

Abstract
The final step of capsidiol biosynthesis is catalyzed by 5-epiaristolochene dihydroxylase (EAH), a cytochrome P450 enzyme that catalyzes the regio- and stereospecific insertion of two hydroxyl moieties into the bicyclic sesquiterpene 5-epiaristolochene (EA). Detailed kinetic studies using EA and the two possible monohydroxylated intermediates demonstrated the release of 1β-hydroxy-EA ((OH)EA) at high EA concentrations and a 10-fold catalytic preference for 1β(OH)EA versus 3α(OH)EA, indicative of a preferred reaction order of hydroxylation at C-1, followed by that at C-3. Sequence alignments and homology modeling identified active-site residues tested for their contribution to substrate specificity and overall enzymatic activity. Mutants EAH-S368C and EAH-S368V exhibited wild-type catalytic efficiencies for 1β(OH)EA biosynthesis, but were devoid of the successive hydroxylation activity for capsidiol biosynthesis. In contrast to EAH-S368C, EAH-S368V catalyzed the relative equal biosynthesis of 1β(OH)EA, 2β(OH)EA, and 3β(OH)EA from EA with wild-type efficiency. Moreover, EAH-S368V converted ~1.5% of these monohydroxylated products to their respective ketone forms. Alanine and threonine mutations at position 368 were significantly compromised in their conversion rates of EA to capsidiol and correlated with 3.6- and 5.7-fold increases in their Km values for the 1β(OH)EA intermediate, respectively. A role for Ile486 in the successive hydroxylations of EA was also suggested by the EAH-I468A mutant, which produced significant amounts 1β(OH)EA, but negligible amounts of capsidiol from EA. The altered product profile of the EAH-I486A mutant correlated with a 3.6-fold higher Km for EA and a 4.4-fold slower turnover rate (kcat) for 1β(OH)EA. These kinetic and mutational studies were correlated with substrate docking predictions to suggest how Ser368 and Ile486 might contribute to active-site topology, substrate binding, and substrate presentation to the oxo-Fe-heme reaction center.
The mevalonate and methylerythritol phosphate pathways are responsible for the biosynthesis of isoprenoids, a diverse group of organic natural products found in animals, plants, fungi, insects, and bacteria. Isoprenoids are further divided into classes of primary and secondary metabolites. Isoprenoids that are primary metabolites include sterols, carotenoids, hormones, and long chain hydrocarbons used to tether particular enzymes to membrane systems, compounds essential for viability. Isoprenoids classified as secondary metabolites include monoterpenes, sesquiterpenes, diterpenes, and triterpenes, and many of these mediate interactions between organisms and their environments (1–5).
Capsidiol is a bicyclic dihydroxylated sesquiterpene produced by several solanaceous plants in response to pathogen or elicitor challenge (6–10) and is considered an important plant defense response because it can prevent the germination and growth of several fungal species (11). Capsidiol biosynthesis is regulated by the expression of two key enzymes (see Scheme 1). 5-Epiaristolochene synthase catalyzes the cyclization of farnesyl diphosphate to the bicyclic intermediate 5-epiaristolochene (EA)1,2 and is responsible for the diversion of farnesyl diphosphate from the mevalonate pathway toward antimicrobial compound biosynthesis (12, 13). Recently, two highly homologous cDNAs for EA dihydroxylase (EAH) were isolated from Nicotiana tabacum and functionally expressed in yeast, and the encoded proteins were characterized as cytochrome P450 enzymes catalyzing the stereo- and regiospecific hydroxylation of EA at C-1 and C-3 (see Scheme 1) (14).
Scheme 1. Biosynthetic pathway for capsidiol in elicitor-treated N. tabacum cells.
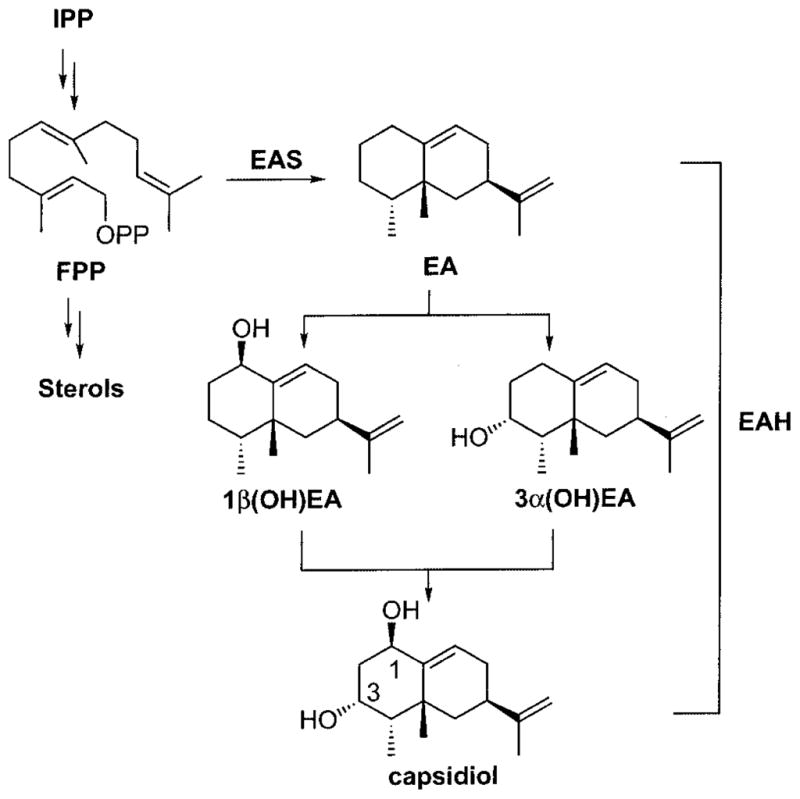
Farnesyl diphosphate (FPP) is diverted from the central mevalonate pathway via the action of a sesquiterpene synthase (5-epiaristolochene synthase (EAS)) to generate EA. Subsequent hydroxylations at C-1 and C-3 catalyzed by EAH yield capsidiol. A preferred hydroxylation order had not been established previously. IPP, isopentenyl pyrophosphate; OPP, pyrophosphate.
Plant P450 enzymes known to catalyze regiospecific monohydroxylation of carbocyclic structures include limonene 3- and 6-hydroxylases (15, 16); cinnamate 4-hydroxylase (17); ferulate 5-hydroxylase (18, 19); taxoid 10β-, 13α-, and 14β-hydroxylases (20–22); the CYP71C subfamily enzymes for 2,4-dihydroxy-7-methoxy-1,4-benzoxazin-3-one biosynthesis in maize and wheat (23, 24); and members of the CYP90 family involved in the biosynthesis of the steroid hormone brassinolide (25, 26). Successive hydroxylation/oxidation reactions catalyzed by plant P450 enzymes have also been documented for tyrosine n-hydroxylase involved in the biosynthesis of the cyanogenic glucoside dhurrin (27), flavonoid 3′,5′-hydroxylase in anthocyanin biosynthesis (28, 29), and several of the enzymes in the gibberellin biosynthetic pathway (30–33). For example, flavonoid 3′,5′-hydroxylase (28, 29) catalyzes the planar insertion of hydroxyl groups into the equivalent meta-positions (C-3′ and C-5′) of the flavonoid B (phenolic) ring and yields 3′-, 5′-, and 3′,5′-hydroxylated reaction products (29). ent-Kaurene oxidase (32) catalyzes the successive oxidation of the methyl substituent of ent-kaurene at C-19 en route to ent-kaurenoic acid. ent-Kaurenoic acid is itself subject to multiple oxidations and a ring contraction through the action of a single P450, CYP88A, or ent-kaurenoic acid oxidase (33). Interestingly, although flavonoid 3′, 5′-hydroxylase and both ent-kaurene and ent-kaurenoic acid oxidases catalyze regiospecific hydroxylations, these enzymes do not exhibit the regio- and stereospecific complexity of EAH. EAH introduces hydroxyl groups at distal sites on opposite faces of the epiaristolochene ring system (see Scheme 1).
A preferred order for the successive hydroxylation of EA has yet to be determined (see Scheme 1). Of the two possible monohydroxylated intermediates of EA, only 3α-hydroxy-EA (3α(OH)EA) has been reported as a minor metabolite accumulating in pepper fruits challenged with fungal spores (34), suggesting that small amounts of this reaction intermediate might be released as a general consequence of catalysis. Consistent with this notion are several observations that 3α(OH)EA can be converted to capsidiol. Whitehead et al. (35) reported a modest conversion of 3α(OH)EA to capsidiol when radiolabeled forms of this intermediate were fed to either control or elicitor-treated callus cultures of tobacco. However, although Ralston et al. (14) documented the ready conversion of 3α(OH)EA to capsidiol by microsomes from yeast overexpressing the EAH gene, they also reported the biosynthesis of capsidiol without the accumulation of either monohydroxylated intermediate, 3α(OH)EA or 1β(OH)EA, in reactions incubated with low concentrations (<1 μM) of the EA substrate. Although 1β(OH)EA has not been reported as a reaction product of EAH or observed in plants exposed to exogenous stimuli, preference for P450-mediated hydroxylations of mono- and diterpenes occurring allylic to carbon–carbon double bonds (16, 20, 21) suggests that the initial hydroxylation catalyzed by EAH could occur at C-1, followed by that at C-3.
This work describes investigations of the successive regio-and stereospecific hydroxylation of EA by EAH. The ready availability of EA, 3α(OH)EA, and 1β(OH)EA greatly facilitated our ability to perform detailed kinetic studies to address the preferred order of hydroxylation. Sequence comparisons with related P450 enzymes and modeling of EAH based upon the mammalian 2C5 steroid hydroxylase structure (36) enabled us to identify amino acid residues potentially providing catalytic specificity to these reactions. These structural hypotheses were then tested by site-directed mutagenesis and kinetic analysis. Together, the results demonstrate a preferred reaction order and provide evidence that Ser368 and Ile486 play a significant role in orchestrating the successive hydroxylation of EA to capsidiol.
EXPERIMENTAL PROCEDURES
Chemicals
Standard laboratory reagents were purchased from Fisher, Sigma, and Aldrich. Authentic standards of EA, 3α(OH)EA, and capsidiol were available from previous work (14). Synthesis of 1β(OH)EA, 3β(OH)EA, EA-1-one, and EA-3-one was conducted as described (37).
Site-directed Mutagenesis
All mutations were engineered into the CYP71D20 (EAH) cDNA using the standard QuikChange protocol (Stratagene) (38). A full-length EAH cDNA (GenBank™/EBI accession number AF368376) inserted into the BamHI/EcoRI restriction sites of the pBluescript II KS(+) vector was used in combination with PfuTurbo DNA polymerase (Stratagene) and the following primer pairs (with the mutation sites underlined): S368A primers, 5′-GACTTCATCCACCGGCTCCACTTTTGGTCC-3′ and 5′-GGACCAAAAGTGGAGCCGGTGGATGAAGTC-3′; S368C primers, 5′-GACTTCATCCACCGTGTCCACTTTTGGTCC-3′ and 5′-GGACCAAAAGTGGACACGGTGGATGAAGTC-3′; S368T primers, 5′-GACTTCATCCACCGACTCCACTTTTGGTCC-3′ and 5′-GGACCAAAAGTGGAGTCGGTGGATGAAGTC-3′; S368V primers, 5′-GACTTCATCCACCGGTTCCACTTTTGGTCC-3′ and 5′-GGACCAAAAGTGGAACCGGTGGATGAAGTC-3′; S368I primers, 5′-GACTTCATCCACCGATTCCACTTTTGGTCC-3′ and 5′-GGACCAAAAGTGGAATCGGTGGATGAAGTC-3′; S368F primers, 5′-GACTTCATCCACCGTTTCCACTTTTGGTCC-3′ and 5′-GGACCAAAAGTGGAAACGGTGGATGAAGTC-3′; I486A primers, 5′-CGAATTATCGGGAATAACTGCTGCTAGAAAGGGTGGCC-3′ and 5′-GGCCACCCTTTCTAGCAGCAGTTATTCCCGATAATTCG-3′. Site-specific mutations were confirmed by automated DNA sequencing performed using the Big-Dye Terminator Cycle sequencing protocol of PerkinElmer Life Sciences in combination with an ABI Prism 310 genetic analyzer (Applied Biosystems, Foster City, CA). Mutated cDNAs were subsequently subcloned from the pBluescript vector into the corresponding BamHI/EcoRI sites of pYeDP60, a standard yeast expression vector providing galactose inducibility for expression of the inserted cDNA (39).
Yeast Expression and Microsome Preparations
Expression vectors harboring wild-type or mutant EAH were introduced into the WAT11 yeast strain, a strain previously engineered with an Arabidopsis NADPH-cytochrome P450 reductase gene (40) and kindly provided by Dr. P. Urban (Centre de Génétique Moléculaire, CNRS, Gif-sur-Yvette, France). Transformants were grown from single colonies; expression of the EAH cDNAs was induced by galactose addition to the growth medium; and microsomes were prepared as described previously (41).
CO Difference Spectra
CO difference spectra were determined to estimate the amount of properly folded wild-type and mutant EAH proteins in microsome preparations. The amount of functional P450 was calculated using an extinction coefficient of 91 mM−1 cm−1 at 450 nm (42). The wild-type and mutant enzymes showed a clear absorption maximum at 450 nm (Supplemental Fig. 1), consistent with properly folded enzymes having the heme cofactor oriented in the correct electron spin state.
EAH Assays
Standard assays were performed in 1-ml glass vials with a final reaction volume of 200 μl containing 4% Me2SO, 2.4 mM NADPH, 100 mM Tris-HCl (pH 7.5), and microsomes at a final concentration of 90 or 180 pmol of EAH protein eq (determined by CO difference spectroscopy)/ml. Substrate concentrations were varied from 2 to 100 μM for EA, 3 to 30 μM for 3α(OH)EA, and 0.5 to 30 μM for 1β(OH)EA. After preincubation of the reaction mixtures at 30 °C for 5 min, the reactions were initiated by the addition of 2.4 mM NADPH and allowed to proceed for 2 min (EA), 1 min (1β(OH)EA), or 5 min (3α(OH)EA), which allowed for 5–20% conversion of substrate to reaction product(s). The reactions were terminated by the rapid addition of 400 μl of ethyl acetate and mixed by vortexing, followed by removal of the ethyl acetate extract. A second ethyl acetate extract was combined with the first, and the combined organic extracts were carefully concentrated on ice under N2 gas, redissolved in 20 μl of ethyl acetate containing 10 ng/μl valencene as an internal standard, and subjected to gas chromatography (GC) and GC/mass spectrometry (MS) analyses. Assays were performed in triplicate for each substrate concentration, and kinetic constants were calculated by a nonlinear regression fit to the Michaelis-Menten equation using EnzymeKinetics Version 1.5 software (Trinity Software). Kinetic constants calculated from Lineweaver-Burk, Eadie-Hofstee, and Hanes-Woolf plots were not significantly different from those derived from nonlinear regression. Optimized reactions were linear with respect to reaction product generation over time and corresponded to a 5–20% conversion of substrate to product, sufficient to generate 1 ng or more of reaction product(s) readily quantified and identifiable by GC/MS.
Reaction Product Analyses
Quantification of the reaction products as well as standards of EA, 1β(OH)EA, 3α(OH)EA, and capsidiol was routinely performed using (+)-valencene (Fluka, Buchs, Switzerland) as an internal standard. Accuracy of the valencene standard was periodically verified relative to a (−)-α-cedrene standard (Fluka). Reaction products (1-μl aliquots) were quantified with an HP-5890 gas chromatograph equipped with an HP-5 capillary column (30 m × 0.25 mm, 0.25-μm phase thickness) and a flame ionization detector as described previously (38). Splitless injections were performed at an injection port temperature of 250 °C with an initial column temperature of 140 °C maintained for 0.5 min. The column temperature was then increased to 230 °C with a 4 °C/min gradient. In addition to comigration and retention time comparisons with authentic standards, reaction products were identified by MS using a Thermo Finnigan DSQ GC/MS system equipped with a Restec Rtx-5 capillary column (30 m × 0.32 mm, 0.25-μm phase thickness). Samples were injected in the splitless mode at 250 °C with an initial oven temperature of 70 °C for 1 min, followed by an 8 °C/min gradient to 230 °C. Mass spectra were recorded at 70 eV, scanning from 35 to 300 atomic mass units, and compared with authentic standards for verification.
Purification of the EAH-S368V Reaction Products
To isolate and identify the EAH-S368V reaction products, ethyl acetate extracts from the in vitro assays were carefully concentrated under an N2 stream and resuspended with hexane such that the final ethyl acetate concentration was <2%. The sample was then applied to a 230–400 mesh silica column (0.5 × 1 cm), and the hydrocarbon fraction was collected with a 2-ml hexane wash. Hydroxylated products were selectively eluted with 2 ml of a 1:19 (v/v) ethyl acetate/hexane solution, followed by 2 ml of a 1:4 (v/v) ethyl acetate/hexane solution. The fractions were carefully reconcentrated under an N2 stream before GC/MS analysis.
Computational Studies
Homology modeling was performed with Modeler Version 6.2 (43–45) by threading the EAH amino acid sequence (GenBank™/EBI accession number AF368376) onto the structural coordinates for the mammalian P450 2C5 protein (Protein Data Bank code 1DT6) (36). Ligands EA, 1β(OH)EA, and 3α(OH)EA were created using Chemdraw Ultra Version 7.0.1, energy-minimized with MOPAC in Chem3D Ultra Version 7.0 (CambridgeSoft Corp., Cambridge, MA), and subsequently used in docking simulation experiments. Docking calculations were conducted with GOLD Version 1.2 software (Genetic Optimization for Ligand Docking, Cambridge Crystallographic Data Centre, Cambridge, United Kingdom) (46–49).
RESULTS
Optimization of the EAH Activity
Numerous preliminary experiments were performed to optimize EAH activity for the conversion of EA to capsidiol. For example, the ability of a miscible organic co-solvent and various detergents to improve EA solubility was assessed. Maximum conversion of EA to capsidiol activity was observed at final concentrations of 2–5% (v/v) Me2SO, but was inhibited at concentrations >10%. Detergents were also evaluated at concentrations ranging from 0.01 to 0.1%. Triton X-100 (v/v), deoxycholate (w/v), and 1-O-octyl β-D-glucopyranoside (w/v) all inhibited EAH activity, whereas CHAPS (w/v) and Tween 20 (v/v) had no effect. Standard assays were therefore performed using EA stock solutions dissolved in Me2SO and assayed at a final Me2SO concentration of 4% (v/v) in all reaction assays. The pH optimum was determined using 100 mM Tris-HCl (pH 7.2–9.2) and phosphate (pH 6.0–7.5) buffering systems. Maximum activity was observed at pH 7.5 with little preference for either the Tris-HCl or phosphate buffer. The optimum NADPH concentration for EAH activity was also determined. The maximum conversion rate for EA to capsidiol was observed at 0.3 mM and remained saturated up to a final concentration of 2.4 mM. The Km for NADPH was subsequently estimated to be 61 ± 5 μM. All subsequent enzyme assays were therefore performed at optimized reaction conditions of 4% (v/v) Me2SO and 2.4 mM NADPH in 100 mM Tris-HCl (pH 7.5).
Kinetic Analysis of EAH Indicates a Specific Reaction Order
To initially address the hydroxylation specificity of EAH (Scheme 1), we tested the enzyme for substrate preference for either of the putative reaction intermediates, 3α(OH)EA and 1β(OH)EA (Fig. 1). Microsomes from WAT11 yeast overexpressing the EAH gene were used as the enzyme source, and the absolute amount of active EAH enzyme was calculated from the carbon monoxide difference spectra. Both 3α(OH)EA and 1β(OH)EA were converted to capsidiol by EAH in NADPH-dependent reactions, but with different efficiencies. The Km for 1β(OH)EA was ~4 times lower and the kcat was ~3 times higher than those for 3α(OH)EA, resulting in a catalytic efficiency (kcat/Km) for 1β(OH)EA ~10 times greater than that for 3α(OH)EA (Table I). The kinetic constants for the hydrocarbon substrate EA were also determined (Fig. 2 and Table I). Although the Km for the hydrocarbon substrate was considerably higher than that for either of the monohydroxylated intermediates, the turnover rates for EA and 1β(OH)EA were similar and significantly greater than that for 3α(OH)EA.
Fig. 1. Concentration-dependent conversion of putative monohydroxylated intermediates to capsidiol by EAH.

3α(OH)EA (■), 1β(OH)EA (▲), or both are potential intermediates in the catalytic conversion of the natural bicyclic hydrocarbon substrate EA to capsidiol. EAH was incubated at the indicated concentrations of substrate for 1 (1β(OH)EA) or 5 (3α(OH)EA) min before identifying and quantifying capsidiol accumulation by GC/MS.
Table I.
Comparison of the EAH kinetic constants for its initial substrate (EA) and two possible monohydroxylated intermediates
| Substrate | Product | Km | kcat | kcat/Km |
|---|---|---|---|---|
| μM | s−1 | s−1/μM | ||
| EA | CD | 19.18 ± 2.90 | 0.493 ± 0.039 | 0.0257 |
| 3α(OH)EA | CD | 6.38 ± 0.42 | 0.200 ± 0.004 | 0.0313 |
| 1β(OH)EA | CD | 1.74 ± 0.08 | 0.582 ± 0.006 | 0.3344 |
Fig. 2. Concentration-dependent conversion of EA to capsidiol by EAH.
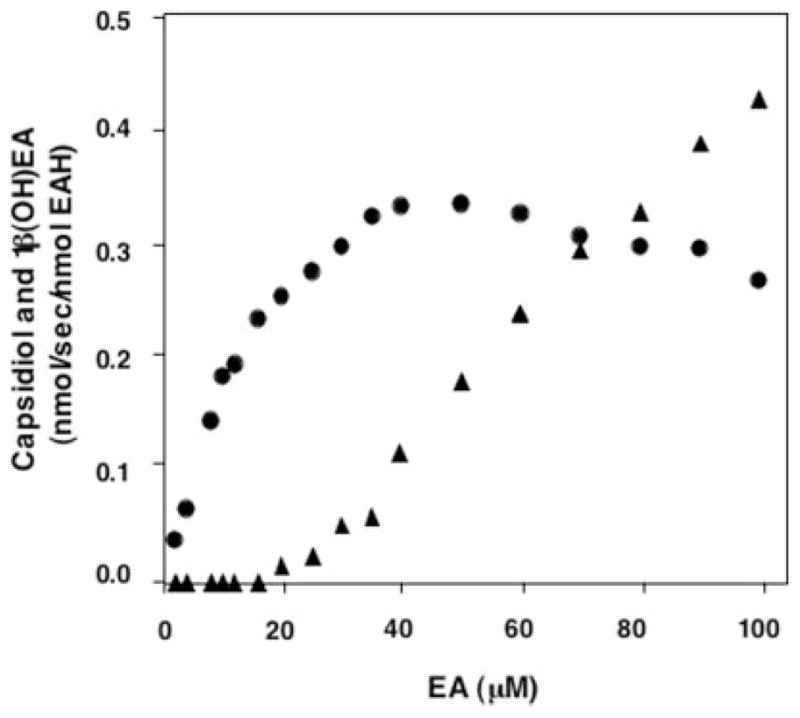
EAH was incubated at the indicated concentrations of EA for 2 min before profiling the reaction products by GC/MS. Capsidiol (●) and 1β(OH)EA (▲) were the only NADPH-dependent reaction products detected.
The kinetic constants reported in Table I are consistent with a catalytic barrier to the successive hydroxylation of the 3α(OH)EA intermediate and suggested that 1β(OH)EA would be the preferred monohydroxylated intermediate. In an effort to capture the release of an initial reaction intermediate, assays were performed with increasing concentrations of EA (2–100 μM), and the reaction product profiles were determined (Fig. 2). Reaction products were observed only after incubations with NADPH and were identified by GC/MS comparisons with authentic standards (Fig. 3). Capsidiol production reached a maximum at ~40 μM EA and gradually decreased after that. Although no monohydroxylated intermediates were detectable at EA concentrations below 16 μM, a proportionate increase in the biosynthesis of 1β(OH)EA was observed at EA concentrations above 20 μM. In contrast, no release of 3α(OH)EA was observed over the entire concentration range investigated.
Fig. 3. GC and MS profiles for the reaction products generated by EAH incubated with EA.

Shown are total ion chromatograms for reaction products generated by EAH in the presence and absence of NADPH (A) and mass spectrum matches for peaks 1 and 2 to authentic standards of 1β(OH)EA and capsidiol (B).
Molecular Analysis of the Dihydroxylase Activities
A combination of computational methods was used to identify structural elements possibly contributing to the successive hydroxylation activity of EAH (Fig. 4). Sequence alignment of EAH with the known substrate recognition sites (SRSs) of closely related P450 enzymes of the CYP71D subfamily (50) revealed several potentially important sites. SRS-5 and SRS-6 were of particular interest given their proximity to the active site in several P450 structures (51) and because specific amino acid positions within these regions have previously been correlated with regio- and stereospecific reaction mechanisms (Fig. 4A) (52–61). A homology model of EAH derived from comparison with the mammalian CYP2C5 structure was constructed (Fig. 4B), and the resultant model suggested that Ser368 and Ile486 project into the active-site cavity oriented toward the heme catalytic site (Fig. 4C).
Fig. 4. Associating amino acids with the regio- and stereospecificity of EAH by amino acid sequence alignment and protein modeling.

Shown is a sequence alignment for select members of the CYP71D subfamily corresponding to SRS-5 and SRS-6 (50), with identical amino acids shaded (A). GenBank™/EBI accession numbers are as follows: CYP71D4 (AJ296346), CYP71D6 (U48434), CYP71D7 (U48435), CYP71D9 (Y10490), CYP71D10 (AF022459), CYP71D11 (AF000403), CYP71D13 (AF124816), CYP71D15 (AF124817), CYP71D16 (AF166332), CYP71D18 (AF124815), CYP71D19 (AF122821), and CYP71D20 (EAH; AF368376). Several of the CYP71D subfamily members have not been functionally characterized, whereas CYP71D13 and CYP71D15 code for (4S)-limonene 3-hydroxylase (15), CYP71D18 for (4S)-limonene 6-hydroxylase (15), CYP71D16 for a diterpene hydroxylase (72), and CYP71D20 for EAH (14). A homology model of EAH was generated using the mammalian P450 2C5 protein (Protein Data Bank code 1DT6) (36) as described under “Experimental Procedures” and is shown here as a ribbon diagram (B). A close-up is shown of the active-site model of EAH showing the spatial location of Ser368 and Ile486 relative to the heme reaction center (C).
To investigate the contributions, if any, of Ser368 and Ile486 to either the first or second hydroxylation reaction as predicted by computational studies, site-specific mutations were generated at each position. The mutant genes were expressed in WAT11 yeast lines, and the absolute amount of newly synthesized microsomal enzyme was calculated from the carbon monoxide difference spectra (Supplemental Fig. 1). Mutant enzymes were then examined for substrate preferences and product specificity by steady-state kinetic analyses (Fig. 5 and Table II). Mutant enzymes harboring smaller amino acid substitutions such as Ala, Thr, Cys, and Val demonstrated reasonable CO binding spectra and were catalytically active. EAH-S368A and EAH-S368T retained their ability to fully convert EA to capsidiol, but the turnover rates for capsidiol formation were 3–13 times lower compared with that of the wild-type enzyme. EAH-S368A and EAH-S368T were also able to convert 1β(OH)EA to capsidiol with kcat values comparable with that of wild-type EAH, but with Km values for 1β(OH)EA 4–6 times greater compared with that of wild-type EAH. Although mutant enzymes containing bulky amino acid substitutions such as Ile and Phe expressed well in yeast and were capable of binding CO, indicative of proper folding, these mutant enzymes were devoid of any hydroxylase activity for EA or 1β(OH)EA.
Fig. 5. Substrate-dependent activities of the EAH Ser368 mutants.

Site-specific mutations corresponding to Ser368 were introduced into the EAH gene as described under “Experimental Procedures”; the mutant genes were expressed in yeast; and yeast microsomes were prepared as the source for each mutant P450 enzyme. CO difference spectroscopy was used to qualify and normalize the amount of properly folded P450 enzyme used in the activity assays. Microsome preparations were incubated at the indicated concentrations of EA or 1β(OH)EA before profiling the reaction products by GC/MS. Except for the EAH-S368V mutant, which generated multiple reaction products, the only NADPH-dependent reaction products observed were capsidiol (△) and 1β(OH)EA (○).
Table II.
Comparison of the kinetic constants for EAH and several site-specific mutants for the initial substrate (EA) and the monohydroxylated intermediate 1β(OH)EA
| Enzyme | Substrate | Product | Km | kcat | kcat/Km |
|---|---|---|---|---|---|
| μM | s−1 | s−1/μM | |||
| Wild-type | EA | CD | 19.18 ± 2.90 | 0.493 ± 0.039 | 0.0257 |
| S368A | EA | CD | 22.67 ± 4.00 | 0.154 ± 0.009 | 0.0068 |
| S368T | EA | CD | 15.18 ± 1.55 | 0.037 ± 0.001 | 0.0024 |
| I486A | EA | CD | ND | ||
| Wild-type | 1β(OH)EA | CD | 1.74 ± 0.08 | 0.582 ± 0.006 | 0.3344 |
| S368A | 1β(OH)EA | CD | 6.20 ± 0.47 | 0.581 ± 0.012 | 0.0937 |
| S368T | 1β(OH)EA | CD | 9.97 ± 1.14 | 0.482 ± 0.019 | 0.0483 |
| S368C | 1β(OH)EA | CD | NA | ||
| S368V | 1β(OH)EA | CD | NA | ||
| I486A | 1β(OH)EA | CD | 21.24 ± 2.84 | 0.132 ± 0.008 | 0.0062 |
| S368C | EA | 1β(OH)EA | 38.63 ± 2.64 | 0.278 ± 0.008 | 0.0072 |
| S368V | EA | 1β(OH)EA | 16.72 ± 2.21 | 0.553 ± 0.025 | 0.0331 |
| I486A | EA | 1β(OH)EA | 68.80 ± 4.04 | 0.293 ± 0.010 | 0.0043 |
EAH-S368A and EAH-S368T possess a Km for the EA substrate that is similar to that of the wild-type enzyme, yet have a significantly higher Km for 1β(OH)EA, suggesting that Ser368 may be more instrumental for the second hydroxylation step rather than the first. Additional mutations at Ser368 provided support for this notion. Both EAH-S368C and EAH-S368V readily catalyzed the biosynthesis of 1β(OH)EA without the generation of any capsidiol when incubated over a wide range of EA concentrations (Fig. 5). Interestingly, the EAH-S368C mutant produced 1β(OH)EA with a catalytic efficiency ~30% that of the wild-type enzyme and only a very negligible amount of EA-1-one (Table II). In contrast, the EAH-S368V mutant produced significant and approximately equal amounts of 1β(OH)EA, 2β(OH)EA, and 3β(OH)EA; a small amount of 3α(OH)EA; another unidentified monohydroxylated compound accounting for 7% of the total products (presumed to be 2α(OH)EA based on retention time and MS); and EA-3-one and EA-1-one at 1.5% (Fig. 6 and Scheme 2). Control microsomes (microsomes from yeast harboring an expression vector not engineered with any EAH gene constructs) were not able to metabolize 1β(OH)EA, but did exhibit a very minor activity (4.6% of that associated with EAH-S368V) for conversion of 3α(OH)EA to the corresponding ketone (data not shown). Overall, the catalytic efficiency of EAH-S368V for the turnover of EA to 1β(OH)EA was slightly superior to the ability of the wild-type enzyme to convert EA to capsidiol (Table II). However, when the biosynthetic rates for all the monohydroxylated and ketone reaction products are taken into account, EAH-S368V exhibited a turnover rate for EA twice that of the wild-type enzyme.
Fig. 6. Reaction product analysis of EAH-S368V incubated with EA.

EAH-S368V was incubated with 40 μM EA, and the total reaction products were fractionated by silica gel chromatography. Total ion chromatograms of the reaction products eluted from the silica gel with 5% ethyl acetate in hexane (A, left panel) are compared with those of authentic standards for EA-3-one and EA-1-one (A, right panel). Mass spectra for peaks 1 and 2 are compared directly with those for the respective ketones (B). Total ion chromatograms of reaction products sequentially eluted from the silica gel with 20% ethyl acetate in hexane (C, left panel) are compared with those of authentic standards for 1β(OH)EA, 3α(OH)EA, and 3β(OH)EA (C, right panel). The mass spectrum for peak 3 matches that for 1β(OH)EA (D), shown previously in Fig. 3. The mass spectra for peaks 5 and 6 are compared directly with those for 3β(OH)EA and 3α(OH)EA (D). However, peak 5 resolves into two nearly equal sized peaks by chiral GC analysis (Supplemental Fig. 2), with peak 5-1 corresponding to 3β(OH)EA and peak 5-2 to 2β(OH)EA (as determined by NMR analysis). RT, retention time.
Scheme 2. Chemical rationalization for the reaction products generated by the EAH-S368V mutant.

Solid arrows refer to reaction products identified by comparison with authentic standards or directly by NMR, whereas the dashed arrow refers to a reaction product structure inferred from GC retention times and MS profiles. The percentages denote the amount of a particular reaction product generated in a typical reaction assay. unk, unknown
Ile486 within SRS-6 was also chosen for mutagenesis because this position exhibits significant variability between other members of the CYP71D subfamily, and the spatial location of its R group is oriented into the active site based on molecular modeling (Fig. 4). Mutation of Ile486 to Ala in EAH also caused a significant reduction in the successive hydroxylation of EA to capsidiol (Fig. 7 and Table II). The EAH-I486A mutant enzyme turned over EA to the monohydroxylated intermediate 1β(OH)EA at approximately one-half the rate of the wild-type enzyme, but produced only negligible amounts of capsidiol from EA. Because an accurate determination of the EA-to-capsidiol conversion was not possible, no kinetic constants for this reaction were calculated. However, the turnover rate for 1β(OH)EA to capsidiol by EAH-I486A had a 12-fold higher Km and a 4-fold lower turnover rate compared to that of the wild-type enzyme (Table II).
Fig. 7. Substrate-dependent activities of the EAH-I486A mutant.

The EAH-I486A mutation was created as described under “Experimental Procedures”; the mutant gene was expressed in yeast; and yeast microsomes were prepared as the source of the EAH-I486A enzyme. CO difference spectroscopy was used to qualify and normalize the amount of properly folded P450 enzyme used in the activity assays. Assays were incubated at the indicated concentrations of EA for 5 min (upper panel) or 1β(OH)EA for 1 min (lower panel) before profiling the reaction products by GC/MS. The only NADPH-dependent reaction products observed were capsidiol (△) and 1β(OH)EA (○). Capsidiol levels were near the detection limits for reactions incubated with EA.
DISCUSSION
The aim of this work was to determine whether successive hydroxylations of EA to capsidiol catalyzed by EAH occur with a preferred reaction order, and if so, if there are structural features of the EAH active site that can be associated with this specificity. Earlier work had demonstrated that EA is readily converted to capsidiol by EAH without the apparent release of any reaction intermediate(s) (14, 41). This in turn prompted speculation that the successive hydroxylations might occur sequentially within a single catalytic cascade terminating with the release of only the dihydroxylated capsidiol product. The relatively large cavities described for the active site of several P450 enzymes (62, 63) lent support to this notion. We therefore suggested that the initial monohydroxylated intermediate might not be released from EAH, but instead might be flipped and/or rotated within the active site in a manner to reposition the intermediate for the second hydroxylation event. The second hydroxylation event would then provide a trigger for product release. The present results clearly refute such speculation and provide strong kinetic evidence for independent and stepwise hydroxylation of EA. EAH readily accepts and catalyzes the conversion of both monohydroxylated intermediates to capsidiol. However, although the turnover rate for 3α(OH)EA to capsidiol is comparable with that for the native hydrocarbon substrate EA, the relative efficiency for 1β(OH)EA conversion is 10-fold greater (Table I). The latter is largely accounted for by a much lower Km for 1β(OH)EA. The accumulation of 1β(OH)EA in reactions initiated with concentrations of EA exceeding 20 μM also clearly demonstrates a specific reaction pathway. When EA concentrations are below 20 μM, EA must be converted first to 1β(OH)EA, which is released but rapidly recaptured and converted to the dihydroxylated product. At concentrations exceeding 20 μM (the approximate Km for EA), EA effectively competes with released 1β(OH)EA for substrate-binding sites. Hence, 1β(OH)EA accumulates in direct proportion to the EA concentration.
The successive hydroxylation activity of EAH exhibits similarities and differences relative to other well characterized multifunctional P450 enzymes. For example, CYP27A1 catalyzes the successive hydroxylation of vitamin D3 (64) and cholesterol (65). Based on the time-dependent appearance of multiple reaction products during in vitro reactions, Sawada et al. (64) concluded that CYP27A1 initially catalyzes hydroxylation at C-25 of vitamin D3, followed by a second hydroxylation at C-24, C-1, or C-26, yielding a complex mixture of a single monohydroxylated product and three or more dihydroxylated ones. No C-1 or C-24 monohydroxylation products were observed, nor products with more than two inserted hydroxyls. Consistent with this complex reaction product profile, the Km for the C-25 monohydroxylated intermediate is equal to that for the original substrate, but the apparent reaction velocity for the successive hydroxylation is only one-tenth that of the initial hydroxylation event. P45011β (66) and P45017α lyase (67) are likewise P450 enzymes catalyzing successive hydroxylation reactions in aldosterone and androgen metabolism, respectively, that generate mono- and dihydroxylated reaction mixtures. Using rapid quench methods, rate constants for the initial hydroxylation step were shown to be ~10-fold greater than the rate constants for the second hydroxylation step, and the dissociation rates of the monohydroxylated intermediates were also competitive with the rate constants for the second hydroxylation step, altogether consistent with the release and accumulation of monohydroxylated intermediates over time. In light of these reports and the current results, our initial observation of complete conversion of EA to a single dihydroxylated product without release of monohydroxylated intermediate (14) can now be attributed to the greater affinity and faster turnover rate of EAH for the monohydroxylated intermediate than for the initial EA substrate.
Previous site-directed mutagenesis studies and the three-dimensional structure of mammalian P450 2C5 (36) provided important frameworks for targeting active-site residues that might play a role in the successive hydroxylation specificity of EAH (68, 69). Using only those sequence regions that map to the active-site cavity of 2C5 and correspond to SRS-5 and SRS-6 (50), sequence alignment of select CYP71D family members revealed that the positions corresponding to amino acids 368 and 486 in EAH are particularly variable (Fig. 4). In fact, previous studies had already documented the functional significance of this variability. Mutagenesis of the position equivalent to amino acid 368 in several members of the CYP2B subfamily was shown to affect the regiospecific hydroxylation of steroids (52, 53). The same position was also reported to play a role in substrate recognition for CYP2A5 (54) and CYP3A4 (55). Swapping this region between two highly homologous plant P450 enzymes, limonene 6-hydroxylase and limonene 3-hydroxylase, followed by reciprocal site-directed mutagenesis, demonstrated that the corresponding position also contributes to the regiospecificity of monoterpene hydroxylation. Exchange of Phe363 with Ile changes the regiospecificity of limonene 6-hydroxylase (CYP71D18) from hydroxylating limonene at C-6 to C-3, but the reciprocal mutation, I363F, completely inactivates limonene 3-hydroxylase activity (56). A similar modification of the corresponding site (position 371) in cinnamate hydroxylase (CYP73A1), another plant-specific P450 family, from Ile to Phe leads to a dramatic decrease in substrate binding and enzymatic activity, but without a major perturbation of protein folding (57).
The position corresponding to amino acid 486 in SRS-6 of EAH has also been extensively evaluated in mammalian hydroxylases. For instance, exchange of Gly478 of CYP2B1 with larger hydrophobic amino acids (Ala, Val, Ile, and Leu) alters the stereospecific hydroxylation of androstenedione (58, 59). In contrast, substitution of Phe for Ile at position 479 of CYP3A4 alters the regiospecific hydroxylation of 7-hexylcoumarin (60). Similarly, substitution of smaller hydrophobic amino acids (Ala, Val, and Leu) for Phe494 in CYP94A2 also alters the regiospecificity of lauric acid hydroxylation (61). Finally, docking putative substrates into molecular models of four phenyl propanoid metabolism P450 enzymes from Arabidopsis thaliana (CYP84A1, CYP75B1, CYP98A3, and CYP73A5) was used to map substrate contact points to positions equivalent to amino acids 368 and 486 (51), but these have yet to be functionally confirmed by site-directed mutagenesis studies.
This work, summarized in Table III, demonstrates that positions 368 and 486 in EAH are both critical for regulating successive hydroxylation reactions as well as dictating regio-and stereospecificity. Substitution of Cys for Ser at position 368 completely abolished the capacity of EAH to generate capsidiol from either EA or 1β(OH)EA, yet the initial hydroxylation reaction of EA to 1β(OH)EA was preserved. Position 486 likewise is critical for the successive hydroxylation reaction. Although the EAH-I486A mutant readily catalyzed the biosynthesis of 1β(OH)EA from EA, it was severely compromised for capsidiol biosynthesis. The latter limitation was in fact correlated with a significant reduction in the binding affinity for the monohydroxylated intermediate.
Table III.
Summary comparisons of mutations at Ser368 and Ile486 on the successive hydroxylation activity of EAH
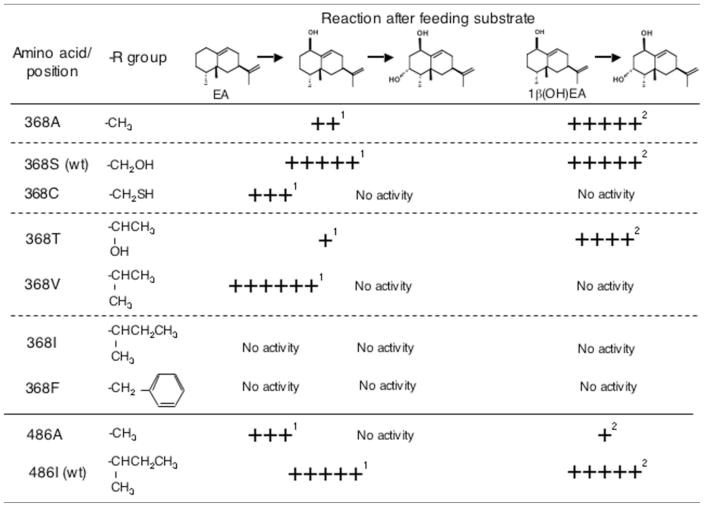 |
Calculated as percent of the wild-type kcat of 0.493.
Calculated as percent of the wild-type kcat of 0.582.
The diversity of hydroxylated products generated by the EAH-S368V mutant underscores its importance in controlling stereo- and regiospecific hydroxylation. Although, like EAH-I486A, this mutant is severely compromised in its ability to convert 1β(OH)EA to capsidiol, it has a total activity 2-fold greater than that of the wild-type enzyme and generates at least five novel hydroxylation products (Fig. 6). These new products represent unique stereospecific products (3β(OH)EA), regiospecific isomers (2α(OH)EA and 2β(OH)EA), and novel successive hydroxylation products (EA-1-one and EA-3-one) that must arise from multiple hydroxylation reactions at C-1 and C-3, respectively (Scheme 2).
Substrate docking experiments using the EAH homology model provide a basis to rationalize the catalytic specificities of EAH and the various Ser368 and Ile486 mutations (Fig. 8). Multiple docking solutions for EA as ligand cluster around two distinct binding modes in which the plane of the bicyclic ring is rotated by ~180°. In binding mode A (Fig. 8A), the β-face of EA is directed toward the heme center, placing C-1 in position for hydroxylation to produce 1β(OH)EA. In binding mode B (Fig. 8B), C-3 is now poised for hydroxylation on the α-face leading to 3α(OH)EA. It is apparent from these binding models that the α-face is more hindered, given the methyl group on C-4. Therefore, one might expect that hydroxylation on the β-face may proceed faster.
Fig. 8. Docking of EA and 1β(OH)EA into a modeled EAH active site.
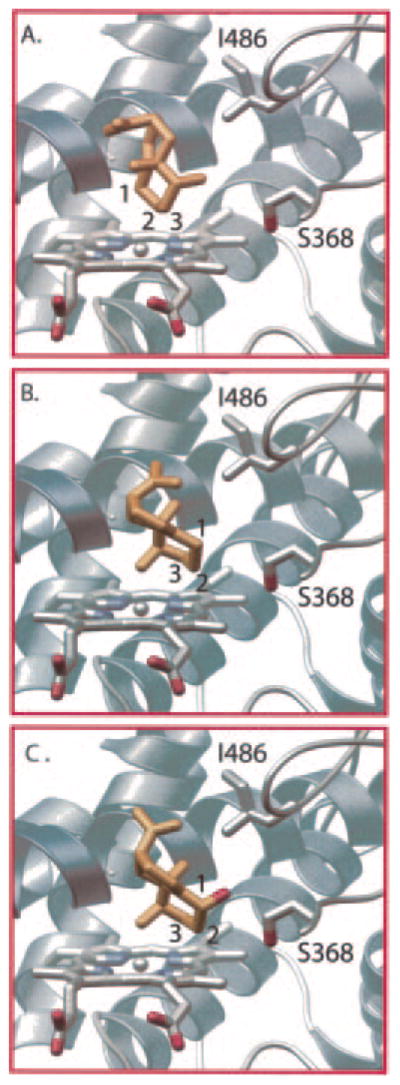
A homology model of EAH was created using the mammalian P450 2C5 (Protein Data Bank code 1DT6) as template and was used for docking experiments with EA and 1β(OH)EA ligands as described under “Experimental Procedures.” Two distinct binding modes of EA that position the β-face of EA toward the heme center for oxidation at C-1 (A) or that place the α-face toward the heme center for C-3 oxidation (B) were predicted from docking experiments. A single binding mode for 1β(OH)EA (C) that poises C-3 near the heme center for oxidation was predicted. The β-hydroxy is directed toward the hydroxyl group of Ser368 for possible hydrogen bonding interactions. Relevant carbon atoms are numbered, and important active-site residues are displayed and labeled.
Docking solutions using 1β(OH)EA as ligand cluster around a single binding mode, placing the β-hydroxy in the vicinity of Ser368 for a potential hydrogen bonding interaction (Fig. 8C). Docking solutions for the 3α(OH)EA ligand produced a single nonproductive binding mode that directs the 3α-hydroxy group toward the heme center and C-1 away (data not shown). A compelling explanation for the greater catalytic efficiency of EAH for 1β(OH)EA than for 3α(OH)EA is that the hydroxyl group of Ser368 anchors the 1β(OH)EA intermediate via a hydrogen bonding network (70) in a proper orientation for the second hydroxylation event. The EAH-S368A mutant may retain successive hydroxylation activity because it accommodates an additional water molecule in the active-site pocket along with the monohydroxylated intermediate, and this water molecule complements the missing hydroxyl function of the Ser or Thr side chain. Confirmation of a hydrogen bonding network or the presence of an additional water molecule in the active site of EAH-S368A will be difficult without direct structural information on the EAH enzyme complexed with substrate analogs (62, 63, 70, 71).
In summary, this work has established a preferred, kinetically discriminated reaction order for the successive hydroxylation of EA by EAH and demonstrated that successive hydroxylation shares important parallels with current models for how regio-and stereospecificity arise in cytochrome P450 enzymes. Sequence alignments of P450 enzymes, especially those representing the SRS regions (50), active-site residues (69), and those alignments between highly homologous but functionally distinct enzymes (56), have proven extremely useful in identifying structural features and amino acids contributing to such specificities of these enzymes. Elucidation of such specificities in the successive hydroxylation reactions of P450 enzymes such as EAH should also benefit from this comparative type of approach.
Supplementary Material
Acknowledgments
We thank Dr. Neil Fannin for valuable assistance with GC and GC/MS analyses and Dr. Balazs Siminszky for measurement of the CO difference spectrum.
Footnotes
The on-line version of this article (available at http://www.jbc.org) contains Supplemental Figs. 1 and 2.
The abbreviations used are: EA, 5-epiaristolochene; EAH, 5-epiaristolochene dihydroxylase; (OH)EA, hydroxy-5-epiaristolochene; GC/MS, gas chromatography/mass spectrometry; CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid; SRS, substrate recognition site.
The proper semi-systematic name for 5-epiaristolochene is 4-epi-eremophila-1(10),11(12)-diene. The common name 5-epiaristolochene relating the structure to (−)-aristolochene (4,5-di-epi-eremophila-9(10),11(12)-diene) is widely used in the literature and is adopted in this work for that reason.
This work was supported by National Institutes of Health Grant GM54029 (to J. P. N. and J. C.) and Grant GM13956 (to R. M. C.).
References
- 1.Stevens KL. Isopentenoids in Plants. Marcel Dekker, Inc; New York: 1984. pp. 63–80. [Google Scholar]
- 2.Kessler A, Baldwin IT. Science. 2001;291:2141–2144. doi: 10.1126/science.291.5511.2141. [DOI] [PubMed] [Google Scholar]
- 3.Gibson RW, Pickett JA. Nature. 1983;302:608–609. [Google Scholar]
- 4.Chappell J. Plant Physiol. 1995;107:1–6. doi: 10.1104/pp.107.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stoessl A, Stothers JB, Ward EWB. Phytochemistry. 1976;15:855–873. [Google Scholar]
- 6.Stoessl A, Unwin CH, Ward EWB. Phytopathology. 1973;63:1225–1231. [Google Scholar]
- 7.Ward EWB, Unwin CH, Stoessl A. Phytopathology. 1973;63:1537–1538. [Google Scholar]
- 8.Guedes MEM, Kuc J, Hammerschmidt R, Bostock R. Phytochemistry. 1982;21:2987–2988. [Google Scholar]
- 9.Watson DG, Rycroft DS, Freer IM, Brooks CJW. Phytochemistry. 1985;24:2195–2200. [Google Scholar]
- 10.Chappell J, Nable R, Fleming P, Andersen RA, Burton HR. Phytochemistry. 1987;26:2259–2260. [Google Scholar]
- 11.Stoessl A, Unwin CH, Ward EWB. Phytopathol Z. 1972;74:141–152. [Google Scholar]
- 12.Vögeli U, Chappell J. Plant Physiol. 1988;88:1291–1296. doi: 10.1104/pp.88.4.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Facchini PJ, Chappell J. Proc Natl Acad Sci U S A. 1992;89:11088–11092. doi: 10.1073/pnas.89.22.11088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ralston L, Kwon ST, Schoenbeck M, Ralston J, Schenk DJ, Coates RM, Chappell J. Arch Biochem Biophys. 2001;393:222–235. doi: 10.1006/abbi.2001.2483. [DOI] [PubMed] [Google Scholar]
- 15.Lupien S, Karp F, Wildung M, Croteau R. Arch Biochem Biophys. 1999;368:181–192. doi: 10.1006/abbi.1999.1298. [DOI] [PubMed] [Google Scholar]
- 16.Wüst M, Little DB, Schalk M, Croteau R. Arch Biochem Biophys. 2001;387:125–136. doi: 10.1006/abbi.2000.2248. [DOI] [PubMed] [Google Scholar]
- 17.Teutsch HG, Hasenfratz MP, Lesot A, Stoltz C, Garnier JM, Jeltsch JM, Durst F, Werck-Reichhart D. Proc Natl Acad Sci U S A. 1993;90:4102–4106. doi: 10.1073/pnas.90.9.4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyer K, Cusumano JC, Somerville C, Chapple C. Proc Natl Acad Sci U S A. 1996;93:6869–6874. doi: 10.1073/pnas.93.14.6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Humphreys JM, Hemm MR, Chapple C. Proc Natl Acad Sci U S A. 1999;96:10045–10050. doi: 10.1073/pnas.96.18.10045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schoendorf A, Rithner CD, Williams RM, Croteau RB. Proc Natl Acad Sci U S A. 2001;98:1501–1506. doi: 10.1073/pnas.98.4.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jennewein S, Rithner CD, Williams RM, Croteau RB. Proc Natl Acad Sci U S A. 2001;98:13595–13600. doi: 10.1073/pnas.251539398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jennewein S, Rithner CD, Williams RM, Croteau R. Arch Biochem Biophys. 2003;413:262–270. doi: 10.1016/s0003-9861(03)00090-0. [DOI] [PubMed] [Google Scholar]
- 23.Frey M, Chomet P, Glawischnig E, Stettner C, Grun S, Winklmair A, Eisenreich W, Bacher A, Meeley RB, Briggs SP, Simcox K, Gierl A. Science. 1997;277:696–699. doi: 10.1126/science.277.5326.696. [DOI] [PubMed] [Google Scholar]
- 24.Nomura T, Ishihara A, Imaishi H, Endo TR, Ohkawa H, Iwamura H. Mol Genet Genomics. 2002;267:210–217. doi: 10.1007/s00438-002-0653-x. [DOI] [PubMed] [Google Scholar]
- 25.Szekeres M, Nemeth K, Koncz-Kalman Z, Mathur J, Kauschmann A, Altmann T, Redei GP, Nagy F, Schell J, Koncz C. Cell. 1996;85:171–182. doi: 10.1016/s0092-8674(00)81094-6. [DOI] [PubMed] [Google Scholar]
- 26.Choe S, Dilkes BP, Fujioka S, Takatsuto S, Sakurai A, Feldmann KA. Plant Cell. 1998;10:231–243. doi: 10.1105/tpc.10.2.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koch BM, Sibbesen O, Halkier BA, Svendsen I, Moller BL. Arch Biochem Biophys. 1995;323:177–186. doi: 10.1006/abbi.1995.0024. [DOI] [PubMed] [Google Scholar]
- 28.Kaltenbach M, Schroder G, Schmelzer E, Lutz V, Schroder J. Plant J. 1999;19:183–193. doi: 10.1046/j.1365-313x.1999.00524.x. [DOI] [PubMed] [Google Scholar]
- 29.Menting J, Scopes RK, Stevenson TW. Plant Physiol. 1994;106:633–642. doi: 10.1104/pp.106.2.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu YL, Li L, Wu K, Peeters AJ, Gage DA, Zeevaart JA. Proc Natl Acad Sci U S A. 1995;92:6640–6644. doi: 10.1073/pnas.92.14.6640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Phillips AL, Ward DA, Uknes S, Appleford NE, Lange T, Huttly AK, Gaskin P, Graebe JE, Hedden P. Plant Physiol. 1995;108:1049–1057. doi: 10.1104/pp.108.3.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Helliwell CA, Poole A, Peacock WJ, Dennis ES. Plant Physiol. 1999;119:507–510. doi: 10.1104/pp.119.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Helliwell CA, Chandler PM, Poole A, Dennis ES, Peacock WJ. Proc Natl Acad Sci U S A. 2001;98:2065–2070. doi: 10.1073/pnas.041588998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Watson DG, Baker FC, Brooks CJW. Biochem Soc Trans. 1983;11:589–590. [Google Scholar]
- 35.Whitehead IM, Threlfall DR, Ewing DF. Phytochemistry. 1989;28:775–779. [Google Scholar]
- 36.Williams PA, Cosme J, Sridhar V, Johnson EF, McRee DE. Mol Cell. 2000;5:121–131. doi: 10.1016/s1097-2765(00)80408-6. [DOI] [PubMed] [Google Scholar]
- 37.Zhao Y, Schenk DJ, Takahashi S, Chappell J, Coates RM. J Org Chem. 2004;69:7428–7435. doi: 10.1021/jo049058c. [DOI] [PubMed] [Google Scholar]
- 38.Rising KA, Starks CM, Noel JP, Chappell J. J Am Chem Soc. 2000;122:1861–1866. [Google Scholar]
- 39.Pompon D, Louerat B, Bronine A, Urban P. Methods Enzymol. 1996;272:51–64. doi: 10.1016/s0076-6879(96)72008-6. [DOI] [PubMed] [Google Scholar]
- 40.Urban P, Mignotte C, Kazmaier M, Delorme F, Pompon D. J Biol Chem. 1997;272:19176–19186. doi: 10.1074/jbc.272.31.19176. [DOI] [PubMed] [Google Scholar]
- 41.Greenhagen BT, Griggs P, Takahashi S, Ralston L, Chappell J. Arch Biochem Biophys. 2003;409:385–394. doi: 10.1016/s0003-9861(02)00613-6. [DOI] [PubMed] [Google Scholar]
- 42.Omura T, Sato R. J Biol Chem. 1964;239:2370–2378. [PubMed] [Google Scholar]
- 43.Marti-Renom MA, Stuart A, Fiser A, Sánchez R, Melo F, Sali A. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 44.Sali A, Blundell TL. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 45.Fiser A, Do RK, Sali A. Protein Sci. 2000;9:1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jones G, Willett P, Glen RC. J Mol Biol. 1995;245:43–53. doi: 10.1016/s0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
- 47.Jones G, Willett P, Glen RC, Leach AR, Taylor R. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 48.Nissink JWM, Murray C, Hartshorn M, Verdonk ML, Cole JC, Taylor R. Proteins. 2002;49:457–471. doi: 10.1002/prot.10232. [DOI] [PubMed] [Google Scholar]
- 49.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Proteins. 2003;52:609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 50.Gotoh O. J Biol Chem. 1992;267:83–90. [PubMed] [Google Scholar]
- 51.Rupasinghe S, Baudry J, Schuler MA. Protein Eng. 2003;16:721–731. doi: 10.1093/protein/gzg094. [DOI] [PubMed] [Google Scholar]
- 52.He Y, Luo Z, Klekotka PA, Burnett VL, Halpert JR. Biochemistry. 1994;33:4419–4424. doi: 10.1021/bi00180a040. [DOI] [PubMed] [Google Scholar]
- 53.Hasler JA, Harlow GR, Szklarz GD, John GH, Kedzie KM, Burnett VL, He YA, Kaminsky LS, Halpert JR. Mol Pharmacol. 1994;46:338–345. [PubMed] [Google Scholar]
- 54.Pelkonen P, Lang MA, Negishi M, Wild CP, Juvonen RO. Chem Res Toxicol. 1997;10:85–90. doi: 10.1021/tx960078m. [DOI] [PubMed] [Google Scholar]
- 55.He YA, He YQ, Szklarz GD, Halpert JR. Biochemistry. 1997;22:8831–8839. doi: 10.1021/bi970182i. [DOI] [PubMed] [Google Scholar]
- 56.Schalk M, Croteau R. Proc Natl Acad Sci U S A. 2000;97:11948–11953. doi: 10.1073/pnas.97.22.11948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schoch GA, Attias R, Le Ret M, Werck-Reichhart D. Eur J Biochem. 2003;270:3684–3695. doi: 10.1046/j.1432-1033.2003.03739.x. [DOI] [PubMed] [Google Scholar]
- 58.Kedzie KM, Balfour CA, Escobar GY, Grimm SW, He YA, Pepperl DJ, Regan JW, Stevens JC, Halpert JR. J Biol Chem. 1991;266:22515–22521. [PubMed] [Google Scholar]
- 59.He YA, Balfour CA, Kedzie KM, Halpert JR. Biochemistry. 1992;31:9220–9226. doi: 10.1021/bi00153a015. [DOI] [PubMed] [Google Scholar]
- 60.Khan KK, Halpert JR. Arch Biochem Biophys. 2000;373:335–345. doi: 10.1006/abbi.1999.1578. [DOI] [PubMed] [Google Scholar]
- 61.Kahn RA, Le Bouquin R, Pinot F, Benveniste I, Durst F. Arch Biochem Biophys. 2001;15:180–187. doi: 10.1006/abbi.2001.2415. [DOI] [PubMed] [Google Scholar]
- 62.Williams PA, Cosme J, Ward A, Angove HC, Matak VD, Jhoti H. Nature. 2003;424:464–468. doi: 10.1038/nature01862. [DOI] [PubMed] [Google Scholar]
- 63.Williams PA, Cosme J, Vinkovic DM, Ward A, Angove HC, Day PJ, Vonrhein C, Tickle IJ, Jhoti H. Science. 2004;305:683–686. doi: 10.1126/science.1099736. [DOI] [PubMed] [Google Scholar]
- 64.Sawada N, Sakaki T, Ohta M, Inouye K. Biochem Biophys Res Commun. 2000;273:977–984. doi: 10.1006/bbrc.2000.3050. [DOI] [PubMed] [Google Scholar]
- 65.Pikuleva I, Javitt NB. Arch Biochem Biophys. 2003;420:35–39. doi: 10.1016/j.abb.2003.09.028. [DOI] [PubMed] [Google Scholar]
- 66.Imai T, Yamazaki T, Kominami S. Biochemistry. 1998;37:8097–8104. doi: 10.1021/bi9802768. [DOI] [PubMed] [Google Scholar]
- 67.Yamazaki T, Ohno T, Sakaki T, Akiyoshi-Shibata M, Yabusaki Y, Imai T, Kominami S. Biochemistry. 1998;37:2800–2806. doi: 10.1021/bi9715066. [DOI] [PubMed] [Google Scholar]
- 68.Lewis DFV. J Inorg Biochem. 2002;91:502–514. doi: 10.1016/s0162-0134(02)00429-4. [DOI] [PubMed] [Google Scholar]
- 69.Kumar S, Scott EE, Liu H, Halpert JR. J Biol Chem. 2003;278:17178–17184. doi: 10.1074/jbc.M212515200. [DOI] [PubMed] [Google Scholar]
- 70.Wester MR, Johnson EF, Marques-Soares C, Dijols S, Dansette PM, Mansuy D, Stout CD. Biochemistry. 2003;42:9335–9345. doi: 10.1021/bi034556l. [DOI] [PubMed] [Google Scholar]
- 71.Scott EE, White MA, He YA, Johnson EF, Stout CD, Halpert JR. J Biol Chem. 2004;279:27294–27301. doi: 10.1074/jbc.M403349200. [DOI] [PubMed] [Google Scholar]
- 72.Wang E, Wang R, DeParasis J, Loughrin JH, Gan S, Wagner GJ. Nat Biotechnol. 2001;19:371–374. doi: 10.1038/86770. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.