the best binding energy -8.25. The 2D plot of hydrogen bond and hydrophobic interactions of bisdemethoxy curcumin at the F-chain active site has shown that the residues CYS59, VAL60 and ILE65 of E-chain interact via hydrophobic interactions and SER22 forms hydrogen bond with bis-demethoxy curcumin at F-chain active site. Three aromatic amino acids PHE405, PHE406 and TRP407 are involved in hydrophobic interactions. ILE492 and GLN494 are crucial for interaction and stabilizing bis-demethoxy curcumin molecule by hydrogen bonds.
Pharmacophore and receptor based drug designing
Since docking at the active site of E chain with curcumin and demethoxy curcumin and at F-chain with demethoxy curcumin strongly support wet experiment conducted by Fang et al [2] [concluded by distance mapping Table 3 (see supplementary material)]. Hence we did simulation to design and synthesize more potent bioactive anticancerous curcumin derivative. In this reference we simulated pharmacophore of E-chain active site for curcumin and pharmacophore of E and F chain active site for demethoxy curcumin. Docking with bis- demethoxy curcumin has not produced any significant interaction with the catalytic residues, therefore, bisdemethoxy curcumin was not considered for further analysis. Pharmacophore of E-chain active site with curcumin molecule has shown three hydrophobic sites, two hydrogen bond acceptor sites and three hydrogen bond donor sites and maximal internal distance between two features was 18.63Å. Pharmacophore of E-chain active site with demethoxycurcumin have shown two hydrophobic sites, two hydrogen bond acceptor sites and four hydrogen bond donor sites and maximal internal distance between two points was 15.13Å (Figure 3). Pharmacophore of F-chain active site with demethoxy curcumin has shown three hydrophobic sites, one hydrogen bond acceptor site and four hydrogen bond donor sites and maximal internal distance between two points was 11.08 Å.
Figure 3.
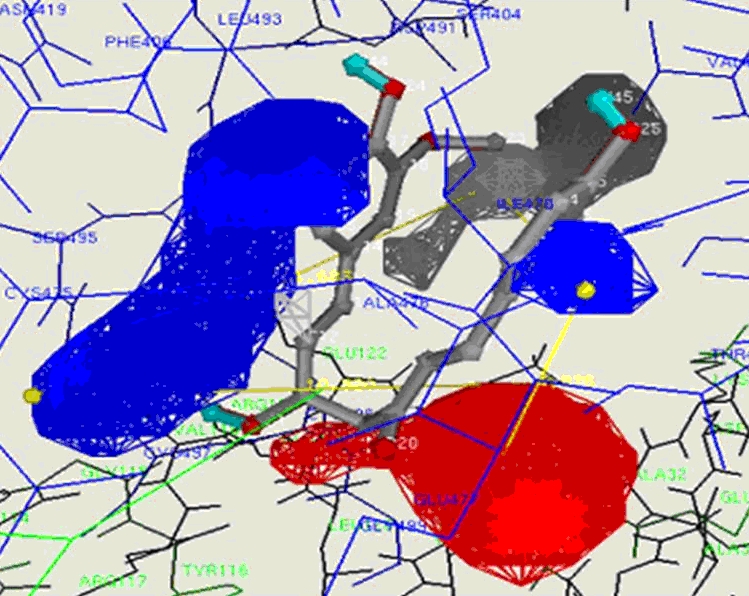
Key site of Interaction of demethoxy curcumin and Contour map [11] of pharmacophore of the F-chain active site: Simulated by ligbuilder programme. [9]
For de-novo design of ligand all parameters (Lipinski rule and ADMET) for drug like molecule were considered. We took curcumin as a seed molecule for E chain active site and demethoxycurcumin as a seed for both E and F chain active sites, growing points were assigned at OH and OCH3 functional groups in all cases. Binding of demethoxy curcumin molecule at E-chain active site was quite precise; hence there was no room to add any substituent. Whereas curcumin at E-chain active site and demethoxy curcumin at F-chain active site have shown big spaces in their hydrophobic pockets so a large hydrophobic moiety could be added to enhance binding and biological activity. The ligand molecule which are being proposed for synthesis are Oc2ccc(/C=C/C(O)CC(=O)/C=C/c1ccc(O)c(OC)c1)nc2OC and Oc2c(cc(/C=C/C(O)CC(=O)/C=C/c1ccc(O)c(OC)c1)nc2OC)CC all being analogs of curcumin molecule. In our docking simulation both chains E and F of thioredoxin reductase were considered together because active site resides at both junctions of chains. Since Fang et al, reported [3] in their mass spectrometric study that it forms 1:2 adduct with Cys-SeCYS residue. Conformation of curcumin molecule at the active site of E-chain is important because Se atom of SeCYS498 is approaching very close to the Sp2 C-11 of main chain of Curcumin molecule. Demethoxycurcumin at both E and F chain active sites is showing more interesting results in comparison to Curcumin itself. In our docking simulation it is clear that demethoxycurcumin molecule is apparently more active than curcumin molecule because the interacting Se atom of catalytic residue SeCys498 and ligand C atoms are very close to each other in both E and F-chain active site. Simulation at the E-chain active site with curcumin and demethoxycurcumin shows the latter to be more potent while bis demethoxy curcumin is not potent because interacting Se atom of receptor and ligand C atom are not as close as it is required for binding. Therefore we can conclude that for greater binding in the pocket of active site at least one methoxy group is necessary [12]. Among all the simulations the demethoxy curcumin molecule has been found to be most bioactive. It has shown greatest docking energy at the E-chain active site and nothing could be added at these functional groups OH, and OCH3. While demethoxy curcumin molecule at the F chain active site has shown greatest binding energy among all simulations of docking at E and F chain active site. Since area and volume of F chain active site is greater than E-chain active site so it provides sufficient space to add larger substituent at the OCH3 and OH functional group.
Conclusion
The present results indicate that synthetic curcumin conjugates could be designed and developed as potent anticancer drugs at TrxR receptor. Our study has shown that curcumin at the E chain and demethoxy curcumin at both E and F chain active sites bind strongly as the Se atom is approaching near to C10, C11 or C12 carbon atom of curcumin molecule where it is already proved that Se atom forms adduct with C13 atom. Therefore, it is possible to predict the design of synthetic curcumin derivatives/ conjugates that may become more potent anti-cancer drugs, which would be in addition to their current anti-cancerous profile.
Supplementary material
Footnotes
Citation:Singh & Misra, Bioinformation 4(5):187-192 (2009)
References
- 1.Garodia P, et al. J Soc Integr Onc. 2007;5:25. doi: 10.2310/7200.2006.029. [DOI] [PubMed] [Google Scholar]
- 2.Aggarwal BB, et al. Anticancer Res. 2003;23:363. [PubMed] [Google Scholar]
- 3.Fang J, et al. J Biol Chem. 2005;280:25284. doi: 10.1074/jbc.M414645200. [DOI] [PubMed] [Google Scholar]
- 4.Arnr ES, Holmgren A. Eur J Biochem. 2000;267:6102. doi: 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- 5.Sandalova T, et al. Proc Natl Acad Sci. 2001;98:9533. doi: 10.1073/pnas.171178698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morris GM, et al. J Comput Aided Mol Des. 1996;10:293. doi: 10.1007/BF00124499. [DOI] [PubMed] [Google Scholar]
- 7.Wallace AC, et al. Prot Eng. 1995;8:127. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 8.McDonald IK, Thornton JM. J Mol Biol. 1994;238:777. doi: 10.1006/jmbi.1994.1334. [DOI] [PubMed] [Google Scholar]
- 9.Wang R, et al. J Mol Model. 2000;6:498. [Google Scholar]
- 10.Sotriffer A, et al. J Med Chem. 2000;43:4109. doi: 10.1021/jm000194t. [DOI] [PubMed] [Google Scholar]
- 11.Sanner MF. J Mol Graph Mod. 1999;17:57. [PubMed] [Google Scholar]
- 12.Tamvakopoulos C, et al. Clin Cancer Res. 2007;13:1269. doi: 10.1158/1078-0432.CCR-06-1839. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.