

Abstract
Protein aggregation, mitochondrial impairment and oxidative stress are common to multiple neurodegenerative diseases. Homeostasis is regulated by a balanced set of anabolic and catabolic responses, which govern removal and repair of damaged proteins and organelles. Macroautophagy is an evolutionarily conserved pathway for the degradation of long-lived proteins, effete organelles and protein aggregates. Aberrations in macroautophagy have been observed in Alzheimer, Huntington, Parkinson, motor neurone and prion diseases. In this review, we will discuss the divergent roles of macroautophagy in neuro-degenerative diseases and suggest a potential regulatory mechanism that could determine cell death or survival outcomes. We also highlight emerging data on neurite morphology and synaptic remodelling that indicate the possibility of detrimental functional trade-offs in the face of neuronal cell survival, particularly if the need for elevated macroautophagy is sustained.
Keywords: autophagy, Beclin 1, cell death, neurite retraction, neuroprotection, PTEN-induced kinase 1
Introduction
While initial studies implicated autophagy as a pro-survival response, enabling eukaryotes to persist during limited periods of starvation, recent studies of neuronal cells suggest a dual ‘double-edged’ role for macroautophagy in regulating cell death and survival. Studies of the nervous system have been particularly illuminating for understanding different roles for autophagy, as neurons are relatively protected from experiencing starvation in vivo. Nevertheless, certain neuronal populations are exquisitely sensitive to inhibition of macroautophagy, indicating an important role for maintaining neuronal cell functionality (reviewed in [1]). Indeed, elevated levels of autophagic vacuoles have been reported in post mortem tissues or animal models of Alzheimer, Huntington, Parkinson, motor neuron and prion diseases (reviewed in [2]). While a growing number of studies have focused on short-term effects of modulating autophagy on cell survival, few have investigated the potential functional impact of elevated or disrupted autophagy, particularly with regard to neuronal physiology. In this review, we focus on the role of autophagy in regulating neuronal cell death, survival and function in neurodegenerative diseases.
Molecular regulation of autophagy
Macroautophagy is the sequestration of cytoplasmic components including organelles for bulk degradation via the lysosome. While microautophagy and chaperone-mediated autophagy also play important roles in cellular homeostasis, we will focus on macroautophagy, hereafter referred to as autophagy, in this review. Autophagy is responsible for the clearance of long-lived proteins, organelles and protein aggregates. Autophagic degradation leads to the recycling of biological macromolecules, which are released for use in biosynthetic pathways [3]. Recently numerous genes have been identified that regulate multiple steps from initiation of autophagic membrane extension to the release of digested macromolecules (reviewed in [4,5]).
The autophagy pathway is comprised of four steps: initiation/nucleation, autophagosome formation, trafficking/maturation and recycling/release of macromolecules [2]. Distinct proteins act concertedly at each step to execute successful autophagic recycling. Initiation begins with the sensing of stress, for example, lack of nutrients or less characterized signals arising from toxins or organelle damage. Classic nutrient stress induces autophagy through the inhibition of the mammalian target of rapamycin, which indirectly results in dephos-phorylation of Atg13 [4], allowing Atg13 to interact with the serine/threonine kinase, Atg1 (UNC-51-like kinases 1 and 2 in mammals) to initiate autophagy in concert with other proteins [4,5]. Starvation also induces autophagy through a class III phosphatidylinositol-3 kinase (PI3K)/Beclin 1 (Atg6) complex [4,5], which is essential for nucleation of site(s) from which the nascent autophagic membrane extends. While these two pathways regulate starvation-induced autophagy, both target of rapamycin-independent and Beclin 1-independent autophagy have been reported [6,7], indicating that initiation of autophagy can proceed independently of one pathway or the other, or involve other mechanisms that remain to be described.
Downstream of initiation, autophagosome formation occurs through two ubiquitin-like conjugation systems [8]. In the Atg12 system, Atg7 and Atg10 act as E1 and E2 ligases, respectively, to conjugate Atg12 to Atg5. This complex localizes to the forming autophagosome through Atg16L [9] and remains until completion of the autophagosome. The second conjugation system regulates the lipidation of Atg8, which has three structural homologues in mammals: MAP1 light chain 3 (LC3), GABARAP and GATE16. Atg7 and Atg3 act as E1 and E2 ligases, respectively, to conjugate LC3 directly to phospholipids in the forming autophagosome membrane. These conjugation systems regulate the elongation, curvature and closure of autophagosome membranes.
After autophagosome formation, maturation of nascent autophagosomes is regulated by small G proteins, and requires acidification and eventual fusion with lysosomes, which may occur through intermediary intersection with the endosome pathway [4]. In any case, delivery to lysosomes appears to be dependent upon microtubules in mammalian cells. Lysosomal hydrolases degrade the contents of autophagic vacuoles, including internalized LC3, and the liberated amino acids are released into the cytosol, a process mediated in yeast by vacuolar effluxers, such as Atg22 [3]. Although not classically considered as a formal step in autophagic degradation, successful recycling also depends upon effective reutilization of the liberated building blocks for bioenergetics or to form new proteins and organelles [2].
Basal autophagy prevents neurodegeneration
While autophagy is required for homeostasis in all cell types, non-dividing cells such as neurons, are particularly sensitive to changes in autophagic degradation [2,10]. As most neurons must survive for the lifetime of the organism, maintenance of organelle function and clearance of aberrant or damaged proteins are critical processes regulated by autophagy.
The generation of conditional autophagy-deficient mice allowed studies showing that mice lacking brain expression of Atg5 or Atg7 rapidly develop neurodegenerative phenotypes in selected neuronal populations early in post-natal life [11,12]. Neuronal cell loss in these mice supports a neuroprotective role for autophagy. At the other end of the spectrum, reduced levels of Beclin 1 have been found in aged humans and in brain tissues from Huntington and Alzheimer disease patients. Thus, age-related autophagy insufficiency may also contribute to certain neurodegenerative diseases [13,14].
Autophagy in clearing protein aggregates
One central theme in neurodegenerative diseases is the near global observation of protein aggregates in degenerating regions of the brain. This supports the concept that autophagy may act as a beneficial aggregate clearance pathway (Figure 1). Indeed, induction of autophagy via rapamycin leads to a decrease in mutant huntingtin aggregates and prevents neurodegeneration in a mouse model of Huntington disease [15]. More recent studies have shown that aggregate-prone proteins, such as alpha-synuclein [16,17], superoxide dismutase 1 [18] and prions [19,20], are also degraded through autophagy. These studies collectively suggest that autophagy modulation may be a therapeutic target for a wide variety of diseases associated with protein aggregation.
Figure 1.

Autophagy reduces neuronal protein aggregates. Aggregate-prone proteins and oligomers, such as amyloid-beta, prions, huntingtin, alpha-synuclein and tau, are engulfed by autophagosomes. After sequestration, autophagosomes deliver the aggregates/preaggregates to the lysosome for degradation. Autophagic clearance thus prevents deposition of further aggregates, inhibiting the formation of histologically visible plaques and intracellular aggregates. Amyloid-beta and alpha-synuclein have each been shown to reduce the efficiency of lysosomal degradation, potentially contributing to an amplifying cycle for further accumulation of protein aggregates. Similarly normal ageing may also involve a decline in lysosomal efficiency.
While autophagy clears certain aggregate-prone proteins, upregulation of autophagy can also contribute to amyloid-beta pathology [21,22], as autophagic vacuoles may represent one site of amyloid-beta generation [21]. Additionally, amyloid-beta may impair lysosomal degradation of autophagosome contents [22]. This late-stage inhibition of autophagy could feed into an amplifying cycle, with increased autophagosome buildup contributing to the generation of more amyloid-beta and formation of plaques. Similarly, mutant alpha-synuclein impairs receptor-mediated lysosomal uptake of protein substrates, and this impairment in chaperone-mediated autophagy induces macroautophagy. However, the efficiency of macroautophagic clearance of aggregates is reduced with ageing or metabolic compromise [23]. In these scenarios, autophagy may be initially induced as a protective response; however, due to defects in completing degradation, it may also contribute to the demise of the cell. While autophagy modulation may be a useful future therapy for clearing aggregates, the underlying processes of protein aggregation and/or lysosomal ageing require further investigation.
Autophagy in oxidative cell damage
While loss of basal autophagy leads to neuronal cell loss, inhibiting the injury-induced elevations in autophagy can also protect against certain types of neuronal insults, particularly in the setting of acute neuronal injuries. For example, the parkinsonian neurotoxin MPP+ was found to cause a form of autophagic neuronal cell death, which was prevented using RNAi-based inhibition of autophagy [6]. Similar observations have been reported in vivo during neonatal hypoxic-ischaemic injury in mice, suggesting that autophagy induction elicited by certain neuronal injuries contributes to cell death [24]. A recent paper proposed that autophagy might also alter the mode of cell death after hypoxic-ischaemic injury in rats, changing the cell death pathway from apoptotic to necrotic when autophagy is inhibited [25].
In a genetic model of recessive parkinsonism, knockdown of the PTEN-induced kinase 1 (PINK1) causes an increase in mitochondrial reactive oxygen species, mitochondrial depolarization and mitochondrial fragmentation prior to degradation by autophagy [26]. In this chronic model, autophagy plays a protective role, and RNAi inhibition of autophagy exacerbates cell death [26]. Furthermore, overexpression of the E3 ligase, parkin, which facilitates autophagic degradation of depolarized mitochondria [27], provides protection in PINK1-deficient cells [26].
One explanation for the difference between the PINK1-deficiency model and the MPP+ model, both of which involve mitochondrial injury and turnover, could be the extent of cellular injury. It would be logical to assume that autophagic recycling of damaged mitochondria would be beneficial only to the point where functioning mitochondria persisted, particularly given the dependence of neurons on mitochondrial respiration. We propose that a large percentage of mitochondria are damaged with MPP+, which may exceed the ability of the cell to successfully recycle/regenerate a necessary level of functional mitochondria. However, in the context of a presumed lower level of damage caused by PINK1 deficiency, the turnover of damaged mitochondria may occur at a rate compatible with mitogenesis, enhancing cell survival.
Pathological vs. physiological autophagy: a threshold hypothesis
While autophagy was initially identified as a cell survival pathway, more recent studies suggest both beneficial and detrimental roles for autophagy. Although the level of cellular damage may determine whether autophagy plays a positive or negative role, whether or not there may also be underlying differences in signalling pathways or larger regulatory mechanisms remain poorly understood. Interestingly, reduced expression of Beclin 1, a central regulator of autophagy nucleation and induction, has been shown in patients with Alzheimer and Huntington diseases [13,14], even though accumulation of autophagosomes is observed in patient Alzheimer disease brain tissues [28] and in a model of Huntington disease [29]. These implicate dysregulation of autophagy in disease pathogenesis or progression.
While autophagy in most contexts is regulated by Beclin 1/class III PI3K function, a non-canonically regulated form of Beclin 1-independent autophagy has been shown to promote cell death in models of oxidative neuronal injury [6,30], in resveratrol-treated cancer cells [31] and during p53 up-regulated modifier of apoptosis (PUMA)-induced mitochondrial perturbation [32]. In these models, intact autophagic degradation was demonstrated; nevertheless, the levels of autophagic vacuoles were unaffected by pharmacologic and molecular inhibition of Beclin 1/PI3K activities. In contrast, RNAi knockdown of core autophagy conjugation proteins inhibited both autophagy and cell death. These data suggest that alternative mechanisms for localized elevations of phosphoinositol-3 phosphate (such as redox inactivation of phosphatases) may operate during cell death-associated autophagy [30]. Alternatively, there may be other as yet unidentified signals originating from damaged mitochondria that serve as alternative nucleation mechanisms.
One such signal may involve mitochondrial targeting of activated extracellular signal-regulated protein kinase (ERK2) [33]. Inhibition of mitogen-activated protein kinase/ERK kinase (MEK) signalling inhibits both Beclin 1-independent [6] and Beclin 1-dependent autophagy [34]. Interestingly, expression of active forms of ERK2 is sufficient to induce autophagic neuronal cell death, and the percent of mitochondrially colocalized LC3 is correlated with the degree of mitochondrial localization of the tagged ERK construct [33]. Mitochondrially derived super-oxide is implicated in redox activation of ERK at mitochondria [35], suggesting the possibility that this pathway can act to sense mitochondrial dysfunction. Redox activation of ERK has been reported to cause decreased mitochondrial respiration [36] and increased mitochondrial autophagy [33], both of which may serve to limit further oxidative damage. Interestingly, a leukaemia study indicates that MEK itself can regulate autophagy through effects on Beclin 1 expression, independently of ERK [37]. Thus, inhibitors of MEK may reduce injury-induced canonical and non-canonical autophagy through different mechanisms. In the leukaemia study, transient MEK/ERK activation induced cytoprotective autophagy, but sustained MEK/ERK activity contributed to cytodestructive levels of autophagy.
In contrast, the beneficial clearance of protein aggregates in multiple other models of neurodegeneration involves classically regulated Beclin 1/PI3K-dependent autophagy. As noted above, fission- and parkin-enhanced mitochondrial autophagy serves a pro-survival function in the context of PINK1 deficiency [26]. To further investigate the regulation of autophagy in this context, we used two PI3K inhibitors, 3-methyladenine (3 mM) and wortmannin (10 μM). Both were able to reverse the increase in GFP-LC3 puncta observed in PINK1 knock-down cells (Figure 2A). RNAi knock-down of Beclin 1 showed similar effects (Figure 2B). These studies further support the concept that physiologically regulated PI3K/Beclin 1-dependent autophagy represents primarily a compensatory response to stress. However, robust activation of autophagy through mechanisms that are not regulated by PI3K/Beclin 1 appears to play a negative role, contributing to cellular injury in MPP+ and the G2019S LRRK2 model of autosomal dominant Parkinson disease [6,30,38].
Figure 2.

PINK1 deficiency induces PI3K/Beclin 1-dependent autophagy. (A) PI3K inhibitors, 3-methyladenine (3MA) or wortmannin (WTN), reduce the number of GFP-LC3-labelled autophagic vacuoles (puncta) induced by PINK1 knock-down in two SH-SY5Y clones, A14 and D14, as compared with V17 vector stable cells (*P < 0.05 vs. vector stable control line; †P < 0.05 vs. respective untreated PINK1 shRNA line; mean ± SE). (B) RNAi using a previously described protocol for human Beclin 1 [6] also reduces the number of GFP-LC3 puncta in PINK1-deficient cells (*P < 0.05 vs. vector stable control line; †P < 0.05 vs. scrambled control siRNA in the respective stable line; mean ± SE).
Collectively, these studies suggest that a hypothetical threshold of autophagy, potentially modulated by PI3K/Beclin 1 dependence, may be a point of divergence between physiological and pathological autophagy (Figure 3). Differences in the eliciting injury [39], the cell type studied [40,41], or the gender of primary isolates [41], may also determine whether autophagy crosses this threshold to promote cell survival or cell death. Thus, a better understanding of PI3K/Beclin 1-independent autophagy mechanisms may lead to therapeutic targets to promote cell survival by inhibiting pathological autophagy while preserving physiological autophagy.
Figure 3.
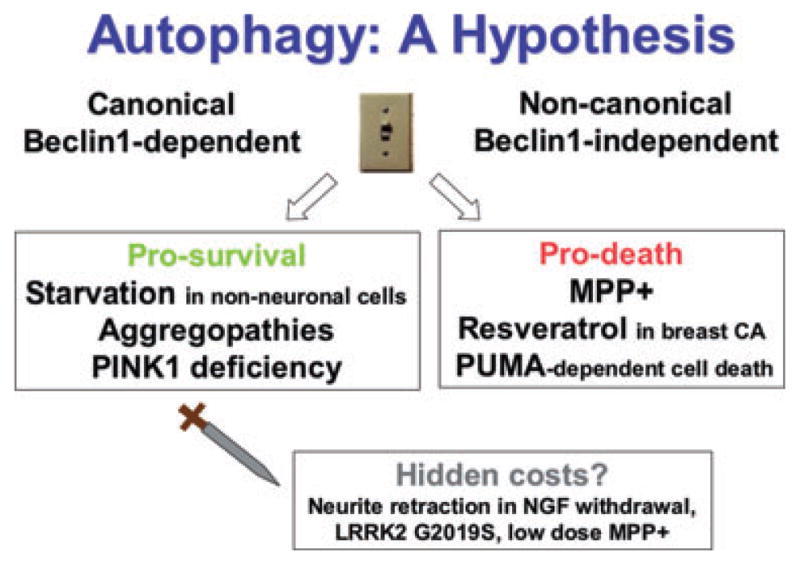
Canonical vs. non-canonical autophagy? In different model systems, induction of autophagy results in divergent outcomes. For example, nitrogen starvation, trophic factor withdrawal, PINK1 deficiency or aggregopathies each induce autophagy that is neuroprotective. However, autophagy appears to contribute to MPP+, resveratrol, hypoxic/ischaemic and p53 up-regulated modifier of apoptosis (PUMA)-dependent cell death. Interestingly, non-canonical autophagy that does not require Beclin 1 has only been reported in a small number of systems, but in each of these, autophagy plays a detrimental role, promoting cell death or neurite retraction. Given that competition for limiting amounts of Beclin 1 has been proposed as a means by which Bcl2 suppresses autophagy [49], we hypothesize that mechanisms that bypass the requirement for Beclin 1 may determine whether or not autophagy is induced to a pro-death level. The role of autophagy is further complicated by considerations of neuronal functional capacity. RNAi studies indicate that Atg7- and LC3-dependent autophagy contributes to the shortening of neurites in both Beclin 1-dependent and independent contexts. While the long-term functional consequences of this remain uncharacterized, it is possible that neuronal survival is enhanced by retraction of neurites, but at a cost to the neural network, contributing to circuit dysfunction in neurodegenerative diseases. Thus, the potential physiologic role(s) of autophagy in neuronal function and plasticity, as well as the functional consequences of autophagy induced in response to pathologic stresses, remain as critically important areas for future investigation.
Autophagy-mediated degeneration: cell survival at a cost?
Until recently, the outcome of autophagy as protective or detrimental has been evaluated using cell death as the major end-point. As autophagy induction promotes neuronal survival, then autophagy is thought to be neuroprotective; however, the functional capacity of neurons undergoing high levels of induced autophagy remains unknown. If neurites and synapses are lost during autophagic recycling, or their functional parameters are significantly altered, is autophagy still neuroprotective?
Multiple studies have recently reported autophagy-mediated neurite retraction in response to varying insults. During acute brain trauma, autophagosomes appear preferentially in neuritic compartments, and these changes could be prevented by antioxidants [42]. In defined culture systems, nerve growth factor (NGF) deprivation, glutamate excitoxicity and the Parkinson disease-linked G2019S mutation in leucine-rich repeat kinase 2 (LRRK2) can each elicit neurite retraction with increased levels of neuritic autophagosomes [38,43,44]. Inhibition of autophagy in the NGF deprivation and LRRK2 G2019S models prevents neurite retraction, suggesting that autophagy plays a role in neurite remodelling [38,44]. Similarly, the parkinsonian neurotoxins MPP+ and 6-OHDA, which increase the number of autophagosomes [6,33], also cause neurite retraction that persists even when cell death is prevented [45,46]. In these models, autophagy induction may help maintain neuronal viability, but the question of whether cell survival might come at a cost to neuritic/synaptic function should be considered (Figure 3).
A related question that needs to be addressed in further studies is whether or not autophagy might play a normal, physiologic role in neuritic/synaptic remodelling, as suggested by the association of LC3-I with axonal constituents [43]. In Caenorhabditis elegans, autophagy appears to regulate levels of GABAA receptors [47], and similar activity-dependent modulation of other synaptic proteins and membrane structures is possible. Interestingly, the E3 ligase parkin, which promotes proteasomal degradation of proteins and autophagic degradation of depolarized mitochondria, has recently been shown to diminish the efficiency of glutamate neurotransmission [48]. Further studies to determine whether autophagy is capable of remodelling mature neurites and synapses, and whether synaptic connections can be re-established after the injury is resolved, remain as exciting, future areas of investigation in neurophysiology and neuropathology.
Summary
Autophagy is a ubiquitous, indispensable process that governs many facets of cellular homeostasis. Neuronal autophagy recycles damaged organelles and degrades long-lived proteins and aggregates, a requirement for healthy cells. Defects in autophagy, either too much or too little, may be one of many components in the pathogenesis of neurodegenerative diseases. Therefore, modulation of autophagy presents an important potential therapeutic target for multiple neurodegenerative diseases. Further understanding of the regulatory mechanisms of neuronal autophagy may yield insights into both normal neurophysiology and pathophysiologic mechanisms underlying common neurodegenerative diseases.
Acknowledgments
The authors’ research is supported in part by funding from the National Institutes of Health (AG026389, R01 supplement AG026389-03S1). C. T. C. is recipient of the AFAR Julie Martin Mid-Career Award in Aging Research sponsored by the Ellison Medical Foundation. S. J. C. was supported in part by F31 NS064728.
References
- 1.Yue Z, Friedman L, Komatsu M, Tanaka K. The cellular pathways of neuronal autophagy and their implication in neurodegenerative diseases. Biochim Biophys Acta. 2009;1793:1496–507. doi: 10.1016/j.bbamcr.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cherra SJ, Chu CT. Autophagy in neuroprotection and neurodegeneration: a question of balance. Future Neurol. 2008;3:309–23. doi: 10.2217/14796708.3.3.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang Z, Huang J, Geng J, Nair U, Klionsky DJ. Atg22 recycles amino acids to link the degradative and recycling functions of autophagy. Mol Biol Cell. 2006;17:5094–104. doi: 10.1091/mbc.E06-06-0479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marino G, Lopez-Otin C. Autophagy: molecular mechanisms, physiological functions and relevance in human pathology. Cell Mol Life Sci. 2004;61:1439–54. doi: 10.1007/s00018-004-4012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–9. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- 6.Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am J Pathol. 2007;170:75–86. doi: 10.2353/ajpath.2007.060524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282:5641–52. doi: 10.1074/jbc.M609532200. [DOI] [PubMed] [Google Scholar]
- 8.Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev. 2001;2:211–16. doi: 10.1038/35056522. [DOI] [PubMed] [Google Scholar]
- 9.Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, Natsume T, Ohsumi Y, Yoshimori T. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci. 2003;116:1679–88. doi: 10.1242/jcs.00381. [DOI] [PubMed] [Google Scholar]
- 10.Chu CT. Autophagic stress in neuronal injury and disease. J Neuropathol Exp Neurol. 2006;65:423–32. doi: 10.1097/01.jnen.0000229233.75253.be. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 12.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 13.Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, Wyss-Coray T. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118:2190–9. doi: 10.1172/JCI33585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shibata M, Lu T, Furuya T, Degterev A, Mizushima N, Yoshimori T, MacDonald M, Yankner B, Yuan J. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J Biol Chem. 2006;281:14474–85. doi: 10.1074/jbc.M600364200. [DOI] [PubMed] [Google Scholar]
- 15.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36 :585–95. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 16.Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem. 2008;283:23542–56. doi: 10.1074/jbc.M801992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–13. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 18.Fornai F, Longone P, Cafaro L, Kastsiuchenka O, Ferrucci M, Manca ML, Lazzeri G, Spalloni A, Bellio N, Lenzi P, Modugno N, Siciliano G, Isidoro C, Murri L, Ruggieri S, Paparelli A. Lithium delays progression of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2008;105:2052–7. doi: 10.1073/pnas.0708022105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aguib Y, Heiseke A, Gilch S, Riemer C, Baier M, Schatzl HM, Ertmer A. Autophagy induction by trehalose counteracts cellular prion infection. Autophagy. 2009;5:361–9. doi: 10.4161/auto.5.3.7662. [DOI] [PubMed] [Google Scholar]
- 20.Heiseke A, Aguib Y, Riemer C, Baier M, Schatzl HM. Lithium induces clearance of protease resistant prion protein in prion-infected cells by induction of autophagy. J Neurochem. 2009;109:25–34. doi: 10.1111/j.1471-4159.2009.05906.x. [DOI] [PubMed] [Google Scholar]
- 21.Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, Mohan PS, Mercken M, Farmery MR, Tjernberg LO, Jiang Y, Duff K, Uchiyama Y, Naslund J, Mathews PM, Cataldo AM, Nixon RA. Macroautophagy – a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol. 2005;171:87–98. doi: 10.1083/jcb.200505082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ling D, Song HJ, Garza D, Neufeld TP, Salvaterra PM. Abeta42-induced neurodegeneration via an age-dependent autophagic-lysosomal injury in Drosophila. PLoS ONE. 2009;4:e4201. doi: 10.1371/journal.pone.0004201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu WH, Dorado B, Figueroa HY, Wang L, Planel E, Cookson MR, Clark LN, Duff KE. Metabolic activity determines efficacy of macroautophagic clearance of pathological oligomeric alpha-synuclein. Am J Pathol. 2009;175:736–47. doi: 10.2353/ajpath.2009.080928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, Waguri S, Kawahara N, Kuida K, Nagata S, Kominami E, Tanaka K, Uchiyama Y. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172:454–69. doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis. 2008;32:329–39. doi: 10.1016/j.nbd.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 26.Dagda RK, Cherra SJ, 3rd, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem. 2009;284:13843–55. doi: 10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–22. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 29.Kegel KB, Kim M, Sapp E, McIntyre C, Castano JG, Aronin N, DiFiglia M. Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J Neurosci. 2000;20:7268–78. doi: 10.1523/JNEUROSCI.20-19-07268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chu CT, Zhu J, Dagda R. Beclin 1-independent pathway of damage-induced mitophagy and autophagic stress: implications for neurodegeneration and cell death. Autophagy. 2007;3:663–6. doi: 10.4161/auto.4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scarlatti F, Maffei R, Beau I, Codogno P, Ghidoni R. Role of non-canonical Beclin 1-independent autophagy in cell death induced by resveratrol in human breast cancer cells. Cell Death Differ. 2008;15:1318–29. doi: 10.1038/cdd.2008.51. [DOI] [PubMed] [Google Scholar]
- 32.Yee KS, Wilkinson S, James J, Ryan KM, Vousden KH. PUMA- and Bax-induced autophagy contributes to apoptosis. Cell Death Differ. 2009;16:1135–45. doi: 10.1038/cdd.2009.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dagda RK, Zhu J, Kulich SM, Chu CT. Mitochondrially localized ERK2 regulates mitophagy and autophagic cell stress: implications for Parkinson’s disease. Autophagy. 2008;4:770–82. doi: 10.4161/auto.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ogier-Denis E, Pattingre S, El Benna J, Codogno P. Erk1/2-dependent phosphorylation of Galpha-interacting protein stimulates its GTPase accelerating activity and autophagy in human colon cancer cells. J Biol Chem. 2000;275:39090–5. doi: 10.1074/jbc.M006198200. [DOI] [PubMed] [Google Scholar]
- 35.Kulich SM, Chu CT. Role of reactive oxygen species in extracellular signal-regulated protein kinase phosphorylation and 6-hydroxydopamine cytotoxicity. J Biosci. 2003;28:83–9. doi: 10.1007/BF02970136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nowak G, Clifton GL, Godwin ML, Bakajsova D. Activation of ERK1/2 pathway mediates oxidant-induced decreases in mitochondrial function in renal cells. Am J Physiol. 2006;291:F840–F855. doi: 10.1152/ajprenal.00219.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang J, Whiteman MW, Lian H, Wang G, Singh A, Huang D, Denmark T. A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1. J Biol Chem. 2009;284:21412–24. doi: 10.1074/jbc.M109.026013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Plowey ED, Cherra SJ, 3rd, Liu YJ, Chu CT. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem. 2008;105:1048–56. doi: 10.1111/j.1471-4159.2008.05217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Singh R, Massey AC, Kane SS, Kaushik S, Grant T, Xiang Y, Cuervo AM, Czaja MJ. Loss of macroautophagy promotes or prevents fibroblast apoptosis depending on the death stimulus. J Biol Chem. 2008;283:4766–77. doi: 10.1074/jbc.M706666200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X, Stolz DB, Shao ZM, Yin XM. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem. 2007;282:4702–10. doi: 10.1074/jbc.M609267200. [DOI] [PubMed] [Google Scholar]
- 41.Du L, Hickey RW, Bayir H, Watkins SC, Tyurin VA, Guo F, Kochanek PM, Jenkins LW, Ren J, Gibson G, Chu CT, Kagan VE, Clark RS. Starving neurons show sex difference in autophagy. J Biol Chem. 2009;284:2383–96. doi: 10.1074/jbc.M804396200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lai Y, Hickey RW, Chen Y, Bayir H, Sullivan ML, Chu CT, Kochanek PM, Dixon CE, Jenkins LW, Graham SH, Watkins SC, Clark RS. Autophagy is increased after traumatic brain injury in mice and is partially inhibited by the antioxidant gamma-glutamylcysteinyl ethyl ester. J Cereb Blood Flow Metab. 2008;28:540–50. doi: 10.1038/sj.jcbfm.9600551. [DOI] [PubMed] [Google Scholar]
- 43.Wang QJ, Ding Y, Kohtz DS, Mizushima N, Cristea IM, Rout MP, Chait BT, Zhong Y, Heintz N, Yue Z. Induction of autophagy in axonal dystrophy and degeneration. J Neurosci. 2006;26:8057–68. doi: 10.1523/JNEUROSCI.2261-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang Y, Fukui K, Koike T, Zheng X. Induction of autophagy in neurite degeneration of mouse superior cervical ganglion neurons. Eur J Neurosci. 2007;26:2979–88. doi: 10.1111/j.1460-9568.2007.05914.x. [DOI] [PubMed] [Google Scholar]
- 45.von Coelln R, Kugler S, Bahr M, Weller M, Dichgans J, Schulz JB. Rescue from death but not from functional impairment: caspase inhibition protects dopaminergic cells against 6-hydroxydopamine-induced apoptosis but not against the loss of their terminals. J Neurochem. 2001;77 :263–73. doi: 10.1046/j.1471-4159.2001.t01-1-00236.x. [DOI] [PubMed] [Google Scholar]
- 46.Bilsland J, Roy S, Xanthoudakis S, Nicholson DW, Han Y, Grimm E, Hefti F, Harper SJ. Caspase inhibitors attenuate 1-methyl-4-phenylpyridinium toxicity in primary cultures of mesencephalic dopaminergic neurons. J Neurosci. 2002;22:2637–49. doi: 10.1523/JNEUROSCI.22-07-02637.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rowland AM, Richmond JE, Olsen JG, Hall DH, Bamber BA. Presynaptic terminals independently regulate synaptic clustering and autophagy of GABAA receptors in Caenorhabditis elegans. J Neurosci. 2006;26:1711–20. doi: 10.1523/JNEUROSCI.2279-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Helton TD, Otsuka T, Lee MC, Mu Y, Ehlers MD. Pruning and loss of excitatory synapses by the parkin ubiquitin ligase. Proc Natl Acad Sci U S A. 2008;105:19492–7. doi: 10.1073/pnas.0802280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]