

Summary
Asparagine-linked glycosylation of polypeptides in the lumen of the endoplasmic reticulum is catalyzed by the hetero-oligomeric oligosaccharyltransferase (OST). OST isoforms with different catalytic subunits (STT3A versus STT3B) and distinct enzymatic properties are coexpressed in mammalian cells. Using siRNA to achieve isoform-specific knockdowns, we show that the OST isoforms cooperate and act sequentially to mediate protein N-glycosylation. The STT3A OST isoform is primarily responsible for cotranslational glycosylation of the nascent polypeptide as it enters the lumen of the endoplasmic reticulum. The STT3B isoform is required for efficient cotranslational glycosylation of an acceptor site adjacent to the N-terminal signal sequence of a secreted protein. Unlike STT3A, STT3B efficiently mediates posttranslational glycosylation of a carboxyl-terminal glycosylation site in an unfolded protein. These distinct and complementary roles for the OST isoforms allow sequential scanning of polypeptides for acceptor sites to insure the maximal efficiency of N-glycosylation.
Introduction
Glycosylation of asparagine residues is an essential protein modification reaction that occurs upon most proteins that enter the secretory pathway in eukaryotic cells. Asparagine-linked oligosaccharides are transferred onto polypeptides in the lumen of the rough endoplasmic reticulum (RER) by the oligosaccharyltransferase (OST) using a dolichol pyrophosphate-linked oligosaccharide (OS-PP-Dol) as the donor substrate. It has long been recognized that the high mannose oligosaccharide (GlcNAc2Man9Glc3) is cotranslationally transferred to the nascent polypeptide as the consensus glycosylation site (sequon, N-X-T/S) enters the RER lumen (Rothman and Lodish, 1977; Chen et al., 1995) by passage through the protein translocation channel (Sec61 complex). Acceptor sites become accessible to the OST active site when there are 65–75 residues of protein between the sequon and the peptidyl-transferase site on the large ribosomal subunit (Whitley et al., 1996; Nilsson et al., 2003; Deprez et al., 2005). This polypeptide length includes 30 residues within the large ribosomal subunit plus roughly 25 residues of extended polypeptide required to span the distance between the cytoplasmic and lumenal faces of the Sec61 complex. The reduced frequency of oligosaccharide addition to glycosylation sequons located less than 75 residues from the C terminus (Gavel and Von Heijne, 1990; Ben-Dor et al., 2004) is thought to be explained by a more rapid passage of the sequon past the OST active site following chain termination, hence, if glycosylated these sites might be modified after the entire protein has been translocated into the ER lumen. With the noteworthy exception of human blood coagulation factor VII (Bolt et al., 2005), posttranslational glycosylation reactions have only been reported in mammalian cells under perturbed conditions when OS-PP-Dol assembly was impaired (Duvet et al., 2002) or when C-terminally truncated substrates were analyzed (Kolhekar et al., 1998). Factor VII is particularly interesting because one sequon is modified cotranslationally, while a second sequon is modified posttranslationally (Bolt et al., 2005).
Mammalian OST is an oligomeric membrane protein consisting of seven to eight nonidentical subunits (reviewed in Kelleher and Gilmore, 2006). Antibodies specific for the cytoplasmic domain of ribophorin I, one of the first identified subunits of the mammalian OST (Kelleher et al., 1992), inhibit protein translocation in vitro by blocking ribosome nascent chain (RNC) targeting to the translocon (Yu et al., 1990). Furthermore, the OST and the Sec61 complex cosediment with RNCs when the RER is solubilized by a nonionic detergent as part of the ribosome-associated membrane protein (RAMP) fraction (Görlich et al., 1992; Shibatani et al., 2005). Although the precise stoichiometry between the OST and the Sec61 complex in uncertain, these studies have led to the concept that a single OST is adjacent to each active translocation channel, thereby facilitating cotranslational N-glycosylation.
STT3 proteins are the active site subunits of the eukaryotic OST (Yan and Lennarz, 2002; Nilsson et al., 2003). Vertebrate, insect and plant genomes encode two STT3 proteins (STT3A and STT3B) (Kelleher et al., 2003; Koiwa et al., 2003; Kelleher and Gilmore, 2006) that are assembled into OST isoforms that differ with respect to the in vitro specific activity and stringency of selecting Dol-PP-GlcNAc2Man9Glc3 in preference to OS-PP-Dol assembly intermediates that lack the terminal glucose residue (Kelleher et al., 2003). STT3A and STT3B mRNAs are widely expressed in human tissues, and both catalytic subunits are coexpressed in tissue culture cell lines.
What advantage is provided to the cell by coexpression of the more active, less selective, STT3B isoform together with the less active, more selective, STT3A isoform? We have tested whether the two OST isoforms have redundant or distinct roles in N-linked glycosylation by siRNA-mediated depletion of STT3A or STT3B. We have found that full glycosylation of polypeptides involves the cooperation of both OST isoforms. Detailed analysis of three glycoprotein substrates revealed that glycosylation is sequential and occurs by a more complex mechanism than previously known. The STT3A isoform is primarily responsible for cotranslational modification of sequons as the nascent polypeptide enters the lumen of the RER. Depletion of STT3A, but not STT3B, causes an induction of the unfolded protein response (UPR) pathway. The STT3B isoform of the OST is less competent for cotranslational glycosylation, but has the capacity to mediate posttranslational modification of skipped glycosylation sites in unfolded proteins.
Results
Isoform-Specific Knockdowns of the OST
Small interfering RNA duplexes (siRNA) were designed to mediate isoform-specific knockdowns of the OST complex. The OST subunit ribophorin I is very stable in HeLa cells (t1/2 = 25 hr; Tsao et al., 1992), hence we anticipated that 3 days of siRNA treatment would be required to achieve significant reductions in STT3A, STT3B, or ribophorin I expression. HeLa cell extracts prepared 72 hr after siRNA transfection were analyzed by protein immunoblotting (Figure 1). The STT3A siRNA reduced STT3A expression to near background levels (<10% of control) without reducing STT3B expression. The previous observation that siRNAs designed to knockdown STT3A cause depletion of STT3B (Wilson and High, 2007; Wilson et al., 2008) is likely explained by an off-target effect. Interestingly, siRNAs specific for STT3A caused an increase in STT3B levels, yet reduced expression of ribophorin I (Figure 1, see quantification). A 7-fold reduction in STT3B expression caused by STT3B siRNA was accompanied by a slight increase in STT3A expression and a decrease in ribophorin I levels.
Figure 1. Effects of siRNA-Mediated Knockdowns on STT3A, STT3B, Ribophorin I, Sec61α, and BiP Expression.
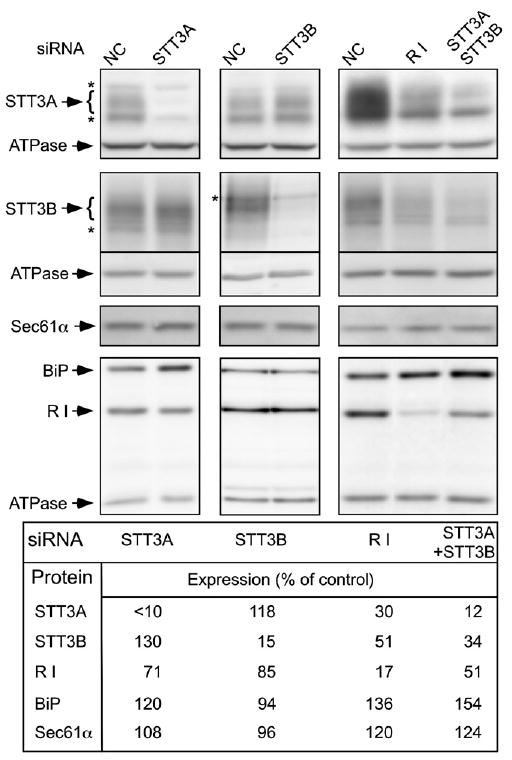
Digitonin-high salt extracts prepared from cells 72 hr after siRNA transfection were processed for immunoblots using antisera for STT3A, STT3B, ribophorin I (R I), BiP, Sec61α, and the α-subunit of the F1F0-ATPase. Asterisks designate nonspecific bands. Similar results were obtained using a second set of each of the siRNAs. The negative control siRNA is abbreviated NC.
The compensatory increases in catalytic subunit expression level are likely explained by elimination of competition between STT3A and STT3B for assembly with the shared set of noncatalytic OST subunits. Since the mammalian OST can be destabilized by loss of the DAD1 subunit (Sanjay et al., 1998), we suspected that STT3A or STT3B depletion destabilizes ribophorin I. Consistent with this explanation, depletion of STT3A plus STT3B led to a 2-fold reduction in ribophorin I content. Furthermore, treatment of cells with siRNA specific for ribophorin I caused a 2- to 3-fold reduction of both STT3A and STT3B (Figure 1). Differences in the extent or duration of ribophorin I depletion might explain why Wilson and colleagues do not observe reductions in STT3A and STT3B content in cells treated with siRNA for ribophorin I (Wilson et al., 2008).
Many glycoproteins are unable to fold when glycosylation is inhibited. The accumulation of defective proteins induces the unfolded protein response (UPR) pathway to cope with the ER protein folding stress (as reviewed by Schroder and Kaufman, 2006). Protein blots were probed with antibodies to BiP, an ER chaperone that is upregulated by the UPR pathway (Figure 1). Elevated levels of BiP were observed whenever STT3A levels were reduced, either directly by transfection with STT3A siRNA or indirectly by transfection with ribophorin I siRNA. The simultaneous depletion of STT3A plus STT3B caused the greatest extent of BiP induction. Depletion of STT3A, STT3B or ribophorin I did not reduce expression of the translocon protein Sec61α.
Digitonin-high salt extracts were assayed for OST activity to correlate subunit loss with enzyme activity. Depletion of ribophorin I, or depletion of STT3A plus STT3B caused a 3- to 4-fold reduction in OST activity (Figure 2A). Isoform-specific depletions had less effect upon OST activity, in part due to the compensatory increase in expression of the other catalytic subunit.
Figure 2. OST Activity and OST Complexes in siRNA-Treated Cells.
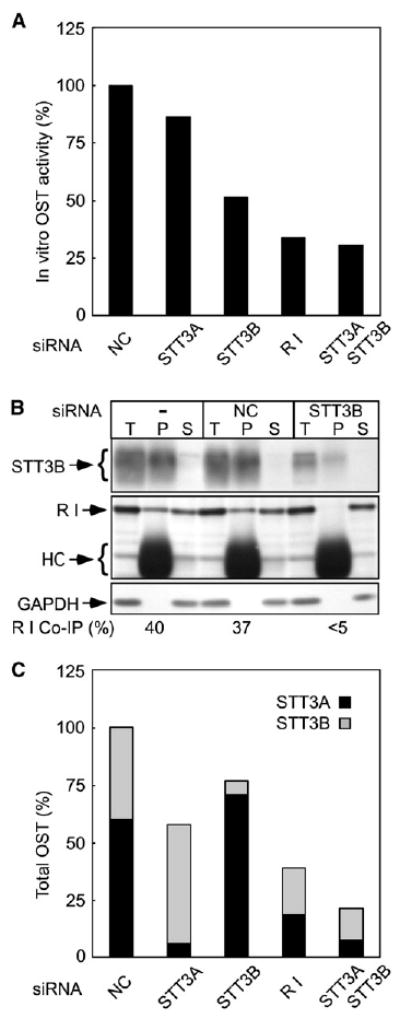
(A) Digitonin-high salt extracts analyzed in Figure 1 were assayed for OST activity. The values shown are averages of duplicate assays normalized to DNA content and expressed relative to negative control siRNA-treated cells.
(B) Native immunoprecipitation using anti-STT3B. Aliquots of the detergent extract (T), immunoprecipitate (P) and supernatant (S) fractions were analyzed on immunoblots. STT3B, R I, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and IgG heavy chain (HC) are labeled.
(C) The relative amount of OST complexes in siRNA-treated cells was estimated using protein immunoblot data (Figure 1) and the STT3A:STT3B ratio (60:40) determined in panel (B).
A co-immunoprecipitation approach was used to determine the STT3A:STT3B ratio in HeLa cells. Antisera raised against the N-terminus of STT3B quantitatively precipitated STT3B from digitonin-high salt extracts under nondenaturing conditions, along with 40 ± 5% of the ribophorin I (Figure 2B). Only traces of ribophorin I were immunoprecipitated when extracts were prepared from STT3B-depleted cells. As unassembled ribophorin I is not detected in cells or tissues (Kelleher and Gilmore, 1997), we can conclude that the STT3A:STT3B ratio in HeLa cells is roughly 60:40. The relative number of total OST complexes (STT3A+STT3B) in the siRNA-treated cells was estimated from the protein immunoblot data in Figure 1 and the 60:40 isoform ratio (Figure 2C). The in vitro OST activity (Figure 2A) is not directly proportional to the calculated cellular content of OST complexes (Figure 2C) due to the higher in vitro activity of the STT3B isoform (Kelleher et al., 2003).
We next asked whether both isoforms of the OST are components of the RAMP fraction. Proteomic analysis has shown that the major components of canine pancreas high-salt RAMP fraction are the Sec61, TRAM and TRAP complexes plus the signal peptidase and the STT3A isoform of the OST (Shibatani et al., 2005). Here, we observed that the STT3B isoform of the OST was more readily extracted than the STT3A isoform from a HeLa cell RAMP fraction under low salt conditions (Figure S1 available with this article online).
Hypoglycosylation of Endogenous Glycoproteins
The effect of OST isoform depletion on total N-linked glycoprotein synthesis was evaluated by capturing pulse-labeled glycoproteins with ConA Sepharose beads. Total radiolabeled glycoproteins were reduced by less than 5% in cells treated with either STT3 siRNA. SDS-PAGE resolution of the ConA eluate revealed several glycoproteins, including p65, that were less intense in extracts from STT3A-depleted cells (Figure 3A, circles) compared to control or STT3B-depleted cells. The reduced intensity of p65 was accompanied by the appearance of three more rapidly migrating proteins that are likely hypoglycosylated p65 variants (Figure 3A, triangle). Endoglycosidase H digestion (Figure S2A), and alkaline fractionation of total cell homogenates prior to ConA binding (Figure S2B) revealed that p65 is a soluble protein with five N-linked glycans. Candidates for p65 were identified using a bioinformatics approach and tested by immunoprecipitation of pulse-labeled cell extracts. Antisera specific for the lysosomal protein saposin D immunoprecipitated p65 (Figure S2C). Saposins A–D are derived by proteolytic processing of prosaposin (Figure 3B), a 65 kD precursor that is comprised of alternating spacer segments and saposin domains (as reviewed by Kishimoto et al., 1992). Prosaposin is also secreted by many cell types (Kishimoto et al., 1992).
Figure 3. Hypoglycosylation of Prosaposin in STT3A-Depleted Cells.
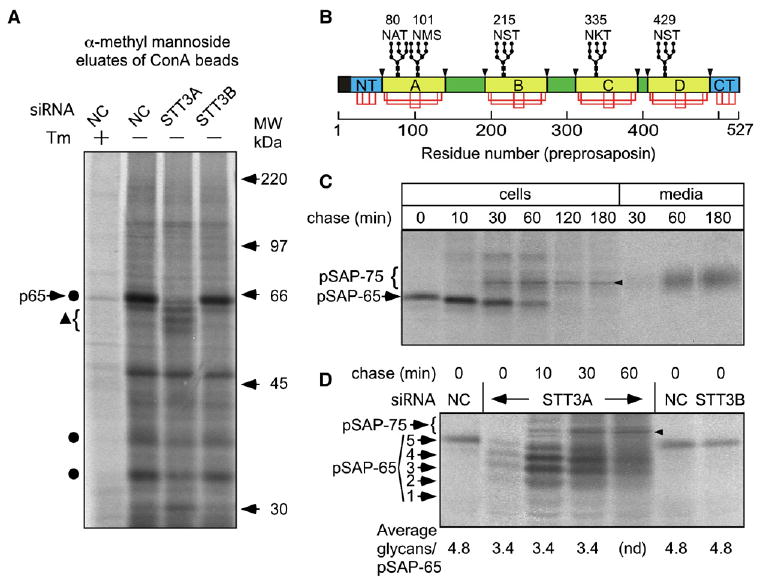
(A, C, and D) HeLa cells treated with the indicated siRNAs were pulse-labeled for 2 min (D) or 5 min (A and C) and chased as indicated (C and D).
(A) One plate of control cells was treated with tunicamycin (Tm) prior to labeling. Equal amounts of 35S-labeled protein were incubated with Con A beads. Glycoproteins were eluted and resolved by SDS-PAGE. Circles designate glycoproteins that decrease in intensity in STT3A-depleted cells. The triangle designates hypoglycosylated variants of p65.
(B) Diagram of preprosaposin showing the signal sequence (black), the homologous N- and C-terminal domains (NT and CT; cyan), the saposin domains (yellow), and spacer segments (green). Glycosylation sites (e.g., 80 NAT) disulfide bonds (red lines) and lysosomal processing sites (arrowheads) are shown. The disulfide-bonding pattern of the invariant cysteine residues in the NT and CT domains is not known.
(C) Pulse-chase labeling of prosaposin in control cells. Intracellular (cells) and secreted (media) prosaposin was recovered by immunoprecipitation. The ER (pSAP-65) and Golgi (pSAP-75) forms of prosaposin are labeled. The arrowhead designates a nonspecific band. (D) Pulse-chase labeling of prosaposin. Glycoforms of pSAP-65 are labeled; fully glycosylated pSAP-75 is indicated by the bracket. The average number of glycans per pSAP-65 was calculated after quantification of the pSAP-65 glycoforms.
Processing of prosaposin (pSAP-65) in control cells was analyzed by pulse-chase labeling. The gel-mobility of pSAP-65 increased in a stepwise manner at the 10 and 30 min chase points (Figure 3C) due to sequential trimming of glucose and mannose residues (Figure S3A). As reported previously (Rijnboutt et al., 1991) transport of pSAP-65 from the ER to the Golgi is rapid. The half time for appearance of prosaposin bearing complex oligosaccharides (pSAP-75) was 33 min. Intact pSAP-75 was first detected in cell culture media at the 30 min chase point, and was as abundant as intracellular prosaposin after a 1 hr chase.
Prosaposin was cotranslationally glycosylated by control cells or STT3B-depleted cells (Figure 3D). The short duration of the pulse (2 min) monitors cotranslational glycosylation since prosaposin synthesis takes roughly 90 s (527 residues at ∼6 residues/sec, (Hershey, 1991)). Prosaposin glycoforms lacking up to four oligosaccharides were synthesized by STT3A-depleted cells, with an average reduction of 1.5 glycans per chain. Notably, the 2-fold decrease in total OST content in these cells did not cause a bimodal distribution of pSAP-65 glycoforms (i.e., 0 or 5 glycans). The average number of glycans on pSAP-65 did not increase during the chase incubation, indicating that skipped sites are not posttranslationally glycosylated. Instead, hypoglycosylated variants of pSAP-65 were trimmed, as shown by the increased gel mobility of each glycoform at the 10 and 30 min chase points. Overlap between hypoglycosylated pSAP-75 and pSAP-65 caused the diffuse smear detected at the 60 min chase point. Experiments conducted using ConA to isolate high-mannose glycoproteins yielded identical results concerning processing of pSAP-65 (Figure S3B).
Cotranslational Glycosylation of a Sequon Near the Signal Peptidase Cleavage Site
Procathepsin C (pCatC) is a major secreted HeLa cell glycoprotein (Vollenweider et al., 1998) that has four glycans (Figure 4A). Procathepsin C chains lacking 1-4 N-linked glycans were synthesized by cells that were treated with siRNA for ribophorin I or siRNAs for STT3A plus STT3B (Figure 4B). The 4-fold reduction in OST content and activity in these siRNA-treated cells did not result in significant synthesis of nonglycosylated pCatC, but instead reduced the average number of glycans per protein by 25% or less. A 2 min pulse, followed by an 8 min chase, showed that pCatC glycosylation was cotranslational even though the cellular concentration of the OST was reduced 4-fold.
Figure 4. Glycosylation of pCatC in siRNA-Treated Cells.
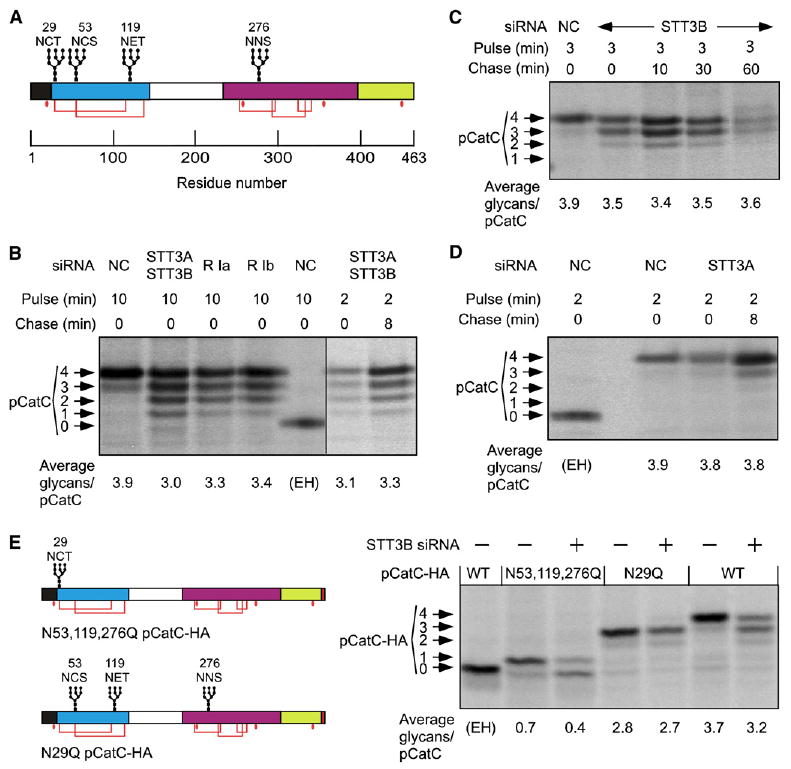
(A) Diagram of the preprocathepsin C showing the signal sequence (black), retained propeptide (cyan), cleaved propeptide, (white), heavy chain (magenta) and light chain (yellow). Glycosylation sites, disulfide bonds (red lines) and free cysteines (red dots) are shown.
(B–D) Cells treated with the indicated siRNAs prior to pulse-chase labeling were used for pCatC immunoprecipitation. Results obtained with two siRNAs for ribophorin I are shown.
(E) Diagrams of pCatC-HA glycosylation site mutants. Cells cotransfected with pCatC-HA constructs and negative control (−) or STT3B (+) siRNAs were pulse labeled for 3 min, and immunoprecipitated using anti-HA. (B, D, and E) As indicated, samples from control cells were digested with Endo H (EH). Procathepsin C with 0-4 N-linked glycans were resolved by SDS-PAGE and the average number of glycans per pCatC chain was calculated.
Depletion of STT3B resulted in the synthesis of pCatC chains lacking 0–2 glycans (Figure 4C). Roughly half the pCatC chains lack one glycan, suggesting that one sequon is typically modified by STT3B. Skipped sites in pCatC were not modified during a 1 hr chase in STT3B-depleted cells. Procathepsin C was immunoprecipitated from STT3A-depleted cells that were pulse-labeled for 2 min and chased for 8 min (Figure 4D). Even when the pulse-labeling period was similar to the biosynthesis time (∼80 s), pCatC had an average of 3.9 glycans in controls cells and 3.8 glycans in STT3A-depleted cells. Taken together, these results indicate that three of the four glycosylation sites in pCatC can be cotranslationally modified by either STT3A or STT3B, while a fourth site is poorly glycosylated in STT3B-depleted cells.
The N-terminal sequon in pCatC is four residues from the signal sequence cleavage site (Figure 4A). Cysteine residues within the N29 and N53 sequons form disulfides with cysteines flanking the third sequon (Horn et al., 2002). We tested the hypothesis that N29 is hypoglycosylated in STT3B-depleted cells by expressing HA-tagged derivatives of pCatC (Figure 4E). As observed for endogenous pCatC, depletion of STT3B caused hypoglycosylation of one site in pCatC-HA. Mutagenesis of the N-terminal sequon (N29Q pCatC-HA) eliminated hypoglycosylation of pCatC in STT3B-depleted cells relative to control cells. Remarkably, pCatC chains that retain the N29 sequon as the only acceptor site were hypoglycosylated in STT3B-depleted cells. Proximity to the signal sequence cleavage site, rather than cysteine within the sequon, is the most likely explanation for the STT3B-dependent modification of the N29 sequon in pCatC.
The STT3B Isoform Mediates Posttranslational N-Glycosylation
Blood coagulation factor VII has two sequons (Figure 5A), one of which (N183) is glycosylated cotranslationally, while the second site (N360) is mainly glycosylated posttranslationally (Bolt et al., 2005). Factor VII pulse-labeled for 45 s consisted of nascent chains (g-nc and nc) plus nonglycosylated (FVII-0), mono-glycosylated (FVII-1) and di-glycosylated (FVII-2) full-length factor VII (Figure 5B). Endo H digestion of the 45 s pulse-labeled sample showed that most nascent chains carry a single glycan which, based upon nascent chain length, is linked to N183. Posttranslational glycosylation of FVII at the N360 site occurred during the chase incubation. The kinetics of FVII glycosylation was analyzed using a more extensive pulse-chase experiment. The time courses for disappearance of FVII-0 and formation of FVII-2 in control cells are well fit by single exponential equations (Figure 5D), with t1/2 values of 8 and 9 min respectively (Table S1). Disappearance of FVII-1 is well fit by a double exponential equation consisting of the parameters that describe consumption of FVII-0 and formation of FVII-2. Pulse-chase labeling of the N360Q Factor VII mutant revealed that 20% of the N183 sites are not cotranslationally glycosylated, and that the majority of skipped N183 sites are not modified during the chase (Figure 5G). Consistent with the detection of FVII-2 at the 0 min chase point in control cells (Figure 5D), roughly one third of the N360 sites were glycosylated at the 0 min chase point in the N183Q FVII mutant (Figure 5H).
Figure 5. Glycosylation of FVII in siRNA-Treated Cells.
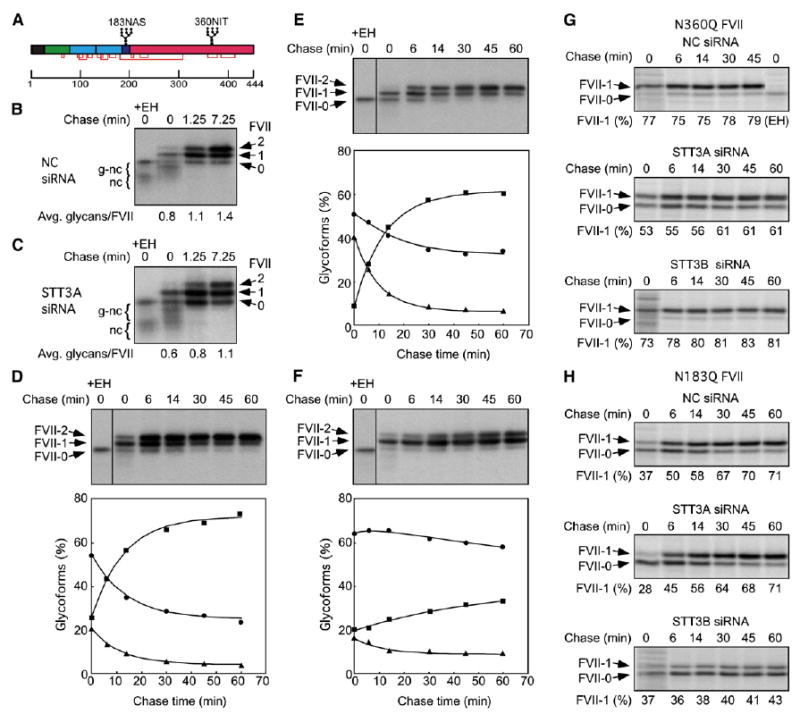
(A) Diagram of the FVII precursor showing the signal sequence (black), the gamma-carboxyglutamate domain (green), two EGF-like domains (cyan), the activation peptide (purple) and the catalytic domain (red). Glycosylation sites and disulfide bonds are shown. Cells cotransfected with wild-type (B–F), N360Q (G), or N183Q (H) FVII plasmids and negative control (B, D, G, and H), STT3A (C, E, G, and H), or STT3B (F–H) siRNAs were pulse labeled for 45 s (B and C) or 2 min (D–H) and chased as indicated. One pulse labeled sample was digested with Endo H (EH). Full-length FVII polypeptides that contain 0, 1, or 2 N-linked oligosaccharides were resolved from glycosylated nascent chains (g-nc) and nonglycosylated and deglycosylated nascent chains (nc). (D–F) The percentages of FVII molecules that have 0 (triangles), 1 (circles) or 2 (squares) N-linked oligosaccharides are shown.
The siRNA-treated cells were transfected with a factor VII expression vector to determine whether a specific OST isoform mediates glycosylation of N183 and N360. Depletion of STT3A increased the percentage of FVII-0 that was detected after pulse labeling for 45 s (Figure 5C) or 2 min (Figure 5E). Nonglycosylated (i.e., Endo H resistant) nascent chains were readily detected in the 45 s pulse-labeled sample. Consumption of FVII-0 and formation of FVII-2 occurred with kinetics that were comparable to that observed for control cells (t1/2 = 6.6 and 8.4 min respectively, Table S1). The plateau value for FVII-1 is elevated relative to control cells (Figure 5E) primarily due to reduced cotranslational glycosylation of the N183 site (Figure 5G). These data indicate that STT3A isoform is important, but not essential, for cotranslational N-glycosylation of Factor VII. The STT3A isoform is completely dispensable for posttranslational glycosylation of N360 (Figure 5H).
Posttranslational glycosylation was greatly reduced in the STT3B-depleted cells, as indicated by the low extent and slow kinetics (t1/2 ∼36 min) of FVII-2 formation (Figure 5F). Inefficient posttranslational modification of the N360 sequon in STT3B-depleted cells was confirmed by pulse-chase analysis of the N183Q mutant (Figure 5H). STT3B depletion did not reduce cotranslational modification of N183, as shown by the low percentage of FVII-0 synthesized during the 2 min pulse label (Figure 5F) and the efficient glycosylation of the N360Q factor VII mutant (Figure 5G). The STT3B isoform is responsible for posttranslational glycosylation of FVII on N360 (Figure 5H) but dispensable for cotranslational glycosylation of FVII on N183 (Figure 5G).
In control and STT3A-depleted cells, posttranslational glycosylation of wild-type factor VII at N360 to yield FVII-2 is more efficient than expected from the analysis of the single site mutants (N183Q and N360Q). If glycosylation of the two sequons is independent, the probability of FVII-2 formation is the product of the single site modification values (0.7 × 0.8 in control cells). Enhanced formation of FVII-2 (e.g., 0.73 instead of 0.56) indicates that posttranslational glycosylation of N360 by the STT3B isoform is positively regulated by cotranslational glycosylation of N183.
Protein Folding Limits Posttranslational Glycosylation by STT3B
Why can certain sequons be modified minutes after protein termination while skipped sites in pSAP-65 are refractory to posttranslational glycosylation? A hydrogen bond between the side chain of serine or threonine and the carboxamide oxygen of asparagine is proposed to be essential for catalysis by the OST (Imperiali et al., 1992), and this conformation cannot be achieved upon folding of the saposin domain. Saposin domains have a conserved N-glycosylation site (N80 in saposin A) located in a turn joining two α-helical segments (Ahn et al., 2006) and are stabilized by a hydrophobic protein core and three invariant disulfide bonds (Figure 6D). To obtain evidence concerning the rate of folding of pSAP-65 and factor VII, we treated detergent lysates of pulse-labeled control cells with the cysteine modification reagent PEG-maleimide. As a control, we examined pegylation of pCatC, a protein with five disulfide bonds and three free cysteine residues (Figure 4A). Only one cysteine (C355) in pCatC is accessible to a bulky reagent like PEG-maleimide in the folded state (Turk et al., 2001; Horn et al., 2002). Pegylation of pulse-labeled pCatC yields a ladder (Figure 6A) as observed previously for other proteins (Lu and Deutsch, 2001), where each PEG adduct increases the apparent molecular weight of pCatC by 10-15 kD. Reduced levels of pCatC with three PEG-Mal adducts at the 30 min chase point suggests that some chains have folded, and have a single exposed cysteine residue.
Figure 6. Disulfide Bond Formation in FVII, pCatC, and pSAP.
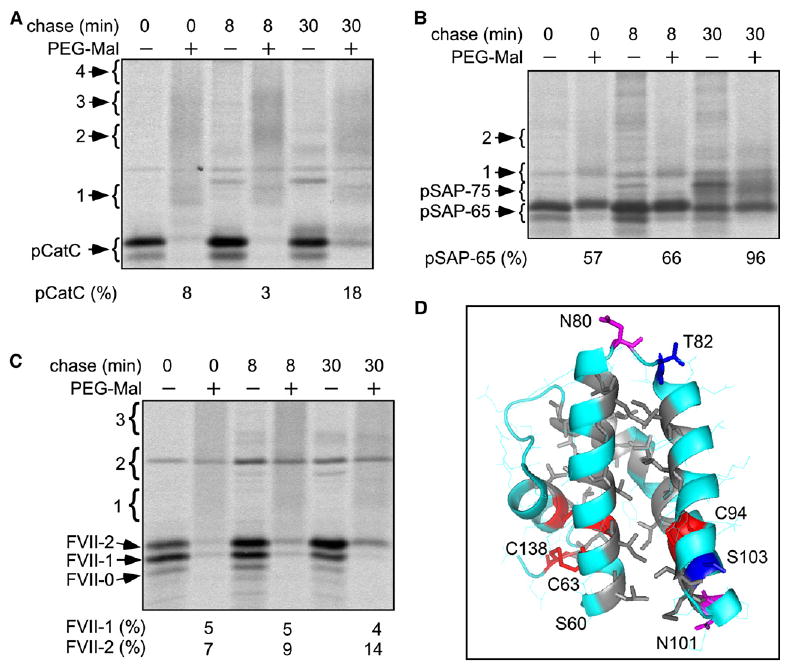
(A–C) Control cells were pulsed for 5 min and chased as indicated. Cell extracts were treated with PEG-maleimide (PEG-Mal) as indicated prior to immnuoprecipitation of pCatC (A), pSAP (B), or FVII (C). Proteins are labeled as in Figure 3–5. Brackets labeled 1–4 designate the migration position of proteins that have 1–4 PEG-Mal adducts. Larger adducts (data not shown) migrated as a smear. The percentage of PEG-Mal resistant pCatC, FVII-1, FVII-2, and pSAP-65 relative to the untreated sample is shown below each panel. (D) The saposin domain is stabilized by three disulfide bonds (in red) and a hydrophobic core (A, I, L, V, and M residues are gray). The distance between the carboxamide oxygen of N80 and the side chain of T82 is 10.9 Å. N101 and T103 are on opposite faces of the third α-helix. The figure was created with MacPyMOL software using the saposin A structure (PDB 2DOB).
Due to the high number of cysteine residues in pSAP-65 (Figure 3B), we focused our analysis on the presence of PEG-Mal resistant pSAP-65 and pSAP-75 (Figure 6B). More than 50% of pSAP-65 chains were PEG-Mal insensitive after the pulse, indicating that pSAP-65 disulfides form rapidly. Although PEG-Mal adducts of pSAP-65 were detected at the 0 and 8 min chase points, these adducts disappeared by the 30 min chase point, and pegylated derivatives of pSAP-75 were not detected.
Formation of disulfide bonds in factor VII was evaluated by comparing the labeling intensity of FVII-0, FVII-1 and FVII-2 in the absence or presence of PEG-Mal (Figure 6C). In contrast to prosaposin, Factor VII was completely susceptible to pegylation after the pulse. Nonglycosylated FVII-0 and monoglycosylated FVII-1 remained sensitive to pegylation for 30 min. Acquisition of PEG-Mal resistance by FVII-2 lagged well behind addition of the second glycan. These results are consistent with the previous conclusion that N360 glycosylation occurs prior to folding of factor VII (Bolt et al., 2005).
Discussion
The objective of this study was to elucidate the in vivo role of the two OST isoforms during protein glycosylation in mammalian cells. Analysis of glycoprotein synthesis in OST isoform-depleted cells revealed that the OST isoforms cooperate in a sequential manner to glycosylate the full range of acceptor sites in glycoproteins.
Nascent Glycoproteins Contact Multiple OSTs
If a single OST complex is adjacent to each active translocation channel, a 40% reduction in OST content in the STT3A-depleted cells should yield translocons that lack an OST complex, thereby resulting in the synthesis of a mixture of fully glycosylated and nonglycosylated proteins. Contrary to this prediction, total glycoprotein synthesis was reduced by less than 5% when STT3A was depleted. Even a 4-fold reduction in OST content in the double-knockdown cells (STT3A + STT3B) led to the synthesis of a heterogeneous mixture of pCatC glycoforms. Remarkably, nonglycosylated pCatC chains were barely detectable. Models that specify localization of a single OST adjacent to each protein translocation channel are incompatible with the observed heterogeneity of hypoglycosylated glycoproteins. Instead, we can conclude that the OST is present in excess relative to active translocons (Figure 7A), and that nascent polypeptides contact multiple OST complexes upon emergence into the ER lumen (Figure 7B). Similar patterns of protein hypoglycosylation were also observed in yeast cells when synthesis of Stt3p was placed under control of the GAL1/GAL10 promoter and STT3 transcription was repressed by growing cells in glucose containing media (Zufferey et al., 1995). An excess of the OST relative to active translocation channels is therefore a conserved property of the endoplasmic reticulum. Quantitative protein immunoblots indicate that canine pancreas microsomes have an excess of OST complexes relative to active protein translocation channels (Guth et al., 2004).
Figure 7. Cooperation between OST Isoforms in N-Linked Glycosylation.
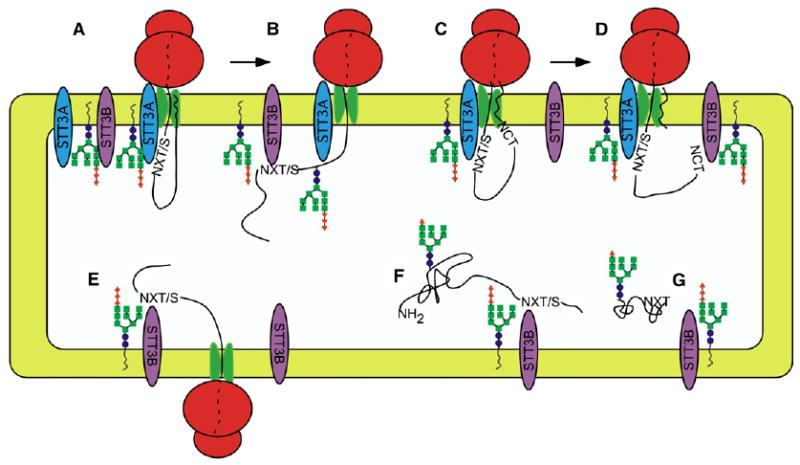
Cotranslational glycosylation of nascent glycoproteins in mammalian cells (A and B). Skipped sites can be cotranslationally glycosylated by STT3B (B). The signal sequence (wavy line) remains bound to the translocon until cleaved (C). Cotranslational glycosylation of an extreme N-terminal glycosylation site by the STT3B isoform (D). Reduced efficiency of cotranslational glycosylation in STT3A-depleted cells (E). Posttranslational glycosylation of a C-terminal sequon by the STT3B isoform (F) is limited by protein folding (G).
Sequential Roles for OST Isoforms in N-Glycosylation
Analysis of single knockdowns revealed that certain glycoproteins like ribophorin I (Figure 1) were not hypoglycosylated. Glycosylation sites that can be efficiently modified by both OST isoforms appear to be common based upon a comparison of total pulse-labeled glycoproteins isolated with ConA beads. However, pulse-chase experiments demonstrated that depletion of STT3A or STT3B had nonequivalent effects upon cotranslational modification of pCatC and pSAP-65. Radiolabeled NYT is used as the acceptor substrate in the in vitro OST assay, so it was formally possible that the more active STT3B isoform prefers NXT sites while the less active STT3A complex prefers NXS sites. This explanation for substrate specificity can be discounted because the STT3A isoform efficiently glycosylates NXT and NXS sites in prosaposin, while the STT3B enzyme glycosylates NXT and NXS sites in pCatC.
Nonidentical phenotypes for STT3 mutants have also been observed in Arabidopsis thaliana. A T-DNA insertion into the A. thaliana STT3A gene, but not the STT3B gene, causes osmotic sensitivity of root tips, and induction of the UPR pathway due to protein hypoglycosylation (Koiwa et al., 2003). In contrast to the viable single mutants, the double mutant (stt3a-1 stt3b-1) is gametophytic lethal, indicating that the plant STT3 proteins have partially overlapping roles.
In vivo pulse-labeling of nascent glycoproteins was essential to allow resolution of cotranslational and early posttranslational glycosylation events. The N183 site in factor VII is an obligatory cotranslational glycosylation site, while the N360 site in primarily posttranslational. Depletion of STT3A reduced the efficiency of N183 glycosylation without altering the kinetics of N360 glycosylation. In contrast, STT3B depletion did not impact N183 glycosylation but reduced the rate and extent of posttranslational glycosylation of N360. Taken together, these results indicate that the in vivo substrate specificity is explained by sequential scanning of the nascent polypeptide for sequons by the STT3A and STT3B isoforms.
What additional evidence supports the conclusion that the STT3A isoform is primarily responsible for cotranslational glycosylation? Depletion of STT3A caused a dramatic decrease in cotranslational glycosylation of pSAP-65. Induction of the UPR pathway upon depletion of STT3A but not STT3B, supports the conclusion that most glycans are added by the STT3A isoform. Efficient cotranslational modification of sequons is explained by localization of the OST adjacent to the translocon, such that roughly 20 residues of nascent polypeptide span the distance between the lumenal face of the Sec61 complex and the OST active site. Experiments using an appropriately positioned cryptic glycosylation site adjacent to a photoreactive lysine derivative allowed efficient crosslinking of a nascent polypeptide to STT3A (Nilsson et al., 2003). A crosslinked product that might correspond to STT3B was conspicuously absent, consistent with a more distal location for STT3B relative to the translocon (Figure 7A).
Accurate donor substrate selection is important for glycoprotein quality control in the ER because the glycan trimming intermediate Glc1Man9GlcNAc2 is recognized by the lectin chaperones calnexin and calreticulin (as reviewed by Helenius and Aebi, 2004). Exquisite selection of Glc3Man9GlcNAc2-PP-Dol in preference to lumenally oriented OS-PP-Dol assembly intermediates is a property of the STT3A isoform (Kelleher et al., 2003). Cotranslational glycosylation of proteins by the STT3A isoform insures that the transferred glycans have the correct structure for the ER quality control pathway.
Since depletion of STT3A did not eliminate cotranslational glycosylation, we can conclude that the STT3B isoform has cotranslational access to skipped glycosylation sites (Figure 7B), thereby enhancing the fidelity of N-glycan addition. Because the STT3B isoform is less proximal to the Sec61 complex, the efficiency of cotranslational glycosylation is impaired when STT3A is depleted (Figure 7E). Cotranslational access of both OST isoforms to substrates explains why many HeLa cell glycoproteins were not hypoglycosylated in STT3A-depleted cells (Figure 3A). The higher turnover rate of the STT3B isoform would favor cotranslational and posttranslational modification of skipped glycosylation sites prior to protein folding.
STT3B-Specific Glycosylation Sites
Two of the three substrates we tested were hypoglycosylated in STT3B-depleted cells. The N360 sequon in factor VII is 84 residues from the C terminus, so chain termination will occur soon after this sequon becomes accessible to the OST. Hence, inefficient cotranslational glycosylation of N360 is not surprising. We predict that sequons located less than 75 residues from the C terminus of a protein constitute a major class of STT3B substrates (Figure 7F).
Hypoglycosylation of procathepsin C in STT3B-depleted cells revealed that STT3B sites are not restricted to the extreme C terminus of the protein, nor are STT3B sites obligatorily post-translational. Disulfide bond formation and N-glycosylation are competing reactions (Allen et al., 1995; Tanaka et al., 2002), hence the N29 site in pCatC must be glycosylated prior to formation of the C30-C118 disulfide. Sequons near the signal sequence cleavage sites of yeast prepro-α-factor and invertase are poorly glycosylated when signal sequence cleavage is blocked (Chen et al., 2001). In the case of pCatC, the proximity between the signal sequence cleavage site and the N29 sequon likely prevents access to the OST active site prior to signal sequence cleavage (Figure 7C). Sequons in lumenal segments of integral membrane proteins are not glycosylated if the asparagine residue is less than 30-40Å from the membrane surface (Nilsson and von Heijne, 1993). Upon cotranslational cleavage of the signal sequence, the N29 site in pCatC may exit the lumenal face of the translocon too rapidly to be modified by the translocon proximal STT3A OST that is scanning the nascent polypeptide for glycosylation sites, but be readily accessible to the translocon distal STT3B isoform (Figure 7D).
Competition between Posttranslational Glycosylation and Protein Folding
Posttranslational glycosylation of proteins by the STT3B isoform of the OST appears to be limited by folding of the polypeptide in the ER lumen (Figure 7F and 7G). As an example of a rapidly folding protein, more than 50% of pSAP-65 chains lack accessible cysteine residues at the end of a 5 min pulse-labeling period. Rapid formation of disulfides in pSAP-65 is consistent with the kinetics of glycan trimming, acquisition of complex glycans, and secretion of pSAP-75. Point mutations (N215H or T217I) in prosaposin that prevent glycosylation at N215 cause a variant form of metachromatic leukodystrophy (Kretz et al., 1990; Wrobe et al., 2000). Folding and transport of N215H prosaposin to the lysosome is normal, but glycan deficient saposin B is less active (Wrobe et al., 2000). Skipped sites in pSAP-65 were not substrates for posttranslational glycosylation (Figure 7G). At the other extreme, posttranslational glycosylation of N360 in FVII is permitted because hypoglycosylated FVII-1 remains unfolded for more than 30 min after the protein enters the ER lumen (Figure 7F).
Posttranslational glycosylation of sequons is not restricted to the mammalian STT3B, but appears to be an ancient and conserved property of the OST. The single subunit Campylobacter jejuni OST (PglB) can glycosylate sequons on a folded protein if the acceptor site is located in a flexible loop (Kowarik et al., 2006). The variant surface glycoprotein (VSG221) of Trypanosoma brucei has two glycosylation sites; one site is cotranslationally glycosylated with a GlcNAc2Man5 glycan, while a second site located near the extreme C terminus is modified posttranslationally with a GlcNAc2Man9 glycan (Ferguson et al., 1986; Manthri et al., 2008). The utilization of different OS-PP-Dol donors for the cotranslational and posttranslational sites might be explained by the presence of three STT3 genes in the T. brucei genome (Manthri et al., 2008). Duplication of the STT3 gene and subsequent divergence of STT3A and STT3B has allowed the evolution of OST isoforms that are optimized for complementary roles in N-glycan addition. We propose that the STT3B isoform of the OST acts after the initial stage of glycoprotein synthesis to enhance sequon occupancy and maximize the efficiency of chaperone-mediated glycoprotein folding and quality control.
Experimental Procedures
Cell Culture and Plasmid or dsRNA Transfection
Two dsRNAs for each target (STT3A, STT3B and R I) were custom synthesized by Dharmacon. Sequences of dsRNAs are in the supplemental materials. Negative control siRNA was purchased from QIAGEN.
HeLa cells (ATCC CCL-13) were cultured in 10 cm2 dishes at 37°C in DMEM (GIBCO), 10% fetal bovine serum with penicillin (100 units/ml) and streptomycin (100 μg/ml). Approximately 2 × 105 HeLa cells were plated and grown for 24 hr prior to transfection with 30 nM dsRNA and Lipofectamine 2000 in OptiMEM using protocols from Dharmacon. Cells were typically assayed 72 hr after transfection. To achieve better depletion of STT3B for pulse-labeling experiments, cells were subjected to a second siRNA transfection after 48–72 hr using 40–60 nM siRNA and were assayed 48 hr after the second transfection.
Construction of factor VII and pCatC-HA glycosylation mutants is described in the supplemental materials. Cells were transfected with the plasmid pcDNA3-FVII, obtained from Dr. Katherine High (Children's Hospital of Philadelphia), 48 hr after treatment with negative control or STT3A siRNAs and were analyzed 24 hr later. After 48–72 hr of treatment of cells with STT3B siRNA, cells were sequentially transfected with STT3B siRNA and pcDNA3-FVII or pcDNA3-pCatC-HA and analyzed 48 hr later. For each plate, 6 μg of plasmid DNA was transfected with Lipofectamine 2000 using the manufacturer's instructions.
In Vitro Oligosaccharyltransferase Assay
Cells at 70% confluency were washed and then scrape-extracted using a digitonin-high salt buffer as described (Kelleher and Gilmore, 1997). An aliquot of the extract was removed to isolate DNA (FlexiGene DNA Kit, QIAGEN), which was quantified (A260) to normalize OST activity relative to cell number. The remainder of the extract was digested with DNase I (50 μg/ml, Sigma) prior to clarification by centrifugation. Clarified extracts were assayed for OST activity as described (Kelleher and Gilmore, 1997) using 10 μM of Nα-Ac-Asn-[125I]Tyr-Thr-NH2 and 0.6 μM porcine pancreas OS-PP-Dol (∼63% Glc3Man9GlcNAc2-PP-Dol).
Antibodies and Native Immunoprecipitation
Sources of antibodies are listed in the supplemental materials. Clarified digitonin extracts from control and STT3B-depleted cells were diluted in buffer to obtain a final concentration of 50 mM Tris-Cl, 150 mM NaCl, 1% digitonin, 1 mM MgCl2, 1 mM MnCl2, 1 mM DTT (native IP buffer). Digitonin samples were incubated overnight with Protein A-Sepharose beads precoated with the STT3B antibody. Immunoprecipitates were washed with native IP buffer containing 0.1% digitonin and eluted with gel loading buffer.
Radiolabeling, Immunoprecipitation, and ConA Fractionation of Glycoproteins
Cell culture medium was replaced with methionine and cysteine free DMEM media (GIBCO), containing 10% dialyzed fetal bovine serum 20 min prior to the addition of 200 μCi/ml of Tran35S label (MP Biomedical). The pulse-labeling period was terminated by the addition of unlabeled methionine (3.75 mM) and cysteine (0.75 mM). Cells from one culture dish at each time point were lysed at 4°C by a 30 min incubation with 1 ml of 50 mM Tris-HCl pH 7.4, 2% Triton X-100, 500 mM NaCl, 5 mM MgCl2, 1× PIC, 200 μM PMSF, and 1 mM DTT.
Cell lysates were clarified by centrifugation (1 min at 30,000 rpm; TLA-100.4 rotor), diluted with 400 μl of 50 mM Tris-HCl, 1× PIC and precleared by incubation for 1-2 hr with a mixture of Protein A and Protein G Sepharose beads (Zymed Laboratories). The precleared lysates were incubated for 16 hr with antibody prebound to a second aliquot of Protein A- and Protein G-Sepharose beads. Beads were washed 7 times with 50 mM Tris-HCl, 500 mM NaCl, 1% Triton X-100, 0.1% SDS, 3 mM EDTA and twice with 10 mM Tris-HCl, 150 mM NaCl before eluting proteins with gel loading buffer. As indicated, immunoprecipitated proteins were digested with Endo H (New England Biolabs). Dry gels were exposed to a phosphor screen (Molecular Dynamics) and scanned in Molecular Imager FX (BioRad) for quantification.
Total radiolabeled glycoproteins were isolated using ConA Sepharose beads. A dish of radiolabeled cells was lysed and proteins denatured at 55°C in 1 ml of 50 mM Tris-HCl (pH 7.4), 0.5% SDS, 20 mM DTT, 1× PIC, 200 μM PMSF. The lysates were diluted 5-fold with 50 mM Tris-HCl, 1.25% Triton X-100, 5 mM MgCl2, 1× PIC, 200 μM PMSF and digested for 15 min at 4°C with 50 μg/ml of DNase I. Extracts were supplemented with 150 mM NaCl, 2 mM MnCl2, 2 mM CaCl2 and incubated with 150 μl of ConA Sepharose 4B (Sigma). The ConA beads were washed with 50 mM Tris-HCl, 0.1% SDS, 1% Triton X-100, 500 mM NaCl, 1 mM MgCl2, 1 mM MnCl2, 1 mM CaCl2 and glycoproteins were eluted during a 1 hr incubation at 37°C with wash buffer containing 1 M methyl α-D-mannopyranoside (Sigma). As a control, cells were treated with tunicamycin (10 μg/ml) for 30 min before radiolabeling.
Modification of Cysteine Residues with PEG-Maleimide
Cells were pulse-labeled and chased prior to lysis in a pH 7.1 phosphate buffer (10 mM NaHPO4, 2 mM KH2PO4, 2.7 mM KCl, 500 mM NaCl, 2% Triton X-100, 1× PIC, 200 μM PMSF) with or without 5 mM PEG-maleimide 5K (Sigma, 63187). After 30 min of incubation at 4°C, lysates were clarified by centrifugation (1 min at 30,000 rpm) and allowed to incubate for 16 hr at 4°C with rotation. Unreacted PEG-maleimide was quenched by 3 hr of incubation with 10 mM DTT. The extracts were diluted with 400 μl of PBS, 1× PIC and processed for immunoprecipitation.
Supplementary Material
Acknowledgments
We are grateful to Dr. Katherine High for providing the plasmid for FVII and Professor Konrad Sandhoff for the SAP D antisera. We also thank members of the Gilmore laboratory for helpful discussions. This work was supported by Public Health Services grant GM 43768 (R.G.) and a fellowship from the Ministerio de Educacion y Ciencia, Spain (C.R.-C).
Footnotes
Supplemental Data: Supplemental Data include Supplemental Experimental Procedures, one table, five figures, and Supplemental References and can be found with this article online at http://www.cell.com/supplemental/S0092-8674(08)01562-6.
References
- Ahn VE, Leyko P, Alattia JR, Chen L, Prive GG. Crystal structures of saposins A and C. Protein Sci. 2006;15:1849–1857. doi: 10.1110/ps.062256606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen S, Naim HY, Bulleid NJ. Intracellular folding of tissue-type plasminogen activator. Effects of disulfide bond formation on N-linked glycosylation and secretion. J Biol Chem. 1995;270:4797–4804. doi: 10.1074/jbc.270.9.4797. [DOI] [PubMed] [Google Scholar]
- Ben-Dor S, Esterman N, Rubin E, Sharon N. Biases and complex patterns in the residues flanking protein N-glycosylation sites. Glycobiology. 2004;14:95–101. doi: 10.1093/glycob/cwh004. [DOI] [PubMed] [Google Scholar]
- Bolt G, Kristensen C, Steenstrup TD. Posttranslational N-glycosylation takes place during the normal processing of human coagulation factor VII. Glycobiology. 2005;15:541–547. doi: 10.1093/glycob/cwi032. [DOI] [PubMed] [Google Scholar]
- Chen W, Helenius J, Braakman I, Helenius A. Cotranslational folding and calnexin binding during glycoprotein synthesis. Proc Natl Acad Sci USA. 1995;92:6229–6233. doi: 10.1073/pnas.92.14.6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, VanValkenburgh C, Liang H, Fang H, Green N. Signal peptidase and oligosaccharyltransferase interact in a sequential and dependent manner within the endoplasmic reticulum. J Biol Chem. 2001;276:2411–2416. doi: 10.1074/jbc.M007723200. [DOI] [PubMed] [Google Scholar]
- Deprez P, Gautschi M, Helenius A. More than one glycan is needed for ER glucosidase II to allow entry of glycoproteins into the calnexin/calreticulin cycle. Mol Cell. 2005;19:183–195. doi: 10.1016/j.molcel.2005.05.029. [DOI] [PubMed] [Google Scholar]
- Duvet S, Op De Beeck A, Cocquerel L, Wychowski C, Cacan R, Dubuisson J. Glycosylation of the hepatitis C virus envelope protein E1 occurs posttranslationally in a mannosylphosphoryldolichol-deficient CHO mutant cell line. Glycobiology. 2002;12:95–101. doi: 10.1093/glycob/12.2.95. [DOI] [PubMed] [Google Scholar]
- Ferguson MA, Duszenko M, Lamont GS, Overath P, Cross GA. Biosynthesis of Trypanosoma brucei variant surface glycoproteins. N-glycosylation and addition of a phosphatidylinositol membrane anchor. J Biol Chem. 1986;261:356–362. [PubMed] [Google Scholar]
- Gavel Y, Von Heijne G. Sequence differences between glycosylated and non-glycosylated Asn-X-Thr/Ser acceptor sites: implications for protein engineering. Protein Eng. 1990;3:433–442. doi: 10.1093/protein/3.5.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guth S, Volzing C, Muller A, Jung M, Zimmermann R. Protein transport into canine pancreatic microsomes: a quantitative approach. Eur J Biochem. 2004;271:3200–3207. doi: 10.1111/j.1432-1033.2004.04252.x. [DOI] [PubMed] [Google Scholar]
- Görlich D, Prehn S, Hartmann E, Kalies KU, Rapoport TA. A mammalian homologue of Sec61p and SecYp is associated with ribosomes and nascent polypeptides during translocation. Cell. 1992;71:489–503. doi: 10.1016/0092-8674(92)90517-g. [DOI] [PubMed] [Google Scholar]
- Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- Hershey JWB. Translational control in mammalian cells. Annu Rev Biochem. 1991;60:717–755. doi: 10.1146/annurev.bi.60.070191.003441. [DOI] [PubMed] [Google Scholar]
- Horn M, Baudys M, Voburka Z, Kluh I, Vondrasek J, Mares M. Free-thiol Cys331 exposed during activation process is critical for native tetramer structure of cathepsin C (dipeptidyl peptidase I) Protein Sci. 2002;11:933–943. doi: 10.1110/ps.2910102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imperiali B, Shannon KL, Unno M, Rickert KW. A mechanistic proposal for asparagine-linked glycosylation. J Am Chem Soc. 1992;114:7944–7945. [Google Scholar]
- Kelleher DJ, Gilmore R. DAD1, the defender against apoptotic cell death, is a subunit of the mammalian oligosaccharyltransferase. Proc Natl Acad Sci USA. 1997;94:4994–4999. doi: 10.1073/pnas.94.10.4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher DJ, Gilmore R. An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology. 2006;16:47–62. doi: 10.1093/glycob/cwj066. [DOI] [PubMed] [Google Scholar]
- Kelleher DJ, Karaoglu D, Mandon EC, Gilmore R. Oligosaccharyltransferase isoforms that contain different catalytic STT3 subunits have distinct enzymatic properties. Mol Cell. 2003;12:101–111. doi: 10.1016/s1097-2765(03)00243-0. [DOI] [PubMed] [Google Scholar]
- Kelleher DJ, Kreibich G, Gilmore R. Oligosaccharyltransferase activity is associated with a protein complex composed of ribophorins I and II and a 48 kD protein. Cell. 1992;69:55–65. doi: 10.1016/0092-8674(92)90118-v. [DOI] [PubMed] [Google Scholar]
- Kishimoto Y, Hiraiwa M, O'Brien JS. Saposins: structure, function, distribution, and molecular genetics. J Lipid Res. 1992;33:1255–1267. [PubMed] [Google Scholar]
- Koiwa H, Li F, McCully MG, Mendoza I, Koizumi N, Manabe Y, Nakagawa Y, Zhu J, Rus A, Pardo JM, et al. The STT3a subunit isoform of the Arabidopsis oligosaccharyltransferase controls adaptive responses to salt/osmotic stress. Plant Cell. 2003;15:2273–2284. doi: 10.1105/tpc.013862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolhekar AS, Quon AS, Berard CA, Mains RE, Eipper BA. Post-translational N-glycosylation of a truncated form of a peptide processing enzyme. J Biol Chem. 1998;273:23012–23018. doi: 10.1074/jbc.273.36.23012. [DOI] [PubMed] [Google Scholar]
- Kowarik M, Numao S, Feldman MF, Schulz BL, Callewaert N, Kiermaier E, Catrein I, Aebi M. N-linked glycosylation of folded proteins by the bacterial oligosaccharyltransferase. Science. 2006;314:1148–1150. doi: 10.1126/science.1134351. [DOI] [PubMed] [Google Scholar]
- Kretz KA, Carson GS, Morimoto S, Kishimoto Y, Fluharty AL, O'Brien JS. Characterization of a mutation in a family with saposin B deficiency: a glycosylation site defect. Proc Natl Acad Sci USA. 1990;87:2541–2544. doi: 10.1073/pnas.87.7.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Deutsch C. Pegylation: a method for assessing topological accessibilities in Kv1.3. Biochemistry. 2001;40:13288–13301. doi: 10.1021/bi0107647. [DOI] [PubMed] [Google Scholar]
- Manthri S, Guther ML, Izquierdo L, Acosta-Serrano A, Ferguson MA. Deletion of the TbALG3 gene demonstrates site-specific N-glycosylation and N-glycan processing in Trypanosoma brucei. Glycobiology. 2008;18:367–383. doi: 10.1093/glycob/cwn014. [DOI] [PubMed] [Google Scholar]
- Nilsson I, Kelleher DJ, Miao Y, Shao Y, Kreibich G, Gilmore R, Von Heijne G, Johnson AE. Photocross-linking of nascent chains to the STT3 subunit of the oligosaccharyltransferase complex. J Cell Biol. 2003;161:715–725. doi: 10.1083/jcb.200301043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson IM, von Heijne G. Determination of the distance between the oligosaccharyltransferase active site and the endoplasmic reticulum membrane. J Biol Chem. 1993;268:5798–5801. [PubMed] [Google Scholar]
- Rijnboutt S, Aerts HM, Geuze HJ, Tager JM, Strous GJ. Mannose 6-phosphate-independent membrane association of cathepsin D, glucocerebrosidase, and sphingolipid-activating protein in HepG2 cells. J Biol Chem. 1991;266:4862–4868. [PubMed] [Google Scholar]
- Rothman JE, Lodish HF. Synchronized transmembrane insertion and glycosylation of a nascent membrane protein. Nature. 1977;269:775–780. doi: 10.1038/269775a0. [DOI] [PubMed] [Google Scholar]
- Sanjay A, Fu J, Kreibich G. DAD1 is required for the function and the structural integrity of the oligosaccharyltransferase complex. J Biol Chem. 1998;273:26094–26099. doi: 10.1074/jbc.273.40.26094. [DOI] [PubMed] [Google Scholar]
- Schroder M, Kaufman RJ. Divergent roles of IRE1alpha and PERK in the unfolded protein response. Curr Mol Med. 2006;6:5–36. doi: 10.2174/156652406775574569. [DOI] [PubMed] [Google Scholar]
- Shibatani T, David LL, McCormack AL, Frueh K, Skach WR. Proteomic analysis of mammalian oligosaccharyltransferase reveals multiple subcomplexes that contain Sec61, TRAP, and two potential new subunits. Biochemistry. 2005;44:5982–5992. doi: 10.1021/bi047328f. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Kitagawa Y, Kadowaki T. Drosophila segment polarity gene product porcupine stimulates the posttranslational N-glycosylation of wingless in the endoplasmic reticulum. J Biol Chem. 2002;277:12816–12823. doi: 10.1074/jbc.M200187200. [DOI] [PubMed] [Google Scholar]
- Tsao YS, Ivessa NE, Adesnik M, Sabatini DD, Kreibich G. Carboxy terminally truncated forms of ribophorin I are degraded in pre-Golgi compartments by a calcium-dependent process. J Cell Biol. 1992;116:57–67. doi: 10.1083/jcb.116.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk D, Janjic V, Stern I, Podobnik M, Lamba D, Dahl SW, Lauritzen C, Pedersen J, Turk V, Turk B. Structure of human dipeptidyl peptidase I (cathepsin C): exclusion domain added to an endopeptidase framework creates the machine for activation of granular serine proteases. EMBO J. 2001;20:6570–6582. doi: 10.1093/emboj/20.23.6570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollenweider F, Kappeler F, Itin C, Hauri HP. Mistargeting of the lectin ERGIC-53 to the endoplasmic reticulum of HeLa cells impairs the secretion of a lysosomal enzyme. J Cell Biol. 1998;194:377–390. doi: 10.1083/jcb.142.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitley P, Nilsson IM, von Heijne G. A nascent secretory protein may traverse the ribosome/endoplasmic reticulum translocase complex as an extended chain. J Biol Chem. 1996;271:6241–6244. doi: 10.1074/jbc.271.11.6241. [DOI] [PubMed] [Google Scholar]
- Wilson CM, High S. Ribophorin I acts as a substrate-specific facilitator of N-glycosylation. J Cell Sci. 2007;120:648–657. doi: 10.1242/jcs.000729. [DOI] [PubMed] [Google Scholar]
- Wilson CM, Roebuck Q, High S. Ribophorin I regulates substrate delivery to the oligosaccharyltransferase core. Proc Natl Acad Sci USA. 2008;105:9534–9539. doi: 10.1073/pnas.0711846105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrobe D, Henseler M, Huettler S, Pascual Pascual SI, Chabas A, Sandhoff K. A non-glycosylated and functionally deficient mutant (N215H) of the sphingolipid activator protein B (SAP-B) in a novel case of metachromatic leukodystrophy (MLD) J Inherit Metab Dis. 2000;23:63–76. doi: 10.1023/a:1005603014401. [DOI] [PubMed] [Google Scholar]
- Yan Q, Lennarz WJ. Studies on the function of the oligosaccharyltransferase subunits: Stt3p is directly involved in the glycosylation process. J Biol Chem. 2002;277:47692–47700. doi: 10.1074/jbc.M208136200. [DOI] [PubMed] [Google Scholar]
- Yu YH, Sabatini DD, Kreibich G. Antiribophorin antibodies inhibit the targeting to the ER membrane of ribosomes containing secretory polypeptides. J Cell Biol. 1990;111:1335–1342. doi: 10.1083/jcb.111.4.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zufferey R, Knauer R, Burda P, Stagljar I, te Heesen S, Lehle L, Aebi M. STT3, a highly conserved protein required for yeast oligosaccharyl transferase activity in vivo. EMBO J. 1995;14:4949–4960. doi: 10.1002/j.1460-2075.1995.tb00178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.