

Abstract
In cancer cells, signal transducer and activator of transcription 3 (Stat3) participates in aberrant growth, survival, angiogenesis, and invasion signals and is a validated target for anti-cancer drug design. We are targeting its SH2 domain to prevent docking to cytokine and growth factor receptors and subsequent signaling. One of the important elements of the recognition sequence, pTyr-Xxx-Xxx-Gln, is glutamine. We incorporated novel Gln mimics into a lead peptide, pCinn-Leu-Pro-Gln-NHBn, and found that a linear, unconstrained side chain and carboxamide are necessary for high affinity, and the benzamide can be eliminated. Replacement of Gln-NHBn with (R)-4-aminopentanamide or 2-aminoethylurea produced inhibitors with equal or greater potency than that of the lead, as judged by fluorescence polarization (IC50 values were 110 and 130 nM, respectively). When Pro was replaced with cis-3,4-methanoproline, the glutamine mimic, (4R,5S)-4-amino-5-benzyloxyhexanamide resulted in an IC50 of 69 nM, the highest affinity Stat3 inhibitor reported to date.
Introduction
Signal transducer and activator of transcription 3 (Stat3) transmits signals from IL-6 family cytokines, epidermal growth factor, plate-derived growth factor, leptin, and vascular epithelial growth factor directly from their receptors on the cell surface to the nucleus.1–5 On binding of these extracellular signaling proteins, Stat3 is recruited to phosphotyrosine residues on their receptors to which it binds via its SH2 domain. Stat3 then becomes phosphorylated on Tyr705, a process known as activation, either by the tyrosine kinase activity of the receptor or that of associated Janus or Src-family kinases. The phosphorylated protein dimerizes via reciprocal interactions between the SH2 domains and pTyr705 residues and the activated complex is translocated to the nucleus where it binds to promoters of genes involved in cell survival, cell cycling, invasion and migration, and angiogenesis. Constitutively activated Stat3 has been detected in tumor samples from numerous cancers.6 Inhibition of Stat3 activity by antisense oligonucleotides, decoy oligonucleotides, and siRNA results in apoptosis and reduced cell growth of tumor cells. Thus Stat3 is target for antitumor drug design.2, 4
We7–11 and others12–21 have been targeting the SH2 domain of Stat3 with phosphopeptides and related peptidomimetics to break up pre-formed dimers and to prevent initial docking to receptors and the subsequent events of activation, dimerization, nuclear transport, and expression of downstream genes. The recognition determinant for this target is pTyr-Xxx-Xxx-Gln.7, 22–24 Of particular importance is the requirement for glutamine three residues C-terminal to the phosphotyrosine, pY+3. From a drug design perspective, the hydrophilic nature of this amino acid is likely to impart specificity for inhibitors of Stat3 since non-Stat SH2 domains typically recognize hydrophobic residues at this position25–30. In a screen of putative receptor docking sites for Stat3, our laboratory found that pTyr-Leu-Pro-Gln-Thr-Val-NH2 (1) was a high affinity ligand and it possessed glutamine at pY+3.7 Extensive studies that probed the interactions between each amino acid and Stat3 were conducted.7–11 Modification of glutamine, for example by side chain N-methylation, conversion to carbamoyl threonine, replacement with methionine sulfoxide, etc., provided information that supported a model of phosphopeptide-Stat3 binding in which the side chain fits tightly into a groove on the surface of the protein at the junction of β strand D and the STAT protein-specific helix, αB’ (Figure 1).31 Direct hydrogen bonds are hypothesized to occur between the NH2 group of Gln and main chain oxygens of Glu638 and Pro639 as well as between the C=O of Gln and the side chain NH2 of Gln644. A water mediated hydrogen bond may also form between the C=O of Gln and the side chain carboxyl group of Glu638. In addition, a hydrogen bond is predicted to exist between the backbone NH of the residue C-terminal to Gln and the side chain hydroxyl group of Tyr640 of Stat3.
Figure 1.

Hydrogen bonding interactions between Ac-pTyr-Leu-Pro-Gln-NHBn and Stat3 from the model described in reference 31. A. Direct hydrogen bonds between the side chain NH2 and the main chain carbonyl groups of Glu638 and Pro639; the side chain C=O of Gln and Gln644; and the NH of benzylamide and Tyr640. B. A water-mediated hydrogen bond between the side chain C=O of Gln and the carboxyl group of Glu638. Peptide 2 is depicted in the green coloring scheme: carbon is green, nitrogen is blue, oxygen is red, and hydrogen is white. Amino acids from Stat3 are depicted in the white coloring scheme with carbon being white and the other elements the same as in the ligand.
Glutamine can be considered to be 4-carboxy-4-aminobutyramide. In this paper, we incorporated a library of novel 4-aminobutyramide (Aba) derivatives into a phosphopeptide to serve as Gln mimics to 1, further understand peptide-protein interactions and 2, reduce the peptidic nature of the inhibitor with the goal of increased stability to proteases and possibly glutaminases. The lead inhibitor for these studies, pCinn-Leu-Pro-Gln-NHBn (2)10, 11 (Chart 1) was chosen for its ease of synthesis and for its high affinity. In 2 the phosphotyrosine of peptide 1 was replaced with 4-phosphoryloxycinnamic acid (pCinn) and the C-terminal Thr-Val-NH2 was substituted with a benzyl group. Conversion of the pTyr to pCinn increased affinity10, 11 and the benzyl group likely fits into a hydrophobic pocket on the surface of Stat3.31 Peptide 2 exhibited an IC50 of 138 nM in a fluorescence polarization assay, as compared to 290 nM for peptide 1.10, 11 The studies described herein revealed that replacement of the C-terminal Gln-NHBn unit of 2 with simpler mimics, 4-aminopentanamide or 2-aminoethylurea, led to inhibitors that were equipotent with the lead. However, when proline was substituted with cis-3,4-methanoproline,8 replacement of the C-terminal CONHBn with the isosteric 1-benzyloxyethyl group resulted in an IC50 value of 69 nM, the highest affinity inhibitor of Stat3 reported to date.
Chart 1.

Structures of starting phosphopeptide inhibitor of Stat3, 1, and the modified lead, 2. The IC50 value of 1 was reported in reference 8 and that of 2 in reference 10
Chemistry32
Peptide synthesis
The novel glutamine mimics were derived from corresponding Nα-Fmoc glutamic acid analogues, the syntheses of which are described below. Phosphopeptides were synthesized manually via attachment of the side chain of the glutamic acid derivatives to Rink amide resin so that upon cleavage from the support the final products possessed glutamine mimics (Scheme 1). The Fmoc protection scheme was used. Couplings were mediated with either diisopropylcarbodiimide (DIPCDI)/1-hydroxybenzotriazole (HOBt) or 1H-Benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate (PyBOP)/HOBt/DIEA. Fmoc removal was accomplished with 3 treatments of 20% piperidine in DMF for 6 min each. All sequences were capped with 4-(di-tert-butylphosphoryl)cinnamic acid.11 Peptides were cleaved from solid supports with TFA:triisopropylsilane:H2O (TFA:TIS:H2O) (95:2.5:2.5)33 and were purified by reverse phase HPLC.
Scheme 1.

General synthesis scheme for inhibitors possessing the new, modified glutamine mimics
For the synthesis of inhibitor 3 in which Gln-NHBn was replaced with Gly-NHBn (Table 1), benzyl amine was attached to FMPB aldehyde resin through reductive amination with NaBH3CN and 1%AcOH/DMF.34 The resulting resin-bound secondary amine was then acylated with Fmoc-Gly-OH using PyBOP/HOBt/DIEA. The remaining amino acids were coupled using DIPCDI/HOBt. Inhibitors 2, 4 and 5 (Table 1) were initiated by coupling Fmoc-Glu-NHBn (50), Fmoc-Asp-NHBn (51), and Fmoc-homoGlu-NHBn (52, homoGlu = homoglutamic acid), respectively, to Rink amide resin via their side chains using PyBOP/HOBt/DIEA. Amino acids 51 and 52 were synthesized as described for 50.8
Table 1.
Effect of the length of the side chain of glutamine analogues in the peptide pCinn-Leu-Pro-NHCH(R)CO-NHBn.
| Peptide | R | IC50 (nM) |
|---|---|---|
| 2 | (CH2)2CONH2 | 138 ± 8 |
| 3 | H | 4,940 ± 1,220 |
| 4 | CH2CONH2 | 874 ± 189 |
| 5 | (CH2)3CONH2 | 1,400 ± 189 |
Synthesis of inhibitors possessing constrained glutamine mimics
(E)-4-Amino-2-butenamide was used as a constrained glutamine mimic in inhibitor 9 (Table 3). The N-Fmoc protected amino acid precursor, 56, was prepared as depicted in Scheme 2. Commercially available Fmoc-aminoethanol (53) was oxidized to the aldehyde (54) by Swern oxidation35 followed by Wittig coupling with PPh3CHCO2tBu to give 55. Hydrolysis of the tert-butyl ester with TFA gave 56, which was attached to Rink amide resin to start the synthesis of 9.
Table 3.
The effect of constrained Gaba analogues on the affinity of pCinn-Leu-Pro-Xxx.
| Peptide | Xxx | IC50 (nM) | Peptide | Xxx | IC50 (nM) |
|---|---|---|---|---|---|
| 8 | 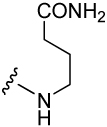 |
574 ± 130 | 11 | 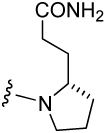 |
3,310 ± 392 |
| 9 | 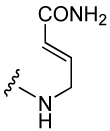 |
1,110 ± 430 | 12 | 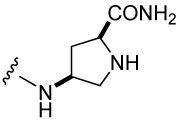 |
3,210 ± 269 |
| 10 | 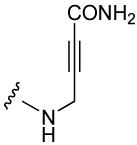 |
1,140 ± 110 | 13 | 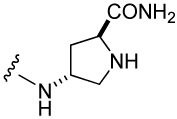 |
3,600 ± 1,270 |
Scheme 2a.

aReagents and conditions. (i) DMSO, oxalyl chloride, DIEA, CH2Cl2, −78°C; (ii) PPh3CHCO2tBu, CH2Cl2; (iii) TFA.
Fmoc-4-aminotetrolic acid (58), used in the synthesis of inhibitor 10 (Table 3), was prepared by protecting the amino group of 4-amino tetrolic acid (57)36 with Fmoc-OSu/10%Na2CO3/1,4-Dioxane (Scheme 3). Throughout the synthesis of inhibitors 9 and 10, Fmoc removal was accomplished with 5% DBU/DMF to avoid Michael addition with piperidine.
Scheme 3a.

aReagents and conditions: (i) Fmoc-OSu, 10% Na2CO3, 1,4-Dioxane.
For inhibitors 12 and 13 (Table 3), commercially available Fmoc-(2S,4S)-4-amino-1-Boc-pyrrolidine-2-carboxylic acid and Fmoc-(2S,4R)-4-amino-1-Boc-pyrrolidine-2-carboxylic acid respectively were coupled to Rink amide resin using DIPCDI/HOBt.
Substitution of the side chain carboxamide of 4-aminobutyramide (Aba)
Inhibitors 8 (Table 3) and 14 (Table 4) were synthesized by initial attachment of Fmoc-4-Abu-OH (Abu = 4-amino butyric acid) and Fmoc-3-aminopropanesulfonyl chloride (59), respectively, to Rink amide resin followed by assembly of remainder amino acids using standard coupling methods. The latter was prepared by treating the sodium salt of Fmoc-3-aminopropane sulphonic acid (60) with SOCl2/DMF (cat)37. Peptide 15 was initiated by coupling of Fmoc-4-Abu-OH to hydroxylamine resin using DIPCDI/HOBt, followed by assembly of the rest of the amino acids. For inhibitor 16, FMPB aldehyde resin was first treated with hydrazine to make the corresponding hydrazone, which was then coupled with Fmoc-4-Abu-OH followed by assembly of remainder of the amino acids. The peptide was cleaved from the resin with 6N HCl/MeCN and purified by HPLC.
Table 4.
Importance of side chain carboxamide group in pCinn-Leu-Pro-Xxx.
| Peptide | Xxx | IC50 (nM) |
|---|---|---|
| 8 | NH(CH2)3CONH2 | 574 ± 130 |
| 14 | NH(CH2)3SO2NH2 | 68,200 ± 23,500 |
| 15 | NH(CH2)3CONHOH | 2,640 ± 45 |
| 16 | NH(CH2)3CONHNH2 | 1,830 ± 770 |
Synthesis of inhibitors possessing 4-alkyl Aba analogues
We demonstrated that during triethylsilane (TES)/Pd-C mediated hydrogenation, the Fmoc group is stable which allowed selective deprotection of benzyl-based protecting groups.38 This observation formed the basis of our strategy for the synthesis of the 4-alkyl substituted Aba analogues used in inhibitors 17–24 (Table 5) and 42–49 (Table 7). A modification of the procedure of Loukas et al.39 was employed for the synthesis of 4-alkyl substituted Aba analogues 64a-i and 68 (Scheme 4). The carboxyl groups of Fmoc-D-amino acids, 61a-i, were converted to their corresponding alcohols (62a-i) via formation of 2-thiopyridyl esters followed by reduction with sodium borohydride. The alcohols were oxidized to aldehydes 63a-i by Swern oxidation,35 and these were elongated by Wittig coupling with Ph3PCHCO2Bn to 64a-i. Concurrent reduction of the double bond and hydrogenolysis of the benzyl group with TES and Pd/C in MeOH gave Fmoc-protected glutamic acid analogues (65a-i) ready for coupling to Rink amide resin. In this scheme, the side chains of D-amino acids end up as carboxyl group replacements in L-Gln analogues. This method was also used to prepare the constrained glutamic acid analogue, Fmoc-3-(2-pyrrolidino) propionate (70), used in the synthesis of 11 (Table 3).
Table 5.
Effect of substitutions at C(4) of Aba in pCinn-Leu-Pro-Xxx.
| Peptide | Xxx | IC50 (nM) | Peptide | Xxx | IC50 (nM) |
|---|---|---|---|---|---|
| 2 | 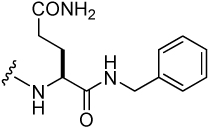 |
138 ± 8 | 24 | 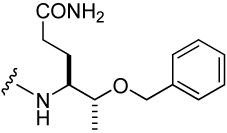 |
272 ± 7 |
| 8 | 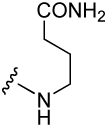 |
574 ± 130 | 25 | 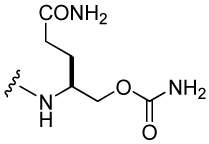 |
615 ± 30 |
| 17 | 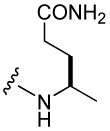 |
110 ± 30 | 26 | 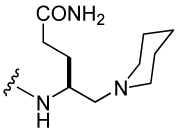 |
1,210 ± 190 |
| 18 | 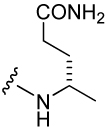 |
1,180 ± 30 | 27 | 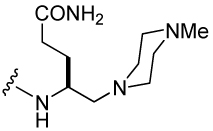 |
1,310 ± 228 |
| 19 | 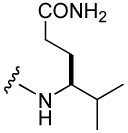 |
1,880 ± 220 | 28 | 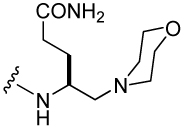 |
1,530 ± 70 |
| 20 | 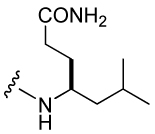 |
4,900 ± 610 | 29 | 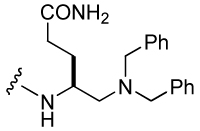 |
1,080 ± 360 |
| 21 | 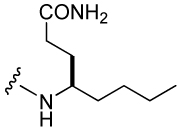 |
1,350 ± 360 | 30 | 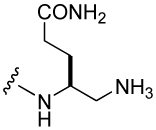 |
1,240 ± 215 |
| 22 | 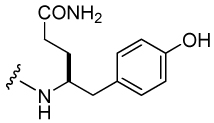 |
848 ± 126 | 31 | 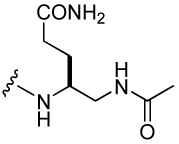 |
589 ± 174 |
| 23 | 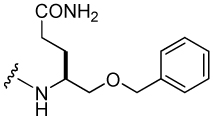 |
294 ± 40 |
Table 7.
Effect of substitution in pCinn-Leu-cis-3,4-methanoPro-Xxx 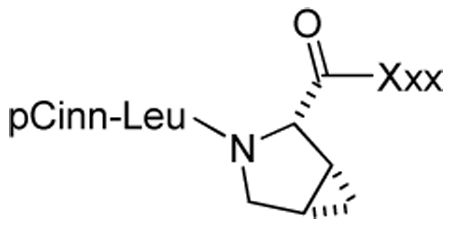
| Peptide | Xxx | IC50 (nM) | Peptide | Xxx | IC50 (nM) |
|---|---|---|---|---|---|
| 40 | 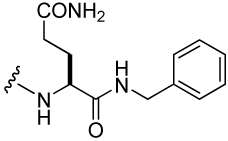 |
68 ± 8 | 45 | 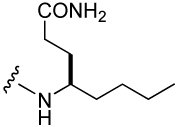 |
699 ± 62 |
| 41 | 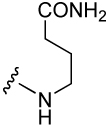 |
303 ± 48 | 46 | 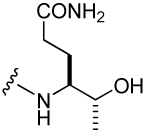 |
437 ± 4 |
| 42 | 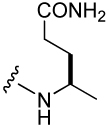 |
175 ± 35 | 47 | 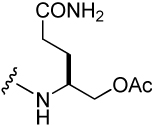 |
290 ± 41 |
| 43 | 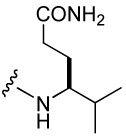 |
511 ± 67 | 48 | 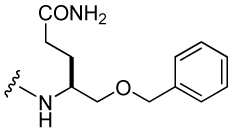 |
94 ± 11 |
| 44 | 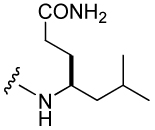 |
1,630 ± 132 | 49 | 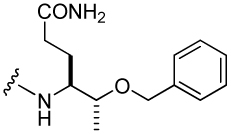 |
69 ± 10 |
Scheme 4a.

aReagents and conditions: (i) a) 2-Mercaptopyridine, DCC, EtOAc; b) NaBH4, THF-H2O; (ii) DMSO, oxalyl chloride, DIEA, CH2Cl2, −78°C; (iii) PPh3CHCO2Bn, CH2Cl2; (iv) TES, Pd/C, MeOH.
Syntheses of inhibitors 26–28 (Table 5) were initiated by coupling of 5-piperidino-4-(9-fluorenylmethoxycarbonylamino)pentanoic acid and analogues (76a-c) to Rink resin. To synthesize 76a-c (Scheme 5), the carboxyl group of Z-L-Glu(OtBu)-OH (71) was reduced to an alcohol (72) via sodium borohydride mediated reduction of the intermediate 2-thiopyridine ester. Intermediate 72 was then transformed to the corresponding iodide (73) by treatment with PPh3/I2/imidazole complex.40 Nucleophilic substitution with piperidine, morpholine and 4-methylpiperazine afforded tert-butyl 5-piperidino-4-benzyloxyaminopentanoate 74a, tert-butyl 5-morpholino-4-benzyloxyaminopentanoate 74b and tert-butyl 5-piperazino-4-benzyloxyaminopentanoate 74c respectively. TES/Pd-C mediated hydrogenation of 74a-c in 2% CHCl3/MeOH followed by Fmoc protection yielded 75a-c. Treatment of 75a-c with neat TFA followed by triturating with ether-hexane gave 76a-c as white powders.
Scheme 5a.

aReagents and conditions: (i) (a) 2-Mercaptopyridine, DCC, EtOAc; (b) NaBH4, THF-H2O; (ii) PPh3/I2/Imidazole/CH2Cl2 (91%); (iii) Piperidine or morpholine or N-methyl piperazine, THF; (iv) (a) TES/Pd-C, 2% CHCl3/MeOH; (b) Fmoc-OSu, 10%Na2CO3, THF; (v) TFA.
For the synthesis of substituted 4-aminomethyl Aba-containing inhibitors (29–31, Table 5), tert-Butyl 4-(9-fluorenyloxycarbonylamino)-5-iodopentanoate (79) was synthesized from Fmoc-Glu(OtBu)-OH using an analogous scheme as for 73 (Scheme 6). Iodide 79 was treated with tetrabutylammonium azide and the resulting azide, 80, was reduced with TES/Pd-C to give the free amine, which was then protected with the Alloc group to give 81. Removal of the side chain tert-butyl group with neat TFA gave 82, which was then attached to Rink amide resin. After addition of the pCinn, Leu, and Pro units, the Alloc group was removed with Pd(PPh3)4/CHCl3/AcOH/NMM41 and the resin was split into three portions. The first was not derivatized and led to inhibitor 30. The second was acetylated with acetic anhydride leading to inhibitor 31. Treatment of the final portion with 3-fold excess of benzaldehyde in the presence of NaCNBH3/AcOH resulted in complete alkylation of the amine, leading to inhibitor 29.
Scheme 6a.

aReagents and conditions: (i) (a) 2-Mercaptopyridine, DCC, EtOAc; (b) NaBH4, THF-H2O; (ii) PPh3/I2/Imidazole/CH2Cl2 (88%); (iii) nBu4NN3/ CH2Cl2 (81%); (iv) (a) TES/Pd-C, 2% CHCl3/MeOH; (b) Allyl chloroformate, DIEA, CH2Cl2 (64%);(v) TFA.
Syntheses of carbamate-containing phosphopeptides
Fmoc-amino alcohols 62a, 62g, and 62h (Scheme 4) and Fmoc-aminoethanol (62j) were converted to the corresponding 4-nitrophenylcarbonates (83a, g, h, and j) by treatment with nitrophenyl chloroformate10 (Scheme 7). These were coupled to Rink amide resin in the presence of DIEA to initiate the synthesis of the inhibitors. After addition of the pCinn, Leu, and Pro residues, cleavage from the resin yielded the carbamate-containing inhibitors 34–37 (Table 6).
Scheme 7a.

aReagents and conditions: (i) nitrophenylchloroformate, Pyridine, CH2Cl2; (ii) PPh3/I2/Imidazole/CH2Cl2(91%); (iii) Bu4NN3/ CH2Cl2 (84%); (iv) TES, Pd/C, 2% CHCl3/MeOH; (v) (a), Fmoc-OSu, NaHCO3; (b), HCl/EtOAc.
Table 6.
Inhibition of Stat3 by carbamates and ureas as glutamine mimics in pCinn-Leu-Pro-Xxx.
| Peptide | Xxx | IC50 (nM) | Peptide | Xxx | IC50 (nM) |
|---|---|---|---|---|---|
| 2 | 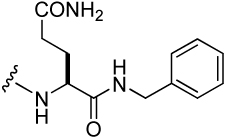 |
138 ± 8 | 36 | 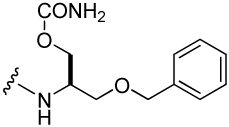 |
454 ± 21 |
| 32 | 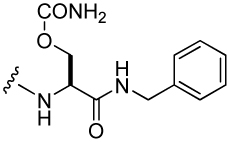 |
379 ± 49a | 37 | 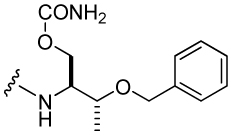 |
6,900 ± 350 |
| 33 | 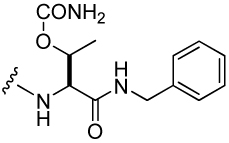 |
850 ± 85a | 38 | 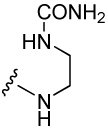 |
131 ± 14 |
| 34 | 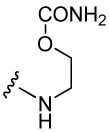 |
360 ± 37 | 39 | 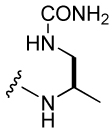 |
1,350 ± 250 |
| 35 | 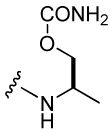 |
797 ± 29 |
From reference 10
Inhibitor 25 (Table 5), possessing a 4-amino-5-carbamoylpentamide on the C-terminus was synthesized by initial attachment of 86 to Rink amide resin, followed by subsequent coupling of remainder of the amino acids. Nitrophenyl carbonate 86 was obtained from reducing the carboxyl group of Fmoc-Gln(Trt) (84) to the corresponding alcohol 85, followed by treatment with 4-nitrophenyl chloroformate (Scheme 8).
Scheme 8a.

aReagents and conditions: (i) (a) 2-Mercaptopyridine, DCC, EtOAc; (b) NaBH4, THF-H2O; (ii) 4-nitrophenylchloroformate, pyridine, CH2Cl2 (62%).
Synthesis of urea containing inhibitors 38 and 39
Syntheses of urea containing inhibitors 38 and 39 (Table 6) started with the coupling of Fmoc aminoethyl nitrophenyl urethanes 90a, j to Rink amide resin to give resin bound ureas. As shown in Scheme 7, commercially available Boc-diaminoethane (91) was treated with Fmoc-OSu followed by HCl in EtOAc to prepare Fmoc-diaminoethane, 89j. Treatment with nitrophenylchloroformate provided the corresponding nitrophenylurethane, 90j. The synthesis of 2-methyl-2-aminonitrophenyl urethane, 90a, was carried out by a different route than that reported by Boeijen et al.42 Fmoc-alaninol, 62a (Scheme 4), was converted to the iodide, 87a, as described for the 4-piperidinomethyl Aba analogues described above, which was then followed by azide substitution with tetrabutyl ammonium azide to give 88a. The use of tetrabutylammonium azide resulted in a higher yield than sodium azide32 for this step. Reduction of 88a with TES/Pd-C38 gave the diamine, 89a, which was converted to the nitrophenyl urethane 90a with nitrophenylchloroformate. The urethane intermediates were added to Rink amide resin to give solid supported ureas. After adding the remainder of the amino acids, cleavage from the resin with TFA gave urea-containing inhibitors 38 and 39.
Synthesis of inhibitor 47
Inhibitor 47 (Table 7) was initiated by coupling of 4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-5-(acetoxy)-pentanoate 93 (Scheme 9) to Rink resin. Hydroxypentanoate 78 (Scheme 6) was acetylated with acetic anhydride and the tert-butyl ester deprotected to give the starting amino acid.
Scheme 9a.

aReagents and conditions: (i) Ac2O, DIEA, DMAP(cat.), CH2Cl2; (ii) TFA
Results
Alteration of the length of the glutamine side chain
To establish the importance of distance of the side chain carboxamide group from the main chain peptide, Gln was replaced with glycine, asparagine, and homoglutamine (Table 1). Substitution with glycine (3), without a side chain, decreased affinity 50 fold, re-iterating the importance of the alkyl carboxamide at pY+3. Peptides containing asparagine (4), with one a carbon contraction, and homoglutamine (5), with a one carbon extension with respect to glutamine, showed reduced activity by 7 and 10-fold, respectively, indicating that the position of the amide group is critical for attaining high affinity. The reduced affinity of 4 is in keeping with a similar result in which Asn substitution for Gln in 1 reduced the ability of the peptide to inhibit DNA binding in an electrophoretic mobility shift assay.7
Main chain interactions at position pY+3
To probe for main chain interactions between the pY+3 residue and Stat3, Gln was either removed (peptide 6) or replaced with Ala (peptide 7). Peptide 7 displayed higher affinity than 6, which is consistent with hydrogen bonding of the peptide bond C-terminal to the pY+3 residue and Tyr640 of Stat3 (Figure 1).31 As in the case of 3, the absence of the Gln side chain accounts for the reduced affinity of 7.
Conformationally constrained Aba analogues
Peptides with properly constrained side chain will exhibit higher affinity due to reduced entropy penalty on binding. The side chain of glutamine has three rotatable C-C bonds. We prepared a series of constrained Aba analogues as models to probe the effect of constraining the Gln side chain (Table 3). Note that Aba (8), exhibiting and IC50 value of 0.57 µM, results in a 3-fold loss of affinity as compared to Gln-NHBn in peptide 2. Incorporation of double and triple bonds in Aba (9 and 10, respectively) reduced affinity ca 2-fold compared to 8. Cyclizing C(4)-N(4) and C(2)-C(4) caused 6-fold reductions compared to Aba (11–13). Taken together with the loss in affinity observed when Gln-Thr-NH2 was replaced with 3-acetamidopyrrolidine, nipecotamide, aminocyclohexane-3-carboxamide, and 4-acetamidopiperazine reported previously,8 we conclude that when bound to Stat3, the alkyl chain of Gln adopts a conformation not readily mimicked by chemical modification.
Importance of the side chain carboxamide group
Previously8 we found that replacement of Gln by the isosteric analogue, methionine sulfoxide, as well as the oxidized version, methionine sulfone, resulted dramatic losses of inhibition. Mono- and dimethylation of the side chain nitrogen of glutamine also reduced affinity. Taken together, these results suggested role for the amide hydrogens in hydrogen-bonding interactions with the protein. To determine if sulfonamide protons could interact with Stat3, the side chain carboxamide of Aba was substituted with a SO2NH2 group (14). This modification resulted in very low affinity (IC50 = 68 µM). In an attempt to mimic the water molecule involved in the water-mediated hydrogen bond observed in the computational model (Figure 1),31 one of the amide protons of Aba was substituted with either a hydroxyl group (peptide 15) or an amino group (peptide 16). In both cases, 3–5 fold-reduced affinity was observed (Table 4). The reduction of affinity may be due to steric crowding in the glutamine binding pocket or loss of intermolecular H-bonding. Thus, a carboxamide group with both amide protons intact is optimal for high affinity.
4-Alkyl Aba analogues
To further probe for interactions between the main chain atoms at pY+3 and Stat3, and to search for new ones, a series of glutamine analogues in which the backbone carboxyl group was substituted with alkyl groups was used to replace Gln-NHBn in peptide 2 (Table 5). A methyl group produced the most potent inhibitor, increasing affinity 5-fold (17) over the hydrogen of Aba (8). Interestingly, compound 17 (IC50 = 110 nM) was equipotent with 2. The configuration of C(4) is important as the S enantiomer of 4-methyl Aba (inhibitor 18) resulted in lower affinity than 17. The larger, simple alkyl substitutions in 19–22 were not tolerated as well as the methyl group. Benzyloxymethyl and benzyloxyethyl groups were appended to C(4) of Aba which resulted in IC50 values of 294 (23) and 272 nM (24) respectively. These groups are nearly isosteric to the CONHBn group in compound 2 and 23 and 24 reduce affinity by ca 2-fold.
Attachment of an amino group to the γ-methyl carbon (30) resulted in an IC50 of 1.2 µM, 10-fold lower affinity than the unsubstituted methyl group. Piperidino-methyl Gaba analogues were synthesized with the long-term goal of solubility of prodrug versions of phosphopeptide inhibitors of Stat3. Compounds 26–28, with IC50 values raging from 1.2 to 1.5 µM, showed almost 10-fold reduced binding affinity compared to the unsubstituted methyl group. The acyclic tertiary amine containing inhibitor (29) also came out with 7-fold decreased affinity. However, acetylation of the amino group of 31 partially restored activity (compound 31). It appears that a charged amine at this position may be deleterious for activity. Addition of a carbamate at the C-terminus, 25, gave an IC50 value of 612 nM, similar to the acetamide 31.
Taken together, these results suggest that the binding surface for the backbone CONH atoms of glutamine of 2 is polar and that the alkyl groups do not make good contact. This is in keeping with the proposed model in which phenolic hydroxyl group of Tyr640 is within hydrogen bonding distance of this group (Figure 1). However, in spite of the polar surface, formal positive charge provided by amines is not tolerated well.
Substitution of glutamine with carbamate and ureas
Previously, we reported the replacement of the γ-methylene group of glutamine with oxygen to give side chain carbamate analogues.10 O-carbamoylserine (32) and O-carbamoylthreonine (33) resulted in 3 and 6-fold losses of affinity compared to peptide 2. To further study this functional group, a series of Aba analogues was converted to 2-aminoethylcarbamates (Table 6). In contrast to the loss in binding of Aba vs Gln-NHBn (2 and 8) discussed above (Table 5), the Aba analogue, 2-aminoethylcarbamate (34) results in affinity equal to that of carbamoylserine benzyl amide (32). Substitution of C(2) of the ethyl group (C(4) of Aba) results in loss of activity, even those possessing benzyloxymethyl and benzylozyethyl groups, 36, and 37 respectively. Replacement of Gln-NHBn with unsubstituted 2-aminoethylurea (38) results in equal affinity, 131 nM. Addition of a methyl group (39) resulted in a 10-fold loss in activity.
Glutamine analogues in peptides containing cis-3,4-methanoproline
In an earlier publication we reported that substitution of proline with cis-3,4-methanoproline (mPro) increased affinity by a factor of two and that hydrocinnamoyl-pTyr-Leu-mPro-Gln-NHBn exhibited and IC50 value of 125 nM in our FP assay.8 mPro is a rather expensive starting material so the studies discussed above were conducted on peptides containing proline. Adding to the cost is the fact commercially available Fmoc-cis-3,4-methanoPro is sold as a racemic mixture of (2S,3R,4S) and (2R,3S,4R) enantiomers and syntheses of peptides with this amino acid produce two diastereomers. Only one half of the material gives the high affinity interaction with Stat3. To test the effect of some of the Gaba analogues discussed above, we substituted mPro for Pro. Only the high affinity diastereomers are reported here.
Incorporation of cis-3,4-methanoPro in peptide 2 resulted in a 2-fold increase in affinity consistent with our previous report8 (40, Table 7). pCinn-Leu-cis-3,4-methanoPro-Gln-NHBn exhibited an IC50 value of 68 nM, the highest affinity Stat3 inhibitor reported reported to date. γ-Alkyl Gaba analogues were also appended onto the C-terminus of pCinn-Leu-cis-3,4-methanoPro and the IC50’s determined by FP. Substituting the benzylamide moiety with hydrogen (compound 41) resulted in a loss of affinity. In contrast to the proline analogues, γ-methyl Gaba (42) was not equipotent to the Gln-NHBn analogue, 40. Larger alkyl groups reduced affinity. The benzyloxymethyl group (48) restored binding to 94 nM. However, the isosteroc benzyloxy ethyl group (49) was equipotent with the benzylamide, exhibiting an IC50 value of 69 nM. Thus compound 49 is a very high affinity peptidomimetic inhibitor of Stat3 in which pTyr is replaced by pCinn, proline is replaced with 3,4-cis-methanoproline, and glutamine is replaced with γ-(2-benzyloxyethyl) Aba. With only one natural amino acid remaining, leucine, we have made great strides in the development of a nonpeptide inhibitor of Stat3.
Conclusions
We have probed the glutamine binding pocket of Stat3 with a series of glutamine and Aba analogues. A non-constrained, linear side chain is necessary for high affinity, as is the carboxamide group. These findings are consistent with the model of Gln fitting into a tight pocket on the surface of the Stat3 SH2 domain and the side chain amide participating in hydrogen bonding interactions.31 Glutamine can be substituted with non-peptide analogues. Replacement of the C-terminal Gln-NHBn unit of pCinn-Leu-Pro-Gln-NHBn with either (R)-4-aminopentamide or 2-aminoethylurea leads to modified peptides with IC50 values of ca 110 nM and 130 nM, respectively, equipotent with the lead inhibitor. However, replacement of proline with cis-3,4-methanoproline and Gln-NHBn with (4R,5S)-4-amino-5-benzyloxyhexamide leads to a modified peptide with enhanced affinity; the IC50 value of 49 was 69 nM. Conversion of these phosphopeptide mimics into cell permeable inhibitors will be reported under separate cover.
Experimental Section
Chemistry
General Nα-protected amino acids were purchased from Advanced Chemtech, NovaBiochem, ChemImpex, or AnaSpec. HOBt was from ChemImpex. Fmoc-cis-3,4-methanoPro was from EMD Biosciences (Formerly NovaBiochem). Rink amide resin was purchased from Advanced Chemtech, loaded between 0.6 and 0.7 mmol/g. Anhydrous DMF for amino acid solutions was from Baker. Other solvents were reagent grade and were used without further purification. Peptides were purified by reverse phase HPLC on a Rainin Rabbit HPLC or a Varian Dynamax HPLC using a Vydac 2.5 × 25 cm Peptide and Protein C18 column or a 2.1 × 25 cm Phenomenex Luna 10 µM C18(2). Gradients of MeCN in H2O (both containing 0.1% TFA) or MeCN in 0.01 M NH4OAc (pH 6.5) at 10 mL/min were employed. Peptides were tested for purity by reverse phase HPLC on an Agilent 1090 HPLC or an Agilent 1100 HPLC using a Vydac 4.6 × 250 mm C18 peptide/protein column or a 4.6 × 250 mm Phenomenex 5 µM C18(2) in two systems, A. 10–50% MeCN in H20 with 0.1% TFA in both solvents and B, 0–40% MeCN in 0.01 M NH4OAc, pH6.5. The flow rate for both was 1.5 mL/min. Peptides were always > 95% pure and were often >98% pure as judged by analytical HPLC. Before evaluation by fluorescence polarization, peptides were dried in vacuo over P2O5 at 37°C for 24 hr. NMR Spectra were recorded on Bruker 300 MHz DPX or 500 MHz DRX spectrometers.
Solid phase peptide synthesis
Manual Method Manual solid phase synthesis was carried out on aliquots of 0.20 gm of Rink resin (0.6 mmol/g). Fmoc groups were removed with 2 × 5 mL of 20% piperidine for 3 and 7 min each. After removing the Fmoc group from the resin, synthesis of inhibitors was initiated by coupling of the three-fold excesses of the new Fmoc-glutamic acid analogues, PyBop, and HOBt, along with six-fold excesses of DIEA in 5 mL of (1:1 v/v) DMF/CH2Cl2. The nitrophenyl carbonates (83 a,h,g,j) and nitrophenyl carbamates (90a,j) were added to resin in three fold excess along with three fold excess of DIEA in 5 mL of (1:1 v/v) DMF/CH2Cl2 as described.10 Reactions were monitored with ninhydrin. After coupling and deprotection steps, resins were washed with 5 × 5 mL of DMF/CH2Cl2. Cleavage was accomplished with three treatments of the resins with 5 mL of TFA: TIS: H2O (95:2.5:2.5)33 for 10 min each. The volumes of the solvents were reduced and the solutions were dropped into ice cold Et2O. After 30 min the precipitates were collected by filtration and were washed 2 × more with Et2O. Crude peptides were dried and peptides were purified by reverse phase HPLC as described in the general methods. All peptides were dried over P2O5 in vacuo at 37° for 24 h before testing. Peptide yields, HPLC retention times and mass spectra are tabulated in Table S1.
Synthesis of Fmoc-Asp-NHBn (51)
Starting with 0.5 g of Fmoc-Asp(tBu)-OH the procedure described by Coleman et al.8 for Fmoc-Glu-NHBn was employed. Yield 0.48 g (89%), white powder. 1H NMR (DMSO-d6, 500 MHz) δ 2.56 (dd, J = 9.0, 16.5 Hz, 1H), 2.27 (dd, J = 5.5, 16.5Hz, 1H), 4.22–4.33 (m, 5H), 4.42 (m, 1H), 7.2–7.35 (m, 7H), 7.43 (t, J = 7.0 Hz, 2H), 7.7 (d, J = 8.0 Hz, 1H), 7.73 (d, J = 7.0 Hz, 2H), 7.9 (d, J = 8.0 Hz, 2H), 8.42 (t, J = 6.0 Hz, 1H). 13C NMR (DMSO-d6, 125 MHz) δ 36.9, 42.6, 47.1, 51.9, 66.3, 120.6, 125.8, 127.1, 127.4, 127.6, 128.1, 128.6, 139.8, 141.2, 144.3, 156.3, 171.2, 172.3. HRMS (M+H) Calc’d: 445.1763; found 445.1772. Anal (C26H24N2O5) C, H, N: Calc’d: C, 70.26; H, 5.44; N, 6.30; found: C, 69.11; H, 5.35; N, 6.24. Note: % C is > 0.4%.
Fmoc-homoGlu-NHBn (52)
Starting with 0.5 g of Fmoc-homoGlu(tBu)-OH the procedure described by Coleman et al.8 for Fmoc-Glu-NHBn was employed. Yield 0.49 g (82%), white powder. 1H NMR (DMSO-d6, 500 MHz) δ 1.56–1.74 (m, 4H), 2.72 (t, J = 7.0 Hz, 2H), 4.1 (m, 1H), 4.26–4.37 (m, 5H), 7.25–7.38 (m, 7H), 7.47 (t, J = 7.5 Hz, 2H), 7.58 (d, J = 8.5 Hz, 1H), 7.78 (d, J = 7.0 Hz, 2H), 7.94 (d, J = 7.5 Hz, 2H), 8.47 (t, J = 5.5 Hz, 1H). 13C NMR (DMSO-d6, 125 MHz) δ 21.6, 31.9, 33.8, 42.1, 47.2, 54.9, 66.1, 120.6, 125.8, 127.2, 127.6, 128.1, 128.7, 139.8, 141.2, 144.3, 144.4, 156.5, 172.3, 174.7. Anal (C28H28N2O5) C, H, N: Calc’d: C, 71.17; H, 5.97; N, 5.93; found C, 70.41; H, 5.92; N, 5.87. HRMS (M+H) Calc’d: 473.2076; found 473.2115. Note: % C is > 0.4%.
Synthesis of 4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-2-butenoic acid (56)
Commercially available Fmoc-Glycinol (53) was oxidized to Fmoc-Glycinal (54) via Swern oxidation.43 A solution of 54 (1.0 g, 3.55 mmol) and tert-butyl (triphenylphosphoranylidene) acetate (1.3 g, 3.91 mmol) in dry CH2Cl2 (20 mL) was stirred for 2 h. The solvent was removed in vacuo and the residue was purified by silica gel column chromatography (15% EtOAc-hexane v/v) to get 55. Yield: 85% (1.20 g). 1H NMR (CDCl3, 500 MHz) δ 1.4 (s, 9H), 3.86 (m, 2H), 4.13 (t, J = 6.5 Hz, 1H), 4.35 (d, J = 6.5 Hz, 2H), 4.9 (m, 1H), 5.76 (d, J = 15.5 Hz, 1H), 6.71 (m, 1H), 7.22 (m, 2H), 7.31 (m, 2H), 7.5 (d, J = 7.5 Hz, 2H), 7.67 (d, J = 7.5 Hz, 2H). 13C NMR (CDCl3, 125 MHz) δ 28.1, 41.7, 47.2, 66.9, 80.7, 120.1, 123.5, 125.0, 127.1, 127.8, 141.4, 142.8, 143.8, 156.2, 165.3. HRMS (M+H) Calc’d: 380.1862; found 380.1856.
Compound 55 (1.0 g) was treated with 5.0 mL of neat TFA for 1 h. The TFA was removed under vacuum and residual acid was removed by the addition and evaporation of toluene (3 × 5 mL). Trituration with ether-hexane resulted in a white precipitate which was collected by filtration and dried over P2O5 yielding 0.81 g of 56 as a white powder, 95%. 1H NMR (DMSO-d6, 500 MHz) δ 3.8 (m. 2H), 4.25 (m, 1H), 4.33 (d, J = 6.5 Hz, 2H), 5.81 (d, J = 15.5 Hz, 1H), 6.76 (m, 1H), 7.34 (m, 2H), 7.42 (m, 2H), 7.66 (t, J = 5.5 Hz, 1H), 7.72 (d, J = 7.5 Hz, 2H), 7.9 (d, J = 7.5 Hz, 2H). 13C NMR (DMSO-d6, 125.0 MHz) δ 41.4, 47.2, 66.0, 120.6, 122.0, 125.6, 127.5, 128.1, 141.2, 144.3, 145.5, 156.6, 167.4. Anal (C19H17NO4) C, H, N: Calc’d: C, 70.58; H, 5.30; N, 4.33. Found C, 70.38; H, 5.56; N, 4.25. HRMS (M+H) Calc’d: 324.1236; Found 324.1164.
Synthesis of 4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-tetrolic acid (58)
4-Aminotetrolic acid (1.0 g, 7.37 mmol) (57), prepared as described by Ahern et al.,36 was treated with 10 mL of 10% Na2CO3 and Fmoc-OSu (2.2 g, 6.6 mmol) in 20 mL of 1,4-dioxane overnight. The solution was washed with 20 mL of EtOAc and the aqueous layer then acidified with conc. HCl. It was the extracted with EtOAc (3 × 30 mL) and the combined organic layers were washed with brine, dried (MgSO4), and evaporated. The residue was triturated with ether-hexane and the precipitate was collected by filtration and dried over P2O5. Yield: 1.3 g (41%). 1H NMR (DMSO-d6, 500 MHz) δ 4.04 (d, J = 5.0 Hz, 2H), 4.29 (t, J = 6.5 Hz, 1H), 4.41 (d, J = 6.5 Hz, 2H), 7.39 (m, 2H), 7.47 (m, 2H), 7.75 (d, J = 7.5 Hz, 2H), 7.93–7.95 (m, 3H). 13C NMR (DMSO-d6, 125 MHz) δ 30.3, 47.1, 66.3, 75.4, 84.7, 120.6, 125.6, 127.6, 128.1, 141.2, 144.2, 154.4, 156.5. Anal (C19H15NO4) C, H, N: Calc’d: C, 71.02; H, 4.71; N, 4.36. Found: C, 70.37; H, 4.67; N, 4.37. HRMS (M+H) Calc’d: 322.1079; Found: 322.1083.
Synthesis of sodium 3-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]propanesulfonate (60)
To a stirred aqueous solution (30 mL) of 3-aminopropanesulfonic acid (1 g, 7.18 mmol) and NaOH (0.3 g, 7.5 mmol), Fmoc-OSu (2.9 g, 8.6 mmol) was added in portions over 10 min. Stirring was continued overnight. Excess Fmoc-OSu was removed by washing with EtOAc (2 × 10 mL). The aqueous layer was lyophilized and the residual solid was triturated with ether-hexane. The product was collected by filtration and dried in vacuo over P2O5. Yield 1.9 g (72%). 1H NMR (DMSO-d6, 500 MHz) δ 1.77 (m, 2H), 2.5 (t, J = 7.5 Hz, 2H), 3.1 (m, 2H), 4.26 (m, 1H), 4.3 (d, J = 7.0 Hz, 2H), 7.37–7.41 (m, 3H), 7.46 (t, J = 7.5 Hz, 2H), 7.74 (d, J = 7.5 Hz, 2H), 7.93 (d, J = 7.5 Hz, 2H). 13C NMR (DMSO-d6, 125 MHz) δ 26.3, 47.2, 49.6, 65.8, 120.6, 125.7, 127.6, 128.1, 141.2, 144.4, 156.6. HRMS (M+H) Calc’d: 384.0882; Found 384.0914.
Synthesis of 3-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]propanesulfonyl chloride (59)37
To a mixture of 60 (1.00 g, 2.6 mmol) and thionyl chloride (10 mL) was added DMF (1 mL) drop-wise. The mixture was heated at reflux for 5 h. The volatiles were removed by evaporation, followed by coevaporation with toluene 3 × 5 mL. Compound 59 was coupled directly to Rink amide resin.
General procedure for N-[(9H-fluoren-9-ylmethoxy)carbonyl]-amino alcohols (62a-i and 67)
A solution of Fmoc-amino acid (5.0 mmol), DCC (6.0 mmol) and 2-mercaptopyridine (5.5 mmol) in 100 mL of EtOAc was stirred for 3 h. The white precipitate was filtered off and the filtrate was concentrated under vacuum. It was then diluted with 50 mL of THF and the solution was added slowly to a suspension of NaBH4 (10.0 mmol) in 20 mL of THF and 10 mL of water at 0 °C. After 30 min, the reaction was quenched by slow addition of ice-cold 5% HCl (aq) (50 mL) and extracted with ether (3 × 150 mL). The combined organic layers were washed with aqueous 10% NaHCO3 (3 × 40 mL), water (2 × 50 mL), and brine (1 × 40 mL). After drying (Na2SO4) and concentration under vacuum the crude residue was purified either by recrystallization from hexane-ether or silica gel column chromatography.
Fmoc-D-Alaninol (62a)
Yield 1.32 g (90%). 1H NMR (CDCl3, 500 MHz) δ 1.07 (m, 3H), 3.43 (m, 1H), 3.55 (m, 1H), 3.73 (m, 1H), 4.11 (m, 1H), 4.33 (m, 2H), 4.75 (brs, 1H), 7.22 (m, 2H), 7.3 (m, 2H), 7.48 (d, J = 7.5 Hz, 2H), 7.66 (d, J = 7.5 Hz, 2H). HRMS (M+H) Calc’d: 298.1443; found: 298.1247.
Fmoc-L-Alaninol (62b)
Yield: 1.4 g (92%). White powder, m.p. 100–102 °C. 1H NMR (CDCl3, 500 MHz) δ 1.07 (m, 3H), 3.43 (m, 1H), 3.55 (m, 1H), 3.72 (m, 1H), 4.11 (m, 1H), 4.33 (m, 2H), 4.78 (brs, 1H), 7.21 (m, 2H), 7.3 (m, 2H), 7.48 (d, J = 7.5 Hz, 2H), 7.66 (d, J = 7.5 Hz, 2H). HRMS (M+H) Calc’d: 298.1443. Found: 298.1356.
Fmoc-D-Valinol (62c)
Yield 1.5 g (91%). White powder, m.p. 126 °C. 1H NMR (CDCl3, 300 MHz) δ 0.93 (m, 6H), 1.84 (m, 1H), 3.64 (m, 2H), 4.2 (t, J = 6.6 Hz, 1H), 4.43 (m, 2H), 4.9 (m, 1H), 7.28–7.42 (m, 4H), 7.58 (d, J = 7.2 Hz, 2H), 7.75 (d, J = 7.2 Hz, 2H). HRMS (M+H) Calc’d: 326.1756; Found: 326.1629.
Fmoc-D-Leucinol (62d)
Yield: 1.55 g (90%). White powder, m.p. 138 °C. 1H NMR (CDCl3, 300 MHz) δ 0.92 (d, J = 5.7 Hz, 6H), 1.32 (m, 2H), 1.63 (m, 1H), 3.46–3.76 (m, 3H), 4.21 (m, 1H), 4.44 (m, 2H), 4.73 (m, 1H), 7.28–7.42 (m, 4H), 7.58 (d, J = 7.5 Hz, 2H), 7.76 (d, J = 7.5Hz, 2H). 13C NMR (CDCl3, 75 MHz) δ 22.1, 23.0, 24.8, 40.4, 47.4, 66.5, 120.0, 125.0, 127.0, 127.7, 141.4, 143.9. HRMS (M+H) Calc’d: 340.1913; Found: 340.1699.
Fmoc-D-norleucinol (62e)
Yield: 1.6 g (92%). White powder, m.p. 140 °C. 1H NMR (CDCl3, 300 MHz) δ 0.92 (m, 3H), 1.34–1.6 (m, 6H), 3.58–3.68 (m, 3H), 4.24 (m, 1H), 4.46 (m, 2H), 4.82 (m, 1H), 7.31–7.45 (m, 4H), 7.61 (d, J = 7.5 Hz, 2H), 7.78 (d, J = 7.5 Hz, 2H). HRMS (M+H) Calc’d: 340.1913; Found: 340.1739.
Fmoc-D-tyrosinol tert-butyl ether (62f)
Yield 2.0 g (92%). m.p. 108 °C. 1H NMR (CDCl3, 300 MHz) δ 1.34 (s, 9H), 2.82 (m, 2H), 3.67 (m, 2H), 3.91 (m, 1H), 4.21 (m, 1H), 4.42 (m, 2H), 4.94 (m, 1H), 6.92 (d, J = 8.4 Hz, 2H), 7.3–7.45 (m, 4H), 7.58 (d, J = 7.5 Hz, 2H), 7.78 (d, J = 7.5 Hz, 2H). HRMS (M+H) Calc’d: 446.2331; Found: 446.2057.
Fmoc-D-serinol benzyl ether (62g)
Yield: 1.61 g (78%). 1H NMR (CDCl3, 300 MHz) δ 3.46–3.83 (m, 5H), 4.2 (t, J = 6.6 Hz, 1H), 4.4 (m, 2H), 4.5 (s, 2H), 5.43 (m, 1H), 7.23–7.41 (m, 9H), 7.58 (d, J = 7.2 Hz, 2H), 7.74 (d, J = 7.2 Hz, 2H). 13C NMR (CDCl3, 75 MHz) δ 47.3, 52.0, 66.8, 70.5, 73.6, 120.0, 125.1, 127.1, 127.7, 128.0, 128.6, 137.6, 141.4, 143.9. HRMS (M+H) Calc’d: 404.1862; Found: 404.1855.
Fmoc-D-threoninol benzyl ether (62h)
Yield: 1.74 g (81%). 1H NMR (CDCl3, 300 MHz) δ 1.22 (d, J = 7.2 Hz, 3H), 2.65 (m, 1H), 3.7 (m, 2H), 3.83 (m, 1H), 4.2 (t, J = 6.6 Hz, 1H), 4.35–4.45 (m, 3H), 4.61 (d, J = 11.4, 1H), 5.32 (m, 1H), 7.28–7.4 (m, 9H), 7.58 (d, J = 7.2 Hz, 2H), 7.73 (d, J = 7.2 Hz, 2H). 13C NMR (CDCl3, 75 MHz) δ 16.2, 47.3, 56.7, 63.9, 66.9, 70.9, 74.4, 120.0, 125.1, 127.1, 127.7, 127.9, 128.0, 128.6, 138.0, 141.4, 144.0. HRMS (M+H) Calc’d: 418.2018; Found: 418.1696.
Fmoc-D-threoninol tert-Butyl ether (62i)
Yield: 1.6 g (82%). 1H NMR (CDCl3, 500 MHz) δ 1.06 (d, J = 6.0 Hz, 3H), 1.11 (s, 9H), 3.52–3.57 (m, 3H), 3.85 (m, 1H), 4.12 (t, J = 7.0 Hz, 1H), 4.26–4.35 (m, 2H), 5.22 (brs, 1H), 7.21 (m, 2H), 7.29 (m, 2H), 7.5 (d, J = 7.5 Hz, 2H), 7.65 (d, J = 7.5 Hz, 2H). 13C NMR (CDCl3, 125 MHz) δ 20.1, 28.7, 47.3, 57.4, 66.9, 74.3, 120.0, 125.1, 127.1, 127.7, 141.4, 143.9, 157.1. HRMS (M+H) Calc’d: 384.2175; Found: 384.1983.
Fmoc-D-Prolinol (67)
Yield: 1.45 g (89%). 1H NMR (CDCl3, 500 MHz) δ 1.6–1.9 (m, 4H), 3.3–3.4 (m, 2H), 3.6 (d, J = 4.5 Hz, 2H), 3.9 (m, 1H), 4.1 (m, 1H), 4.2 (t, J = 6.5 Hz, 1H), 4.3–4.44 (m, 2H), 7.24–7.35 (m, 4H), 7.54 (d, J = 7.5 Hz, 2H), 7.7 (d, J = 7.5 Hz, 2H). 13C NMR (CDCl3, 125 MHz) δ 24.0, 28.4, 47.2, 60.5, 63.5, 66.1, 67.5, 120.0, 125.1, 127.1, 127.8, 141.4, 144.0, 156.8. HRMS (M+H) Calc’d: 324.1600; Found: 324.1588.
General procedure for the synthesis of N-[(9H-fluoren-9-ylmethoxy)carbonyl]-amino aldehydes (63a-i and 68)
Fmoc-Amino aldehydes were synthesized by Swern oxidation of corresponding Fmoc-D-amino alcohols35. To a solution of oxalyl chloride (8.0 mmol) in 30 mL of dry CH2Cl2, stirred at −78 °C under argon was added DMSO (16.0 mmol) via syringe dropwise with vigorous stirring. After 20 min, a solution of Fmoc-D-amino alcohol (5.0 mmol) in 20 mL of dry CH2Cl2, was added while maintaining the bath temperature at −78 °C. Stirring was continued further for 30 min. Dry and distilled DIEA (30 mmol) was then added using a syringe and the reaction mixture then allowed to warm to room temperature without removing bath. The reaction mixture then quenched with 20 mL of ice-cold water and extracted with CH2Cl2 (3 × 80 mL). The combined organic layers were washed with 1N HCl (3 × 30 mL), brine (1 × 30 mL) and dried (MgSO4) and concentrated under vacuum. The crude aldehydes were used immediately for the next step without characterization.
General procedure for synthesis of N-[(9H-fluoren-9-ylmethoxy)carbonyl] γ-amino- α,β -unsaturated benzyl esters (64a-i, 69)
A mixture of Fmoc- amino aldehyde (4.0 mmol) and benzyl (triphenylphosphoranylidene)-acetate (4.4 mmol) in dry CH2Cl2 (20 mL) was stirred for 3h. The progress of reaction was monitored by thin layer chromatography. After completion of reaction, the solvent was removed in vacuo and the residue was purified by silica gel chromatography using EtOAc in hexane.
Benzyl (R)-4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-(E)-2-pentenoate (64a)
Prepared in Mandal et al. (2007).38
Benzyl (S)-4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-(E)-2-pentenoate (64b)
Yield: 1.52 g (87%). White powder, m.p 130–131 °C. 1H NMR (CDCl3, 500 MHz) δ 1.18 (m, 3H), 4.1 (t, J = 7.0 Hz, 1H), 4.35 (m, 2H), 4.65 (brs, 1H), 5.1 (s, 2H), 5.84 (d, J = 16.0 Hz, 1H), 6.81 (d, J = 15.0 Hz, 1H), 7.2–7.3 (m, 9H), 7.48 (d, J = 7.0 Hz, 2H), 7.65 (d, J = 7.0 Hz, 2H). 13C NMR (CDCl3, 125 MHz) δ 20.2, 47.3, 66.4, 120.0, 120.2, 125.0, 127.1, 127.7, 128.3, 128.4, 128.6, 135.8, 141.4, 143.8, 143.9, 149.3, 155.4, 166.0. HRMS (M+H) Calc’d: 428.1862; Found: 428.1876.
Benzyl (R)-5-methyl-4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-(E)-2-hexenoate (64c)
Yield: 1.55g (85%). White powder, m.p 141 °C. 1H NMR (CDCl3, 300 MHz) δ 0.94 (m, 6H), 1.89 (m, 1H), 4.23 (m, 2H), 4.48 (m, 2H), 4.75 (d, J = 9.0 Hz, 1H), 5.21 (s, 2H), 5.96 (d, J = 15.9 Hz, 1H), 6.93 (m, 1H), 7.31–7.42 (m, 9H), 7.60 (d, J = 7.2 Hz, 2H), 7.77 (d, J = 7.2 Hz, 2H). 13C NMR (CDCl3, 75 MHz) δ 18.0, 18.9, 32.2, 47.3, 66.4, 120.0, 121.5, 124.9, 127.1, 127.7, 128.3, 128.6, 135.8, 141.4, 143.8, 147.4, 155.8, 165.9. Anal (C29H29NO4) C, H, N: Calc’d: C, 76.46; H, 6.42; N, 3.07; Found: C, 75.74; H, 6.45; N, 3.32. HRMS (M+H) Calc’d: 456.2175; Found: 456.1540.
Benzyl (R)-6-methyl-4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-(E)-2-heptenoate (64d)
Yield: 1.6 g (82%). White powder, m.p. 118 °C. 1H NMR (CDCl3, 300 MHz) δ 0.94 (d, J = 6.3 Hz, 6H), 1.42 (m, 2H), 1.67 (m, 1H), 4.22 (t, J = 6.3 Hz, 1H), 4.41–4.5 (m, 3H), 4.65 (m, 1H), 5.2 (s, 2H), 5.96 (d, J = 15.3 Hz, 1H), 6.88 (m, 1H), 7.31–7.42 (m, 9H), 7.60 (d, J = 7.2 Hz, 2H), 7.77 (d, J = 7.2 Hz, 2H). 13C NMR (CDCl3, 75 MHz) δ 22.7, 24.7, 43.7, 47.3, 50.4, 66.4, 120.0, 120.4, 124.9, 127.1, 127.7, 128.3, 128.4, 128.6, 135.9, 141.4, 143.8, 148.9, 155.6, 166.0. Anal (C30H31NO4) C, H, N: Calc’d: C, 76.73; H, 6.65; N, 2.98; Found: C, 76.56; H, 6.71; N, 3.02. HRMS (M+H) Calc’d: 470.2331; Found: 470.2342.
Benzyl (R) 4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-(E)-2-octenoate (64e)
Yield: 1.52 g (78%), White powder, m.p. 116 °C. 1H NMR (CDCl3, 300 MHz) δ 0.93 (m, 3H), 1.33 (m, 4H), 1.53 (m, 2H), 4.23 (m, 1H), 4.35 (m, 1H), 4.48 (m, 2H), 4.7 (m, 1H), 5.21 (s, 2H), 5.96 (d, J = 15.6 Hz, 1H), 6.9 (m, 1H), 7.3–7.43 (m, 9H), 7.6 (d, J = 7.5 Hz, 2H), 7.78 (d, J = 7.5 Hz, 2H). 13C NMR (CDCl3, 75.0 MHz) δ 13.9, 22.3, 27.7, 34.2, 47.3, 52.0, 66.4, 120.0, 120.6, 124.9, 127.1, 127.7, 128.3, 128.4, 128.6, 135.9, 141.4, 143.8, 143.9, 148.7, 155.7, 166.0. Anal (C30H31NO4) C, H, N: Calc’d: C, 76.73; H, 6.65; N, 2.98; Found: C, 76.61; H, 6.71; N, 2.99. HRMS (M+H) Calc’d: 470.2331; Found: 470.2338.
Benzyl (R)-5-(4-tert-butoxyphenyl)-4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-(E)-2-pentenoate (64f)
Yield: 1.9 g (81%). White powder, m.p. 82°C. 1H NMR (CDCl3, 300 MHz) δ 1.33 (s, 9H), 2.87 (m, 2H), 4.2 (t, J = 6.6 Hz, 1H), 4.4 (m, 2H), 4.63–4.74 (m, 2H), 5.2 (s, 2H), 5.88 (d, J = 15.9 Hz, 1H), 6.91–6.94 (m, 3H), 7.02–7.1 (m, 2H), 7.30–7.42 (m, 10H), 7.55 (m, 2H), 7.77 (d, J = 7.5 Hz, 2H). 13C NMR (CDCl3, 75 MHz) δ 28.8, 47.2, 66.4, 66.7, 78.4, 120.0, 121.1, 124.2, 124.9, 127.1, 127.7, 128.2, 128.3, 128.6, 129.8, 135.8, 141.3, 143.7, 147.7, 154.5, 155.5, 165.8. Anal (C37H37NO5) C, H, N: Calc’d: C, 77.19; H, 6.48; N, 2.43; Found: C, 77.09; H, 6.46; N, 2.54. HRMS (M+H) Calc’d: 576.2750; Found: 576.2092.
Benzyl (S)-5-benzyloxy-4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-(E)-2-pentenoate (64g)
Yield: 1.53 g (72%). 1H NMR (CDCl3, 500 MHz) δ 3.5 (m, 2H), 4.15 (t, J = 7.0 Hz, 1H), 4.4–4.6 (m, 2H), 4.5 2 (m, 1H), 5.15 (s, 2H), 5.34 (d, J = 8.0 Hz, 1H), 6.0 (d, J = 15.5 Hz, 1H), 6.93 (m, 1H), 7.24–7.34 (m, 14 H), 7.54 (d, J = 7.0 Hz, 2H), 7.7 (d, J = 7.0 Hz, 2H). 13C NMR (CDCl3, 125 MHz) δ 47.3, 52.00, 66.5, 70.9, 73.4, 120.1, 122.0, 122.1, 125.1, 127.2, 127.8, 128.1, 128.2, 128.4, 128.5, 128.6, 128.7, 136.0, 137.5, 141.4, 143.9, 144.0, 146.2, 146.3, 155.9, 165.9. Anal (C34H31NO5) C, H, N: Calc’d: C, 76.53; H, 5.86; N, 2.62. Found: C, 76.42; H, 5.96; N, 2.66. HRMS (M+H) Calc’d: 534.2280. Found: 534.2291.
Benzyl (4S, 5S)-5-benzyloxy-4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-(E)-2-hexenoate (64h)
Yield: 1.55 g (68%), white powder. 1H NMR (CDCl3, 300 MHz) δ 1.22 (d, J = 5.7 Hz, 3H), 3.7 (m, 1H), 4.2 (t, J = 6.6 Hz, 1H), 4.3–4.45 (m, 4H), 4.58 (d, J = 11.7 Hz, 1H), 5.2 (s, 2H), 5.98 (d, J = 15.3 Hz, 1H), 6.98 (m, 1H), 7.28–7.38 (m, 14H), 7.58 (d, J = 7.5Hz, 2H), 7.74 (d, J = 7.5Hz, 2H). 13C NMR (CDCl3, 75 MHz) δ 16.5, 47.3, 66.3, 66.9, 71.1, 75.1, 120.0, 121.7, 125.0, 127.0, 127.7, 127.8, 127.9, 128.3, 128.5, 128.6, 135.9, 137.7, 141.3, 143.8, 143.9, 147.2, 165.8. Anal (C35H33NO5) C, H, N: Calc’d: C, 76.76; H, 6.07; N, 2.56. Found: C, 76.61; H, 6.06; N, 2.55. HRMS (M+H) Calc’d: 548.2437. Found: 548.2451.
Benzyl (4S, 5S)-5-tert-butoxy-4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-(E)-2-hexenoate (64i)
Yield: 1.4 g (65%) as an oil. 1H NMR (CDCl3, 500 MHz) δ 0.99–1.06 (m, 12H), 3.72 (m, 1H), 4.14 (m, 1H), 4.2 (m, 1H), 4.35–4.42 (m, 2H), 5.1 (s, 2H), 5.16 (d, J = 9.0 Hz, 1H), 5.87 (d, J = 15.5 Hz, 1H), 6.9 (m, 1H), 7.22–7.3 (m, 9H), 7.51 (d, J = 7.0 Hz, 2H), 7.66 (d, J = 7.0 Hz, 2H). 13C NMR (CDCl3, 125 MHz) δ 20.4, 28.6, 47.3, 57.5, 66.4, 66.9, 68.2, 74.3, 120.0, 121.2, 125.1, 127.1, 127.8, 128.2, 128.3, 128.6, 136.0, 141.4, 143.8, 147.9, 156.4, 166.0. HRMS (M+H) Calc’d: 514.2593. Found: 514.1597.
N-[(9H-Fluoren-9-ylmethoxy)carbonyl] benzyl 3-pyrrolidin-2-yl-(E)-propenoate (69)
Yield: 1.2 g (62%) as an oil. 1H NMR (CDCl3, 500 MHz) δ 1.7–1.96 (m, 4H), 3.36 (m, 2H), 4.1 (m, 0.5H), 4.14–4.2 (m, 1H), 4.3 (m, 0.5H), 4.4 (m, 1.5H), 4.48 (m, 0.5H), 5.57 (d, J = 15.5 Hz, 0.5H), 5.8 (d, J = 15.5 Hz, 0.5H), 6.6 (m, 1H), 6.68 (m, 0.5H), 6.8 (m, 0.5H), 7.1–7.3 (m, 9H), 7.42 (m, 1H), 7.5 (m, 1H), 7.6–7.68 (m, 2H). 13C NMR (CDCl3, 125 MHz) δ 22.6, 23.5, 30.8, 31.7, 46.5, 46.8, 47.3, 57.9, 58.2, 66.5, 67.5, 120.0, 120.4, 120.7, 125.0, 127.1, 127.7, 128.5, 135.8, 141.3, 143.7, 143.9, 147.9, 148.1, 155.1, 155.4, 158.2, 158.6, 166.1, 166.3. HRMS (M+H) Calc’d: 454.2018. Found: 454.1068.
General Procedure for the synthesis of 4-alkyl substituted 4-(9-fluorenylmethyloxycarbonylamino) butyric acids
To a stirred suspension of Fmoc protected γ-amino-α,β-unsaturated benzyl esters (2.0 mmol) and 10% Pd-C (10% by wt) in 15 mL of methanol-THF(4:1 v/v), was added TES (20.0 mmol) dropwise under argon atmosphere. The reaction started with evolution of hydrogen gas. After completion of reaction (TLC), the mixture was filtered through celite and the solvent was removed in vacuo. The crude product was purified either by triturating with ether-hexane or by short silica gel column chromatography, eluting with 2% hexane-EtOAc, followed by hexane-EtOAc-MeOH (3:6:1 v/v/v).
(R)-4-[[(9H-Fluoren-9-ylmethoxy)carbonyl]amino]-2-pentanoic acid (65a)
Yield: 0.6 g (88%) Prepared in Mandal et al (2006).38 White powder, m.p. 140–141 °C.
(S)-4-[[(9H-Fluoren-9-ylmethoxy)carbonyl]amino]-2-pentanoic acid (65b)
Yield: 0.59 g (87%). White powder, m.p. 128–130 °C. 1H NMR (DMSO-d6, 500 MHz) δ 0.94 (d, J = 6.5 Hz, 3H), 1.52 (m, 2H), 2.1 (m, 2H), 3.41 (m, 1H), 4.1 (m, 1H), 4.14–4.23 (m, 2H), 7.05 (d, J = 8.5 Hz, 1H), 7.22 (m, 2H), 7.31 (m, 2H), 7.59 (d, J = 7.5 Hz, 2H), 7.78 (d, J = 7.5 Hz, 2H). 13C NMR (DMSO-d6, 125.0 MHz) δ 21.3, 31.0, 31.7, 46.4, 47.3, 65.5, 120.6, 125.5, 127.5, 128.1, 141.2, 144.4, 156.1, 174.8. HRMS (M+H) Calc’d. 340.1549; Found 340.1566.
(S)-5-Methyl-4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-2-hexanoic acid (65c)
Yield: 0.66 g (89%). White powder, m.p. 120 °C. 1H NMR (CDCl3, 300 MHz) δ 0.9 (m, 6H), 1.5–1.78 (m, 3H), 2.36 (t, J = 7.5 Hz, 2H), 3.5 (m, 1H), 4.22 (m, 1H), 4.42 -4.6 (m, 3H), 7.3–7.43 (m, 4H), 7.60 (d, J = 7.5 Hz, 2H), 7.77 (d, J = 7.5 Hz, 2H). 13C NMR (CDCl3, 75 MHz) 17.8, 19.0, 27.4, 31.1, 32.5, 47.5, 56.1, 66.4, 1120.0, 125.0, 127.0, 127.7, 141.4, 143.9, 156.6, 178.8. Anal (C22H25NO4) C, H, N: Calc’d: C, 71.91; H, 6.86; N, 3.81. Found: C, 71.60; H, 6.82; N, 3.80. HRMS (M+H) Calc’d: 368.1862. Found: 368.1276.
(S)-6-Methyl-4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-2-heptanoic acid (65d)
Yield: 0.68 g (88%). White powder, m.p. 150 °C. 1H NMR (CDCl3, 300 MHz) δ 0.91 (d, J = 6.3 Hz, 6H), 1.24 (m, 2H), 1.53–1.62 (m, 2H), 1.85 (m, 1H), 2.35 (t, J = 7.2 Hz, 2H), 3.73 (m, 1H), 4.2 (m, 1H), 4.44–4.5 (m, 3H), 7.31–7.42 (m, 4H), 7.60 (d, J = 7.2 Hz, 2H), 7.77 (d, J = 7.2 Hz, 2H). 13C NMR (CDCl3, 75.0 MHz) δ 22.2, 23.0, 24.8, 30.8, 45.0, 47.4, 49.1, 66.3, 119.95, 125.0, 127.0, 127.7, 141.4, 143.9, 156.3, 178.2. Anal (C23H27NO4) C, H, N: Calc’d: C, 72.42; H, 7.13; N, 3.67. Found: C, 72.14; H, 7.25; N, 3.80. HRMS (M+H) Calc’d: 382.2018. Found: 382.1545.
(R) 4-[[(9H-Fluoren-9-ylmethoxy)carbonyl]amino]-2-octanoic acid (65e)
Yield: 0.67 g (87%). White powder, 1H NMR (DMSO-d6, 500 MHz) δ 0.85 (s, 3H), 1.2–1.4 (m, 6H), 1.53 (m, 1H), 1.66 (m, 1H), 2.18 (m, 2H), 3.4 (m, 1H), 4.22 (m, 1H), 4.3 (m, 2H), 7.1 (d, J = 8.0 Hz, 1H), 7.3–7.43 (m, 4H), 7.7 (d, J = 7.0 Hz, 2H), 7.9 (d, J = 7.0 Hz, 2H). 13C NMR (DMSO-d6, 125 MHz) δ 14.0, 22.5, 27.9, 30.5, 30.8, 35.3, 47.4, 51.0, 66.4, 120.0, 125.0, 127.1, 127.7, 141.4, 143.9, 156.4, 178.2. HRMS (M+H) Calc’d: 382.2018; Found: 382.1975. Anal (C23H27NO4) C, H, N: Calc’d: C, 72.42; H, 7.13; N, 3.67. Found: C, 72.65; H, 7.25; N, 4.19.
(R)-5-(4-tert-butoxyphenyl)-4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-2-pentanoic acid (65f)
Yield: 0.86 g (89%). White powder, m.p.127 °C. 1H NMR (CDCl3, 500 MHz) δ 1.22 (s, 9H), 1.53 (m, 1H), 1.76 (m, 1H), 2.3 (m, 2H), 2.6 (m, 2H), 3.8 (m, 1H), 4.3 (m, 2H), 4.6 (d, J = 9.0 Hz, 1H), 6.8 (d, J = 8.0 Hz, 2H), 6.9 (d, J = 8.0 Hz, 2H), 7.2–7.3 (m, 4H), 7.5 (d, J = 7.0 Hz, 2H), 7.7 (d, J = 7.0 Hz, 2H). 13C NMR (CDCl3, 75 MHz) δ 29.1, 30.8, 40.8, 47.3, 51.9, 66.5, 78.4, 120.0, 124.1, 125.0, 127.1, 127.7, 129.8, 132.1, 141.3, 143.8, 154.0, 156.1, 178.3. Anal (C30H33NO5) C, H, N: Calc’d: C, 73.90; H, 6.82; N, 2.87. Found: C, 72.71; H, 6.69; N, 2.95. HRMS (M+H) Calc’d: 488.2437; Found: 488.2423
(S)-5-benzyloxy-4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-2-pentanoic acid (65g)
Yield: 0.69 g (77%). White powder. 1H NMR (DMSO-d6, 500 MHz) δ 1.58 (m 1H), 1.80 (m, 1H), 2.24 (m, 2H), 3.38 (m, 2H), 3.67 (m, 1H), 4.23 (m, 1H), 4.3–4.37 (m, 2H), 4.48 (s, 2H), 7.23–7.35 (m, 8H), 7.42 (m, 2H), 7.71 (d, J = 7.0 Hz, 2H), 7.89 (d, J = 7.5 Hz, 2H), 12.1 (s, 1H). 13C NMR (DMSO-d6, 125 MHz) δ 26.9, 30.7, 47.2, 50.3, 65.7, 72.3, 72.4, 120.5, 120.6, 125.7, 127.4, 127.6, 127.8, 128.0, 128.2, 128.5, 128.8, 138.9, 141.2, 144.4, 156.4, 174.7. Anal (C27H27NO5) C, H, N: Calc’d: C, 72.79; H, 6.11; N, 3.14. Found: C, 72.76; H, 6.23; N, 3.09. HRMS (M+H) Calc’d: 446.1967; Found: 446.1984.
(4S, 5S)-5-Benzyloxy-4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-2-hexanoic acid (65h)
Yield: 0.75 g (82%). White powder. 1H NMR (DMSO-d6, 500 MHz) δ 1.07 (d, J = 6.9 Hz, 3H), 1.63 (m, 1H), 1.8 (m, 1H), 2.2–2.3 (m, 2H), 3.53 (m, 1H), 3.62 (m, 1H), 4.22 (m, 1H), 4.26–4.31 (m, 3H), 4.5 (m, 2H), 7.2 (d, J = 5.4 Hz, 1H), 7.27–7.43 (m, 9 H), 7.72 (d, J = 7.5 Hz, 2H), 7.9 (d, J = 7.5 Hz, 2H), 12.04 (s, 1H). 13C NMR (DMSO-d6, 125 MHz) δ 15.6, 24.9, 31.0, 47.3, 53.9, 65.7, 70.4, 76.3, 120.6, 125.7, 127.5, 127.7, 127.8, 128.1, 128.6, 129.2, 139.4, 141.2, 144.4, 156.7, 174.8. Anal (C28H29NO5) C, H, N: Calc’d: C, 73.18; H, 6.36; N, 3.05. Found: C, 72.52; H, 6.38; N, 3.01. HRMS (M+H) Calc’d: 460.2124; Found: 460.2141.
(4S, 5S)-5-tert-butoxy-4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-2-hexanoic acid (65i)
Yield: 0.71 g (83%) as an oil. 1H NMR (CDCl3, 500 MHz) δ 1.03 (d, J = 6.0 Hz, 3H), 1.12 (s, 9H), 1.65–1.76 (m, 2H), 2.31 (t, J = 7.0 Hz, 2H), 3.48 (m, 1H), 3.6 (m, 1H), 4.14 (m, 1H), 4.34–4.41 (m, 2H), 4.5 (m, 1H), 5.0 (d, J = 9.5 Hz, 1H), 7.25 (m, 2H), 7.32 (m, 2H), 7.53 (d, J = 7.0 Hz, 2H), 7.68 (d, J = 7.5 Hz, 2H), 10.1 (s, 1H). 13C NMR (CDCl3, 125 MHz) δ 20.0, 27.7, 28.7, 31.0, 47.4, 56.0, 66.7, 68.4, 73.9, 120.0, 125.1, 127.1, 127.7, 141.4, 143.8, 157.2, 178.9. HRMS (M+Na) Calcd: 448.2100. Found: 448.2134.
N-[(9H-fluoren-9-ylmethoxy)carbonyl] 3-pyrrolidin-2-yl-propionic acid (70)
Yield: 0.59 g (80%) as an oil. 1H NMR (CDCl3, 500 MHz) δ 1.53–2.07 (m, 6H), 2.36 (m, 1H), 3.3–3.45 (m, 2.5H), 3.95 (m, 0.5H), 4.0 (m, 1H), 4.35–4.46 (m, 1H), 4.58 (m, 1H), 7.28–7.38 (m, 4H), 7.77 (m, 2H), 7.74 (m, 2H), 9.4 (m, 2H). 13C NMR (CDCl3, 125 MHz) δ 22.8, 23.6, 28.9, 29.4, 30.2, 31.2, 46.4, 47.3, 57.1, 66.8, 67.4, 119.95, 125.0, 127.1, 127.7, 141.4, 143.9, 155.8, 178.5. HRMS (M+H) Calcd: 366.1705. Found: 366.1651.
Synthesis of tert-butyl 4-(benzyloxycarbonylamino)-5-hydroxy-pentanoate (72)
To a stirred solution of Z-Glu(OtBu)-OH (5.0 g, 14.8 mmol) and DCC (3.46 g, 16.8 mmol) in 150 mL of EtOAc, was added 2-mercaptopyridine (1.81 g, 16.33 mmol). Stirring was continued for 4h at which time the precipitate was filtered off and the solvent was removed under vacuum. The residue was dissolved in 100 mL of 1,4-dioxane, cooled to 0 °C and treated with NaBH4 (2.0 g). When the reaction was finished, as monitored by TLC, approximately after 30 min, it was then quenched slowly by adding 2% KHSO4 (aq). The mixture was extracted with Et2O (3 × 150 mL). The combined organic layers were washed with 5% NaHCO3 (2 × 50 mL), brine, dried (MgSO4), and concentrated under vacuum. The crude product was purified by silica gel column chromatography eluting with 35% EtOAc/hexane (v/v) to give 4.2 gm of 72 as colorless oil. Yield 88%. 1H NMR (CDCl3, 500 MHz) δ 1.45 (s, 9H), 1.77 (m, 1H), 1.87 (m, 1H), 2.34 (m, 2H), 2.76 (brs, 1H), 3.6 (m, 1H), 3.65–3.7 (m, 2H), 5.1 (s, 2H), 5.25 (d, J = 6.5Hz, 1H), 7.36 (s, 5H). 13C NMR (CDCl3, 125 MHz) δ 28.0, 28.1, 66.8, 80.9, 128.0, 128.2, 128.4, 128.6, 136.4, 156.6, 173.3. HRMS (M+H) Calc’d: 324.1811; Found 324.0416
Synthesis of tert-butyl 4-(benzyloxycarbonylamino)-5-iodo-pentanoate (73)
A solution of PPh3 (4.8 g, 18.6 mmol), I2 (2.4 g, 18.6 mmol), and imidazole (2.1 g, 31.0 mmol) in dry CH2Cl2 (30 mL) was stirred for 30 min under inert atmosphere at room temperature. To this solution, a solution of 62 (2.0 g. 6.2 mmol) in 15 mL of dry CH2Cl2 was added and stirring continued for 3 h. The solvent was removed and the crude product was purified by silica gel chromatography eluting 15 % EtOAc-hexane (v/v). A white solid was obtained (2.45 g, 91%). 1H NMR (CDCl3, 500 MHz) δ 1.45 (s, 9H), 1.85 (m, 2H), 2.3 (m, 2H), 3.33 (m 1H), 3.42 (m, 1H), 3.5 (m, 1H), 5.1 (brs, 3H), 7.37 (s, 5H). 13C NMR (CDCl3, 125 MHz) δ 28.1, 30.0, 31.8, 66.9, 80.8, 127.9, 128.1, 128.2, 128.3, 128.5, 128.7, 136.3, 155.7, 172.3. HRMS (M+H) Calc’d: 434.0828; Found 434.0541.
Synthesis of tert-butyl 4-(benzyloxycarbonylamino)-5-(1-piperidino)-pentanoate (74a)
A solution of 73 (1.0 gm, 2.3 mmol) in 10 mL of dry THF was treated with piperidine (0.7 mL, 6.92 mmol) overnight under argon. The reaction mixture was diluted with 100 mL of EtOAc and was washed with water and brine. After drying (MgSO4) and concentration under vacuum the crude product was purified by silica gel chromatography eluting with 50% EtOAc/hexane (v/v) to give desired N-alkylated product. Yield: 0.74 g, 82%. 1H NMR (CDCl3, 500 MHz) δ 1.45 (s, 9H), 1.53–1.58 (m, 4H), 1.7 (m, 1H), 1.93 (m, 1H), 2.28–2.40 (m, 8H), 3.73 (m, 1H), 5.03 (brs, 1H), 5.12 (s, 2H), 7.37 (s, 5H). 13C NMR (CDCl3, 125.0 MHz) δ 24.3, 26.0, 28.0, 28.1, 28.3, 32.0, 54.9, 66.5, 80.3, 127.8, 128.1, 128.4, 128.6, 129.1, 136.8, 156.4, 172.9. Anal (C22H34N2O4) C, H, N: Calc’d: C, 67.66; H, 8.78; N, 7.17. Found: C, 67.89; H, 8.91; N, 7.21. HRMS (M+H) Calc’d: 391.2597; Found: 391.0992.
tert-butyl 4-(benzyloxycarbonylamino)-5-(4-morpholiino)-pentanoate (74b)
The same procedure as for 74a was used. Yield 0.76 g (84 %). 1H NMR (CDCl3, 500 MHz) δ 1.45 (s, 9H), 1.67 (m, 1H), 1.94 (m, 1H), 2.3–2.48 (m, 8H), 3.65 (s, 4H), 3.77 (m, 1H), 4.9 (brs, 1H), 5.11 (s, 2H), 7.36 (s, 5H). 13C NMR (CDCl3, 125 MHz) δ 28.0, 28.1, 32.0, 48.4, 53.9, 62.8, 66.9, 67.3, 80.4, 127.9, 128.1, 128.4, 128.6, 136.6, 156.4, 172.8. Anal (C21H32N2O5) C, H, N: Calc’d: C, 64.26; H, 8.22; N, 7.14. Found: C, 63.85; H, 8.26; N, 7.07. HRMS (M+H) Calc’d: 393.2389; Found: 393.1541.
tert-butyl 4-(benzyloxycarbonylamino)-5-(4-methyl-1-piperazino)-pentanoate (74c)
The same procedure as for 74a was used. Yield 0.73 g (78%). 1H NMR (CDCl3, 500 MHz) δ 1.44 (s, 9H), 1.67 (m, 1H), 1.92 (m, 1H), 2.27 (s, 3H), 2.3–2.57 (m, 12H), 3.74 (m, 1H), 4.94 (m, 1H), 5.13 (s, 2H), 7.36 (s, 5H). 13C NMR (CDCl3, 125 MHz) δ 28.0, 28.1, 32.0, 45.4, 45.9, 46.4, 48.6, 53.2, 55.0, 62.1, 66.5, 80.4, 127.9, 128.1, 128.4, 128.5, 128.6, 136.7, 156.4, 172.8.` Anal (C22H35N3O4) C, H, N: Calc’d: C, 65.16; H, 8.70; N, 10.36. Found: C, 64.50; H, 8.71; N, 10.05. HRMS (M+H) Calc’d: 406.2706; Found: 406.1604.
Synthesis of tert-butyl 4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-5-(1-piperidino)-pentanoate (75a)
To a suspension of 74a (1.0 g, 2.56 mmol) and 10 % Pd-C (0.2 g) in 2% CHCl3/MeOH(20 mL), TES (4.2 mL, 26.0 mmol) was added dropwise under argon atmosphere. After completion (TLC monitoring), the catalyst was filtered off through celite and the filter cake washed with 10 mL of methanol. The combined filtrates were concentrated under vacuum and residual methanol was removed by the addition and evaporation of toluene (2 × 10 mL). The residue was dissolved in 10 mL of 10% Na2CO3 and 20 mL of 1,4-dioxane. Fmoc-OSu (1.3 g, 3.84 mmol) was added and the mixture was stirred overnight. The mixture was extracted with EtOAc (2 × 40 mL) and the combined organic layers were washed with water followed by brine. The organic part was dried (MgSO4) and concentrated under vacuum. The crude product was purified by silica gel column chromatography eluting with 10% methanol-chloroform (v/v) to give desired product. Yield 0.94 g, 76 %. 1H NMR (CDCl3, 500 MHz) δ 1.3–1.4 (m, 10H), 1.65 (m, 1H), 1.7–1.87 (m, 6H), 2.00 (m, 1H), 2.26 (m, 2H), 2.52 (m, 1H), 2.62 (m, 1H), 2.81 (m, 1H), 3.35 (m, 1H), 3.4 (m, 1H), 3.65 (m, 1H), 4.1–4.2 (m, 4H), 4.3 (m, 1H), 6.9 (d, J = 9.5 Hz, 1H), 7.23 (m, 2H), 7.3 (m, 2H), 7.54 (m, 2H), 7.66 (d, J = 7.5 Hz, 2H). 13CNMR (125 MHz, CDCl3) δ 22.0, 22.4, 28.0, 28.7, 31.1, 45.5, 47.0, 51.6, 55.6, 59.2, 67.4, 81.0, 119.8, 125.3, 125.5, 127.1, 127.6, 127.7, 141.3, 143.7, 144.1, 156.8, 172.1. HRMS (M+H) Calc’d: 479.2910; Found: 479.1051.
tert-Butyl 4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-5-(1-morpholino)pentanoate (75b)
The same procedure as for 75a was employed Yield: 0.91 g (74%). 1H NMR (CDCl3, 500 MHz) δ 1.35 (s, 9H), 1.66 (m, 1H), 1.77 (m, 1H), 2.25 (m, 2H), 2.78–2.86 (m, 3H), 3.41 (m, 2H), 3.67 (m, 1H), 3.91 (m, 4H), 4.12 (m, 2H), 4.21–4.3 (m, 2H), 6.37 (d, J = 9.5 Hz, 1H), 7.24 (m, 2H), 7.31 (m, 2H), 7.52 (m, 2H), 7.68 (d, J = 7.5 Hz, 2H). 13C NMR (CDCl3, 125 MHz) δ 28.0, 28.1, 30.9, 45.2, 46.9, 51.1, 53.9, 60.3, 63.6, 67.5, 81.2, 114.8, 117.1, 119.9, 125.2, 125.3, 127.1, 127.7, 127.8, 141.2, 141.3, 143.5, 144.1, 156.7, 162.1, 162.3, 172.0. HRMS (M+H) Calc’d: 481.2702; Found: 481.1066.
tert-Butyl [[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-5-(4-methy1-1-piperazino)-pentanoate (75c)
The same procedure as for 75a was employed. Yield: 0.78 g (64%). 1H NMR (500 MHz, CDCl3) δ 1.35 (s, 9H), 1.6 (m, 1H), 1.75 (m, 1H), 2.22 (m, 2H), 2.74 (s, 3H), 2.88 (m, 1H), 3.13 (m, 1H), 3.33–3.5 (m, 8H), 3.97 (m, 1H), 4.11 (t, J = 6.5 Hz, 1H), 4.28 (d, J = 7.0 Hz, 2H), 5.7 (d, J = 9.0 Hz, 1H), 7.23 (m, 2H), 7.32 (m, 2H), 7.5 (m, 2H), 7.68 (m, 2H). 13C NMR (125 MHz, CDCl3) δ 27.8, 28.0, 28.3, 31.2, 43.3, 46.5, 46.9, 50.6, 60.6, 67.3, 81.2, 112.7, 115.0, 117.3, 119.9, 125.11, 127.2, 127.7,127.9, 141.3, 143.4, 144.1, 156.8, 162.1, 162.4, 162.7, 171.9. HRMS (M+H) Calc’d: 494.3019; Found: 494.2492.
Synthesis of 4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-5-(1-piperidino)-pentanoic acid (76a)
Compound 75a (0.7 g) was treated with 95% TFA/CH2Cl2 for 1h and the solvent was removed under vacuum. Traces of TFA were removed by addition and evaporation of toluene (2 × 10 mL). The product then triturated with ether -hexane to give a white solid (0.56 g, 90%). 1H NMR (DMSO-d6, 500 MHz) δ 1.36 (m, 1H), 1.56–1.76 (m, 7H), 2.21 (m, 2H), 2.88 (m, 2H), 3.05 (m, 1H), 3.16 (m, 1H), 3.37 (m, 1H), 3.5 (m,1H), 3.88 (m, 1H), 4.25 (m,1H), 4.32 (m, 1H), 4.52 (m, 1H), 7.34 (m, 2H), 7.42 (m, 3H), 7.7 (d, J = 7.5 Hz, 2H), 7.9 (d, J = 7.5 Hz, 2H), 9.24 (s, 1H). 13C NMR (DMSO-d6, 125 MHz) δ 21.6, 22.5, 28.3, 30.2, 46.2, 47.3, 52.4, 53.6, 59.6, 65.8, 120.6, 125.5, 125.6, 127.5, 128.1, 141.3, 144.2, 144.3, 156.5, 158.5, 158.7, 174.3. HRMS (M+H) Calc’d: 423.2284. Found 423.2716.
4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-5-(1-morpholino)-pentanoic acid (76b)
The same procedure as for 76a was employed. Yield: 0.56 g, (90%). 1H NMR (DMSO-d6, 500 MHz) δ 1.61 (m, 1H), 1.74 (m, 1H), 2.21 (m, 2H), 3.1–3.48 (m, 6H), 3.73 (m, 2H), 3.91 (m, 3H), 4.24–4.32 (m, 2H), 4.5 (m, 1H), 7.34 (m, 2H), 7.43 (m, 3H), 7.7 (d, J = 7.5 Hz, 2H), 7.9 (d, J = 7.5 Hz, 2H), 9.9 (brs, 1H). 13C NMR (DMSO-d6, 125 MHz) δ 28.1, 30.2, 45.8, 47.2, 60.0, 63.5, 65.9, 120.6, 125.5, 125.6, 127.5, 128.1, 141.3, 144.2, 144.3, 156.5, 158.6, 158.8, 174.3. HRMS (M+H) Calc’d: 425.2076. Found 425.2117.
4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-5-(4-methy1-1-piperazino)-pentanoic acid (76c)
The same procedure as for 76a was employed. Yield: 0.45 g (72%). 1H NMR (DMSO-d6, 500 MHz) δ 1.55 (m, 1H), 1.78 (m, 1H), 2.22 (m, 2H), 2.66 (m, 2H), 2.8 (s, 3H), 2.9–3.5 (m, 8H), 3.71 (m, 1H), 4.24–4.3 (m, 2H), 4.41 (m, 1H), 7.25 (d, J = 8.0 Hz, 1H), 7.34 (m, 2H), 7.42 (m, 2H), 7.7 (m, 2H), 7.9 (d, J = 7.5 Hz, 2H). 13C NMR (DMSO-d6, 125 MHz) δ 28.1, 30.5, 42.5, 47.3, 47.4, 49.6, 65.7, 115.6, 117.9, 120.6, 125.6, 127.5, 128.1, 141.3, 144.3, 144.4, 156.5, 158.6, 158.9, 159.2, 159.4, 174.6. HRMS (M+H) Calc’d: 438.2393. Found 438.2433.
Synthesis of tert-butyl 4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-5-hydroxy-pentanoate (78)
Fmoc-Glu(OtBu)-OH (77) (4.0 g, 9.4 mmol) was treated as described for the preparation of 72 to give 78. Yield: 3.2 g (83%) NMR same as in ref44. 1H NMR (500 MHz, CDCl3) δ 1.37 (s, 9H), 1.71 (m, 1H), 1.8 (m, 1H), 2.25 (m, 2H), 3.5 (m, 1H), 3.6 (m, 2H), 4.13 (m, 1H), 4.33 (m, 2H), 5.12 (brs, 1H), 7.24 (m, 2H), 7.33 (m, 2H), 7.52), 7.52 (d, J = 7.5 Hz, 2H), 7.68 (d, J = 7.5 Hz, 2H). 13C NMR (CDCl3, 125 MHz) δ 25.7, 28.1, 31.9, 47.3, 53.0, 64.7, 66.7, 81.0, 120.0, 125.1, 127.1, 127.7, 141.3, 143.9, 155.9, 173.3. HRMS (M+H) Calc’d: 412.2124; Found: 412.2132.
Synthesis of tert-butyl 4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-5-iodo-pentanoate (79)
Followed the same procedure as described for 73 starting with 2.0 g (4.86 mmol) of 78. Yield: 2.3 g (88%). 1H NMR (500 MHz, CDCl3) δ 1.38 (s, 9H), 1.78 (m, 2H), 2.2–2.3 (m, 2H), 3.24 (m, 1H), 3.35 (m, 1H), 3.4 (m, 1H), 4.15 (m, 1H), 4.3 (m, 1H), 4.4 (m, 1H), 4.95 (d, J = 7.5 Hz, 1H), 7.25 (m, 2H), 7.33 (m, 2H), 7.53 (d, J = 7.0 Hz, 2H), 7.7 (d, J = 7.5 Hz, 2H). 13C NMR (CDCl3, 125 MHz) δ 28.1, 29.9, 31.8, 47.3, 50.5, 66.8, 80.9, 120.0, 125.0, 125.1, 127.1, 127.7, 141.3, 143.8, 155.7, 172.4. HRMS (M+H) Calc’d: 522.1141; Found: 522.1157.
Synthesis of tert-butyl 4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-5-azido-pentanoate (80)40
To a solution of 79 (2.0 g, 3.84 mmol) in 30 mL of dry CH2Cl2, nBu4NN3 (3.3 g, 11.5 mmol) was added in portions. After completion of the reaction (ca 8 h, monitored by TLC), the solvent was removed in vacuo and the product was purified by silica gel chromatography (10% EtOAc-hexane v/v). Yield: 1.45 g (86%). 1H NMR (500 MHz, CDCl3) δ 1.37 (s, 9H), 1.73 (m, 2H), 2.2–2.3 (m, 2H), 3.35 (m, 2H), 3.71 (m, 1H), 4.13 (m, 1H), 4.3–4.4 (m, 2H), 4.93 (d, J = 8.0 Hz, 1H), 7.24 (m, 2H), 7.33 (m, 2H), 7.51 (d, J = 7.5 Hz, 2H), 7.68 (d, J = 7.5 Hz, 2H). 13C NMR (CDCl3, 125.0 MHz) δ 28.1, 32.0, 46.8, 51.2, 54.8, 66.8, 80.9, 120.1, 125.1, 127.1, 127.7, 127.8, 141.3, 143.8, 155.9, 172.5. HRMS (M+H) Calc’d: 437.2189; Found: 437.2208.
Synthesis of tert-butyl 4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-5-(allyloxycarbonyl)amino-pentanoate (81)
To a stirred suspension of 80 (1.0 g, 2.3 mmol) and 10% Pd-C (0.1 g) in 15 mL of 1% CHCl3 in MeOH, TES (4.0 mL) was added dropwise under argon atmosphere. After completion ~ 20 min, the mixture was filtered through celite and the filtrate then concentrated under vacuum. The residue was dissolved in 15 mL of CH2Cl2 and was treated with allylchloroformate (0.5 mL, 4.6 mmol) and DIEA (1.2 mL, 6.9mmol) for 3 hr at 0 °C. The solution was transferred to a separatory funnel with an additional 50 mL of CH2Cl2 and was washed with 5% HCl (2 × 15 mL), 5% NaHCO3 (20 mL), brine and dried (MgSO4). The solvent was removed and the crude product was purified by silica gel chromatography (25% EtOAc-hexane, v/v) yielding 81 as a white powder. Yield: 0.73 g (64%). 1H NMR (CDCl3, 500 MHz) δ 1.46 (s, 9H), 1.75 (m, 1H), 1.84 (m, 1H), 2.34 (m, 2H), 3.28–3.36 (m, 2H), 3.73 (m, 1H), 4.22 (m, 1H), 4.35–4.45 (m, 2H), 4.55 (m, 2H), 5.17–5.3 (m, 4H), 5.90 (m, 1H), 7.33 (m, 2H), 7.42 (m, 2H), 7.61 (d, J = 7.0 Hz, 2H), 7.78 (d, J = 7.0 Hz, 2H). 13C NMR (CDCl3, 125 MHz) δ 27.3, 28.1, 32.0, 45.1, 47.3, 52.0, 65.7, 66.7, 80.8, 117.7, 120.0, 125.1, 127.1, 127.7, 132.8, 141.3, 143.9, 156.6, 156.9, 172.7. HRMS (M+H) Calc’d: 495.2495 Found 495.2478. Anal (C28H35N2O6) C, H, N: Calc’d: C, 68.00; H, 6.93; N, 5.66. Found C, 67.93; H, 6.88; N, 5.62.
Synthesis of 4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-5-(allyloxycarbonyl)amino-pentanoic acid (82)
Compound 81 (0.6 g, 1.2 mmol) was treated with neat TFA for 1 h. The TFA was removed under vacuum and the residue was triturated with ether-hexane to form a white solid. The product was collected by filtration and dried in vacuo over P2O5. Yield: 0.44 g (82%). 1H NMR (DMSO-d6, 500 MHz) δ 1.5 (m, 1H), 1.7 (m, 1H), 2.2 (m, 2H), 3.03 (m, 2H), 3.5 (m, 1H), 4.19–4.3 (m, 3H), 4.45 (m, 2H), 5.14 (dd, J = 2.0, 17.0 Hz, 1H), 5.26 (dd, J = 2.0, 28.5 Hz, 1H), 5.87 (m, 1H), 7.15 (d, J = 14.5, 1H), 7.23 (t, J = 20.0 Hz, 1H), 7.32 (m, 2H), 7.42 (m, 2H), 7.68 (d, J = 12.0 Hz, 2H), 7.88 (d, J = 12.0 Hz, 2H). 12.04 (brs, 1H). 13C NMR (DMSO-d6, 125 MHz) δ 27.3, 30.7, 44.6, 47.3, 50.8, 64.7, 65.7, 117.3, 120.6, 125.7, 127.5, 128.1, 134.2, 141.2, 144.3, 144.4, 156.4, 156.6, 174.6. HRMS (M+H) Calc’d: 439.1869; Found 439.1836. Anal (C24H27N2O6) C, H, N: Calc’d: C, 65.74; H, 5.98; N, 6.39. Found C, 65.56; H, 5.93; N, 6.28.
Synthesis of tert-butyl 4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-5-acetoxy-pentanoate (92)
Acetic anhydride (1.4 mL, 14.6 mmol) was added to a solution of 78 (1.0 g, 2.43 mmol), DIEA (0.85 mL, 4.86 mmol) and DMAP (0.03 g, 0.24 mmol) in dry CH2Cl2 (15 mL). After completion of the reaction (TLC), 10 mL of water was added and the mixture extracted with CH2Cl2 (3 × 30 mL). The combined organic layers were washed with 5 % HCl (2 × 20 mL) and saturated NaHCO3 (2 × 20 mL). After drying (MgSO4) and concentration under vacuum, the crude product was purified by silica gel column chromatography eluting with 25 % EtOAc-hexane (v/v) to give 1.0 g (93%) of 92. 1H NMR (CDCl3, 500 MHz) δ 1.37 (s, 9H), 1.64–1.77 (m, 2H), 1.98 (s. 3H), 2.23 (m, 2H), 3.84 (m, 1H), 4.00 (m, 2H), 4.13 (m, 1H), 4.34 (m, 2H), 4.88 (d, J = 9.0 Hz, 1H), 7.24 (m, 2H), 7.32 (m, 2H), 7.5 (d, J = 7.5 Hz, 2H), 7.68 (d, J = 7.5 Hz, 2H). 13C NMR (CDCl3, 125.0 MHz) δ 28.1, 31.9, 46.8, 47.2, 50.4, 66.0, 66.6, 80.8, 119.9, 120.1, 125.0, 127.0, 127.2, 127.6, 127.8, 141.3, 143.9, 143.8, 156.0, 170.9, 172.5. HRMS (M+H) Calc’d: 454.2230; Found 454.2240.
Synthesis of 4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-5-acetoxy-pentanoic acid (93)
0.5 g of 92 was treated with 2 mL of neat TFA for 1 h. The TFA was removed in vacuo and residual acid was removed by addition and evaporation of toluene (2 × 5 mL). Trituration with ether- hexane resulted in a white solid which was collected by filtration and dried in vacuo over P2O5. Yield: 0.4 g (90%). 1H NMR (CDCl3, 500 MHz) δ 1.77 (m, 1H), 1.89 (m, 1H), 2.1 (s, 3H), 2.42 (m, 2H), 3.96 (m, 1H), 4.06 (m, 1H), 4.14 (m, 1H), 4.22 (m, 1H), 4.46 (m, 2H), 4.95 (d, J = 8.5 Hz, 1H), 7.33 (m, 2H), 7.42 (m, 2H), 7.6 (m, 2H), 7.78 (d, J = 7.5 Hz, 2H). 13C NMR (CDCl3, 125 MHz) δ 26.6, 30.4, 31.6, 47.2, 50.1, 66.0, 66.7, 119.8, 120.0, 120.1, 125.0, 127.0, 127.2, 127.6, 127.9, 141.3, 143.8, 156.2, 171.0, 177.9. HRMS (M+Na) Calc’d: 420.1423 Found 420.1442.
General procedure for 4-Nitrophenyl-Fmoc-amino carbonates: Synthesis of 4-Nitrophenyl 2-[(9H-fluoren-9-ylmethoxy)carbonyl]aminoethyl carbonate (83j)
4-Nitrophenyl chloroformate (1.6 g, 7.8 mmol) in 10 mL of dry CH2Cl2 was added drop wise to a stirred solution of Fmoc-aminoethanol (2.0 g, 7.06 mmol) and pyridine (0.9 mL, 10.6 mmol) in 20 mL of dry CH2Cl2 at 0 °C under an atmosphere of argon. Upon completion (TLC), the solution was transferred to a separatory funnel with an additional 20 mL of CH2Cl2. The organic solution was washed with 10% HCl (3 × 30 mL), 10% Na2CO3 (3 × 30 mL), and brine (50 mL) and was dried (MgSO4). The crude mixture was purified by silica gel chromatography eluting with 40% EtOAc-hexane to give 2.6 g (82%) of desired product as a white solid. 1H NMR (300 MHz, CDCl3) δ 3.6 (m, 2H), 4.25–4.48 (m, 5H), 5.2 (m, 1H), 7.28–7.45 (m, 6H), 7.6 (d, J = 7.2 Hz, 2H), 7.8 (d, J = 7.5 Hz, 2H), 8.28 (d, J = 8.4 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 39.9, 47.2, 66.9, 68.2, 120.0, 121.6, 121.7, 124.9, 125.3, 125.5, 127.0, 127.7, 141.4, 143.8, 145.5, 152.4, 155.4. Anal.Calcd for C24H20N2O7: C, 64.28; H, 4.50; N, 6.25; O, 24.98; Found C, 63.91; H, 4.42; N, 6.36. HRMS (M+H) Calc’d: 454.2230; Found 454.2240.
4-Nitrophenyl 2-[(9H-fluoren-9-ylmethoxy)carbonyl]amino-3-benzyloxybutyl carbonate (83g)
The procedure for 83j was used. Compound 62g (1.0 g, 2.4 mmol) yielded 1.1 g of 83g (77%). 1H NMR (CDCl3, 500 MHz) δ 1.16 (d, J = 6.0 Hz, 3H), 3.68 (m, 1H), 3.97 (m, 1H), 4.11 (m, 1H), 4.20 (d, J = 6.5 Hz, 2H), 4.27–4.33 (m, 3H), 4.55 (m, 1H), 5.16 (d, J = 9.5 Hz, 1H), 7.17–7.27 (m, 11H), 7.47 (d, J = 7.0 Hz, 2H), 7.64 (d, J = 7.0Hz, 2H), 8.08 (d, J = 8.5 Hz, 2Hz). 13C NMR (CDCl3, 125 MHz) δ 16.0, 47.3, 54.1, 67.1, 68.6, 70.8, 72.0, 120.1, 121.8, 125.1, 125.3, 127.2, 127.8, 128.0, 128.1, 128.6, 137.8, 141.4, 143.8, 145.4, 152.4, 155.5, 156.8. HRMS (M+H) Calc’d: 449.1349; Found 449.1357.
4-Nitrophenyl 2-[(9H-fluoren-9-ylmethoxy)carbonyl]amino-3-benzyloxy propyl carbonate (83f)
The procedure for 83j was used. Compound 62f (1.0 g 2.47 mmol) yielded 1.2 g of 83g (81%). 1H NMR (CDCl3, 300 MHz) δ 3.64 (m, 2H), 4.25 (m, 2H), 4.46 (m, 4H), 4.58 (s, 2H), 5.27 (d, J = 8.4 Hz, 1H), 7.31–7.45 (m, 13H), 7.61 (d, J = 7.5 Hz, 2H), 7.79 (d, J = 7.5 Hz, 2H), 8.24 (m, 2H). 13C NMR (CDCl3, 75 MHz) δ 47.2, 49.5, 67.0, 68.1, 68.5, 73.5, 120.1, 121.7, 125.0, 125.3, 127.1, 127.8, 128.1, 128.6, 137.5, 141.4, 143.8, 145.4, 152.3, 155.4, 156.2. Anal (C32H28N2O8) C, H, N: Calc’d: C, 67.60; H, 4.96; N, 4.93. Found C, 67.62; H, 4.86; N, 4.96. HRMS (M+H) Calc’d: 569.1924; Found 569.1937.
4-Nitrophenyl 2-[(9H-fluoren-9-ylmethoxy)carbonyl]amino-propyl carbonate (83a)
The procedure for 83j was used. Compound 62a (1.0 g 3.36 mmol) yielded 1.3 g of 83a (84%). 1H NMR (CDCl3, 500 MHz) δ 1.20 (s, 3H), 4.09–4.22 (m 4H), 4.36 (m, 2H), 4.78 (brs, 1H), 7.22–7.34 (m, 6H), 7.51 (d, J = 7.5 Hz, 2H), 7.69 (d, J = 7.5 Hz, 2H), 8.17 (d, J = 9.0 Hz, 2H). 13C NMR (CDCl3, 125.0 MHz) δ 17.3, 45.8, 47.2, 66.8, 71.6, 120.1, 121.7, 124.9, 125.3, 127.1, 127.8, 141.4, 143.8, 145.5, 152.5, 155.4, 155.7. Anal (C25H22N2O7) C, H, N: Calc’d: C, 64.93; H, 4.80; N, 6.06. Found C, 64.78; H, 4.69; N, 6.04. HRMS (M+H) Calc’d: 463.1505; Found 463.1507.
Synthesis of N-triphenylmethyl 4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-5-hydroxypentanamide (85)
Starting with 2.0 g (3.27 mmol) of Fmoc-Gln(Trt)-OH (84), the same procedure as described above for the preparation of amino alcohols 62 a-i was followed to give 85. Yield : 1.2 g (63%) 1H NMR (CDCl3, 500 MHz) δ 1.71 (m, 1H), 1.83 (m, 1H), 2.2–2.34 (m, 2H), 3.36–3.44 (m, 2H), 3.58 (m, 1H), 4.22 (t, J = 6.5 Hz, 1H), 4.43 (d, J = 6.5 Hz, 2H), 5.46 (d, J = 8.5 Hz, 1H), 7.1 (s, 1H), 7.24–7.32 (m, 17H), 7.4–7.42 (m, 2H), 7.61 (d, J = 7.5 Hz, 2H), 7.77 (d, J = 7.5 Hz, 2H). HRMS (M+H) Calc’d: 597.2753; Found: 597.2763.
Synthesis of 4-nitrophenyl [N-triphenylmethyl 4-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-pentanamide-5-yl] carbonate (86)
To a stirred solution of 85 (1.0 g, 1.67 mmol) and pyridine (0.27 mL, 3.34 mmol) in 20 mL of dry CH2Cl2 at 0 °C, a solution nitrophenyl chloroformate (0.37 g, 1.84 mmol) in 10 mL of dry CH2Cl2 was added dropwise. The reaction was slowly warmed up to room temperature and stirred for 4h. It was then diluted with an additional 100 mL of CH2Cl2 and transferred to a separatory funnel. The organic layer was washed with water (20 mL), brine (20 mL) and dried (MgSO4). The solvent was removed and the concentrated crude product was purified by silica gel chromatography (20% EtOAc-hexane, v/v). Yield: 0.79 g (62%). 1H NMR (CDCl3, 500 MHz) δ 1.74 (m, 1H), 1.8 (m, 1H), 2.26 (m, 2H), 3.88 (m, 1H), 4.1–4.15 (m, 3H), 4.33 (m, 1H), 4.40 (m, 1H), 5.08 (d, J = 8.5 Hz, 1H), 6.7 (s, 1H), 7.1–7.22 (m, 19H), 7.25–7.31 (m, 2H), 7.47 (d, J = 7.5 Hz, 2H), 7.64 (d, J = 7.5 Hz, 2H), 8.11 (d, J = 9.0 Hz, 2H). 13C NMR (CDCl3, 125 MHz) δ 26.6, 33.4, 47.4, 49.7, 66.6, 70.7, 96.2, 120.0, 120.1, 121.6, 121.8, 125.0, 125.3, 127.0, 127.2, 127.7, 127.9, 128.1, 128.6, 128.8, 141.4, 143.7, 143.8, 144.6, 145.4, 152.4, 155.4, 156.4, 171.1. HRMS (M+H) Calc’d: 762.2815; Found 762.3604.
Supplementary Material
Table 2.
Probes of potential interactions of the backbone atoms at pY+3 and Stat3.
| Peptide | Sequence | IC50 (nM) |
|---|---|---|
| 2 | pCinn-Leu-Pro-Gln-NHBn | 138 ± 8 |
| 6 | pCinn-Leu-Pro-NH2 | 11,400 ± 1,600 |
| 7 | pCinn-Leu-Pro-Ala-NH2 | 7,800 ± 730 |
Acknowledgements
We are grateful to the National Cancer Institute (CA096652) and the M. D. Anderson Cancer Center University Cancer Fund, for support of this work. Partial support by a Grant from the Center for Targeted Therapy of The University of Texas M. D. Anderson Cancer Center and the Texas Institute of Drug and Diagnostic Development at The University of Texas at Austin is acknowledged. We also acknowledge the NCI Cancer Center Support Grant CA016672 for the support of our NMR facilities and the M. D. Anderson Cancer Center Chemistry Core Facility for some of the mass spectrometry. Funding as an Odyssey Fellow (Z.R.) was supported by the Odyssey Program and the Cockrell Foundation Award for Scientific Achievement at UTMDACC.
Abbreviations
- Aba
4-aminobutyramide
- 4-Abu
4-aminobutyric acid
- DIEA
diisopropylethylamine
- DIPCDI
diisopropylcarbodiimide
- Fmoc
9-fluorenylmethoxycarbonyl
- FMPB
4-(4-formyl-3-methyoxyphenoxy)butyryl
- HOBt
1-hydroxybenzotriazole
- homoGlu
homoglutamic acid
- Met(O)
methionine sulfoxide
- Met(O2)
methionine sulfone
- mPro
cis-3,4-methanoproline
- pCinn
4-phosphoryloxy cinnamic acid
- PyBOP
1H-Benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate
- SAR
structure activity relationship
- TES
triethylsilane
- TIS
triisopropylsilane
Footnotes
Supporting Information Available. Mass spectral and HPLC characterization of peptides 3–49.
References
- 1.Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell. Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 2.Darnell JE., Jr. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer. 2002;2:740–749. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- 3.Bromberg J, Darnell JE., Jr The role of STATs in transcriptional control and their impact on cellular function. Oncogene. 2000;19:2468–2473. doi: 10.1038/sj.onc.1203476. [DOI] [PubMed] [Google Scholar]
- 4.Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat. Rev. Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 5.Cirillo D, Rachiglio AM, La Montagna R, Giordano A, Normanno N. Leptin signaling in breast cancer: an overview. J. Cell. Biochem. 2008;105:956–964. doi: 10.1002/jcb.21911. [DOI] [PubMed] [Google Scholar]
- 6.Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19:2474–2488. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 7.Ren Z, Cabell LA, Schaefer TS, McMurray JS. Identification of a high-affinity phosphopeptide inhibitor of Stat3. Bioorg. Med. Chem. Lett. 2003;13:633–636. doi: 10.1016/s0960-894x(02)01050-8. [DOI] [PubMed] [Google Scholar]
- 8.Coleman DR, Ren Z, Mandal PK, Cameron AG, Dyer GA, Muranjan S, Campbell M, Chen X, McMurray JS. Investigation of the binding determinants of phosphopeptides targeted to the SRC homology 2 domain of the signal transducer and activator of transcription 3. Development of a high-affinity peptide inhibitor. J. Med. Chem. 2005;48:6661–6670. doi: 10.1021/jm050513m. [DOI] [PubMed] [Google Scholar]
- 9.Coleman DR, IV, Kaluarachchi K, Ren Z, Chen X, McMurray JS. Solid phase synthesis of phosphopeptides incorporating 2,2-dimethyloxazolidine pseudoproline analogs: evidence for trans Leu-Pro peptide bonds in Stat3 inhibitors. Int. J. Pept. Res. Ther. 2008;14:1–9. [Google Scholar]
- 10.Mandal PK, Heard PA, Ren Z, Chen X, McMurray JS. Solid-phase synthesis of Stat3 inhibitors incorporating O-carbamoylserine and O-carbamoylthreonine as glutamine mimics. Bioorg. Med. Chem. Lett. 2007;17:654–656. doi: 10.1016/j.bmcl.2006.10.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mandal PK, Limbrick D, Coleman DR, Dyer GA, Ren Z, Birtwistle JS, Xiong C, Chen X, Briggs JM, McMurray JS. Conformationally constrained peptidomimetic inhibitors of signal transducer and activator of transcription 3: evaluation and molecular modeling. J. Med. Chem. 2009;52:2429–2442. doi: 10.1021/jm801491w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turkson J, Ryan D, Kim JS, Zhang Y, Chen Z, Haura E, Laudano A, Sebti S, Hamilton AD, Jove R. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J. Biol. Chem. 2001;276:45443–45455. doi: 10.1074/jbc.M107527200. [DOI] [PubMed] [Google Scholar]
- 13.Turkson J, Kim JS, Zhang S, Yuan J, Huang M, Glenn M, Haura E, Sebti S, Hamilton AD, Jove R. Novel peptidomimetic inhibitors of signal transducer and activator of transcription 3 dimerization and biological activity. Mol. Cancer Ther. 2004;3:261–269. [PubMed] [Google Scholar]
- 14.Siddiquee KA, Gunning PT, Glenn M, Katt WP, Zhang S, Schrock C, Sebti SM, Jove R, Hamilton AD, Turkson J. An oxazole-based small-molecule Stat3 inhibitor modulates Stat3 stability and processing and induces antitumor cell effects. ACS Chem Biol. 2007;2:787–798. doi: 10.1021/cb7001973. [DOI] [PubMed] [Google Scholar]
- 15.Gunning PT, Katt WP, Glenn M, Siddiquee K, Kim JS, Jove R, Sebti SM, Turkson J, Hamilton AD. Isoform selective inhibition of STAT1 or STAT3 homo-dimerization via peptidomimetic probes: structural recognition of STAT SH2 domains. Bioorg. Med. Chem. Lett. 2007;17:1875–1878. doi: 10.1016/j.bmcl.2007.01.077. [DOI] [PubMed] [Google Scholar]
- 16.Gunning PT, Glenn MP, Siddiquee KA, Katt WP, Masson E, Sebti SM, Turkson J, Hamilton AD. Targeting protein-protein interactions: suppression of Stat3 dimerization with rationally designed small-molecule, nonpeptidic SH2 domain binders. Chembiochem. 2008;9:2800–2803. doi: 10.1002/cbic.200800291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shao H, Cheng HY, Cook RG, Tweardy DJ. Identification and characterization of signal transducer and activator of transcription 3 recruitment sites within the epidermal growth factor receptor. Cancer Res. 2003;63:3923–3930. [PubMed] [Google Scholar]
- 18.Shao H, Xu X, Mastrangelo MA, Jing N, Cook RG, Legge GB, Tweardy DJ. Structural requirements for signal transducer and activator of transcription 3 binding to phosphotyrosine ligands containing the YXXQ motif. J. Biol. Chem. 2004;279:18967–18973. doi: 10.1074/jbc.M314037200. [DOI] [PubMed] [Google Scholar]
- 19.Dourlat J, Valentin B, Liu WQ, Garbay C. New syntheses of tetrazolylmethylphenylalanine and O-malonyltyrosine as pTyr mimetics for the design of STAT3 dimerization inhibitors. Bioorg. Med. Chem. Lett. 2007;17:3943–3946. doi: 10.1016/j.bmcl.2007.04.107. [DOI] [PubMed] [Google Scholar]
- 20.Chen J, Nikolovska-Coleska Z, Yang CY, Gomez C, Gao W, Krajewski K, Jiang S, Roller P, Wang S. Design and synthesis of a new, conformationally constrained, macrocyclic small-molecule inhibitor of STAT3 via 'click chemistry'. Bioorg. Med. Chem. Lett. 2007;17:3939–3942. doi: 10.1016/j.bmcl.2007.04.096. [DOI] [PubMed] [Google Scholar]
- 21.Gomez C, Bai L, Zhang J, Nikolovska-Coleska Z, Chen J, Yi H, Wang S. Design, synthesis, and evaluation of peptidomimetics containing Freidinger lactams as STAT3 inhibitors. Bioorg. Med. Chem. Lett. 2009;19:1733–1736. doi: 10.1016/j.bmcl.2009.01.091. [DOI] [PubMed] [Google Scholar]
- 22.Stahl N, Farruggella TJ, Boulton TG, Zhong Z, Darnell JE, Jr, Yancopoulos GD. Choice of STATs and other substrates specified by modular tyrosine-based motifs in cytokine receptors. Science. 1995;267:1349–1353. doi: 10.1126/science.7871433. [DOI] [PubMed] [Google Scholar]
- 23.Gerhartz C, Heesel B, Sasse J, Hemmann U, Landgraf C, Schneider-Mergener J, Horn F, Heinrich PC, Graeve L. Differential activation of acute phase response factor/STAT3 and STAT1 via the cytoplasmic domain of the interleukin 6 signal transducer gp130. I. Definition of a novel phosphotyrosine motif mediating STAT1 activation. J. Biol. Chem. 1996;271:12991–12998. doi: 10.1074/jbc.271.22.12991. [DOI] [PubMed] [Google Scholar]
- 24.Wiederkehr-Adam M, Ernst P, Muller K, Bieck E, Gombert FO, Ottl J, Graff P, Grossmuller F, Heim MH. Characterization of phosphopeptide motifs specific for the Src homology 2 domains of signal transducer and activator of transcription 1 (STAT1) and STAT3. J. Biol. Chem. 2003;278:16117–16128. doi: 10.1074/jbc.M300261200. [DOI] [PubMed] [Google Scholar]
- 25.Sawyer TK. Src homology-2 domains: structure, mechanisms, and drug discovery. Biopolymers. 1998;47:243–261. doi: 10.1002/(SICI)1097-0282(1998)47:3<243::AID-BIP4>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 26.Muller G. Peptidomimetic SH2 domain antagonists for targeting signal transduction. Top. Curr. Chem. 2001;211:17–59. [Google Scholar]
- 27.Shakespeare WC. SH2 domain inhibition: a problem solved? Curr. Opin. Chem. Biol. 2001;5:409–415. doi: 10.1016/s1367-5931(00)00222-2. [DOI] [PubMed] [Google Scholar]
- 28.Garcia-Echeverria C. Antagonists of the homology 2 (SH2) domains of Grb2, Src, Lck and ZAP-70. Curr. Med. Chem. 2001;8:1589–1604. doi: 10.2174/0929867013371905. [DOI] [PubMed] [Google Scholar]
- 29.Metcalf CA, III, Sawyer T. Src homology-2 domains and structure-based, small-molecule library approaches to drug discovery. In: Makriyannis A, Biegel D, editors. Drug Discovery Strategies and Methods. New York, NY: Marcel Dekker, Inc.; 2004. pp. 23–59. [Google Scholar]
- 30.Garcia-Echeverria C. Inhibitors of signaling interfaces: targeting Src homology 2 domains in drug discovery. Protein Tyrosine Kinases. 2006:31–52. [Google Scholar]
- 31.McMurray JS. Structural basis for the binding of high affinity phosphopeptides to Stat3. Biopolymers. 2008;90:69–79. doi: 10.1002/bip.20901. [DOI] [PubMed] [Google Scholar]
- 32.A preliminary account of the syntheses of 63c-h and 64c-h was presented a the 20th American Peptide Symposium, June 2007 Mandal PK, McMurray JS. Application of triethylsilane and palladium-charcoal-induced reductions in the synthesis of Fmoc-glutamic acid analogues. Adv. Exp. Med. Biol. 2009;611:181–182. doi: 10.1007/978-0-387-73657-0_83.
- 33.Pearson DA, Blanchette M, Baker ML, Guindon CA. Trialkylsilanes as scavengers for the trifluoroacetic acid deblocking of protecting groups in peptide synthesis. Tetrahedron Lett. 1989;30:2739–2742. [Google Scholar]
- 34.Mazurov A. Solid phase synthesis of 5,6,7,8-tetrahydro-1H-imidazo[4,5-g]quinoxalin-6-ones. Tetrahedron Lett. 2000;41:7–10. [Google Scholar]
- 35.Wen JJ, Crews CM. Synthesis of 9-fluorenylmethoxycarbonyl-protected amino aldehydes. Tetrahedron: Asymmetry. 1998;9:1855–1858. [Google Scholar]
- 36.Ahern DG, Lassiter AG, Filer CN. An improved synthesis of 4-aminotetrolic acid. Synth. Commun. 2002;32:665–667. [Google Scholar]
- 37.Humljan J, Gobec S. Synthesis of N-phthalimido [beta]-aminoethanesulfonyl chlorides: the use of thionyl chloride for a simple and efficient synthesis of new peptidosulfonamide building blocks. Tetrahedron Lett. 2005;46:4069–4072. [Google Scholar]
- 38.Mandal PK, McMurray JS. Pd-C-induced catalytic transfer hydrogenation with triethylsilane. J. Org. Chem. 2007;72:6599–6601. doi: 10.1021/jo0706123. [DOI] [PubMed] [Google Scholar]
- 39.Loukas V, Noula C, Kokotos G. Efficient protocols for the synthesis of enantiopure gamma-amino acids with proteinogenic side chains. J. Pept. Sci. 2003;9:312–319. doi: 10.1002/psc.458. [DOI] [PubMed] [Google Scholar]
- 40.Mondal S, Fan E. Mild and efficient synthesis of Fmoc-protected amino azides from Fmoc-protected amino alcohols. Synlett. 2006:306–308. [Google Scholar]
- 41.Kates SA, De La Torre BG, Eritja R, Albericio F. Solid-phase N-glycopeptide synthesis using allyl side-chain protected Fmoc-amino acids. Tetrahedron Lett. 1994;35:1033–1034. [Google Scholar]
- 42.Boeijen A, van Ameijde J, Liskamp RM. Solid-phase synthesis of oligourea peptidomimetics employing the Fmoc protection strategy. J. Org. Chem. 2001;66:8454–8462. doi: 10.1021/jo010656q. [DOI] [PubMed] [Google Scholar]
- 43.Debaene F, Da Silva JA, Pianowski Z, Duran FJ, Winssinger N. Expanding the scope of PNA-encoded libraries: divergent synthesis of libraries targeting cysteine, serine and metallo-proteases as well as tyrosine phosphatases. Tetrahedron. 2007;63:6577–6586. [Google Scholar]
- 44.Jobron L, Hummel G. Solid-Phase Synthesis of New S-Glycoamino Acid Building Blocks. Org. Let. 2000;2:2265–2267. doi: 10.1021/ol006019o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.