

Abstract

C14,15-Dihydro-C22,25-epi north unit of cephalostatin 1 has been synthesized in eleven operations from a commercially available hecogenin acetate via multiple reductions and oxidations. The key transformations include: i) CrVI-catalyzed E-ring opening, ii) C17 hydroxylation, and iii) a base-triggered cyclization cascade.
The cephalostatins and ritterazines are structurally unique marine natural products that display extreme cytotoxicity against various human cancers.1 The natural targets cephalostatin 1, cephalostatin 7, cephalostatin 12, ritterazine M, and ritterazine K have been synthesized by us,2 while we and others3 have also been active in the synthesis and testing of analogs. The forty-five members of the cephalostatin and ritterazine family, along with the growing number of analogs and related monosteroidal glycosides afforded the basis for elucidating some of the structure activity relationships (SAR) and common pharmacophore of these potent cytotoxins:4 (1) “polarity match” consisting of polar north domains and less polar south domains with a connecting pyrazine moiety; (2) bis-spiroketals as prooxocarbenium moieties; (3) C17 (north) and C23'(south) hydroxyl group; and (4) Δ14 olefin moiety.
Semiempirical calculations for rationalizing the SAR of the bis-steroidal pyrazines revealed a strong correlation between bioactivity and enthalpy of oxacarbenium ion formation.5 Our efforts for calculation-guided design and synthesis of cephalostatin analogs led to the finding of the hyperactive C25-epi-ritterostatin GN1N (2), which is ~100 times more cytotoxic than ritterostatin GN1N (1) thereby being more potent than cephalostatin 1 (3, Figure 1), the most potent member of the cephalostatin family. Simply by comparing these three compounds (1, 2, and 3), most organic chemists would consider that C25-epi-cephalostatin 1 (4) would be a logical one to prepare. Calculations5 also support that the C25-epi-cephalostatin 1 (4) should be in the hyperactive class.
Figure 1.

Effect of C25 Stereochemistry on the Cytotoxicity of Cephalostatin Analogs
In conjunction with our quest to achieve an efficient second generation synthesis of the north unit of cephalostatin analogs,6 we have developed a “Red-Ox” strategy where multiple oxidations/reductions are employed as key transformations to deliver the target hemispheres. Herein, we report a progress toward the synthesis of C25-epi north 1 (6) from hecogenin acetate 5 via “Red-Ox” modifications (Scheme 1).
Scheme 1.
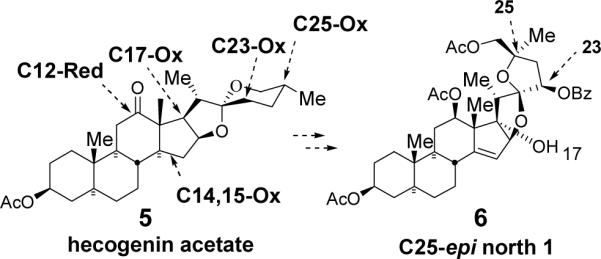
“Red-Ox” Strategy
Our “Red-Ox” strategy synthesis of the C25-epi north 1 6 started from commercially available plant-derived hecogenin acetate 5 (Scheme 2). Borohydride reduction of hecogenin acetate 5 at −78 °C followed by acetylation afforded rockogenin acetate 7 in a nearly quantitative yield. The action of t-BuNO2/BF3●OEt27 on 5/6 spiroketal 7 regioselectively delivered C23 oxime 8, a masked ketone, which was then hydrolyzed in the presence of acid to unveil ketone 9. Obtaining a workable stereoisomeric excess at C23 relied on (S)-CBS reduction8 (C23R (axial OH) 78 % 10; C23S (equatorial OH) 13 %). Regio- and stereoselective triethylsilane reduction of 5/6 spiroketal 10 resulted in the formation of F-ring opened diol 11 in 94 % yield. Selective tosylation of the primary alcohol in the presence of the secondary alcohol using catalytic 1,4-diazabicyclo[2,2,2]octane (DABCO) followed by C23 benzoylation furnished 12, which was then subjected to a sequential iodination and DBU-mediated E2 elimination to give terminal olefin 13a.
Scheme 2.

With olefin 13a in hand, we investigated C25,26-oxyfunctionalization using Sharpless asymmetric dihydroxylation.9 As expected from our previous studies,10 stereoselective dihydroxylation of the olefin moiety was especially difficult. Reasonable excess of C25R stereoisomer was obtained only when using (DHQ)2PHAL ligand and C23 substituted substrate (Table 1). Stereochemistry at C25 was unambiguously determined by a single crystal X-ray crystallography. The diol 14a was subjected to sequential protection of the primary alcohol with an acetyl group and the tertiary alcohol with the trifluoroacetyl group to provide 15 (Scheme 2).
Table 1.
Asymmetric Dihydroxylation of the C25-olefin
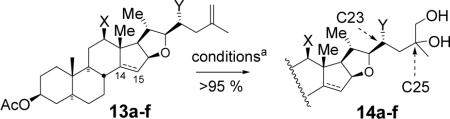
| substrate | X | Y | C14-15 | ligand | product | 25R:25S |
|---|---|---|---|---|---|---|
| 13a | OAc | OBz | 14α-H | (DHQ)2PHAL | 14a | 5:1 |
| 13b | OAc | OBz | 14α-H | (DHQD)2PHAL | 14b | 1:3 |
| 13c | OBz | H | 14α-H | (DHQ)2PHAL | 14c | 1:1 |
| 13d | OBz | H | 14α-H | (DHQD)2PHAL | 14d | 1:1 |
| 13e | OBz | H | Δ 14 | (DHQ)2PHAL | 14e | 1:5 |
| 13f | OBz | H | Δ 14 | (DHQD)2PHAL | 14f | 2:1 |
aOsO4 (2 mol %), ligand (10 mol %), K3Fe(CN)6 (3 equiv), K2CO3 (3 equiv), t-BuOH/H2O (1:1), 0 °C.
Having established the requisite stereochemistry at C12, 23, and 25, we next turned to C17 hydroxylation (Scheme 3). For this transformation, opening of the E-ring was required. While there are a number of tetrahydrofuran ring opening methods,11 steroidal E-ring of 15 was found inert to those conditions so that starting material was recovered in most cases. However, our recently developed CrVI-mediated C–H oxidation12 protocol smoothly effected E-ring opening to deliver diketone 16 in 84 % yield. After extensive experimentations, formation of C17-OH 18 was finally achieved by TMSI/hexamethyldisilazane-mediated13 thermodynamic silylenol ether 17 formation followed by Rubottom oxidation by TFMDO14 generated in situ. The C17-OH group was introduced in a stereoselective manner. The use of different bases other than hexamethyldisilazane or in situ generated TMSI resulted in no formation of the silylenolether 17. Removal of trifluoroacetyl protecting group of 18 with 1,8-diazabicyclo[5,4,0]undec-7-ene triggered a cyclization cascade to form hemiacetal 19 as a single stereoisomer. Reduction of the hemiacetal 19 with excess triethylsilane and TMSOTf at −78 °C delivered C14,15-dihydro-C22,25-epi north 1 (20) in 89 % yield.15
Scheme 3.

In summary, we have developed an efficient synthetic route for C14,15-dihydro-C22,25-epi north 1 (20) wherein CrVI-catalyzed E-ring opening, stereoselective C17 hydroxylation, and a cyclization cascade are used as key reactions. Dihydro-22,25-epi north 1 (20) was prepared in 11 operations and 4 % overall yield from hecogenin acetate. The presented results illustrate that the “Red-Ox”-based synthesis provides a facile and efficient access to cephalostatin analogs from hecogenin acetate 5. Further synthetic efforts to convert C17-hyroxy-C16,22-diketone 18 into C25-epi north 1 (6) and to make cephalostatin analogs containing C14,15-dihydro-C22,25-epi north 1 hemisphere 20 are in progress and results will be reported in due course.
Supplementary Material
Acknowledgment
This investigation was generously supported by funds provided by the National Institute of Health (CA 60548). We are grateful to Daniel Jamieson (Purdue University) for assistance with collecting 13C NMR data. We acknowledge Arlene Rothwell and Karl Wood (Purdue University) for providing MS data.
Footnotes
Supporting Information Available: General experimental procedure and NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Cephalostatin Support Studies. 35. For 34, see: Lee, J. S.; Cao, H.; Fuchs, P. L. J. Org. Chem. 2007, 72, 5820.
References
- 1.(a) Pettit GR, Inoue M, Herald DL, Krupa TS. J. Am. Chem. Soc. 1988;110:2006. [Google Scholar]; (b) Pettit GR, Inoue M, Kamano Y, Herald DL. Chem. Commun. 1988:1440. [Google Scholar]; (c) Pettit G, Xu JP, Schimidt JM. Bioorg. & Med. Chem. Lett. 1995;5:2027. [Google Scholar]; (d) Fukuzawa S, Matsunaga S, Fusetani N. J. Org. Chem. 1994;59:6164. doi: 10.1021/jo970091r. [DOI] [PubMed] [Google Scholar]; (e) Fukuzawa S, Matsunaga S, Fusetani N. J. Org. Chem. 1995;60:608. doi: 10.1021/jo970091r. [DOI] [PubMed] [Google Scholar]; (f) Fukuzawa S, Matsunaga S, Fusetani N. Tetrahedron. 1995;51:6707. [Google Scholar]; (g) Fukuzawa S, Matsunaga S, Fusetani N. J. Org. Chem. 1997;62:4484. doi: 10.1021/jo970091r. [DOI] [PubMed] [Google Scholar]; (h) Fukuzawa S, Matsunaga S, Fusetani N. Tetrahedron Lett. 1996;37:1447. [Google Scholar]
- 2.(a) LaCour TG, Guo C, Boyd MR, Fuchs PL. Org. Lett. 2000;2:33. doi: 10.1021/ol991153y. [DOI] [PubMed] [Google Scholar]; (b) Lee S, Fuchs PL. Org. Lett. 2002;4:317. doi: 10.1021/ol016572l. [DOI] [PubMed] [Google Scholar]; (c) LaCour TG, Guo C, Bhandaru S, Boyd MR, Fuchs PL. J. Am. Chem. Soc. 1998;120:692. [Google Scholar]; (d) Kim S, Sutton SC, Guo C, LaCour TG, Fuchs PL. J. Am. Chem. Soc. 1999;121:2056. [Google Scholar]
- 3.(a) Heathcock CH, Smith SC. J. Org. Chem. 1994;59:6828. [Google Scholar]; (b) Jautelat R, Muller-Fahrnow A, Winterfeldt E. Chem. Eur. J. 1999;5:1226. [Google Scholar]; (c) Basler S, Brunck A, Jautelat R, Winterfeldt E. Helv. Chim. Acta. 2000;83:1854. and references therein. [Google Scholar]; (d) Phillips ST, Shair MD. J. Am. Chem. Soc. 2007;129:6589. doi: 10.1021/ja0705487. [DOI] [PubMed] [Google Scholar]; (e) Taber DF, Taluskie KV. J. Org. Chem. 2006;71:2797. doi: 10.1021/jo052656m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Betancor C, Freire R, Perez-Martin I, Prange T, Suárez E. Org. Lett. 2002;4:1295. doi: 10.1021/ol025580e. [DOI] [PubMed] [Google Scholar]; (g) Taber DF, Joerger J-M. J. Org. Chem. 2008;73:4155. doi: 10.1021/jo800454w. [DOI] [PubMed] [Google Scholar]; (h) Taber DF, DeMatteo PW, Taluskie KV. J. Org. Chem. 2007;72:1492. doi: 10.1021/jo061935m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Pettit GR, Tan R, Xu JP, Ichihara Y, Williams MD, Boyd MR. J. Nat. Prod. 1998;61:953. doi: 10.1021/np9800405. and references therein. [DOI] [PubMed] [Google Scholar]; (b) Fukuzawa S, Matsunaga S, Fusetani N. J. Org. Chem. 1997;62:4484. doi: 10.1021/jo970091r. and references therein. [DOI] [PubMed] [Google Scholar]; (c) Yu W, Jin Z. J. Am. Chem. Soc. 2001;123:3369. doi: 10.1021/ja004098t. [DOI] [PubMed] [Google Scholar]
- 5.LaCour TG. Ph.D. Thesis. Purdue University; 2001. [Google Scholar]
- 6.(a) Li W, LaCour TG, Boyd MR, Fuchs PL. J. Am. Chem. Soc. 2002;124:4548. doi: 10.1021/ja017323v. [DOI] [PubMed] [Google Scholar]; (b) Lee S, Fuchs PL. Org. Lett. 2002;4:313. doi: 10.1021/ol0165894. [DOI] [PubMed] [Google Scholar]; (c) Lee JS, Fuchs PL. J. Am. Chem. Soc. 2005;127:13122. doi: 10.1021/ja0531935. [DOI] [PubMed] [Google Scholar]; (d) Lee JS, Fuchs PL. Org. Lett. 2003;5:2247. doi: 10.1021/ol034551g. [DOI] [PubMed] [Google Scholar]
- 7.(a) Gonzalez AG, Frerire R, Garcia-Estrada MG, Suarez E. Tetrahedron. 1972;28:1289. [Google Scholar]; (b) Barton DHR, Sammes PG, Taylor MV, Werstiuk E. J. Chem. Soc. C. 1970:1977. [Google Scholar]; (c) Lee S, Fuchs PL. Can. J. Chem. 2006;84:1442. [Google Scholar]; (d) Betancor C, Freire R, Perez-Martin I, Prange T, Suarez E. Org. Lett. 2002;4:1295. doi: 10.1021/ol025580e. [DOI] [PubMed] [Google Scholar]
- 8.(a) Corey EJ, Bakshi RK, Shibata S. J. Am. Chem. Soc. 1987;109:5551. [Google Scholar]; (b) Corey EJ, Roberts BE. J. Am. Chem. Soc. 1997;119:12425. [Google Scholar]
- 9.Sharpless KB, Amberg W, Bennani YL. J. Org. Chem. 1992;57:2768. [Google Scholar]
- 10.(a) Jeong JU, Guo C, Fuchs PL. J. Am. Chem. Soc. 1999;121:2071. [Google Scholar]; (b) Kim SK, Sutton SC, Guo C, LaCour TG, Fuchs PL. J. Am. Chem. Soc. 1999;121:2056. [Google Scholar]
- 11.(a) Tenaglia A, Terranova E, Waegell B. J. Org. Chem. 1992;57:5523. [Google Scholar]; (b) Carlsen P, Katsuki T, Martin VS, Sharpless KB. J. Org. Chem. 1981;46:3936. [Google Scholar]; (c) Scarborough RM, Smith AB. J. Am. Chem. Soc. 1980;102:3904. [Google Scholar]
- 12.(a) Lee S, Fuchs PL. J. Am. Chem. Soc. 2002;124:13978. doi: 10.1021/ja026734o. [DOI] [PubMed] [Google Scholar]; (b) Lee S, Fuchs PL. Org. Lett. 2004;6:1437. doi: 10.1021/ol049712a. [DOI] [PubMed] [Google Scholar]
- 13.(a) Hoeger CA, Okamura WH. J. Am. Chem. Soc. 1985;107:268. [Google Scholar]; (b) Miller RD, McKean DR. Synthesis. 1979:730. [Google Scholar]
- 14.Mello R, Fiorentino M, Fusco C, Curci R. J. Am. Chem. Soc. 1989;111:6749. [Google Scholar]
- 15.The C22 stereochemistry was determined by comparing 1H and 13C NMR spectra of 20 with those of 14,15-dihydro-17-deoxy-22,25-epi north 1, of which structure was solved by a single crystal X-ray crystallography (see Supporting Information).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.