

Abstract
Background
Atopic dermatitis (AD) is a chronic inflammatory dermatosis now increasingly linked to mutations that alter the structure and function of the stratum corneum (SC). Activators of peroxisome proliferators-activated receptor (PPAR)α, β/δ, γ and liver-X-receptor (LXR) regulate epidermal protein and lipid production, leading to superior barrier function. Additionally, some of these activators exhibit potent anti-hyperplastic and anti-inflammatory activity in irritant contact dermatitis and acute allergic contact dermatitis mouse models.
Objective
We evaluated the efficacy of PPAR/LXR activation in a hapten (oxazolone [Ox])-induced atopic dermatitis-like model (Ox-AD) in hairless mice.
Methods
Ox-AD was established with ten Ox challenges (every-other day) on the flank. After the establishment of Ox-AD, twice daily topical application with individual PPAR/LXR activators was then performed for 4 days, with continued Ox challenges every other day. The efficacy of topical PPAR/LXR activators to reduce parameters of Ox-AD was assessed physiologically, morphologically and immunologically.
Results
Certain topical activators of PPARα, PPARβ/δ, and LXR, but not activators of PPARγ, reversed the clinical dermatosis, significantly improved barrier function, and increased SC hydration in Ox-AD mice. In addition, the same activators, but again not PPARγ, largely reversed the immunologic abnormalities in Ox-AD mice, including the elevated TH2 markers, such as tissue eosinophil/mast cell density, serum TARC levels, density of CRTH2-positive lymphocytes (but not serum IgE levels), and reduced IL-1α and TNFα activation, despite on-going hapten challenges.
Conclusion
These results suggest that topical applications of certain activators/ligands of PPARα, β/δ, and LXR could be useful for the treatment of AD in humans.
Keywords: atopic dermatitis, barrier function, LXR, mouse model, PPARα, PPARβ/δ, PPARγ, TH2 cells
Key Message
-
#
Certain activators of PPARs and LXR display beneficial effects in an animal model of atopic dermatitis.
-
#
Thus, PPARα, β/δ, and LXR activators could prove useful for the treatment of atopic dermatitis in humans.
INTRODUCTION
Though long considered a primary immunologic disorder, atopic dermatitis (AD) exhibits prominent abnormalities in permeability barrier function that we and others have suspected play a role in disease pathogenesis1-4. Notably, even the uninvolved skin of atopics exhibits abnormal water permeability5-7. These suspicions have been confirmed by recent molecular genetic investigations, which have identified a strong association between inherited mutations in the gene that encodes the corneocyte structural protein, filaggrin, and AD8-10. Moreover, AD is characterized by a deterioration in other epidermal protective functions, including SC cohesion11, antimicrobial defense12, and decreased SC hydration6, 7, which further complicates disease management.
Despite compelling evidence for a primary, barrier-based abnormality, therapy for human AD is still largely directed at downstream immunologic abnormalities. While topical glucocorticoids can be beset with unacceptable side effects, topical immunomodulators are only moderately effective, and could result in long-term risks13, 14. Thus, there is a strong need for alternate therapies that are not only safe and effective, but also directed at correcting the barrier dysfunction that ‘drives’ AD.
Activators of PPARα, β/δ, γ and LXRα/β, display potent, largely-positive effects on epidermal structure and function, including upregulation of filaggrin [rev. in 15]. Moreover, they display substantial anti-inflammatory activity in murine models of both irritant and acute allergic contact dermatitis16, 17, and they potently reverse epidermal hyperplasia and normalize epidermal differentiation in hyperproliferative murine disease models18. Because the endogenous activators of these receptors are naturally-occurring lipids that can be generated within the epidermis (e.g., free fatty acids, eicosanoids, and oxygenated sterols), these nuclear hormone receptors could represent key regulators of epidermal homeostasis15. Since human AD exhibits primary abnormalities in epidermal barrier function, resulting in downstream epidermal hyperplasia, aberrant differentiation, and TH2-dominant reactions, the PPAR/LXR activators, in theory, possess a profile of activity that suggests potential utility in AD. We recently described a hapten-induced AD-like model that recapitulates a large spectrum of the epidermal and immunologic abnormalities of AD in humans19, including a prominent TH2 infiltrate. Hence, we evaluated here several PPAR/LXR activators in this model, identifying which classes of agents demonstrate apparent clinical benefit, and the extent to which these activators reverse the structural, functional, and immunologic abnormalities in affected mice. Our results show that certain activators of LXRα/β, PPARα and PPARβ/δ display broad efficacy, while PPARγ activators exhibited little activity in this AD model.
METHODS
Animals and Materials
Female hairless mice (hr/hr), aged 6 – 8 weeks old, were purchased from Charles River laboratories (Wilmington, MA, USA) and fed mouse diet (Ralston-Purina Co., St Louis, MO, USA) and water ad libitum. WY14643 (PPAR α activator), clofibrate (PPAR α activator), T0901317 (LXR activator), 22(R)-hydroxycholesterol (LXR activator), clobetasol propionate, and oxazolone were purchased from Sigma Chemical Co. (St. Louis, MO, USA). GW7647 (PPAR α activator), GW0742 (PPAR β/δ activator), GW1929 (PPARγ activator) and GW3965 (LXR activator) were purchased from TOCRIS Bioscience (Ellisville, MO, USA). Ciglitazone (PPARγ activator) was purchased from Cayman Chemical (Ann Arbor, MI, USA). GW1514 (PPAR β/δ activator) was a gift from Dr. Timothy Willson (Glaxo-SmithKline, Triangle Park, NC, USA). Rabbit anti-mouse antibody against the prostaglandin D receptor, CRTH2/DP2, was from Cayman Chemical (Ann Arbor, MI, USA). Goat anti-mouse antibody against IL-1 α was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit anti-human antibody against CD3 was purchased from Dako (Glostrup, Denmark). Biotinylated goat anti-rabbit IgG antibody and biotinylated horse anti-goat antibody were purchased from Vector Laboratories (Burlingame, CA, USA). Biotinylated monoclonal antibody against proliferating cell nuclear antigen (PCNA) was purchased from CalTag Laboratories (Burlingame, CA, USA).
Experimental Protocols and Functional Studies
All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the San Francisco Veterans Administration Medical Center and performed in accordance with their guidelines. Animals were sensitized by a single topical treatment with 50 μl of 1% oxazolone. After one week, they were treated topically with 60 μl of 0.1% oxazolone to both flanks once every other day for an additional four weeks (total of 12 challenges). After the 10th challenge, when the phenotype of AD-like, chronic allergic dermatitis was established, the therapeutic effects of activators of nuclear hormone receptors were performed as follows: 1h after the 11th challenge, twice daily application of activators (20 μl) of 10mM WY14643, 10mM GW7647, 1mM clofibrate, 4mM GW1514, 10mM GW0742, 10mM ciglitazone, 10mM GW1929, 10mM T0901317, 10mM GW3965, 10mM 22(R)-hydroxycholesterol, and 0.05% clobetasol propionate in vehicle (propylene glycol:ethanol, 7:3) were performed for 4 days; the 12th challenge with oxazolone was performed 1h before the first application of the activator or vehicle on that day. Topical clobetasol, a super-potent, class 1, topical glucocorticoid (GC), with proven efficacy in human AD, served as a positive control, while another Ox-AD group was treated with vehicle alone. Basal TEWL was measured with an electrolytic water analyzer (Meeco, Warrington, PA, USA) and SC hydration assessed as capacitance, was measured with a Corneometer CM820 (Courage & Khazaka, Germany), as described previously20. SC surface pH was measured with a flat, glass surface electrode from Mettler-Toledo (Giessen, Germany), attached to a pH meter (PH900; Courage & Khazaka, Cologne, Germany), as described previously21. These physiological measurements were performed immediately before the 11th challenge and 48h after the 12th challenge with oxazolone. Skin samples were collected 48h after the 12th challenge with Ox(day 5). Blood samples were collected immediately before the 11th challenge with Ox and 48h after the 12th challenge (i.e. after liposensor treatments).
Immunohistochemistry
Immunohistochemical staining for CRTH2 and IL-1 α was performed, as described previously22. Briefly, 5 μm paraffin sections were incubated with the primary antibodies overnight at 4°C. After washes × 3, sections were incubated with the secondary antibody for 30 min. Staining was detected with the ABC-peroxidase kit from Vector Lab. To detect proliferating cells by PCNA staining, 5μm paraffin sections were incubated with the biotinylated monoclonal antibody against the Ki-67 antigen overnight at 4°C, and staining was detected by the ABC-peroxidase method. Sections were examined with a light microscope Carl Zeiss (Jena, Germany), and digital images were captured with AxioVision software (Carl Zeiss Vision, Munich, Germany).
Quantitative Morphology
The density of CRTH2-positive cells, eosinophils assessed in hematoxylin & eosin stained sections, mast cells detected by toluidine blue stain, in an area of 220μm × 170 μm were counted in more than 15 fields in the dermis of each sample. The thickness of epidermal nucleated layers was measured with AxioVision software (Carl Zeiss Vision, Munich, Germany) in hematoxylin & eosin sections; measurements were performed in more than 15 fields at intervals of 100μm in each sample. The number of PCNA-positive cells, observed within a 50μm length of epidermis, was counted in more than 10 sites for each sample; data are reported as the mean ± SEM.
Serum IgE and TARC Measurements
Blood samples were collected from mice tails before and at the end of the therapeutic protocols described above. Serum IgE and TARC concentrations were determined by ELISA with a mouse IgE quantitation kit from Bethyl Laboratories (Montgomery, TX, USA) and Quantikine® for mouse CCL17/TARC immunoassay from R&D system (Minneapolis, MN, USA), according to the manufacturer’s instructions.
Electron Microscopy
Skin biopsies of both vehicle- and Ox-treated mice were fixed in Karnovsky’s fixative overnight, and post-fixed with either 0.25% ruthenium tetroxide or 1% aqueous osmium tetroxide, containing 1.5% potassium ferrocyanide, as described previously23. Ultrathin sections were examined using an electron microscope (Zeiss 10A, Carl Zeiss, Thornwood, NY), operated at 60 kV.
Zymographic Assessment of Enzyme Activity
Serine protease (SP) activity was assessed in freshly-obtained skin samples by in situ zymography, as described previously24. Five (5) μm frozen sections were incubated with BODIPY-FI0-casein for 2 h at 37°C. After 3x washing with 1% Tween-20, sections were counter-stained with propidium iodide, and examined with the confocal microscope, as above.
Statistical Analysis
The two-tailed Student’s t-test was employed to determine significant differences between two groups. A further ANOVA analysis was calculated, followed by an alpha-corrected post hoc test (Bonferroni), when three or more groups were compared. Data are expressed as mean ± SEM.
RESULTS
Macroscopic and Histologic Response of Ox-AD to Liposensor Activators
We showed previously that sensitization of hairless mice with the hapten, oxazolone (Ox), followed by 10 challenges over 20/21 days, leads to a dermatosis with global epidermal and immunologic features of human AD (Ox-AD mice)19. Here, we applied two or three different activators of PPARα, β/δ, γ, and LXR, twice daily for four days to Ox-AD mice with established dermatitis (i.e., having received 10 prior challenges). Ox-AD mice also continued to receive regularly-scheduled, Ox challenge doses (every-other day x2) during the treatment phase. Ox-AD mice treated with two different synthetic activators of LXR (but not the endogenous activator, 22-rOH cholesterol); two different synthetic ligands of PPARα (but not clofibrate); and two different synthetic ligands of PPARβ/δ showed grossly (clinically) improved dermatitis, despite ongoing Ox challenges (Fig. 1A; see supplemental Fig. 1S in the Online for clinical effects of other activators). Likewise, dermatitis cleared in clobetasol-treated mice, but cutaneous atrophy (telangiectasia, fine wrinkling) became evident by day five of treatment. In contrast, two different, synthetic PPARγ activators (ciglitazone and GW1929) largely failed to improve Ox-AD mice (Fig. 1A; Fig. 1S in the Online).
Figure 1. A: PPARα, β/δ, and LXR Activators Reverse Murine Ox-AD.
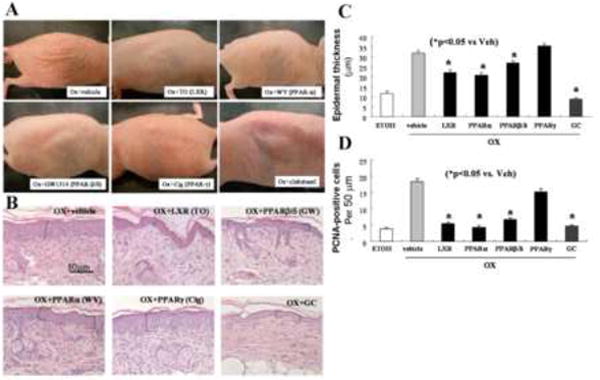
Gross appearance after applications of ligands for LXR(10mM T0901317), PPARα(10mM WY14643), β/δ(4mM GW1514), and γ (10mM ciglitazone), and glucocorticoid (GC; 0.05% clobetasol). As a vehicle control (Ox+vehicle), propylene glycol and ethanol (7:3) alone was applied. B: Histological appearance after treatment with ligands for LXR, PPARα, β/δ and γ, and the glucocorticoid (GC), clobetasol. (H&E staining) C: Quantitative changes in epidermal hyperplasia, in LXR, PPARα, β/δ, and γ ligands, and the glucocorticoid (GC), clobetasol - treated mice were assessed in coded, randomized micrographs (see Methods). D: PCNA-positive cells counts (per 50 μm) were quantitated as described in Methods. ETOH: Normal skin in which ethanol was applied instead of Ox (n=30 measurements each from 3 separate samples for epidermal hyperplasia assessment; and 22-27 measurements each for PCNA assessment).
The macroscopic (clinical) response to the activators was mirrored by changes in histology. While Ox-AD mice treated with vehicle alone showed both prominent epidermal hyperplasia and a dense inflammatory infiltrate, the histologic appearance of LXRα/β, PPARα, and PPARβ/δ activator-treated (but not PPARγ-treated) Ox-AD mice improved (Fig. 1B), with the greatest reductions in both epidermal hyperplasia and inflammation occurring in mice treated with the two synthetic LXR activators (TO901317 and GW3965), and with the two synthetic PPARα activators (WY14643 and GW7647), and the two synthetic PPARβ/δ activators (GW1514 and GW0742). (Quantitative data on epidermal hyperplasia and in PCNA immunostaining are in Figs. 1C&D, supplemental Fig. 2S). However, Ox-AD mice treated with the PPARγ activator, ciglitazone, demonstrated no decline in either epidermal thickness or inflammation, while in contrast, clobetasol-treated mice showed the greatest reduction in inflammation and epidermal thickness (Fig. 1C), consistent with the clinically-apparent atrophy produced by this drug (c.f., Fig. 1A). Together, these results show that activators of LXRα/β, PPARα, and PPARβ/δ normalize or improve epidermal hyperplasia and histologic evidence of inflammation in the established dermatitis of Ox-AD mice.
PPAR/LXR Activators Normalize Epidermal Structure and Function
Ox-AD mice, like humans with AD, display characteristic abnormalities in epidermal structure and function19. While barrier function deteriorates, indicated by elevated transcutaneous water loss (TEWL) rates, SC hydration declines, and surface pH increases, approaching neutrality, as in human AD. Therefore, we next assessed whether the PPAR/LXR activators reverse these functional abnormalities in Ox-AD mice. In parallel with the clinical and histologic findings, Ox-AD mice treated with the two synthetic LXRα/β ligands (but not 22ROH-chol), two of the three PPARα ligands (but not clofibrate), and the two PPARβ/δ activators, demonstrated significant reductions in both TEWL (Fig. 2A) and surface pH (Fig. 2C; Fig. 3S in the Online for functional data for additional ligands). Although the changes in TEWL were largest in Ox-AD mice treated with LXR, PPARα, and PPARβ/δ activators, only the LXR activators and one PPARα activator (GW7647) significantly increased SC hydration (towards normal) (Fig. 2B; Fig. 3S in the Online). Pertinently, clobetasol also normalized barrier function, increased SC hydration, and lowered SC pH, despite the above-described evidence of cutaneous atrophy (Figs. 1, 2, Fig. 2S in the Online). In contrast, PPARγ activators improved none of the functional parameters (Figs. 2A-C; Figs. 2S&3S in the Online). These results demonstrate that certain PPAR/LXR activators, but not PPARγ activators, normalize and/or improve epidermal function in Ox-AD mice.
Figure 2. Ligands of LXR, PPARα, β/δ, and Clobetasol Normalize Epidermal Function.
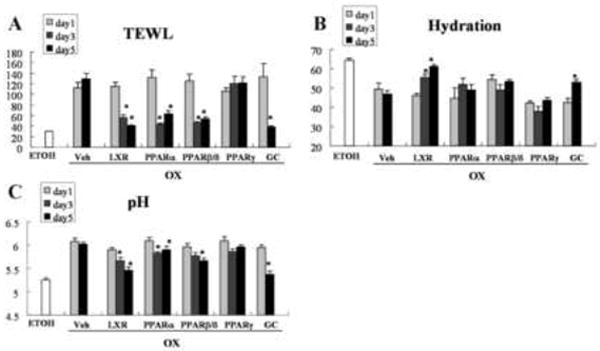
Epidermal barrier function, assessed as transepidermal water loss (TEWL), surface pH, and SC hydration were measured and topical applications of the LXR, PPARα, β/δ and PPARγ ligands, as well as the glucocorticoid (GC), clobetasol propionate, and the vehicle (Veh) were performed as described in Methods. ETOH = control group normal mice. Each experiment was repeated twice and representative data are displayed. For TEWL and SC hydration, n=24 measurements from 5 different animals in each group; for pH n=16 measurements from the same animals. * p<0.05 (day 1 vs. either day 3 or day 5).
The structural basis for the permeability barrier abnormality in Ox-AD mice (and in human AD) in part reflects a failure of lamellar bodies to undergo complete exocytosis25, resulting in a paucity of extracellular lamellae19, 26. The further, selective decline of ceramides in human AD has been ascribed either to diminished activity of Cer-generating enzymes (i.e. β-glucocerebrosidase and acidic sphingomyelinase)27, 28, or to accelerated destruction of Cer precursors29, 30. Our prior studies support the first mechanism (i.e., that a pH-induced increase in serine protease activity could account for abnormal lamellar bilayers in Ox-AD mice both by degrading lipid-processing enzymes, and by blockade of lamellar body secretion)24, 31. Thus, we assessed whether the PPAR/LXR ligands normalized 1) lamellar body secretion; and 2) the post-secretory processing of secreted ceramide precursors (Figs. 3A-E). Treatment with the synthetic LXR activator (T0901317) restored normal quantities of secreted lamellar body contents to the SC interstices (Fig. 3E vs. C&D), and these secreted contents then demonstrated a normal sequence of progressive maturation into mature lamellar bilayers (Figs. 3B vs. A).
Figure 3. Topical LXR Ligand Normalizes Lamellar Body Secretion and Lamellar Bilayer Structure in Ox-AD Mice.
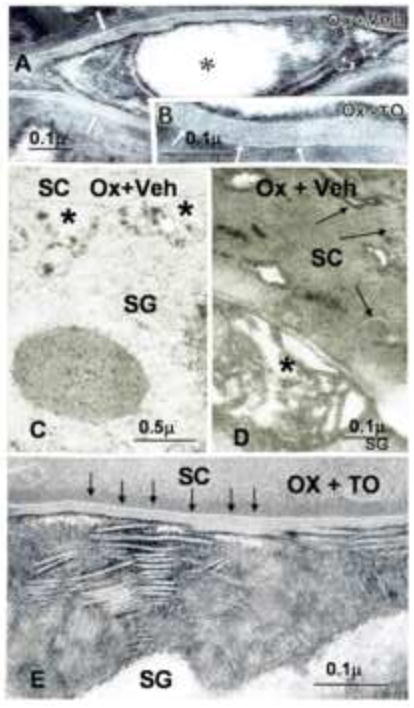
Stratum corneum of oxalozone (Ox) + vehicle (Veh)-treated skin sites demonstrate incomplete formation of lamellar bilayers (Panel A, open arrows and asterisks), while Ox + LXR ligand (TO; TO901317)-treated sites display both normal quantities of secreted lipid (Panel E) and normal organization of lamellar bilayers (Panels B&E, arrows). Decreased lamellar body secretion is indicated by paucity of lamellae at stratum granulosum (SG)-SC interface (Panel C, asterisks). As a result of decreased secretion, abundant lamellar body contents remain entombed in corneocytes (Panel D, arrows). A, B, D, E, ruthenium tetroxide post-fixation; C, osmium tetroxide post-fixative. Mag bars (A) = 0.2 μm (B) = 0.1 μm. (C) = 0.5 μm; (D) = 0.1 μm; (E) = 0.1 μm.
Both the LXR activator (T0901317) and the PPARα activator (WY16463), that provoked the greatest declines in SC pH (c.f., Fig. 2C), reduced serine protease activity, while the LXR activator further restricted activity to a narrow, but intense band at the SG-SC interface (Fig. 4 – data for WY compound not shown). The reduction of serine protease activity normalized the lamellar body secretion31, providing a biochemical basis for the restoration of normal lamellar membrane structures by certain liposensor activators.
Figure 4. Topical LXR Ligand Reduces Serine Protease Activity.
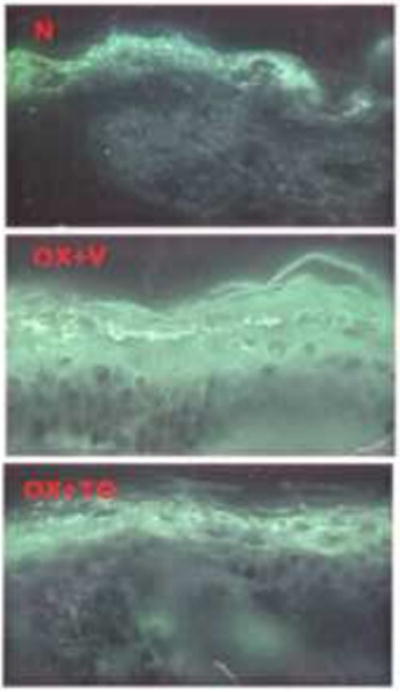
Serine protease activity, assessed zymographically, in frozen sections of normal+ethanol vehicle (N), Ox+vehicle (V), and Ox+ LXR ligand, TO901317 (TO).
PPAR/LXR Activators Decrease Inflammation, Including TH2 Immunophenotype, in Ox-AD Mice
Prior studies have shown that LXRα/β, as well as PPARα, -β/δ, and/or -γ activators, improve inflammation in several different models of inflammatory dermatoses; i.e., psoriasiform hyperplasia, irritant contact dermatitis; and hapten (Ox)-induced, acute allergic contact dermatitis15; however, at least some of the anti-inflammatory effects of PPARγ activators were receptor-independent, because they occurred even in PPARγ-deficient (ko) mice32. Therefore, we next assessed whether the PPAR/LXR activators improve inflammatory/immunologic parameters in Ox-AD mice, assessing various immune end-points five days after twice-daily, liposensor-activator treatment in parallel with ongoing Ox challenges. We first assessed two general indicators of inflammation; i.e., tissue eosinophilia and mast cell density/degranulation. The LXRα (T0901317), PPARα (WY16463), PPARβ/δ (GW1514), and PPARγ (ciglitazone) activators, as well as the super-potent, topical glucorticoid, clobetasol, each lowered tissue eosinophilia levels significantly (LXR and PPARγ shown as Fig. 5A). Likewise, the same activators (except the PPARγ activator) decreased mast cell density and the extent of mast cell degranulation (except the PPARγ activator) (Fig. 5B). These observations were confirmed by quantitative assessment in randomized, coded micrographs (Figs. 5C&D). Together, these results show that certain PPARα, β/δ, and LXR activators decrease both cutaneous eosinophilia and mast cell density in Ox-AD mice.
Figure 5. Topical PPAR/LXR Activators Decrease Tissue Eosinophilia and Mast Cells in Ox-AD Mice.
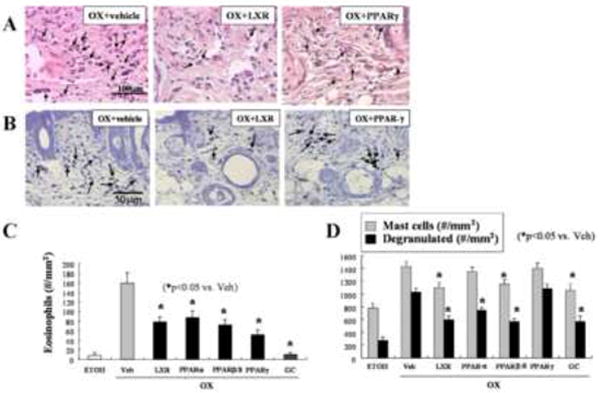
Eosinophils were stained in paraffin-embedded with hematoxylin & eosin (Panel A, arrows), while mast cells were stained with toluidine blue. (Panel B, arrows). Panels C&D: Quantitation of tissue eosinophils (C) and mast cells numbers (and degranulated mast cell numbers) per mm2 (D) was performed as described in Methods.
We next assessed more certain parameters of TH2-mediated inflammation in PPAR/LXR-treated Ox-AD mice, using serum TARC levels and the density of CRTH2-positive cells as markers for TH2 immune status. The LXRα/β (T0901317), PPARα (WY16463), and PPARβ/δ (GW1514) activators, as well as clobetasol, significantly reduced the levels of both of these markers (Figs. 6A-C). In contrast, a PPARγ activator (ciglitazone) reduced neither CRTH2 cell density nor serum TARC. Yet, neither the topical PPAR/LXR activators nor topical clobetasol significantly reduced serum IgE levels in Ox-AD mice (Fig. 6D). Together, these results show that the PPAR/LXR activators exhibit potent anti-inflammatory activities against specific components of the immune response in Ox-AD mice.
Figure 6. PPARα, β/δ, and LXR Ligands Normalize TH2 Inflammation in Ox-AD Mice.
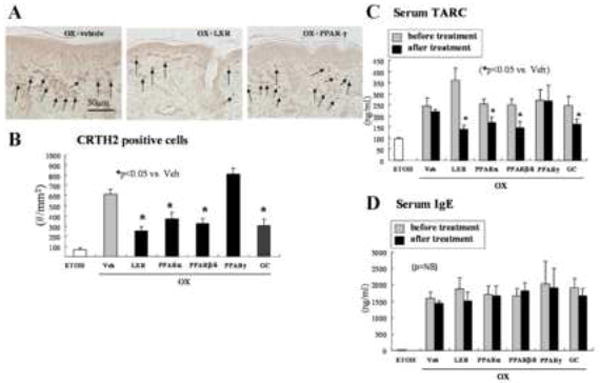
Skin samples and blood were collected as described in Methods for CRTH2 immunostaining (A), CRTH2-positive cell counts (B), serum TARC (C) and IgE level (D). ETOH: Ethanol-treated sites in normal mice, as control for Ox treatment. CRTH2: n=30; TARC: n=6-7; IgE: n=3-5.
PPAR/LXR Activators Decrease Generation of IL-1α and TNFα
Barrier disruption leads to increased production of epidermal cytokines, a pro-inflammatory mechanism linked to the barrier-initiated (‘outside-inside’) pathogenesis of AD [rev. in 33]. We next assessed whether PPAR/LXR treatment decreased cytokine generation in parallel with improved barrier function in Ox-AD mice. Constitutive levels of IL-1α and TNFα were very low in normal epidermis, but immunostaining increased in both the epidermis and dermis of Ox-AD mice treated with vehicle (Veh) alone or the PPARβ/δ activator GW1514 (Fig. 4S in the Online). While PPAR/LXR activators of all the receptors tested other than PPARβ/δ, including in this case, the PPARγ activator (ciglitazone), reduced IL-1α and TNFα immunostaining, the LXR activator (TO901317) was most effective, and comparable to clobetasol (Figs. 4S in the Online). These results show that topical applications of certain PPAR/LXR activators reduce epidermal cytokine production.
DISCUSSION
We show here that certain activators of the liposensor subclass of class II nuclear hormone receptors, in particular those for LXR, PPARα, and PPARβ/δ, improve multiple parameters of the AD-like dermatosis in a hapten-induced mouse model. A very recent study also showed that a PPARα activator prevented the emergence of inflammation in another murine model of AD34. Since the present model recapitulates virtually all of the known clinical, structural, functional, lipid biochemical, and immunologic abnormalities of human AD19, their efficacy suggests that these agents may hold promise for the treatment of human AD. Interestingly, not all of the activators were effective; i.e., neither the LXR activator, 22rOH-cholesterol, nor the PPARα activator, clofibrate, demonstrated benefits. The apparent lack of benefit of 22rOH-cholesterol could also be due to the fact that this naturally-occurring compound could be metabolized further into inactive species. Alternatively, at an applied dose of 10 mM, 22rOH cholesterol could act as a bulk lipid that destabilizes extracellular lamellar bilayers35. Moreover, neither of the two PPARγ activators displayed broad efficacy, despite a recent report that a PPARγ activator, rosiglitazone, displays some anti-inflammatory activity in human AD36. Moreover, it remains possible that PPARγ activators could be effective when administered systemically, rather than topically. Furthermore, it remains to be determined whether results from this AD mouse model, which may not be a strict analogue of human AD, necessarily will predict efficacy for human AD. For example, many cases of human AD are of different etiology (e.g., inherited filaggrin deficiency), and can be complicated by colonizing microbial pathogens, which further aggravate the barrier abnormality in human AD37, but are not known to exacerbate dermatitis in the mouse analogues. Hence, empiric testing of individual activators of these receptors will be needed to determine whether the activators are delivered transdermally, and agents are of optimal benefit.
PPARs and LXR activators likely improve barrier function by at least two parallel mechanisms - stimulation of epidermal differentiation and lipid production [rev. in 15]. Since increased epidermal lipid production likely generates additional endogenous activators of these nuclear hormone receptors, this process can be viewed as a type of feed-forward mechanism that coordinately regulates generation of both the corneocytes and the extracellular matrix of the SC.
According to the ‘outside-inside’ view of disease pathogenesis37, 38, inherited abnormalities in proteins important for the barrier predispose to the development of AD. Conversely, normalization of barrier function would, in turn, reduce the two major drivers of inflammation in AD: 1) Cytokine generation, originating from perturbed corneocytes, as a signal that upregulates homeostatic repair mechanisms, should decline. Indeed, our results show that both IL-1α and TNFα levels decline after PPAR and LXR activator treatment; 2) while the second mechanism is self-evident, it has not yet been experimentally verified; i.e., improved permeability barrier function would simultaneously reduce the transdermal penetration of pro-inflammatory xenobiotes, including haptens and microbial pathogens. Of course, there is evidence in other cell types that certain PPARs, particularly PPARδ activators, exert anti-inflammatory effects on macrophages and T cells15. Whether this mechanism was operative in these studies was not, however, assessed. Pertinently, both topical retinoids and 1,25 dihydroxyvitamin D3 analogues aggravate human AD (perhaps by activating epidermal pro-inflammatory cytokines39, or by further aggravating the barrier abnormality). Notably, they also accentuate disease expression in the Ox-AD model (Hatano, Y. and Elias, P.M., unpublished observations). The lack of efficacy of retinoids and vitamin D analogues in human AD is readily explicable by a comparison of the divergent activities shown by synthetic retinoids, vitamin D analogues, and the PPAR/LXR on epidermal structure and function (Table 1S in the Online). Notably, both retinoids and vitamin D analogues impair barrier function, and stimulate epidermal proliferation in vivo, while retinoids (but not vitamin D analogues) also impede epidermal differentiation. Furthermore, it should be noted that the predicted activity profile of PPAR/LXR (and retinoids/vitamin D analogues) will be dependent upon the expression levels of their respective receptors, as well as their heterodimerizing partner, RXR, which could independently influence ligand activity in AD.
A final key question remains: AD certainly is not the only dermatosis that is attributed to inherited mutations that alter barrier function. At least three other disorders (i.e., epidermolytic hyperkeratosis due to K1/K10 mutations; loricrin keratoderma; and transglutaminase 1-deficient lamellar ichthyosis) all display barrier abnormalities, but no known propensity to develop AD. Could the more coherent SC in these disorders restrict antigen access?
Since these studies demonstrate broad and potent anti-inflammatory properties in yet another mouse disease model, i.e., hapten-induced AD, the PPAR/LXR activators could be effective in a variety of other dermatologic settings. Yet, these studies did not examine all of the anti-inflammatory mechanisms by which these agents could work. While they could exert direct effects on leukocytes and macrophages, as has been shown for some of the liposensors agents in other clinical settings [cited in 15], our studies suggest alternatively or additionally, that the liposensor activators reduce inflammation by first normalizing permeability barrier function. As a result of, or at least in parallel with a return of barrier function to normal, the amplitude of cytokine generation declined in response to treatment with the liposensor activators. Likely, the downstream signal cascade that follows cytokine production subsequently declines, which should, in turn, decrease the downstream signaling of chemokines and adhesion molecules that lead to inflammation (‘outside-inside’ paradigm for the pathogenesis of inflammatory dermatoses)1, 2, 4. Since all of the liposensor activators (except PPARγ) improved barrier function in Ox-AD mice, normalization of barrier function alone could account for decreased inflammation by downregulation of the cytokine cascade, as has been shown for other approaches that correct barrier function followed by a decrease in cytokine signalling26, 40, 41. If the principal anti-inflammatory mechanism is secondary to restored barrier function, then it is interesting to speculate whether topical therapy might be substantially more effective than systemic therapy with these same agents. Yet, reduction in inflammation also could improve barrier function34, since a ‘vicious cycle’ is operative in the pathogenesis of AD38, 41, 42.
Supplementary Material
Acknowledgments
We are indebted to Ms. Joan Wakefield for superb editorial assistance.
Declaration of all sources of funding: These studies were supported by NIH grants AR19098, AG028492, AI059311, and the Medical Research Service, Department of Veterans Affairs.
Abbreviations
- AD
atopic dermatitis
- CRTH
chemoattractant receptor-homologous molecule expressed on TH2
- IgE
immunoglobin E
- LXR
liver X receptor
- Ox
oxazolone
- PPAR
peroxisome proliferator-activated receptor
- SC
stratum corneum
- SG
stratum granulosum
- TARC
thymus & activation-related chemokine
- IL
interleukin
- TNF
tumor necrosis factor
- RXR
retinoic X receptor
- PCNA
proliferating cell nuclear antigen
- GC
glucocorticoid
- TEWL
transepidermal water loss
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Elias PM, Wood LC, Feingold KR. Epidermal pathogenesis of inflammatory dermatoses. Am J Contact Dermat. 1999;10:119–26. [PubMed] [Google Scholar]
- 2.Elias PM, Feingold KR. Does the tail wag the dog? Role of the barrier in the pathogenesis of inflammatory dermatoses and therapeutic implications. Arch Dermatol. 2001;137:1079–81. [PubMed] [Google Scholar]
- 3.Elias P, Wood L, Feingold K. Relationship of the epidermal permeability barrier to irritant contact dermatitis. Immunol Allergy Clin North America. 1997;17:417–30. [Google Scholar]
- 4.Taieb A. Hypothesis: from epidermal barrier dysfunction to atopic disorders. Contact Dermatitis. 1999;41:177–80. doi: 10.1111/j.1600-0536.1999.tb06125.x. [DOI] [PubMed] [Google Scholar]
- 5.Seidenari S, Giusti G. Objective assessment of the skin of children affected by atopic dermatitis: a study of pH, capacitance and TEWL in eczematous and clinically uninvolved skin. Acta Derm Venereol. 1995;75:429–33. doi: 10.2340/0001555575429433. [DOI] [PubMed] [Google Scholar]
- 6.Sugarman JL, Fluhr JW, Fowler AJ, Bruckner T, Diepgen TL, Williams ML. The objective severity assessment of atopic dermatitis score: an objective measure using permeability barrier function and stratum corneum hydration with computer-assisted estimates for extent of disease. Arch Dermatol. 2003;139:1417–22. doi: 10.1001/archderm.139.11.1417. [DOI] [PubMed] [Google Scholar]
- 7.Proksch E, Jensen JM, Elias PM. Skin lipids and epidermal differentiation in atopic dermatitis. Clin Dermatol. 2003;21:134–44. doi: 10.1016/s0738-081x(02)00370-x. [DOI] [PubMed] [Google Scholar]
- 8.Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 2006;38:441–6. doi: 10.1038/ng1767. [DOI] [PubMed] [Google Scholar]
- 9.Weidinger S, Illig T, Baurecht H, Irvine AD, Rodriguez E, Diaz-Lacava A, et al. Loss-of-function variations within the filaggrin gene predispose for atopic dermatitis with allergic sensitizations. J Allergy Clin Immunol. 2006;118:214–9. doi: 10.1016/j.jaci.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 10.Irvine AD, McLean WH. Breaking the (un)sound barrier: filaggrin is a major gene for atopic dermatitis. J Invest Dermatol. 2006;126:1200–2. doi: 10.1038/sj.jid.5700365. [DOI] [PubMed] [Google Scholar]
- 11.Cork MJ, Robinson DA, Vasilopoulos Y, Ferguson A, Moustafa M, MacGowan A, et al. New perspectives on epidermal barrier dysfunction in atopic dermatitis: gene-environment interactions. J Allergy Clin Immunol. 2006;118:3–21. doi: 10.1016/j.jaci.2006.04.042. quiz 2-3. [DOI] [PubMed] [Google Scholar]
- 12.Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, et al. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med. 2002;347:1151–60. doi: 10.1056/NEJMoa021481. [DOI] [PubMed] [Google Scholar]
- 13.Patel TS, Greer SC, Skinner RB., Jr Cancer concerns with topical immunomodulators in atopic dermatitis: overview of data and recommendations to clinicians. Am J Clin Dermatol. 2007;8:189–94. doi: 10.2165/00128071-200708040-00001. [DOI] [PubMed] [Google Scholar]
- 14.Weischer M, Rocken M, Berneburg M. Calcineurin inhibitors and rapamycin: cancer protection or promotion? Exp Dermatol. 2007;16:385–93. doi: 10.1111/j.1600-0625.2007.00555.x. [DOI] [PubMed] [Google Scholar]
- 15.Schmuth M, Jiang YJ, Dubrac S, Elias PM, Feingold KR. Thematic Review Series: Skin Lipids. Peroxisome proliferator-activated receptors and liver X receptors in epidermal biology. J Lipid Res. 2008;49:499–509. doi: 10.1194/jlr.R800001-JLR200. [DOI] [PubMed] [Google Scholar]
- 16.Sheu MY, Fowler AJ, Kao J, Schmuth M, Schoonjans K, Auwerx J, et al. Topical peroxisome proliferator activated receptor-alpha activators reduce inflammation in irritant and allergic contact dermatitis models. J Invest Dermatol. 2002;118:94–101. doi: 10.1046/j.0022-202x.2001.01626.x. [DOI] [PubMed] [Google Scholar]
- 17.Fowler AJ, Sheu MY, Schmuth M, Kao J, Fluhr JW, Rhein L, et al. Liver X receptor activators display anti-inflammatory activity in irritant and allergic contact dermatitis models: liver-X-receptor-specific inhibition of inflammation and primary cytokine production. J Invest Dermatol. 2003;120:246–55. doi: 10.1046/j.1523-1747.2003.12033.x. [DOI] [PubMed] [Google Scholar]
- 18.Komuves LG, Hanley K, Man MQ, Elias PM, Williams ML, Feingold KR. Keratinocyte differentiation in hyperproliferative epidermis: topical application of PPARalpha activators restores tissue homeostasis. J Invest Dermatol. 2000;115:361–7. doi: 10.1046/j.1523-1747.2000.00076.x. [DOI] [PubMed] [Google Scholar]
- 19.Man MQ, Hatano Y, Lee SH, Man M, Chang S, Feingold KR, et al. Characterization of a hapten-induced, murine model with multiple features of atopic dermatitis: structural, immunologic, and biochemical changes following single versus multiple oxazolone challenges. J Invest Dermatol. 2008;128:79–86. doi: 10.1038/sj.jid.5701011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi EH, Man MQ, Wang F, Zhang X, Brown BE, Feingold KR, et al. Is endogenous glycerol a determinant of stratum corneum hydration in humans? J Invest Dermatol. 2005;125:288–93. doi: 10.1111/j.0022-202X.2005.23799.x. [DOI] [PubMed] [Google Scholar]
- 21.Hachem JP, Behne M, Aronchik I, Demerjian M, Feingold KR, Elias PM, et al. Extracellular pH Controls NHE1 expression in epidermis and keratinocytes: implications for barrier repair. J Invest Dermatol. 2005;125:790–7. doi: 10.1111/j.0022-202X.2005.23836.x. [DOI] [PubMed] [Google Scholar]
- 22.Demerjian M, Man MQ, Choi EH, Brown BE, Crumrine D, Chang S, et al. Topical treatment with thiazolidinediones, activators of peroxisome proliferator-activated receptor-gamma, normalizes epidermal homeostasis in a murine hyperproliferative disease model. Exp Dermatol. 2006;15:154–60. doi: 10.1111/j.1600-0625.2006.00402.x. [DOI] [PubMed] [Google Scholar]
- 23.Hou SY, Mitra AK, White SH, Menon GK, Ghadially R, Elias PM. Membrane structures in normal and essential fatty acid-deficient stratum corneum: characterization by ruthenium tetroxide staining and x-ray diffraction. J Invest Dermatol. 1991;96:215–23. doi: 10.1111/1523-1747.ep12461361. [DOI] [PubMed] [Google Scholar]
- 24.Hachem JP, Man MQ, Crumrine D, Uchida Y, Brown BE, Rogiers V, et al. Sustained serine proteases activity by prolonged increase in pH leads to degradation of lipid processing enzymes and profound alterations of barrier function and stratum corneum integrity. J Invest Dermatol. 2005;125:510–20. doi: 10.1111/j.0022-202X.2005.23838.x. [DOI] [PubMed] [Google Scholar]
- 25.Fartasch M. Epidermal barrier in disorders of the skin. Microsc Res Tech. 1997;38:361–72. doi: 10.1002/(SICI)1097-0029(19970815)38:4<361::AID-JEMT4>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 26.Chamlin SL, Kao J, Frieden IJ, Sheu MY, Fowler AJ, Fluhr JW, et al. Ceramide-dominant barrier repair lipids alleviate childhood atopic dermatitis: changes in barrier function provide a sensitive indicator of disease activity. J Am Acad Dermatol. 2002;47:198–208. doi: 10.1067/mjd.2002.124617. [DOI] [PubMed] [Google Scholar]
- 27.Cui CY, Kusuda S, Seguchi T, Takahashi M, Aisu K, Tezuka T. Decreased level of prosaposin in atopic skin. J Invest Dermatol. 1997;109:319–23. doi: 10.1111/1523-1747.ep12335839. [DOI] [PubMed] [Google Scholar]
- 28.Kusuda S, Cui CY, Takahashi M, Tezuka T. Localization of sphingomyelinase in lesional skin of atopic dermatitis patients. J Invest Dermatol. 1998;111:733–8. doi: 10.1046/j.1523-1747.1998.00370.x. [DOI] [PubMed] [Google Scholar]
- 29.Higuchi K, Hara J, Okamoto R, Kawashima M, Imokawa G. The skin of atopic dermatitis patients contains a novel enzyme, glucosylceramide sphingomyelin deacylase, which cleaves the N-acyl linkage of sphingomyelin and glucosylceramide. Biochem J. 2000;350(Pt 3):747–56. [PMC free article] [PubMed] [Google Scholar]
- 30.Hara J, Higuchi K, Okamoto R, Kawashima M, Imokawa G. High-expression of sphingomyelin deacylase is an important determinant of ceramide deficiency leading to barrier disruption in atopic dermatitis. J Invest Dermatol. 2000;115:406–13. doi: 10.1046/j.1523-1747.2000.00072.x. [DOI] [PubMed] [Google Scholar]
- 31.Hachem JP, Houben E, Crumrine D, Man MQ, Schurer N, Roelandt T, et al. Serine protease signaling of epidermal permeability barrier homeostasis. J Invest Dermatol. 2006;126:2074–86. doi: 10.1038/sj.jid.5700351. [DOI] [PubMed] [Google Scholar]
- 32.Mao-Qiang M, Fowler AJ, Schmuth M, Lau P, Chang S, Brown BE, et al. Peroxisome-proliferator-activated receptor (PPAR)-gamma activation stimulates keratinocyte differentiation. J Invest Dermatol. 2004;123:305–12. doi: 10.1111/j.0022-202X.2004.23235.x. [DOI] [PubMed] [Google Scholar]
- 33.Elias P, Hatano Y, Williams M. Basis for the barrier abnormality in atopic dermatitis: ‘outside-inside-outside’ pathogenic mechanisms. J Allergy Clin Immunol. 2008 doi: 10.1016/j.jaci.2008.01.022. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Staumont-Salle D, Abboud G, Brenuchon C, Kanda A, Roumier T, Lavogiez C, et al. Peroxisome proliferator-activated receptor alpha regulates skin inflammation and humoral response in atopic dermatitis. J Allergy Clin Immunol. 2008;121:962–8. e6. doi: 10.1016/j.jaci.2007.12.1165. [DOI] [PubMed] [Google Scholar]
- 35.Mao-Qiang M, Feingold KR, Elias PM. Inhibition of cholesterol and sphingolipid synthesis causes paradoxical effects on permeability barrier homeostasis. J Invest Dermatol. 1993;101:185–90. doi: 10.1111/1523-1747.ep12363729. [DOI] [PubMed] [Google Scholar]
- 36.Behshad R, Cooper KD, Korman NJ. A retrospective case series review of the peroxisome proliferator-activated receptor ligand rosiglitazone in the treatment of atopic dermatitis. Arch Dermatol. 2008;144:84–8. doi: 10.1001/archdermatol.2007.22. [DOI] [PubMed] [Google Scholar]
- 37.Elias PM, Hatano Y, Williams ML. Basis for the barrier abnormality in atopic dermatitis: outside-inside-outside pathogenic mechanisms. J Allergy Clin Immunol. 2008;121:1337–43. doi: 10.1016/j.jaci.2008.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elias PM, Steinhoff M. “Outside-to-inside” (and now back to “outside”) pathogenic mechanisms in atopic dermatitis. J Invest Dermatol. 2008;128:1067–70. doi: 10.1038/jid.2008.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li M, Messaddeq N, Teletin M, Pasquali JL, Metzger D, Chambon P. Retinoid X receptor ablation in adult mouse keratinocytes generates an atopic dermatitis triggered by thymic stromal lymphopoietin. Proc Natl Acad Sci U S A. 2005;102:14795–800. doi: 10.1073/pnas.0507385102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wood LC, Elias PM, Sequeira-Martin SM, Grunfeld C, Feingold KR. Occlusion lowers cytokine mRNA levels in essential fatty acid-deficient and normal mouse epidermis, but not after acute barrier disruption. J Invest Dermatol. 1994;103:834–8. doi: 10.1111/1523-1747.ep12413597. [DOI] [PubMed] [Google Scholar]
- 41.Hatano Y, Uchida Y, Man M, Crumrine D, Chang S, Mauro T, et al. Maintenance of a acidic stratum corneum prevents emergence of murine atopic dermatitis. J Invest Dermatol. doi: 10.1038/jid.2008.444. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Elias PM, Williams ML, Holleran WM, Jiang YJ, Schmuth M. Pathogenesis of permeability barrier abnormalities in the ichthyoses: inherited disorders of lipid metabolism. J Lipid Res. 2008;49:697–714. doi: 10.1194/jlr.R800002-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.