

Abstract
Cell-penetrating peptides (CPPs) can enter many types of cells and have become useful tools for introducing a variety of cargo such as exogenous peptides, proteins, and nucleic acids into cultured cells in vitro. Tat CPPs derived from the HIV-1 Tat protein are the most widely used among the arginine-rich CPPs. Even though CPPs hold considerable promise for drug delivery, poor biological stability and high in vivo clearance may limit their effectiveness for delivering cargo. Therefore, we utilize a retro-inverso form of a Tat peptide, R.I.-CKTat9, which is proteolytically stable. In the current study, the cellular entry mechanism of this arginine-rich CPP is investigated. Fluorescently labeled R.I.-CKTat9 entered HeLa cells in a concentration- and energy-dependent manner demonstrating both diffuse and punctate (vesicular) appearance inside the cells. The labeled R.I.-CKTat9 colocalized with labeled transferrin in the punctate structure, suggesting that the peptide enters HeLa cells by clathrin-dependent endocytosis. Incubation of cells with an isotonic/high K+ buffer (KPBS) or an NH4Cl solution abolished the diffuse but not the punctate fluorescence, suggesting that membrane potential plays a critical role. This result also suggests that the flux originates from the endosome, not the extracellular space, and relies on the acidity of the endosome. Impairment of clathrin-mediated endocytosis by RNAi with clathrin heavy chain function and endocytosis inhibitors greatly reduced or completely abolished both diffuse and punctate fluorescence, further supporting a single route of endocytosis and subsequent endosomal escape. Since cells in the mitotic (M) phase shut down endocytosis but maintain plasma membrane potential, this property was used to further confirm the endocytic mechanism. Direct measurement of plasma membrane potential confirmed its persistence in M phase arrested HeLa cells. Consistent with our working hypothesis, these cells did not show any vesicular nor diffuse fluorescence of labeled R.I.-CKTat9, providing compelling evidence for the sequential steps of endocytosis and endosomal escape. Binding of labeled R.I.-CKTat9 to the surface of HeLa cells at 0 °C was reduced under the mildly acidic conditions of early endosomes, suggesting an acidity-dependent endosomal escape mechanism. Overall, these results indicate that both endocytosis and membrane potential are required for R.I.-CKTat9 entry into HeLa cells and suggest that translocation occurs at the endosomal membrane.
Keywords: HIV-1 Tat cell penetrating peptide, protein transduction domain, cell penetration mechanism
INTRODUCTION
Cell-penetrating peptides (CPPs), also known as protein transduction domains (PTDs), are a collection of different families of short peptides believed to enter cells by penetrating cell membranes1–6. CPPs have attracted a great deal of interest in the hope of enhancing gene therapy, vaccine development, and drug delivery7–9. The widely used Tat CPPs are derived from the basic domain of HIV-1 Tat protein10–12. Mutagenesis studies have demonstrated that the critical feature of the basic domain for cell penetration is the guanidine group of the arginine residues and the positive charge. Other factors, such as a potential nuclear localization signal within the domain, sequence, chirality, or the linear configuration, are not essential10–14. Indeed, small-molecule mimics of arginine-rich CPP that carry several guanidine groups per molecule can carry conjugated cargos into the cytoplasm and nucleus of cultured cells as efficiently as Tat CPP15. Tat CPP-assisted cargo delivery has been reported in a variety of experimental protocols2–9. Tat CPPs have been shown using microscopy, flow cytometry, agent uptake, or cell functionality to ferry various cargos (e.g., peptides, fusion proteins, nucleic acids, microspheres, liposomes, polymers, imaging agents, phage, etc) into mammalian cells in vitro. Although there is little doubt that Tat CPPs are capable of delivering many cargo types into the cytosol/nucleus compartment of mammalian cells, not all in vitro Tat CPP-assisted cargo delivery attempts have been successful. Furthermore, limited in vivo studies have yielded mixed results (reviewed in16, 17). One possible reason is that peptides have poor biological stability and high rates of clearance. However, it is also generally recognized that the lack of clarity in the mechanism of cell penetration18 not to mention the complexity of drug delivery19 contribute significantly to the poor in vitro - in vivo correlation.
The fundamental cell-penetration mechanism of Tat CPPs remains unclear. It was reported that the cell fixation method used in some of the previous studies led to artifactual uptake of CPPs or oligonucleotides20–23. This and the lack of distinction between internalization and cell surface binding constituted a large part of the early supporting evidence for the cell energy- and cell type-independence. At present, whether Tat CPPs enter the cytosol/nucleus compartment by directly penetrating the plasma membrane, by endocytosis and endosomal escape, or by both remain important unanswered questions. Further, in the case of endocytosis and endosomal escape, the endocytic pathway that is taken has also not been clearly established. We have proposed the use of a proteolytically stable CPP, R.I.-Tat916. Since it is widely known that the cell-penetrating mechanism is dependent upon the type of CPP used, the scope of the present discussion is limited to Tat CPPs and Tat CPP-cargos.
There are studies favoring an endocytic mechanism, although the reported endocytic pathway varies. For example, it was shown that fluorescently labeled Tat CPPs or full-length Tat protein, which can be viewed as a Tat CPP fusion protein, underwent clathrin-mediated endocytosis before entering the cytoplasm24–27. Endosomal acidification was required for endosomal escape. Fluorescently labeled polyarginine (a Tat CPP analog) took a similar route, first assuming punctate (vesicular) and then diffuse (cytoplasmic and nuclear) appearance28–29. Interestingly, both polyarginine and a Tat CPP induced clathrin-mediated endocytosis that not only internalized the peptides but also other receptors (TNFR1, 2, and EGFR). These receptors are known to take this route of endocytosis30. In HeLa cells, a fluorescently labeled Tat CPP was partially colocalized with transferrin23. However, a rhodamine-tagged Tat CPP fused to a 17 amino acid cargo peptide did not co-localize with transferrin, but with cholera toxin, a lipid raft marker. The removal of cholesterol from the plasma membrane abolished the internalization of the fusion peptide, suggesting a lipid raft-dependent endocytic mechanism31. The latter case was in agreement with a study on a Tat CPP-EGFP fusion protein, which implicated the caveolar endocytosis pathway32. Yet, chemical inhibitors suggested a third endocytosis pathway, macropinocytosis, for the uptake of fluorescently labeled Tat CPP33 or polyarginine34. Another study showed that the function of Cre protein fused to a Tat CPP did not depend on clathrin- or caveolae-mediated endocytosis, but on macropinocytosis35. Recent literature suggests that a tagged Tat CPP could enter a cell using multiple pathways, including different endocytic pathways36–38. It is clear that differences in the Tat CPP-carried cargo, cell type, and experimental procedure probably account for the reported mechanistic differences in endocytosis pathway(s).
Another possible cell entry mechanism is direct cell penetration. Based on the adaptive partitioning of polyarginine into a lipid phase and the correlation between the deduced cell membrane potential and the amounts of a Tat CPP and a polyarginine associated with cells, a membrane potential-driven direct plasma membrane penetration hypothesis known as the membrane potential hypothesis has been proposed39. Based on cell fractionation40 or the cytosolic reductive environment41, it was concluded that Tat CPPs and polyarginines entered cells by direct penetration. In confocal microscopy studies, it was argued that, since the uptake of fluorescently tagged Tat CPPs and a polyarginine into cells persisted at a low temperature and was fast, cell entry did not involve endocytosis42–44.
In order to better understand the roles of endocytosis and membrane potential in the cellular entry of R.I.-Tat9, the effects of membrane potential depolarization and endocytosis-inhibition were examined in HeLa cells. The results show that membrane potential plays a critical role. The results further suggest that the Tat CPP enters HeLa cells predominately through clathrin-mediated endocytosis and escapes from endosomes into the cytosol/nucleus compartment.
MATERIALS AND METHODS
Materials
Transferrin, Hoechst 33258, 2-(4-amidinophenyl)-1H-indole-6-carboxamidine (DAPI), 2-deoxyglucose, sodium azide, sucrose, NH4Cl, HEPES, and monodansylcadaverine (MDC) were purchased from Sigma (St. Louis, MO), Alexa Fluor 488-transferrin, Rhodamine-Dextran 10000, Rho-transferrin, trypan blue solution, and tissue culture reagents from Invitrogen (Carlsbad, CA), chc-2 siRNA from Dharmacon (Lafayette, CO 80026), and Lab-Tek II 4-chamber slides and chamber #1.5 coverglass systems from Nalge Nunc International Corp (Naperville, IL). siRNA transfection reagent INTERERin was purchased from Genesee Scientific (San Diego, CA). All other common chemicals and supplies were commercially available.
Peptide synthesis and labeling
The synthesis, characterization, and derivation of the R.I.-CKTat-9 peptide in the retro-inverso form (dCdK-dRdRdRdQdRdRdKdKdR or R.I.-CK-Tat57–49) have been published by us16.
Fluorescence and confocal microscopies
HeLa cells for fluorescence microscopy were seeded in Lab-Tek II 4-chamber slides or 24-well plates. For confocal microscopy, cells were seeded in chamber cover glasses. Cells were used when they reached a confluence of ~ 50%. They were incubated with the labeled R.I.-CKTat9 &/or transferrin (Tf ) in 20 mM HEPES-buffered (pH 7.4) Hank’s balanced saline solution (HBSS) for the specified time at the specified temperature, washed with PBS once, stained with 0.5 × trypan blue in PBS (1× is the undiluted 0.4 % concentration) at room temperature for 2 min, and washed with PBS twice. For microscopy examining the membrane potential effect, cells grown in 24-well plates were used. The incubation and one time PBS wash were done in the plates, the cells were detached with a brief trypsinization, followed by trypan blue staining/PBS wash and transferring to a regular slide for microscopy. The fluorescence microscopy examining the effect of M phase was performed similarly using cells arrested in M phase after 100 nM nocodazole treatment for 16 hours, mitotic shake-off, and attachment to poly-D-lysine-treated 24-well plates. The chamber slides, chamber coverglasses, or regular slides were examined immediately or put on ice before microscopy. Fields with heavy trypan blue stained cells were excluded from observation. The fluorescence microscopy has been described before16 and in this study an additional inverted Olympus IX71 fluorescence microscope was similarly used. The confocal microscopy was done on a Leica TCS SP2 Spectral Confocal Microscope using the XYZ mode and a 0.25- or 0.40-micron section height. A mid cell section is shown in each image. The fluorescence channels for different dyes were well separated using sequential scanning to ensure no bleach-through from one channel to another. Cell energy depletion was achieved with a 30 min pre-incubation of 0.25% NaN3/50 mM 2-deoxyglucose in HBSS, followed by an incubation with the same incubation buffer that in addition contained a labeled Tat CPP. The pair of special buffers (KPBS and PBS) used for the membrane potential elimination is as follows. PBS: 136.9 mM NaCl/2.7 mM KCl/30 mM Na2HPO4/1.76 mM KH2PO4/0.1% glucose/pH7.4. KPBS: the same as PBS except that KCl had replaced NaCl.
Tat CPP uptake
To test the effect of membrane potential, HeLa cells in four wells per group grew in 96-well plates at ~ 70% confluence were washed with PBS or KPBS and incubated with 10 μM of Flu-R.I.-CKTat9 in PBS or KPBS, respectively. After incubation at 37 °C for 30′, the wells were washed with PBS or KPBS. The PBS or KPBS in each well was replaced with 50 μl/well of 1 N NaOH to lyse the cells. The lysate was neutralized with 50 μl/well of 1 N HCl next day. The fluorescence in each 100 μl/well of total lysate was read in a plate reader at 485/535 nm. The average reading of a separate blank group of four wells, which was incubated with PBS without the labeled Tat CPP but subjected to the same lysis/neutralization process, was used as a blank for the plate reader data. A portion of the total 100 μl of the neutralized lysate in each well was used to determine total cellular protein amount per well using the BioRad Protein Assay reagent. The fluorescence level in each well was normalized by its protein amount to correct for unevenness in cell number between wells. Student t-test was used to estimate the statistical difference in fluorescence units/μg cellular protein between PBS and KPBS groups. To quantify the difference in labeled Tat CPP uptake between HeLa cells in interphase and M phase, procedure similar to above for the PBS and KPBS pair was done using attached interphase cells and cells arrested in M phase after nocodazole-treatment (100 nM/16h), mitotic shake-off, and pre-attachment to poly-D-lysine-treated 24-well plates. Parallel to each uptake experiment, a corresponding microscopy session was performed to evaluate the percentage of cells arrested in M phase and the extent of artifactual uptake by dead cell/cell debris (cf. Fig. 5E). The uptake was normalized against total cellular protein to minimize the nocodazole cytotoxic effect.
Fig. 5.

HeLa cells in the M phase do not take up Alexa488-Tf (green) or Rhodamine-labeled R.I.-CKTat9 (red). In A, B, and C, a cluster of untreated HeLa cells were incubated with a live DNA dye Hoechst 33258 (blue)/Alexa488-Tf (green)/15 μM Rho-R.I.CKTat9 (red) for 40 min, followed by wash and live fluorescence microscopy for the blue, green, and red fluorescence, respectively. The long arrow indicates a cell clearly at the anaphase stage of the M phase while the two short arrows indicate the two other cells possibly in M phase as well (A). In contrast to neighboring interphase cells, the three arrow-indicated cells did not take up Alexz488-Tf (B) and Rho-R.I.CKTat9 (C). D and E are merged blue and red fluorescence images of cells untreated and treated, respectively, with 100 nM nocodazole for 16 hours, incubated with Hoechst 33258/10 μM Rho-Tat for 40 min, and washed. Cells arrested in M phase were made attached after 1 hour pre-attachment in the presence of nocodazole to poly-D-lysine-treated 24-well plates. D shows untreated interphase HeLa cells having taken up considerable but varying amount of Rho-Tat. E shows that most nocodazole-treated cells were arrested in the prophase stage of the M phase and these cells did not take up Rho-R.I.CKTat9. Note, the authors deliberately chose a field to be presented in E to illustrate some treated cells (arrow-indicated) that began to exit the telophase stage of the M phase (chromosomes de-condensed) and began to take up Rho-R.I.CKTat9. Also shown is the unavoidable cell debris and necrotic cells that artificially took up Rho-R.I.CKTat9 (*-indicated). There were also some apparently apoptotic cells with membrane blebs and degraded DNA that did not take up Rho-R.I.CKTat9 (not shown).
RNAi of the clathrin heavy chain
The target sequence of chc-2 siRNA is UAAUCCAAUUCGAAGACCAAU, a part of the clathrin heavy chain mRNA sequence. chc-2 siRNA was synthesized as Option C siRNA by Dharmacon, Inc. The transfection procedure was basically the same as described by Motley, A. et al45 with a scaling down to fit our 24-well culture system using HeLa cells. A critical improvement of this study over the published protocol45 was the use of the siRNA transfection reagent INTERERin, which allowed for less cytotoxicity and a 15-fold reduction in siRNA amount from 150 to 10 pmol per well (or chamber). The double-transfected HeLa cells were used at 96-hour post the start of the first transfection.
Treatments that inhibit the clathrin-mediated endocytosis
HeLa cells grew in chamber coverglasses or chamber slides at about half confluence were washed with PBS once, pre-incubated at 37 °C for 20 min in a CO2 incubator with no inhibitor (control), or one of the three inhibitors (0.3 M sucrose, 50 mM NH4Cl, or 400 μM MDC) in 20 mM HEPES-buffered DMEM medium without serum. The pre-incubation media were replaced with the same medium containing in addition 0.35 μM Alexa488-Tf/7 μM Rho-R.I.-CKTat9 for confocal microscope, or 7 μM Flu-R.I.-CKTat9 for fluorescence microscope, and the cells were incubated at 37 °C for 30 min more. The cells were then washed and stained as described above for the two types of microscopy.
Electrophysiological recordings
Measurements of the resting membrane potential in HeLa cells were obtained using the current-clamp mode of whole-cell patch clamp technique. Voltage was recorded with an Axoclamp 200 amplifier (Molecular Devices), filtered at 1 kHz, and digitized with a Power1401 interface (Cambridge Electronic Design). The external bath solution was HBSS supplemented with 20 mM HEPES, pH7.4. The pipette solution contained (in mM) 105 K-methanesulfonate, 17.5 KCl, 10 HEPES, 0.2 EGTA, and 8 NaCl. The pH of the internal solution was set to 7.3 with KOH. Electrodes were coated with sylgard and fire polished (Narishige MF-83). Resistances ranged from 4 to 6 MΩ. Pipette offset current was zeroed immediately before contacting the cell membrane. Recordings were made at room temperature (19–22°C) and considered acceptable when they met the following criteria: stable membrane potential, low noise level, and discernible membrane time constant upon step current injection. Interphase recording was done on cells at the end of G2 phase of shake-off cells without nocodazole-treatment and pre-attachment to poly-D-lysine-treated culture dishes, and M phase recording on shake-off cells arrested in M phase after 16 hour nocodazole-treatment and pre-attachment to poly-D-lysine-treated culture dishes. In parallel, the described phases of the cell cycle were verified simultaneously on separate cells using microscopy after nuclear staining with live DNA dye Hoechst 33258; at least 98% of all live cells counted were in the designated phases.
pH-dependent binding
pH incubation buffers at 7.4, 7.0, 6.5, 6.0, and 5.5 were made as described by us46. pH incubation buffer at 5.0 was made the same way except that MES was used in place of HEPES. Near confluent HeLa cells in 24-well plates were incubated with Flu-R.I.-CKTat9 at 0 °C for 1 hour in different pH buffers and washed with the same buffers pre-chilled on ice. The monolayers of cells were lysed with 1 N NaOH overnight and neutralized with 1 N HCl. Part of each lysate was transferred to a 96-well plate, mixed with 0.1 volume of 1 M HEPES at pH 7.4, and read in a fluorescence plate reader. The remaining part was used for total cellular protein determination using the BioRad Protein Assay reagent. The fluorescence values were converted to the absolute amounts of the peptide in pico mole, using a standard curve constructed with pure Flu-R.I.-CKTat9 in different pH buffers, corrected for different pH values by undergoing through the same incubation/lysis/neutralization process. The amount of converted peptide was normalized to cellular protein content. Each data point is the mean of at least three wells +/− s.d.
RESULTS
Fluorescently labeled R.I.-CKTat-9 enters HeLa cells in a cell energy- and concentration-dependent manner
The proteolysis-resistant retro inverso-form (R.I.) of a Tat CPP, CKTat9, was used in order to exclude the influence of proteolytic degradation from the interpretation of the anticipated results. There was no difference in intracellular distribution patterns between fluorescently labeled R.I.- and L-CKTat9 (data not shown). R.I.-CKTat9 (Acetyl-dC-dK-dR-dR-dR-dQ-dR-dR-dK-dK-dR-CONH2) was synthesized with two extra D-form residues of dC and dk (italic) at the N-terminus of R.I.-Tat9 (underlined) (total 11 residues; the corresponding L-Tat9 is the peptide Tat(49–57) in the Tat protein). The dk (bold) was used for fluorescently labeling at the ε-amine whereas the dC was reserved for future cargo-conjugation. Fluorescent tags attached were 5(6)-Carboxyfluorescein or Rhodamine.
HeLa cells were incubated with 1 to 10 μM of 5(6)-Carboxyfluorescein-R.I.-CKTat9 (Flu-R.I.-CKTat9) for 40 min. It has been previously determined that the active uptake at 40 min represents greater than half of its saturation level16. Appearance of fluorescence was concentration-dependent but it varied between cells (cf. Fig. 4: Ctrl/Flu-R.I.CKTat for variation). At concentrations below 3 μM, the punctate (vesicular) fluorescence predominated (Fig. 1: 2μM), at concentrations above 7 μM (Fig. 1: 10μM and Fig. 2: 10μM/PBS/Flu-R.I.-CKTat9) the diffuse (cytosolic/nuclear) fluorescence predominated, and at 7 μM both the punctate and the diffuse fluorescence were apparent (Fig. 1:7μM/confocal), which is in agreement with the observations made by others42–44. Similar results were obtained with rhodamine-R.I.-CKTat9 (Rho-R.I.-CKTat9) (see Fig. 3 & 4). Confocal images (Fig. 1: 7μM/confocal & DIC) indicate that the punctate and diffuse fluorescence are within the HeLa cells. Cell energy-depletion at 37 °C virtually abolished both the punctate and diffuse fluorescence (Fig. 1: 10μM/e-dpltn), as did low temperature (0 °C) incubation (not shown). This result indicates that the entry into these two cellular compartments occurs by an active cellular process.
Fig. 4.

Endocytosis inhibitors greatly reduce the punctate and the diffused fluorescence of labeled R.I.-CKTat9. The procedure is described in the text. Control/Tf & Control/Tat, Sucrose/Tf & Sucrose/Tat, NH4Cl/Tf & NH4Cl/Tat are confocal green and red channel images for Alexa488-Tf and Rho-R.I.CKTat9, respectively, in the absence or presence of each of the inhibitors, respectively. Ctrl/Flu-R.I.CKTat and MDC/Flu-R.I.CKTat are fluorescence microscopic images in the absence and presence of MDC, respectively. Note the variation in green fluorescence intensity between control cells.
Fig. 1.
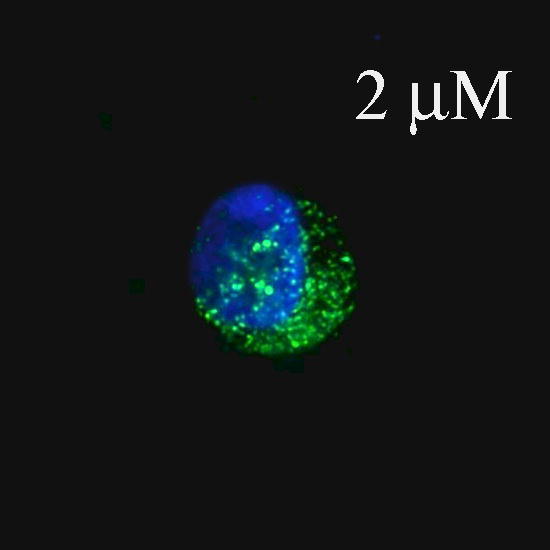
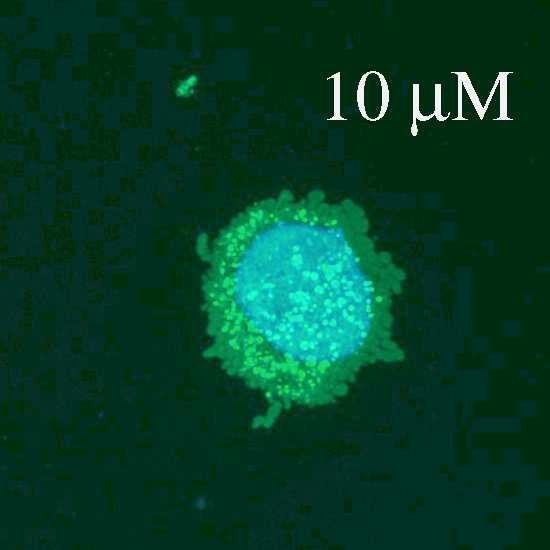
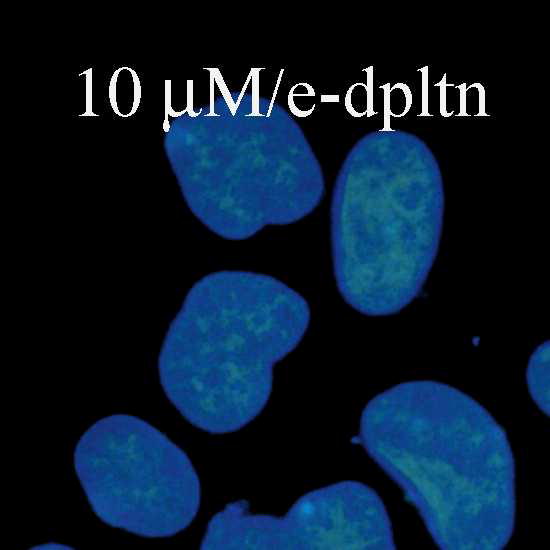

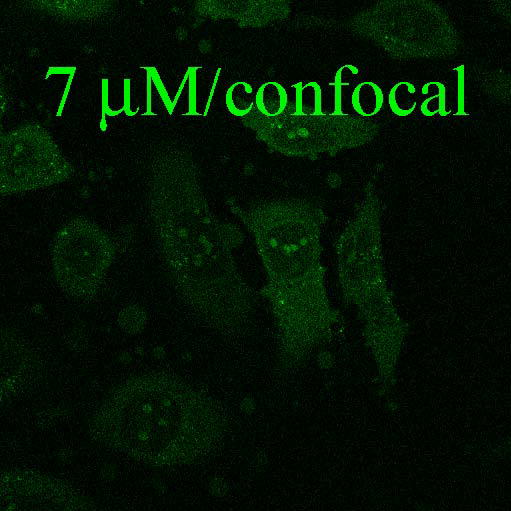
Fluorescence and confocal microscopic images of live HeLa cells incubated with Flu-R.I.-CKTat9 (green) and the live DNA dye Hoechst 33258 (blue). 2μM, 10μM, & 10μM/e-dpltn: merged fluorescence microscopic images of Flu-R.I.-CKTat9 and Hoechst 33258; cells were incubated with 2 and 10 μM of Flu-R.I.-CKTat9 without cell energy-depletion or 10 μM of Flu-R.I.-CKTat9 with cell energy-depletion, respectively. 7μM/DIC & 7μM/confocal: DIC and green channels of confocal microscopic images, respectively, of cells incubated with 7μM of Flu-R.I.-CKTat9. Note, to better show punctate and diffuse fluorescence, single cells at high magnification after trypsinization are presented in the first two images. For multi cell images, see Fig. 4 & 5. (Readers may want to inspect some of the images at higher magnifications of the online version from ACS/MP web site: http://pubs3.acs.org/acs/journals/toc.page?incoden=mpohbp).
Fig. 2.

KPBS incubation eliminates the diffuse, but not the punctate, green Flu-R.I.-CKTat9. Live HeLa cells were incubated with 0.35 μM Rho-Tf & 10 μM of Flu-R.I.-CKTat9 in PBS or KPBS for 40 min and washed. The live cells were observed under a confocal microscope. 10μM/PBS/Flu-R.I.-CKTat9 and 10μM/KPBS/Flu-R.I.-CKTat9: green channel for Flu-R.I.-CKTat9 in PBS and KPBS, respectively; 10 μM/PBS/Rho-Tf and 10 μM/KPBS/Rho-Tf: red channel for Rho-Tf in PBS or KPBS, respectively; 10 μM/PBS/merged and 10 μM/KPBS/merged: merged green and red channel images of PBS and KPBS incubations, respectively.
Fig. 3.

Effective clathrin heavy chain RNAi eliminated both the punctate and the diffused intracellular fluorescence of Flu-R.I.-CKTat9 in live HeLa cells. HeLa cells were double transfected with the negative control BCRP siRNA or the clc-2 siRNA, incubated with 10 μg/ml of DAPI and 0.35 μM Alexa 488-Tf for the fluorescence microscopy (top two images, respectively, captured with a 63× objective) or 10 μg/ml of DAPI, 75 μM Rhodamine-Dextran (Dex), and 10 μM of Rho-R.I.-CKTat9 (FTat) for the confocal microscopy (middle and bottom six images, respectively), washed, and examined microscopically. BCRP RNAi/Alexa488 Tf shows abundant punctate Tf fluorescence, which is absent in Clthr RNAi/Alexa488 Tf. BCRP RNAi-DAPI, BCRP RNAi-Dex, and BCRP RNAi-FTat are confocal blue, red, and green channel images, respectively, of the same middle section of a field captured using a 40× objective, so are Clthr RNAi-DAPI, Clthr RNAi-Dex, and Clthr RNAi-FTat. Comparison between the BCRP and the Clathrin RNAi images shows that Clathrin heavy chain RNAi eliminated both the punctate and the diffuse fluorescence of the labeled Tat CPP but had no effect on the labeled Dex fluorescence. Note the clathrin RNAi section had almost twice as many cells as that of the BCRP RNAi section as revealed by the DAPI staining of nuclei.
KPBS incubation eliminates the diffuse but not the punctate Flu-R.I.-CKTat9 and the peptide is colocalized with Rho-transferrin
Animal cells maintain a lower Na+ concentration, a higher K+ concentration, and a lower voltage potential (i.e., the membrane potential) compared to the extracellular space. It is primarily the leaky K+ channels and the K+ gradient that determine the membrane potential47. The membrane potential is lost (depolarized) when the cells are incubated with an isotonic/high K+ buffer such as KPBS, in which K+ has replaced Na+. Rothbard, J. B et al have shown that incubating three types of cells with a high potassium buffer similar to KPBS reduces the cell-associated fluorescence by more than 90% of a labeled polyarginine as analyzed by flow cytometry39. However, the high potassium buffer did not inhibit the endocytosis of a fluorescently labeled transferrin into one of the tested cell types. The authors proposed a mechanism of membrane potential-driven, direct plasma membrane translocation for the observation. Using HeLa cells and with our labeled Tat CPP, we observed a similar KPBS effect; i.e., KPBS reduces the cell-associated fluorescence by greater than 90% but did not affect endocytosis. In our case, the diffuse but not the punctate fluorescence was eliminated. Fig. 2 shows that, in cells incubated with Rho-Tf & 10 μM of Flu-R.I.-CKTat9 in KPBS, there is only green punctate fluorescence of Flu-R.I.-CKTat9 (Fig. 2: 10 μM/KPBS/Flu-R.I.-CKTat9) as opposed to punctate and diffused fluorescence in the control cells in PBS (Fig. 210 μM/PBS/Flu-R.I.-CKTat9). Quantitative uptake experiments showed that total cell-associated fluorescence in cells incubated with KPBS was 44.9% of that in the control cells incubated with PBS (p < 0.006). Since the amount of Flu-R.I.-CKTat9 bound at cell surface is negligible (cf. Fig. 1: 10μM/e-dpltn), the total cell-associated fluorescence consists primarily of fluorescence in the endosomes and the cytosol/nucleus. The uptake data therefore indicates that at 10 μM, the labeled Tat CPP is distributed slightly more in the cytosol/nucleus than in the endosomes and KPBS treatment eliminates the cytosol/nucleus portion. The effect of KPBS persisted at the highest tested concentration of 50 μM Flu-R.I.-CKTat9 (not shown). Although binding to cell surface was low, it could be adequately measured. There was no difference in the amount of cell surface-bound Flu-R.I.-Tat9 at 0 °C between in KPBS and in PBS (not shown), indicating that the effect of KPBS is not based on binding.
Additionally, using confocal microscopy, it was found that in KPBS incubated cells nearly 100% of the green punctate structures (Fig. 2: 10 μM/KPBS/Flu-R.I.-CKTat9) colocalized with the red Rho-Tf (Fig. 2: 10 μM/KPBS/Rho-Tf) (colocalization is shown in Fig. 2: 10 μM/KPBS/merged; see also Fig. 4). However, not all of the red punctate Rho-Tf fluorescence colocalized with the green punctate structures. This is also true with the cells incubated with PBS (Fig. 2: 10 μM/PBS/merged). Since the receptor for Tf (TfR) undergoes constitutive recycled endocytosis, the result suggests that Flu-R.I.-CKTat9 is taken up into endosomes by the clathrin-mediated endocytosis just as Rho-Tf is but it does not undergo recycling. Overall, the results suggest that the flux into the cytosol/nucleus compartment requires an intact membrane potential, but the origin of the flux cannot be pinpointed. Three possible origins of the flux are: the extracellular fluid, the topologically equivalent endosomal lumen where a membrane potential persists after endosome formation, or both. Rothbard, J. B et al favored the extracellular space but did not rule out the other two possibilities39. As shown later, the endosomal lumen is likely to be the origin.
RNAi with the clathrin heavy chain eliminates both the diffuse and the punctate fluorescence of Rho-R.I.-CKTat9
In order to understand the origin of the flux, the suspected clathrin-mediated endocytosis mechanism was shut down to see whether the diffuse fluorescence remained. An siRNA specific for the clathrin heavy chain (chc-2 siRNA) was used. This siRNA had been shown to knock down expression to undetectable levels and abolish Tf internalization45. Following a published protocol45, HeLa cells were double transfected with chc-2 siRNA. At 96-h post the first transfection, the cells were incubated with Alexa488-Tf and the membrane-permeable DNA dye DAPI for fluorescence microscopy or 10 μM of Fluo-R.I.-CKTat9 plus the fluid-phase endocytosis marker Rhodamine-Dextran 10000 (R-Dex) and DAPI for the confocal microscopy. The cells were washed and examined under a fluorescence microscope or a confocal microscope, respectively. An siRNA for the mechanistically irrelevant transporter BCRP was used as a negative control for the clathrin heavy chain RNAi. In the top two fluorescence microscopic images of Fig. 3, the BCRP RNAi (BCRP RNAi/Alexa488 Tf) did not affect the endocytosis of labeled Tf, while the clathrin heavy chain RNAi (Clthr RNAi/Alexa488 Tf) almost completely abolished punctate fluorescence due to the labeled Tf, leaving only weak cell surface-bound fluorescence. This result confirms the effectiveness of the clathrin RNAi as reported by the original authors who optimized and validated the RNAi45. In the bottom six confocal microscopic images of Fig. 3, there was no difference between BCRP RNAi (BCRP RNAi/Dex) and clathrin heavy chain RNAi (Clthr RNAi/Dex) in the endocytosis of R-Dex, suggesting that fluid-phase endocytosis was not affected and hence that the clathrin RNAi was specific for clathrin-mediated endocytosis. Since Tf is a prototypical substrate of the clathrin-mediated endocytosis, this result indicates that this endocytosis pathway had been almost completely knocked down in our chc-2 siRNA-transfected cell. In contrast to the negative control of BCRP RNAi (BCRP RNAi/Tat), the clathrin heavy chain RNAi almost completely abolished both the punctate and the diffused fluorescence of Fluo-R.I.-CKTat9 (Clthr RNAi/Tat). The result thus supports the single route of Tat CPP entry into the cytosol from the endosomes that follows the clathrin-mediated endocytosis pathway. A quantitative analysis should further strengthen the conclusion. However, due to the artifact of fluorescent staining of dead cell debris on live cells as a result of cytotoxicity of both double transfection and clathrin heavy chain RNAi, (e.g., the bright spots in the image of Fig. 3/Clthr RNAi/Alexa488 Tf), such quantitative analysis could not be satisfactorily performed. To overcome this problem, we separately developed a functional assay of Tat CPP internalization using a derivative of HeLa cells and functionally and quantitatively showed that the clathrin heavy chain RNAi eliminated the Tat CPP function that occurs in the nucleus (unpublished), further supporting the conclusion drawn from the clathrin heavy chain RNAi presented in this study.
Chemical inhibitors of the clathrin-mediated endocytosis (hypertonic sucrose and MDC) greatly reduce both the diffuse and the punctate Rho-R.I.-CKTat9, while the vesicular pH-neutralizing agent NH4Cl only eliminates the diffuse Rho-R.I.-CKTat9
Chemical inhibitors of clathrin-mediated endocytosis were also used to assess the origin of the flux into the cytosol/nucleus compartment. Chemical inhibitors have the advantage of faster action than genetic inhibitors, usually within an hour as compared to several days for siRNA. As a result, cell injury and death were greatly reduced, minimizing artifactual uptake of labeled R.I.-Tat9. In addition, using different means to inhibit the endocytosis pathway minimizes the possibility that membrane potential has been compromised.
HeLa cells were incubated with Alexa488-Tf (green) and 10 μM of Rho-R.I.-CKTat9 (red) for confocal microscopy examination, or 10 μM of Flu-R.I.-CKTat9 (green) alone for the fluorescence microscopy examination, in the absence (controls) or the presence of one of the three inhibitors (sucrose, NH4Cl, or MDC), washed, and examined under a confocal, or a fluorescence, microscope. The confocal result in Fig. 4 shows that Alexa488-Tf is localized only in the cytosolic punctate (vesicular) structures (Control/Tf) when no inhibitors were present whereas the Rho-R.I.-CKTat9 took on both a diffuse and punctate appearance (Control/Tat). Hypertonic sucrose treatment greatly reduced the punctate Alexa488-Tf (Sucrose/Tf) and the punctate and diffused Rho-R.I.-CKTat9 (Sucrose/Tat). In contrast, NH4Cl treatment had no effect on the punctate Alexa488-Tf (NH4Cl/Tf) and the punctate Rho-R.I.-CKTat9 (NH4Cl/Tat) but completely abolished the diffused Rho-R.I.-CKTat9 (NH4Cl/Tat). Using fluorescence microscopy, Flu-R.I.-CKTat9 is both diffuse and punctate (ctrl/Flu-R.I.CKTat9) in the absence of MDC but both types of fluorescence are greatly reduced in the presence of MDC (MDC/Flu-R.I.CKTat9). Since both hypertonic sucrose48 and MDC49 are known to block the formation of the clathrin-coated vesicles and NH4Cl to neutralize the acidity of endosomes/lysosomes, the results suggest that Tat CPP enters HeLa cell by the clathrin-mediated endocytosis, escapes to the cytosol and the escape requires endosomal acidity. Although the chemical inhibition results are in good agreement with the RNAi result, the chemical inhibition mechanisms are generally poorly understood and their effects on membrane potential are not known.
HeLa cells arrested in M phase shut down endocytosis but maintain plasma membrane potential
These cells fail to show both the punctate and the diffused fluorescence of labeled Tat CPP. Mammalian cells shut down endocytosis during the M (mitotic) phase as membrane traffic is inhibited (reviewed in 50). However, the plasma membrane potential is maintained throughout a cell cycle since it is critical for many cellular activities even though it fluctuates by about two-fold (reviewed in 51). The lowest potential generally occurs in the G1 phase and the potential remains high in the rest S, G2, and M phases. Therefore, the M phase provides a natural way to shut down endocytosis without the liability of collapsing membrane potential (depolarization) as RNAi or chemical inhibition does. We serendipitously discovered this approach by observing that HeLa cells in M phase did not take up fluorescently labeled transferrin (green) and R.I.-CKTat9 (red) (Fig. 5A, B, C). This observation strongly suggests that flux of R.I.-CKTat9 into the cytosol/nucleus compartment requires endocytosis.
To use this approach, the persistence of membrane potential in M phase was then verified by electrophysiological recording on spherical HeLa cells arrested in M phase after exposure to 100 nM nocodazole for 16 h, mitotic shake-off, and attachment to poly-D-lysine-coated culture dishes. In parallel fluorescence microscopy, DNA staining showed that about 98% of these cells had DNA condensed into chromosomes, mostly at the prophase stage of the M phase (cf. Fig. 5E). The voltages of 12 such cells arrested in M phase ranged from −15 to −44 mV (average −25 +/− s.e.m. 2.6 mV). As a control, spherical cells flushed from flasks not treated with nocodazole were used and then attached to poly-D-lysine-coated culture dishes. These cells were presumably at the end of G2 phase on the verge of entering the prophase of the M phase. The voltage of 14 such cells was in the range of −23 to −46 (average −39.7 +/− s.e.m. 1.8 mV). In parallel fluorescence microscopy, more than 98% of these presumably G2 cells exhibited interphase nuclear morphology (not shown). In view of the fact that some of the cells arrested in M phase might have sustained physiological trauma during the 16h nocodazole treatment and hence had lower potential than those cells naturally transiting through a normal M phase, the conclusion is made that HeLa cells arrested in M phase maintained membrane potential similar to those in other phases of the cell cycle. The resting membrane potential recorded in these experiments, though slightly lower, are comparable to those reported by others52, 53 for HeLa cells (−45 to −55 mV). In multiple sessions of fluorescence microscopy it was verified that, in contrast to interphase cells (Fig. 5D), HeLa cells arrested in M phase (Fig. 5E) did not take up Rho-R.I.CKTat9. To quantify the difference in uptake between the two cell cycle phases, three uptake experiments were performed, each in parallel to a corresponding microscopy session, using untreated cells in the presumed G2 phase and cells arrested in the M phase after nocodazole-treatment, mitotic shake-off, and attachment to poly-D-lysine-treated 24-well plates. The amount of uptake was normalized against total cellular protein to minimize the nocodazole cytotoxic effect. The percentages of uptake of cells in M phase relative to that of cells in G2 phase (control) were 9.8 +/− 1.5% (n=2, mean +/− s.d.), 34.4% (n=1), and 45 +/− 7% (n=3, mean +/− s.d.). The two experiments with higher than 9.8 % of control values had many more dead cells and cell debris resulting in artifactual uptake of some of the labeled Tat CPP (cf. Fig. 5E). Taking into account the artifactual uptake, the uptake by a very small percentage of cells that were out of M phase, and the current microscopic evidence, we conclude that HeLa cells in M phase shut down the uptake to an extent that is almost all-or-none. This compelling evidence supports a R.I.-Tat9 entry pathway though endocytosis and endosomal escape rather than by direct plasma membrane crossing since HeLa cells in M phase shut down endocytosis while their maintaining membrane potential.
The amount bound to Flu-R.I.-CKTat9 to HeLa cell surface starts to drop at mild acidic pH
In an attempt to understand the role of acidity on endosomal escape, the dissociation of bound labeled R.I.-CKTat9 molecules from the HeLa cell surface was found to start at pH 6.5 (Fig. 6), implying an endosomal escape mechanism. The 7-fold drop in binding is particularly noteworthy. Fluorescently labeled R.I.-CKTat9 bound to cell surface is easy to measure but is not usually seen in microscopy. This discrepancy is seemingly due to a difference in kinetics. Binding is measured on ice when the energy-dependent entry step is stopped but the binding step is permitted, which is in contrast to uptake at 37 °C that allows for fast entry but little accumulation on cell surface.
Fig. 6.

Low pH reduces Flu-R.I.-CKTat9 binding to HeLa cells. HeLa cells in 24-well plates were incubated with Flu-R.I.-CKTat9 at 0 °C for 1 hour in the buffers of different pH values and washed with the same buffers pre-chilled on ice. The cells were lysed with 1 N NaOH, neutralized, measured for fluorescence and protein amount, and analyzed as described in the Experimental section. Each data point is the mean of three wells +/− s. d.
DISCUSSION
The cytosol and the nucleus of an animal cell constitute a single intracellular compartment that is continuous through thousands of nuclear pores. Molecules smaller than 25–40 kDa generally diffuse through these pores. The lumen of all of the other membrane-bound (vesicular) organelles, including the endosomes, the lysosomes, the ER, the Golgi apparatus, and the nuclear envelope, constitutes another compartment that is topologically equivalent to the extracellular space. These organelles communicate with each other through vesicular trafficking and with the extracellular space through endocytosis and exocytosis. The value of CPPs lies in their ability to deliver hydrophilic cargos into the intracellular compartment. How CPPs, alone or with cargos, get into the intracellular compartment is still not fully understood. Specifically, the relative roles of membrane potential and endocytosis need to be delineated.
Terrone et al showed in 2003 that a labeled polyarginine peptide entered large unilamellar lipid vesicles in a membrane potential-dependent manner28. In 2005 Rothbard et al first described the importance of membrane potential in cell penetration39. Surprisingly, few reports have since examined the effect of KPBS-depolarization on Tat CPP cellular entry. Working with a labeled retro inverso Tat CPP-HA2 fusion peptide, Tunnemann et al38 showed that KPBS-treatment eliminated both the diffuse and the vesicular fluorescence. The current results are similar to Rothbard’s in that KPBS-treatment affected the diffuse, but not the vesicular, fluorescence of our labeled Tat CPP. Strikingly, in all three cases KPBS treatment affected diffuse fluorescence, suggesting that membrane potential is necessary for Tat CPP entry into the intracellular compartment. Since, in two of the three cases endocytosis occurred in the presence of KPBS, the alternative interpretation that KPBS abolishes endocytosis is not likely. Another possible alternate interpretation is that KPBS disrupts the first binding step at the cell surface. However, this is not likely either since no difference in the amount of cell surface-bound Tat CPP at 0 °C between in PBS and KPBS was found in this study. However, whether membrane potential is required at the plasma or endosomal membranes, or both remained a significant question. The first scenario is the simplest in interpretation. The current results unexpectedly support the second scenario; i.e., a sequential route of endocytosis and endosomal escape with the membrane potential being required at the second step (Fig 7.).
Fig. 7.

Summary of the results from this study showing a sequential entry route consisting of two steps. The experimental treatments indicated in each box block an entry step indicated by a neighboring “X”.
Fig. 7 shows that any means of blocking clathrin-mediated endocytosis (e.g., cell energy-depletion, 0 °C, RNAi, sucrose, MDC, or M phase of the cell cycle) abolishes or greatly reduces CPP entry into both topological compartments, whereas treatment eliminating the plasma membrane potential (KPBS) or neutralizing endosomal acidity (NH4Cl) abolishes only entry into the intracellular (cytosol/nucleus) compartment. Additionally, a labeled R.I.-CKTat9 colocalized with a labeled Tf. Therefore, the current results support and extend previous conclusions that Tat CPPs enter the intracellular compartment predominantly, if not exclusively, by clathrin-mediated endocytosis and subsequent endosomal escape23–27, 30. Multiple treatments were used to minimize the likelihood of compromising membrane potential. Exploiting the effect of the M phase is the most compelling evidence of the current studies since by directly measuring plasma membrane potential it was excluded as a confounding factor and alternate explanation of the results. To our knowledge, endocytosis inhibition using the M phase is novel and circumvents transfection often associated with RNAi and the use of dominant negative mutants. Overall, the current results strongly support the sequential route of entry and the important role of membrane potential. At least in the case of R.I.-Tat9, this represents a deviation from the membrane potential hypothesis39. The requirement of an intact membrane potential for entry into the cytosol/nucleus compartment may be universal for arginine-rich CPPs but requires further study. Recently we confirmed that an IGFBP-3-derived basic peptide is a CPP, whose entry into several cell types also depends on membrane potential (to be published).
Another significant finding of this study is the pH-dependence of binding (Fig. 6) that led to the hypothesis that might explain the effect of KPBS. Reducing ligand binding by lowering pH is not a new concept. However, it is remarkable that the mildly acidic pH (pH ~ 6.5) at which dissociation begins (Fig. 6) is consistent with the low affinity binding at neutral pH16. Our hypothesis makes a reasonable assumption that, during endosome formation (i.e., pinching off), the cell surface negatively charged non-specific receptors for the Tat CPP and the positively charged Tat CPP (i.e., the two binding parties) remain the same. In other words, the cell surface binding-pH dependence to the lumen of the endosome can be applied to this situation. In many other aspects, our hypothesis is based on known facts. At practical nM to μM concentrations, a positively charged Tat CPP binds to the mammalian cell surface non-specific high capacity/low affinity receptors16, such as the negatively charged proteoglycans25. The binding is followed by further clustering (i.e., avidity-enhanced concentration)54 on the cell surface into the coated pits and clathrin-mediated endocytosis ensues30. Even without clustering, a small fraction of adsorbed molecules may follow the clathrin-mediated pathway55, 56. An endosome thus formed concentrates a Tat CPP more than 1000-fold from the extracellular fluid47 as evidenced by the bright punctate fluorescence in microscopy. As soon as an endosome is pinched off, clathrin-uncoating47 and V-ATPase assembly/activation57 take place. On the early endosome, the V-ATPase quickly reduces the luminal pH to ~ 6.258, at which the majority of bound Tat CPP molecules are set free (Fig. 6), resulting in a free luminal peptide concentration in the range of hundreds of μMs or higher. At this high concentration, charge-charge repulsion16 becomes the driving force to push the peptide molecules across the endosomal membrane. The hypothesis need not invoke a path of endocytosis into the later endosome and lysosome, whose pH is at about 5.3 and 5.0, respectively58. The hypothesis may also explain the fast appearance of the diffused fluorescence, since an early endosome takes only a minute or so to form47 and the accompanying Tf can recycle back to the extracellular space with a half-life of as short as a few minutes59. The effect of NH4Cl is easily explained by the removal of the driving force. The effect of KPBS can be explained by the loading of high concentration of K+ into early endosomes, which creates an opposite potential to dampen the H+-pumping into the lumen by the V-ATPase58; i.e., KPBS reduces the driving force indirectly. Put in another way, membrane potential is required for the maintenance of a suitable environment in which endosomal acidification can occur. It should be mentioned that a similar but less comprehensive endosomal escape mechanism has been proposed based on a study using non-Tat CPPs and large unilamellar vesicles60.
Our findings differ with some reports showing various routes of entry or even entry at low temperatures61. There also appears to be a threshold concentration for diffused labeled Tat CPPs or polyarginine to appear within the cytosol. However, the threshold appears much higher for Tat-CPPs (this work and references37, 43–44) than for polyarginine42–43, 61. The reasons for these differences are not clear at this point. One potential reason is the difference in Tat CPPs, labels, or the various and different experimental parameters such as the cell line used or many different Tat CPP concentrations that were studied in the experiments. Since there could be multiple competing entry pathways available in a cell36–38; slight differences in the property of a labeled Tat CPP and/or some experimental parameters could influence transport and favor entry via one pathway over another pathway. Understanding the relationships between the properties of a particular molecule and the transport pathway is important. In the current studies, clathrin-mediated endocytosis may have become the predominant pathway. The other possible reason for these differences is that different mechanistic tools with poor specificities (such as endocytosis inhibitors) were used. This was discussed in detail in a recent review of the role of macropinocytosis in cell penetration36. With better understanding of these relationships, some conflicting results are likely to be resolved. For example, a differentiation-dependent reduction in endocytosis62 can explain why MDCK cells do not take up Tat CPP while some endocytosis-competent cells readily take up Tat CPPs (reference in62). It is noteworthy that this mechanistic insight would caution that cell type-dependent cell penetration (or cargo-dependent cell penetration, etc) wouldn’t automatically imply different underlying mechanisms. In this case, HeLa cells and undifferentiated MDCK cells could well share a common first endocytic step62. In other words, as far as cellular entry is concerned, the differentiation-dependent shutdown of endocytosis in MDCK cells could be no different from the M phase-dependent shutdown in HeLa cells. Consequently, similar observations made with MDCK62 cells may be considered another piece of supporting evidence suggesting that the predominant route is endocytosis with subsequent endosomal escape.
In conclusion, a hypothesis for endosomal escape is proposed, which could explain the results and may be further experimentally tested. The current results provide ample evidence for a dominating, if not single, route of R.I.-Tat9 CPP entry into the cytosol/nucleus via clathrin-mediated endocytosis and its subsequent endosomal escape. The results also confirm and clarify the importance of the role of membrane potential in R.I.-Tat9 uptake. It remains to be seen if and how the current findings are applicable to other Tat CPPs, Tat CPP with cargos, non-Tat CPPs, and other cell types.
Acknowledgments
This work was supported by NIH grant AI 51214. We thank Dr. C. Gardner for her assistance in confocal microscopy at the Environmental and Occupational Health Science Institute/Cancer Institute of New Jersey Core Facility.
ABBRIEVIATIONS
- Alexa488-Tf
Alexa Fluor 488-transferrin
- CPP
cell penetrating peptide
- DIC
differential interference contrast
- Flu-R.I.-CKTat9
5(6)-Carboxyfluorescein-R.I.-CKTat9
- MDC
monodansylcadaverine
- Rho-Dex
Rhodamine-Dextran 10000
- Rho-R.I.-CKTat9
rhodamine-R.I.-CKTat9
- Rho-Tf
Rho-transferrin
- R.I
retro inverso
- RNAi
RNA interference
- siRNA
small interfering RNA
- Tf
transferrin
- TfR
transferrin receptor
References
- 1.Torchilin VM. Protein- and peptide-mediated transduction: mechanisms and implications for drug delivery. Adv Drug Deliv Rev. 2005;57:489–490. [Google Scholar]
- 2.Langel U, editor. Cell-penetrating peptides: processes and applications. CRC Press LLC; Boca Raton, FL: 2002. [Google Scholar]
- 3.Fischer PM, Krauz E, Lane DP. Cellular delivery of impermeable effector molecules in the form of conjugates with peptides capable of mediating membrane translocation. Bioconj Chem. 2001;12:825–841. doi: 10.1021/bc0155115. [DOI] [PubMed] [Google Scholar]
- 4.Hawiger J. Noninvasive intracellular delivery of functional peptides and proteins. Curr Opin Chem Biol. 1999;3:89–94. doi: 10.1016/s1367-5931(99)80016-7. [DOI] [PubMed] [Google Scholar]
- 5.Lindgren M, Hallbrink M, Prochiantz A, Langel U. Cell-penetrating peptides. Trends Pharmacol Sci. 2000;21:99–103. doi: 10.1016/s0165-6147(00)01447-4. [DOI] [PubMed] [Google Scholar]
- 6.Chen J, Li G, Lu J, Chen L, Huang Y, Wu H, Zhang J, Lu D. A novel type of PTD, common helix-loop-helix motif, could efficiently mediate protein transduction into mammalian cells. Biochem Biophys Res Commu. 2006:931–940. doi: 10.1016/j.bbrc.2006.06.173. [DOI] [PubMed] [Google Scholar]
- 7.Morris MC, Chaloin L, Heitz F, Divita G. Translocating peptides and proteins and their use for gene delivery. Curr Opin Biotech. 2000;11:461–466. doi: 10.1016/s0958-1669(00)00128-2. [DOI] [PubMed] [Google Scholar]
- 8.Nori A, Kopecek J. Intracellular targeting of polymer-bound drugs for cancer chemotherapy. Adv Drug Delev Rev. 2005;57:609–636. doi: 10.1016/j.addr.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 9.Wadia JS, Dowdy SF. Modulation of cellular function by TAT mediated transduction of full length proteins. Curr Protein Pept Sci. 2003;4:97–104. doi: 10.2174/1389203033487289. [DOI] [PubMed] [Google Scholar]
- 10.Fawell S, Seery J, Daikh Y, Moore C, Chen L, Pepinsky B, Barsoum J. Tat-mediated delivery of heterologous proteins into cells. Proc Natl Acad Sci USA. 1994;91:664–668. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vives E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem. 1997;272:16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 12.Park J, Ryu J, Kim K, Lee HJ, Bahn JH, Han K, Choi EY, Lee KS, Kwon HY, Choi SY. Mutational analysis of a human immunodeficiency virus type 1 Tat protein transduction domain which is required for delivery of an exogenous protein into mammalian cells. J Gen Virol. 2002;83:1173–1181. doi: 10.1099/0022-1317-83-5-1173. [DOI] [PubMed] [Google Scholar]
- 13.Wender PA, Mitchell DJ, Pattabiraman K, Pelkey E, Steinman L, Rothbard J. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc Natl Acad Sci USA. 2000;97:13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Futaki S. Membrane-permeable arginine-rich peptides and the tanslocation mechanisms. Adv Drug Deliv Rev. 2005;57:547–558. doi: 10.1016/j.addr.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 15.Okuyama M, Laman H, Kingsbury SR, Visintin C, Leo E, Eward KL, Stoeber K, Boshoff C, Williams GH, Selwood DL. Small-molecule mimics of an α-helix for efficient transport of proteins into cells. Nat Methods. 2007;4:153–158. doi: 10.1038/nmeth997. [DOI] [PubMed] [Google Scholar]
- 16.Zhang X, Li W, Pooyan S, Su Y, Gardner CR, Leibowitz MJ, Stein S, Sinko P. Quantitative assessment of the cell penetrating properties of RI-Tat9: evidence for a cell type-specific barrier at the plasma membrane of epithelial cells. Mol Pharmaceut. 2004;1:145–155. doi: 10.1021/mp034014y. [DOI] [PubMed] [Google Scholar]
- 17.Wadia JS, Dowdy SF. Transmembrane delivery of protein and peptide drugs by TAT-mediated transduction in the treatment of cancer. Adv Drug Delev Rev. 2005;57:579–596. doi: 10.1016/j.addr.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 18.Brooks H, Lebleu B, Vives E. Tat peptide-mediated cellular delivery: back to basics. Adv Drug Delev Rev. 2005;57:559–577. doi: 10.1016/j.addr.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 19.Chen L, Harrison SD. Cell-penetrating peptides in drug development: enabling intracellular targets. Biochem Societ Trans. 2007;35:821–825. doi: 10.1042/BST0350821. [DOI] [PubMed] [Google Scholar]
- 20.Lundberg M, Johansson M. Positively charged DNA-binding proteins cause apparent cell membrane translocation. Bioche Biophys Res Commun. 2002;291:367–371. doi: 10.1006/bbrc.2002.6450. [DOI] [PubMed] [Google Scholar]
- 21.Pichon C, Monsigny M, Roche AC. Intracellular localization of oligonucleotides: influence of fixative protocols. Antisense Nucleic Acid Drug Dev. 1999;9:89–93. doi: 10.1089/oli.1.1999.9.89. [DOI] [PubMed] [Google Scholar]
- 22.Leifert JA, Harkins S, Whitton JL. Full-length proteins attached to the HIV tat protein transduction domain are neither transduced between cells, nor exhibit enhanced immunogenicity. Gene Ther. 2002;9:1422–1428. doi: 10.1038/sj.gt.3301819. [DOI] [PubMed] [Google Scholar]
- 23.Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, Chernomordik LV, Lebleu B. Cell-penetrating peptides. A reevaluation of the mechanism of celllualr uptake. J Biol Chem. 2003;278:585–590. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 24.Potocky TB, Menon AK, Gellman SH. Cytoplasmic and nuclear delivery of a TAT-derived peptide and a β-peptide after endocytic uptake into HeLa cells. J Biol Chem. 2003;278:50188–50194. doi: 10.1074/jbc.M308719200. [DOI] [PubMed] [Google Scholar]
- 25.Richard JP, Melikov K, Brooks H, Prevot P, Lebleu B, Chernomordik LV. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J Biol Chem. 2005;280:15300–15306. doi: 10.1074/jbc.M401604200. [DOI] [PubMed] [Google Scholar]
- 26.Fischer R, Kohler K, Fotin-Mleczek M, Brock R. A stepwise dissection of the intracellular fate of cationic cell-penetrating peptides. J Biol Chem. 2005;280:12625–12635. doi: 10.1074/jbc.M311461200. [DOI] [PubMed] [Google Scholar]
- 27.Vendeville A, Rayne F, Bonhoure A, Bettache N, Montcourrier P, Beaumelle B. HIV-1 Tat enters T cells using coated pits before translocating from acidified endosomes and eliciting biological responses. Mol Biol Cell. 2004;15:2347–2360. doi: 10.1091/mbc.E03-12-0921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Terrone D, Sang SLW, Roudaia L, Silvius JR. Penetratin and related cell-penetrating cationic peptides can translocate across lipid bilayers in the presence of a transbilayer potential. Biochem. 2003;42:13787–13799. doi: 10.1021/bi035293y. [DOI] [PubMed] [Google Scholar]
- 29.Fuchs SM, Raines RT. Pathway for polyarginine entry into mammalian cells. Biochem. 2004;43:2438–2444. doi: 10.1021/bi035933x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fotin-Mleczek M, Welte S, Mader O, Duchardt F, Fischer R, Hufnagel H, Scheurich P, Brock R. Cationic cell-penetrating peptides interfere with TNF signaling by induction of TNF receptor internalization. J Cell Sci. 2005;118:3339–3351. doi: 10.1242/jcs.02460. [DOI] [PubMed] [Google Scholar]
- 31.Jones SW, Christison R, Bundell K, Voyce CJ, Brockbank SMV, Newham P, Lindsay MA. Characterization of cell-penetrating peptide-mediated peptide delivery. Brit J Pharmacol. 2005;145:1093–1102. doi: 10.1038/sj.bjp.0706279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fittipaldi A, Giacca M. Transcellular protein transduction using the Tat protein of HIV-1. Adv Drug Deliv Rev. 2005;57:597–608. doi: 10.1016/j.addr.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 33.Kaplan IM, Wadia JS, Dowdy SF. Cationic TAT peptide transduction domain enters cells by macropinocytosis. J Control Rel. 2005;102:247–253. doi: 10.1016/j.jconrel.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 34.Nakase I, Niwa M, Takeuchi T, Somomura K, Kawabata N, Koike Y, Takehashi M, Tanaka S, Ueda K, Simpson JC, Jones AT, Sugiura Y, Futaki S. Cellular uptake of arginine-rich peptides: roles for macropinocytosis and actin rearragenment. Mol Ther. 2004;10:1011–1022. doi: 10.1016/j.ymthe.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 35.Wadia JS, Stan RV, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion protein after lipid raft macropinocytosis. Nat Med. 2004;10:310–315. doi: 10.1038/nm996. [DOI] [PubMed] [Google Scholar]
- 36.Jones AT. Macropinocytosis: search for an endocytic identity and a role in the uptake of cell penetrating peptides. Membr Trafic. 2007;11:670–684. doi: 10.1111/j.1582-4934.2007.00062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duchardt F, Fotin-Mleczek M, Schwarz H, Fischer R, Brock R. A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic. 2007;8:848–866. doi: 10.1111/j.1600-0854.2007.00572.x. [DOI] [PubMed] [Google Scholar]
- 38.Tunnemann G, Martin RM, Haupt S, Patsch C, Edenhofer F, Gardoso MC. Cargo-dependent mode of uptake and bioavailability of TAT-containing proteins and peptides in living cells. FASEB J. 2006;20:1775–1784. doi: 10.1096/fj.05-5523com. [DOI] [PubMed] [Google Scholar]
- 39.Rothbard JB, Jessop TC, Wender PA. Adaptive translocation: the role of hydrogen bonding and membrane potential in the upkake of guanidinium-rich transporters into cells. Adv Drug Deliv Rev. 2005;57:495–504. doi: 10.1016/j.addr.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 40.Zaro JL, Shen W-C. Quantitative comparison of membrane transduction and endocytosis of oligopeptides. Biochem Biophys Res Commun. 2003;307:241–247. doi: 10.1016/s0006-291x(03)01167-7. [DOI] [PubMed] [Google Scholar]
- 41.Hallbrink M, Floren A, Elmquist A, Pooga M, Bartfai T, Langel U. Cargo delivery kinetics of cell-penetrating peptides. Biochim Biophys Acta. 2001;1515:101–109. doi: 10.1016/s0005-2736(01)00398-4. [DOI] [PubMed] [Google Scholar]
- 42.Mitchell DJ, Kim DT, Steinman L, Fathman CG, Rothbard JB. Polyarginine enters cells more efficiently than other polycationic homopolymers. J Pept Res. 2000;56:318–325. doi: 10.1034/j.1399-3011.2000.00723.x. [DOI] [PubMed] [Google Scholar]
- 43.Thoren PEG, Persson D, Isakson P, Goksor M, Onfelt A, Norden B. Uptake of analogs of penetratin, Tat(48–60), and oligoargine in live cells. Biochem Biophys Res Commun. 2003;307:100–107. doi: 10.1016/s0006-291x(03)01135-5. [DOI] [PubMed] [Google Scholar]
- 44.Ziegler A, Pierluigi N, Durrenberger M, Seelig J. The cationic cell-penetrating peptide CPPTAT derived from the HIV-1 protein TAT is rapidly transported into living fibroblasts: optical, biophysical, and metabolic evidence. Biochem. 2005;44:138–148. doi: 10.1021/bi0491604. [DOI] [PubMed] [Google Scholar]
- 45.Motley A, Bright NA, Seaman NJ, Robinson MS. Clathrin-mediated endocytosis in AP-2-depleted cells. J Cell Biol. 2003;162:909–918. doi: 10.1083/jcb.200305145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Su Y, Zhang X, Sinko P. Human organic anion-transporting polypeptide OATP-A (SLC21A3) acts in concert with P-glycoprotein and multidrug resistance protein 2 in the vectorial transport of Saquinavir in Hep G2 cells. Mol Pharmaceutics. 2004;1:49–56. doi: 10.1021/mp0340136. [DOI] [PubMed] [Google Scholar]
- 47.The membrane potential in animal cells depends mainly on K+ leak channels and the K+ gradient across the plasma membrane. In Molecular Biology of the Cell. Alberts, B., Bray, D., Lewis, J., Raff, M., Roberts, K., Watson, J.D., Eds,; Garland Publishing, Inc.: New York & London, 1994; 524–525.
- 48.Heuser JE, Anderson RGW. Hypertonic media inhibit receptor-mediated endocytosis by blocking clathrin-coated pit formation. J Cell Biol. 1989;108:389–400. doi: 10.1083/jcb.108.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davies PJA, Davies DR, Levitzki A, Maxfield FR, Milhaud P, Willingham MC, Pastan IH. Transglutaminase is essential in receptor-mediated endocytosis of α2-macroglobulin and polypeptide hormones. Nature. 1980;283:162–167. doi: 10.1038/283162a0. [DOI] [PubMed] [Google Scholar]
- 50.Warren G. Membrane partitioning during cell division. Annu Rev Biochem. 1993:323–348. doi: 10.1146/annurev.bi.62.070193.001543. [DOI] [PubMed] [Google Scholar]
- 51.Boonstra J, Mummery CL, van Zoelen EJJ, van der Saag PT, de Laat SW. Monovalent cation transport during the cell cycle. Antican Res. 1982;2:265–274. [PubMed] [Google Scholar]
- 52.Szabo I, Brutsche S, Tombola F, Moschioni M, Satin B, Telford JL, Rappuoli R, Montecucco C, Papini E, Zoratti M. Formation of anion-selective channels in the cell plasma membrane by the toxin VacA of Helicobacter pylori is required for its biological activity. EMBO J. 1999;18:5517–5527. doi: 10.1093/emboj/18.20.5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stein MA, Mathers DA, Yan H, Baimbridge KG, Finlay BB. Enteropathogenic Escherichia coli markedly decreases the resting membrane potential of Caco-2 and HeLa human epithelial cells. Infect Immun. 1996;64:4820–4825. doi: 10.1128/iai.64.11.4820-4825.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ziegler A, Seelig J. Binding and clustering of glycosaminoglycans: a common property of mono- and multivalent cell-penetrating compounds. Biophys J. 2008;94:2142–2149. doi: 10.1529/biophysj.107.113472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Raub TJ, Roberts RM. Cell surface glycoproteins of CHO cells II. Surface distribution and pathway of internalization. Exp Cell Res. 1986;165:92–106. doi: 10.1016/0014-4827(86)90535-5. [DOI] [PubMed] [Google Scholar]
- 56.Haung M, Ma Z, Khor E, Lim L-Y. Uptake of FITC-Chitosan nanoparticles by A549 cells. Pharm Res. 2002;19:1488–1494. doi: 10.1023/a:1020404615898. [DOI] [PubMed] [Google Scholar]
- 57.Inoue T, Wang Y, Jefferies K, Si J, Hinton A, Forgac M. Structure and regulation of the V-ATPase. J Bioenerg Biomem. 2005;37:393–398. doi: 10.1007/s10863-005-9478-8. [DOI] [PubMed] [Google Scholar]
- 58.Grabe M, Oster G. Regulation of organelle acidity. J Gen Physiol. 2001;117:329–343. doi: 10.1085/jgp.117.4.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maxfield FR, McGraw TE. Endocytic recycling. Nat Rev Mol Cell Biol. 2004;2:121–128. doi: 10.1038/nrm1315. [DOI] [PubMed] [Google Scholar]
- 60.Magzoub M, Pramanik A, Graslund A. Modeling the endosomal escape of cell-penetrating peptides: transmembrane pH gradient driven translocation across phospholipid bilayers. Biochem. 2005;44:14890–14897. doi: 10.1021/bi051356w. [DOI] [PubMed] [Google Scholar]
- 61.Fretz MM, Penning NA, Al-Taei S, Futaki S, Takeuchi T, Nakase I, Storm G, Jones AT. Temperature-, concentration- and cholesterol-dependent translocation of L- and D-octa-arginine across the plasma and nuclear membrane of CD34+ leukemia cells. Biochem J. 2007;403:335–342. doi: 10.1042/BJ20061808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Foerg C, Ziegler U, Fernandez-Carneado J, Giralt E, Merkle HP. Differentiation restricted endocytosis of cell penetrating peptides in MDCK cells cooresponds with activities of Rho-GTPases. Pharm Res. 2007;24:628–642. doi: 10.1007/s11095-006-9212-1. [DOI] [PubMed] [Google Scholar]