

Abstract
The E. coli maltose binding protein (MBP) has been utilized as a translational fusion partner to improve the expression of foreign proteins made in E. coli. When located N-terminal to its cargo protein, MBP increases the solubility of intracellular proteins and improves the export of secreted proteins in bacterial systems. We initially explored whether MBP would have the same effect in the methylotrophic yeast Pichia pastoris, a popular eukaryotic host for heterologous protein expression. When MBP was fused as an N-terminal partner to several C-terminal cargo proteins expressed in this yeast, proteolysis occurred between the two peptides, and MBP reached the extracellular region unattached to its cargo. However, in two of three instances, the cargo protein reached the extracellular region as well, and its initial attachment to MBP enhanced its secretion from the cell. Extensive mutagenesis of the spacer region between MBP and its C-terminal cargo protein could not inhibit the cleavage although it did cause changes in the protease target sites in the fusion proteins, as determined by mass spectrometry. Taken together, these results suggested that an uncharacterized P. pastoris protease attacked at different locations in the region C-terminal of the MBP domain, including the spacer and cargo regions, but the MBP domain could still act to enhance the secretion of certain cargo proteins.
Keywords: Pichia pastoris, recombinant expression, fusion protein, maltose binding protein, proteolysis, secretion
INTRODUCTION
Pichia pastoris is a methylotrophic yeast that has been genetically engineered to express heterologous proteins valued for industrial, pharmaceutical and basic research purposes [1, 2]. In the past ten years, over 700 proteins from bacteria to humans have been produced in this yeast. The use of P. pastoris as a foreign host offers many advantages, as compared to other expression systems [3]. First, P. pastoris does not have the endotoxin problem associated with bacteria, or the viral contamination problem of proteins produced in animal cell cultures. Second, it is almost as easy to manipulate on the molecular genetic level as Saccharomyces cerevisiae [4, 5]. Third, the yeast can express proteins at high levels, intracellularly or extracellularly, at fairly low costs. Fourth, unlike prokaryotes, it is capable of performing many eukaryotic posttranslational modifications, such as glycosylation, disulfide bond formation, and proteolytic processing [3]. Finally, unlike S. cerevisiae, P. pastoris is able to reach high cell densities in fermenters because of its preference for respiratory growth over fermentation [6, 7].
A valuable option in the P. pastoris expression system is the availability of secretion signal peptides that can be attached to the recombinant protein, causing it to be exported out of the cell. The 85 amino acid S. cerevisiae MATα prepro signal peptide has been the most successful in this role and is the one commonly included in commercially available vectors [3, 8]. The MATα signal sequence consists of a nineteen amino acid signal (pre) sequence followed by a sixty-six residue (pro) sequence containing three consensus N-linked glycosylation sites and a dibasic Kex2 endopeptidase processing site. This peptide is essential for the targeting of the protein through the endoplasmic reticulum (ER), Golgi apparatus and ultimately to the plasma membrane [9–12]. In most cases the signal peptide is efficiently removed from the foreign protein prior to exit from the yeast cell. Because P. pastoris secretes few of its own proteins, the secreted recombinant protein is the major polypeptide species found in the extracelluar growth medium [3]. Thus, this programmed export acts as a valuable step in the purification of the heterologous protein, making it a convenient host in terms of time and resources.
Despite these strengths, there are limitations to the P. pastoris system. Foremost, some recombinant proteins, which are engineered to be secreted, get “stuck” inside the yeast cell and fail to arrive at their extracellular destination [13–15]. Furthermore, these retained proteins are often misfolded and degraded intracellularly, creating a major obstacle [16]. This is a common problem in E. coli and other bacteria as well, but in these prokaryotic systems the export of several proteins has been improved by an in frame translational fusion with a partner protein [17, 18]. Fusion partners, such as the E. coli maltose binding protein (MBP), are believed to assist in the proper folding of the desired protein so that the hybrid can arrive at its extracelluar destination [19, 20]. The partner protein acts as an in-frame escort for the foreign cargo protein, leading to improved yields in many cases. These fusion partners usually serve a second function besides secretion assistance: aiding in purification by affinity chromatography [21]. After the escort domain binds to a matrix and the hybrid protein is purified, the partner is removed by protease treatment, liberating the desired cargo protein [22].
While several fusion partners are available for bacterial expression, relatively few exist for yeast [23, 24]. The cellulose binding domain has been shown to be an affinity tag and secretion enhancer in S. cerevisiae; however, it is problematic in P. pastoris [25, 26]. Bacterial Hsp70 has been recently demonstrated to improve secretion efficiency of one recombinant peptide in P. pastoris, but it does not have properties of a purification tag [27]. A commonly used translation fusion partner, which consistently helps with both localization and purification, does not exist yet for P. pastoris. The hexahistidine (His6) motif is often used to tag and purify heterologous proteins secreted from P. pastoris due to its affinity to certain metals, but this peptide is believed to have no positive effect on the secretion efficiency and sometimes is not effective in affinity purification [18, 28]. Thus, a fusion partner which could serve to a) increase secretion efficiency and b) speed purification of recombinant proteins would be highly desirable.
We initially hypothesized that the secretion of some foreign proteins by P. pastoris would be enhanced by fusing them in frame to the E. coli maltose binding protein (MBP), the product of the malE gene. For nearly 20 years, MBP has been utilized in a commercially available kit (New England Biolabs, Beverly, MA) as a translational fusion partner to improve the expression of foreign proteins made in E. coli [20, 29, 30]. MBP can be used for either: 1) secretion into the periplasm when associated with a signal peptide or, 2) intracellular expression if no signal peptide is present [19]. The foreign protein of interest is fused C-terminal to the MBP protein. Inserted between N-terminal MBP and the C-terminal cargo protein is a recognition site for a commercially available protease. The fusion is expressed by the bacterial cells, purified using amylose column chromatography, and the isolated peptide is then digested with a specific protease to release the desired recombinant protein for a variety of downstream uses [31].
Adopting a similar strategy for protein expression to P. pastoris, we created constructs containing the MATα prepro sequence and a MBP fused to a variety of C-terminal cargo proteins for expression in the yeast system. We wished to determine whether MBP could improve the export of the proteins, which by themselves displayed different levels of secretion efficiency. Although our findings indicate that proteolysis of fusion proteins can be a significant obstacle within the yeast’s secretory mechanism, MBP could act as an escort to increase the secretion of some recombinant proteins in P pastoris.
MATERIALS AND METHODS
Strains, Media and Reagents
P. pastoris strain yJC100 (wild type) is a derivative of the original wild-type P. pastoris strain NRRL Y11430 (North Regional Research Laboratories, US Department of Agriculture, Peoria, IL) and has been described previously [1]. yJC100 cells were cultured in either YPD medium (1% yeast extract, 2% peptone, and 2% glucose), BMGY (buffered complex medium with 1% glycerol, 2% peptone, and 1% yeast extract) or BMMY (buffered complex medium with 0.5% methanol, 2% peptone, and 1% yeast extract) [5]. Cultivation of P. pastoris strains was at 30°C unless otherwise indicated.
Recombinant DNA manipulations were carried out in the Escherichia coli strain TOP10 (Invitrogen Corp., Carlsbad, CA). TOP10 cells were cultured in LB medium (0.5% yeast extract, 1% glucose, and 0.5% NaCl) at 37°C. Antibiotics were added to LB medium at the following final concentrations: 100 µg/ml ampicillin and 25 µg/ml Zeocin for plasmid selection. Recombinant DNA methods, including bacterial transformation, were performed essentially as described [32]. Plasmid DNA was purified from E. coli cultures using a QIAprep Spin Miniprep Kit (Qiagen, Chatsworth, CA). PCR products were purified with the QIAprep PCR Cleanup Kit (Qiagen, Chatsworth, CA) prior to restriction digestion. Restriction enzymes were purchased from MBI Fermentas (Hannover, MD). DNA digested with restriction enzymes was resolved in TBE agarose gels. DNA fragments were purified from agarose gels by using the Geneclean II kit (Qbiogene, Carlsbad, CA). Chromosomal DNA from P. pastoris transformants was prepared using the OmniPrep™ kit from GenoTechnology, Inc. (St. Louis, MO). Oligonucleotides were synthesized by Sigma Genosys (Plano, TX). All mutated sites and ligation junctions in newly synthesized vectors were confirmed by DNA sequencing (Geneway Research, Hayward, CA).
Construction of vectors
The malE gene and Factor Xa recognition site (FXa) was amplified from the plasmid pMAL-c2X (New England Biolabs, Beverly, MA) using the primers 5’MalEPstI CGGCTGCAGTGAAAATCGAAG and 3’MalERI CGTGAATTCCCCTTCCCTCG. The resulting PCR fragment was restricted with the enzymes PstI and EcoRI and ligated into the corresponding sites of pPICZαB (Invitrogen Corp, Carlsbad, CA) to yield pJV1 (pPICZαB-MBP-FactorXa). To create pJV1F, the malE gene and Factor Xa site was amplified from the plasmid pMAL-c2X using the oligos 5’MalEPstI CGGCTGCAGTGAAAATCGAAG and 3’Malrev3RI CCACGTGAATTCCTTCCCTCGATCCC. The resulting PCR fragment was restricted with PstI and EcoRI and ligated into the corresponding sites of pPICZαB to yield a plasmid which expresses MBP-Factor Xa fused to myc and His6 tags under the control of the AOXI promoter. Sequences encoding myc and His6 tags are provided in the pPICZαB vector. To create pJV2 (pPICZαB-MBP-FactorXa-FKBP12-myc-His6), initially, the sequence for FKBP12 was amplified from pDT4 (gift of David Thomas, University of the Pacific) and cloned into pMAL-c2X using the primers Mbpfkbfo TTGGATCCGGCGTGCAGGTGGAGACTATC and Mbpfkbrev TTAAGCTTTTATTCCAGTTTTAGAAGCTC, digested with BamHI and HindIII, and cloned into the BamHI and HindIII sites of pMAL-c2X. Subsequently the malE-FKBP12 fusion was amplified using the oligos 5’MalEPstI CGGCTGCAGTGAAAATCGAAG and Fkbp12NotI CGGCGGCCGCGACTTCCAGTTTTAGAAGCTCC. The resulting PCR fragment was restricted with the enzyme PstI and NotI and ligated into the corresponding sites of pPICZαB to yield a plasmid which expresses a secreted MBP and Factor Xa site fused to FKBP12 with myc and His6 tags under the control of the AOXI promoter (pJV2). pJV3 was made as follows: the gene for C. elegans spe-9 was amplified from pAS12 (gift of Andrew Singson, Rutgers University) using the oligos Espefo TAGAATTCGACTGTGCAAAAATCAG and Esperev TAGTCGACTCAAACCAGTGGCAACGAAAATTC. The PCR product was digested and cloned into the EcoRI and SalI sites of pMAL-c2X. Subsequently the malE-spe9 fusion was amplified using the oligos 5’MalEPstI CGGCTGCAGTGAAAATCGAAG and Spe9NotIR GCGGCGGCCGCCAGTGGCAACGAAAATTC. The resulting PCR fragment was restricted with the enzyme PstI and NotI and ligated into the corresponding sites of pPICZαB to yield a plasmid, pJV3, which expresses a secreted MBP fused to Spe-9 protein with myc and His6 tags under the control of the AOXI promoter. pJV4 was created by first amplifying the EGFP coding sequence from pEGFP-C3 (Clontech, Mountain View, CA) with the primers 5’EGFPEcoRI CCGGAATTCGGTGAGCAAGGGC and 3’EGFPNotI GCTGGCGGCCGCCTTGTACAGCTCGTC. The resulting PCR product was digested with EcoRI and NotI enzymes and inserted into the respective sites of pJV1F to create a MBP-FXa-EGFP-myc-His6 fusion.
To create pAW1, the malE gene was amplified with the enterokinase proteolytic (EK) site from the plasmid pMAL-c2E (New England Biolabs, Beverly, MA) using the oligos 5’MalE-Fred CCCTGCAGTGAAAACTGAAGAAGGTAAAC and 3’MalE-Wilma CCGAATTCGGTACCTTGTCATCGTCATC. The resulting PCR fragment was restricted with the enzymes PstI and EcoRI and ligated into the corresponding sites of pPICZαB to yield pAW1 (pPICZαB-MBP-EK-myc-His6). pAW2 was constructed by using PCR to amplify the FKBP12 coding sequence from pDT4 with the primers 5mbpEfkbp12R1 CCGGAATTCCATGCTCGAGGGC and Fkbp12NotI CGGCGGCCGCGACTTCCAGTTTTAGAAGCTCC. The resulting PCR fragment was restricted with the enzyme EcoRI and NotI and ligated into the corresponding sites of pAW1 to yield a plasmid which expresses a secreted MBP fused to an enterokinase protease recognition site followed by FKBP12 with myc and His6 tags under the control of the AOXI promoter. pAW3 was constructed by using PCR to amplify the EGFP coding sequence from pEGFP-C3 with the primers 5’EGFPEcoR1 CCGGAATTCGGTGAGCAAGGGC and 3’EGFP-Not1 GCTGGCGGCCGCCTTGTACAGCTCGTC. The resulting PCR fragment was restricted with the enzymes EcoR1 and NotI and ligated into the corresponding sites of pAW1 to yield a plasmid which expresses a secreted MBP fused to an enterokinase protease recognition site followed by EGFP coding sequence with myc and His6 tags under the control of the AOXI promoter.
pKS1 was created by amplifying the EGFP coding sequence from pEGFP-C3 with the primers 5’EGFPEcoRI CCGGAATTCGGTGAGCAAGGGC and 3’EGFPNotI GCTGGCGGCCGCCTTGTACAGCTCGTC. The resulting PCR product was digested with EcoRI and NotI enzymes and inserted into the respective sites of pPICZαB to yield a plasmid which expresses a secreted EGFP fused to C-terminal myc and His6 tags under the control of the AOXI promoter. pCA2 was constructed by amplifying the FKBP12 coding sequence from pDT4 with the primers FKBP12Ecor1f CGGAATTCCCATGCTCGAGGGCGTGCAGG and FKBP12Not1r CGGCGGCCGCGACTTCCAGTTTTAGAAGCTCC. The PCR product was digested with the enzymes EcoR1 and NotI and ligated into the corresponding sites of pPICZαB to yield a plasmid which expresses a secreted FKBP12 fused to C-terminal myc and His6 tags under the control of the AOXI promoter. pCH1 (pPICZαB -HRP) was kindly provided by Frances H. Arnold (California Institute of Technology). The plasmid expresses a secreted version of the HRP (horseradish peroxidase) protein from the AOX1 promoter. pHL2 was created by amplifying the HRP coding sequence of pCH1 with the primers HarryHRPfoEcoR1 AAGAATTCCCCAGTTAACCCCTACATTC and HarryHRPrevNot1 TTGGCGGCCGCAGAGTTGCTGTTGACCAC, digesting the PCR product with enzymes EcoR1 and NotI, and inserting it into the respective sites of pJV1F. The resultant pHL2 expresses a secreted MBP fused to a Factor Xa protease recognition site followed by HRP coding sequence with myc and His6 tags under the control of the AOXI promoter.
Site directed mutagenesis
Site directed mutagenesis was performed with the QuikChange Site-directed Mutagenesis Kit (Stratagene, La Jolla, CA) according to manufacturer’s instructions to construct pHL-IEGV, pHL-ΔIEGR, pHL-ΔpolyN, and pHL-ΔEA.
Pichia pastoris transformation
Transformations of P. pastoris were done by the electrotransformation method as described [33]. Plasmids were linearized within the AOXI promoter with restriction enzymes, purified with the QIAprep Spin PCR Purification Kit (Qiagen, Valencia, CA) and electroporated into competent yJC100 cells. Transformed cells were allowed to recover for one hour in 1 mL of a 50% 1M sorbitol/50% YPD solution at 30°C and then plated on selective medium. For P. pastoris, Zeocin was added to a final concentration of 100 µg/ml for selection of transformants. Transformed colonies were then purified by streaking for isolated colonies on selective medium.
Colony PCR
The rapid colony PCR method, used to determine the presence of the recombinant genes in cells, has been previously described [34].
Small scale MBP fusion expression
Cultures were first grown overnight in YPD medium to stationary phase. On the second day, the optical density was measured, and 1.0 OD600 units of each culture were pelleted for 5 minutes at 2000×g at room temperature. The cells were suspended in 5 ml of BMGY medium in a 50 ml conical centrifuge tube. On the third day, the optical density was measured, and 10 OD600 units of each culture were pelleted for 5 minutes at 2000×g at room temperature. The cells were suspended in 5 mls of BMMY medium in a 50 ml conical centrifuge tube. The cultures were induced for 48 and 72 hours at 30°C with shaking (225 rpm), adding methanol to 0.5% every 24 hours to compensate for losses from evaporation and metabolism. At each time point, the OD600 of each culture was measured, and 1.0 ml was spun down at 10,000×g for 1 minute to separate cells from extracellular supernatant. The supernatants were transferred to a new microcentrifuge tube, and Protease Arrest protease inhibitor (G Biosciences, St. Louis, MO) was added. Pellets and supernatants were immediately frozen and stored at −80°C. This protocol was also scaled up to 20 fold (100 ml BMMY) for larger preparations of MBP fusion peptides.
Concentration of samples
Cell cultures were harvested by centrifugation (5000 rpm, 4°C, 10 minutes) to remove cell debris. The resulting supernatant was then filtered through a 0.22 µm low binding membrane (Corning, Lowell, MA). The filtered supernatant was added to the Macrosep 10K Centrifugal Device (Pall Life Science, Ann Arbor, MI) and centrifuged at 5000×g for 30 minutes at 4°C. This procedure was repeated until the desired volume was reached (concentrate 50 times). The above procedure was repeated with MBP column buffer (200 mM NaCl, 20 mM Tris-HCl, 1 mM EDTA, pH 7.4) to exchange buffer (MBP column buffer: concentrated supernatant = 10:1).
Purification of MBP fusion proteins
Purification with amylose magnetic beads (New England Biolabs, Beverly, MA) was performed according to the manufacturer’s instructions. Briefly, for small scale purification, 100 µl of bead suspension, corresponding to 1 mg of beads, was added to a 1.5 ml sterile microcentrifuge tube. 500 µl MBP column buffer was added to the tube and mixed completely. The tube was applied to the magnet for 1 minute to pull beads to the side of the tube, and the buffer was removed. The beads were washed again with 500 µl of MBP column buffer. Then 250 µl of concentrated cell culture supernatant was added to the beads and mixed thoroughly. The mixture was incubated at 4°C with gentle rocking for 1 hour. The magnetic beads were collected by holding the tube near the magnet, and the unbound fractions were removed. Three successive washes with 500 µl of MBP column buffer were performed. Then 50 µl of MBP elution buffer (MBP column buffer containing 10 mM maltose) was added to the bead pellet. The tube was vortexed and incubated for 10 minutes at 4°C with gentle rocking. The tube was applied to the magnet, and the eluted MBP-fusion protein was pipetted into a clean microcentrifuge tube. An additional 50 µl of elution buffer was added to the beads and the elution step was repeated. The elution supernatants were pooled.
Large scale purification with amylose resin was carried out as follows. Amylose resin (20 ml) slurry was added into a 50 ml conical tube. The tube was centrifuged at 600×g for 1 minute at 4°C and the supernatant was removed. Then the resin was washed three times, each time with 33 ml of MBP column buffer and centrifugation at 600×g for 1 min at 4°C. MBP column buffer (20 ml) and concentrated, buffer-exchanged MBP-fusion protein (5 ml) was added to the amylose resin. The conical tube containing the amylose resin was vortexed and incubated at 4°C with gentle rocking for 4 hours. The resin suspension was transferred to the column. Non-specific proteins were washed away with 6 column volumes of MBP column buffer. Protein elution was achieved with 2.5 column volumes of MBP elution buffer, and 1 ml fractions were collected. The protein concentration was determined by spot western with anti-MBP antibody.
Mass spectrometry analysis
The proteins were purified by amylose resin followed by a subsequent SDS-PAGE gel. The gel bands were excised, reduced with DTT and alkylated with acrylamide (Cys-S-β-propionamide) and finally digested with trypsin or Glu-C. The peptide pool was loaded onto a C18 column for nano-reverse phase HPLC-MS-analysis (LCQ Deca XP+, Thermo, San Jose, CA).
The purified protein from the amylose magnetic beads was desalted with a spin column (Pierce, Rockford, IL, USA) and then dried by speed vacuum. Trypsin (Promega, Madison, WI) digestion of proteins was performed in 40 µl of 25 mM ammonium bicarbonate (pH 7.8) with an enzyme/substrate ratio of 1:20 (w/w). The digest was carried out at 37°C overnight. The endoproteinase Asp-N (Sigma, Saint Louis, MO) digestion of protein was performed in 40 µl of 100 mM Tris-HCl buffer (pH 8.5) using an enzyme/substrate ratio of 1:100 (w/w) at 37 °C overnight. The digestion was desalted with a C18 ZipTip (Millipore, Bedford, MA, USA) according to the manufacturer’s procedure. Briefly, the C18 ZipTip pipette tip was equilibrated with MeCN and 0.1% TFA for 3 cycles each. Then the peptides were adsorbed onto the C18 ZipTip by aspirating and dispensing the sample 10 cycles for the maximum binding. Finally, the C18 ZipTip was washed with 0.1% TFA for 3 cycles, and the peptide mixture was eluted with 50% MeCN/0.1% TFA in 10 µl for 10 cycles.
MALDI-TOF analysis was performed on a Kratos/Shimadzu AXIMA CFR MALDI-TOF and a MALDI-QIT-TOF (Shimadzu Biotech, Pleasanton, CA) mass spectrometer operating in positive-ion reflectron mode. A nitrogen laser (337 nm) was used to ionize the samples. Volumes of 1 µl of the samples to be analyzed were mixed with 1 µl of α-cyano-4-hydroxycinnamic acid (CHCA, saturated in 50% MeCN in 0.1% TFA) or 1 µl of 2,5-dihydroxybenzoic acid (DHB, saturated in 50% MeCN/0.1% TFA), and the resulting mixture was applied to the target and air-dried. Calibration was performed externally with the Sigma Peptide and Protein MALDI-MS Calibration Kit (Sigma, Saint Louis, MO).
Polyacrylamide gel electrophoresis and western analysis
Protein concentrations were determined using the Pierce (Rockford, IL) Bicinchoninic Acid (BCA) Protein Assay kit with bovine serum albumin as a standard. The spot western technique has been described in detail [5]. Either equivalent amounts of protein or volumes of extracellular medium from equivalent numbers of cells were loaded onto stacked sodium dodecyl sulfate-polyacrylamide gels (SDS-PAGE) for electrophoresis [32]. For samples of supernatant, volumes of extracellular medium corresponding to approximately 0.25 OD600 of cells per lane (i.e. 25 µl of medium from cells at OD600 10) were loaded. For intracellular extracts, 50 µg of total protein were loaded into each lane. Gels were either stained for total protein visualization using Silver Snap II (Pierce, Rockford, IL) stain or analyzed by western analysis. For westerns, proteins were transferred onto nitrocellulose membrane using a Mini Trans-Blot Electrophoretic Transfer Cell (Bio-Rad, Hercules, CA) according to the manufacturer’s instructions. Immunoblots were visualized with the Western-Star Protein Detection Kit (Tropix, Bedford, MA). MBP-2 protein was purchased from New England Biolabs, and the horseradish peroxidase was acquired from Sigma. The anti-myc, anti-FKBP12 and anti-EGFP antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) while the anti-MBP antibodies were acquired from New England Biolabs. Anti-HRP antibodies were purchased from Jackson ImmunoResearch (West Grove, PA). Secondary antibodies, conjugated to alkaline phosphatase, were purchased from Tropix (Bedford, MA). Chemiluminescent signals resulting from alkaline phosphatase activity were captured with a ChemiImager 5500 (Alpha Innotech, San Leandro, CA).
RESULTS
High level expression and proteolysis of MBP fusions
MBP, maltose binding protein, is frequently used in E. coli expression systems as a fusion partner for improved expression and purification of a recombinant cargo protein. We tested the feasibility of using MBP in Pichia pastoris for the same purpose. First, pJV1 (MBP-FXa) was designed to check if MBP by itself can be secreted effectively in the P. pastoris expression system. In this construct, MBP and the Factor Xa site (Ile-Glu-Gly-Arg) was fused in frame with the Saccharomyces cerevisiae MATα prepro peptide under the control of the inducible alcohol oxidase 1 (AOX1) promoter (Figure 1). As was the case with all our recombinant strains in this work, the transformed cells underwent colony PCR analysis to ensure that the yeast cells contained the entire expression cassette. Cells harboring this construct were grown on glycerol to accumulate biomass and then shifted to methanol-containing medium to induce the expression and secretion of the MBP peptide. Aliquots of extracellular medium from equivalent numbers of cells were removed after specific time periods. The silver-stained SDS-PAGE of these samples indicated that the protein present in the 48 hour supernatant of yeast cells with pJV1 (Figure 2A, lane 3) was similar in size compared to the standard MBP-2 peptide purchased from New England Biolabs, which was 42.5 kDa (Figure 2A, lane 1). The approximate 45 kDa size of MBP-FXa peptide from pJV1 suggested that the MATα prepro peptide had been removed prior to export while the band intensity indicated that it had undergone efficient secretion, estimated at 50 µg/mL. Western blot of pJV1 cell culture supernatant with anti-MBP antibody confirmed the identity of MBP (Figure 2B, lane 3). Strains containing the empty plasmid (pPICZαB) did not express the protein as expected (Figure 2B, lane 2).
Figure 1. Diagram of Representative Constructs Generated for Fusion Protein Studies.

Constructs were made with combinations of six elements: the MATα prepro peptide (MATα), E.coli maltose binding protein (MBP), a protease site for Factor Xa (F) or enterokinase (E), a cargo protein (CARGO), and a myc tag (✦) for antibody detection.
Figure 2. Silver stain and western analysis of pJV1 (MBP-FXa) and pJV2 (MBP-FXa-FKBP12-myc-His6) expression.

Strains containing the expression plasmids were grown in BMGY glycerol medium and then induced in BMMY methanol medium, as described in the Methods. Aliquots of culture supernatant, corresponding to equivalent numbers of cells (based on OD600), were subjected to SDS-PAGE and then visualized with either (A) silver stain or (B) western blot. Westerns utilized a primary rabbit anti-MBP antibody followed by a secondary goat anti-rabbit antibody. Lane M: molecular weight marker; lane 1: MBP-2 standard; lane 2: pPICZαB supernatant; lane 3: pJV1 supernatant; lane 4: pJV2 supernatant.
Our secondary experiments were aimed at determining if the MBP could serve as a fusion partner to improve the secretion of recombinant cargo proteins. These peptides by themselves are secreted at different levels of efficiency; thus they were suitable to test the ability of MBP to act as an escort for secretion. We constructed pJV2 to express MBP-FXa-FKBP12-myc-His6 (expected molecular weight of 58 kDa), which is illustrated (Figure 1). FKBP12, a human protein associated with immunoregulation and basic cellular processes involving protein folding and trafficking, interacts with several intracellular signal transduction proteins and was the object of research in our lab in the past [35]. The linker region between MBP and FKBP12 is the Factor Xa recognition sequence. This site is specifically recognized by the human Factor Xa protease, which is used to facilitate the removal of MBP from the cargo protein after fusion protein purification with amylose resin in a bacterial expression system. Also the pJV2 fusion peptide contained C-terminal myc and His6 tags as the sequences encoding myc and His6 tags are provided in the pPICZαB vector. To our surprise, the protein present in pJV2 cell culture supernatant, which should be MBP-FXa-FKBP12-myc-His6, had a molecular mass of approximately 45 kDa as opposed to the expected 58 kDa (Figure 2A, lane 4). MBP-specific western blot analysis indicated that the protein contained MBP at least as a part of its structure (Figure 2B, lane 4). Furthermore, when the blots were probed with anti-myc or anti-FKBP12 antibodies, no peptide was detected in the extracellular fractions of cells expressing pJV2 (data not shown). The band identified by our MBP antibody was about the same molecular weight as the commercially available 42.5 kDa MBP-2 and the MBP-FXa peptide expressed from pJV1 (Figure 2B). Varying culture parameters (growth at 22°C, utilizing medium without peptone and yeast extract supplements, utilizing growth medium buffered to pH 5 and 7) did not affect this phenomenon. These results suggested that the secreted fusion protein expressed from pJV2 (MBP-FXa-FKBP12-myc-His6) experienced proteolysis either inside or outside the cell during secretion. Based on the size of the secreted protein, proteolysis occurred possibly in the MBP/Factor Xa/FKBP12 junction region.
Cleavage of MBP fusions regardless of the identity of the cargo proteins
We hypothesized that the proteolysis was associated with the cargo protein FKBP12 because the Factor Xa sequence has not been proteolyzed by P. pastoris in other published reports [36, 37]. Therefore, two new constructs were designed, which were pJV3 (MBP-FXa-Spe9-myc-His6) and pJV4 (MBP-FXa-EGFP-myc-His6), as illustrated in Figure 1. The C. elegans spe-9 gene encodes a sperm transmembrane protein that contains EGF-like repeats and plays an important role in fertilization [38]. Enhanced green fluorescent protein (EGFP) is a useful tool for looking at gene expression [39]. Strains were grown on glycerol and then induced on methanol to trigger the expression of the MBP fusions, as done previously. As shown in Figure 3, MBP-specific western blot analysis of the cell culture supernatant for both fusion proteins indicated bands of comparable size to those expressed from pJV1 (MBP-FactorXa, lane 4) and pJV2 (MBP-FXa-FKBP12-myc-His6, lanes 5 and 6). In relation to other fusion proteins, the pJV3 (MBP-FXa-Spe-9-myc-His6, lane 7) construct was secreted less efficiently overall, possibly due to the fact that cargo protein Spe-9 by itself was hard to express and secrete. This Spe-9 fusion was degraded to a greater degree and to a smaller size compared to the pJV2 peptide. Although the cell culture supernatant of pJV4 (MBP-FXa-EGFP-myc-His6, lane 8) showed the presence of a minor species at 75 kDa, which corresponded to the expected size of the full-length protein MBP-FXa-EGFP-myc-His6, the major species of protein was around 45 kDa in the cell culture supernatant, corresponding to MBP alone. In addition, a 29 kDa band, detected by anti-EGFP antibodies, was also found at low levels in the pJV4 cell culture supernatant (data not shown).
Figure 3. Western analysis of pJV2 (MBP-FXa-FKBP12-myc-His6), pJV3 (MBP-FXa-Spe9-myc-His6) and pJV4 (MBP-FXa-EGFP-myc-His6) proteins with anti-MBP antibody.

Lane 1: MBP-2 standard; lane 2: pPICZαB supernatant; lane3: empty; lane 4: pJV1 supernatant; lane 5: pJV2 supernatant; lane 6: pJV2 concentrated supernatant; lane 7: pJV3 supernatant; lane 8: pJV4 supernatant.
Because the cargo proteins are different in the pJV2 (MBP-FXa-FKBP12-myc-His6), pJV3 (MBP-FXa-Spe-9-myc-His6) and pJV4 (MBP-FXa-EGFP-myc-His6) expression strains but the all strains produced a similar proteolyzed MBP fragment (Figure 3), we hypothesized that the cleavage must occur at or in close proximity of the Factor Xa recognition site IEGR or in the C-terminal MBP region. However, computer analysis of the MBP protein sequence indicated that no known P. pastoris protease sites (i.e. Kex2, Ste13) are located in this region (www.expasy.ch/tools). Second, provided the C-terminal processing is due to the same protease in all cases, the difference in the pJV3 and pJV4 results indicated that the cleavage rate depends on the cargo protein itself.
Localization of the proteolytic event
Proteolytic degradation is a common problem encountered in the expression of recombinant peptides in large scale fermentation in P. pastoris [6, 14, 40]. To begin to understand the nature of this MBP associated degradation, we desired to determine if the MBP proteolysis occurred inside or outside the cell. Therefore, both intracellular and extracellular fractions were isolated from cultures expressing the pJV2 product, MBP-FXa-FKBP12-myc-His6 fusion. MBP-specific western blot analysis of the intracellular and extracellular fractions from the yeast strain harboring the pJV2 fusion protein indicated that the major protein species in the cell extract was the larger intact fusion protein while only minor portions were proteolyzed (Figure 4, lane 1). On the other hand, the cell culture supernatant contained primarily the smaller, degraded protein and only minor quantities of the intact fusion protein (Figure 4, lane 4). These results suggested that the proteolysis started inside the cell and was completed during secretion.
Figure 4. Western analysis of pJV2 (MBP-FXa-FKBP12-myc-His6) supernatant and cell extract with anti-MBP antibody.

Volumes of culture supernatant, corresponding to equivalent numbers of cells (based on OD600), were analyzed in supernatant lanes while equivalent masses of cell extract were examined in cell extract lanes. Lane 1: pJV2 cell extract; lane 2: pPICZαB cell extract; lane 3: pPICZαB supernatant; lane 4: pJV2 supernatant; lane 5: MBP-2 standard
Purification and determination of the cleavage site in the pJV2 (MBP-FXa-FKBP12-myc-His6) fusion
To gain further insight into the proteolysis mechanism, the terminal amino acid sequence in the processed fusion protein was elucidated. Here, the pJV2 (MBP-FXa-FKBP12-myc-His6) was used as the model protein. The extracellular medium containing the proteolyzed MBP-FXa-FBP12-myc-His6 was subjected to identical purification strategy used to isolate MBP fusions from E. coli cellular extracts. This procedure, based on amylose magnetic bead binding, worked well for the P. pastoris-derived gene product, confirming that the MBP moiety was correctly folded and functional (data not shown). Furthermore, when the purified MBP fragment was digested with PNGaseF, an enzyme which removes N-linked carbohydrate groups from proteins, its mobility did not change significantly, suggesting that glycosylation was minimal for MBP in the P. pastoris secretory network (data not shown).
To identify the proteolysis site, the purified pJV2 peptide was then digested with trypsin. The tryptic peptide pool was extracted by the C18 ZipTip and applied to the MALDI-TOF analysis. Based on the mass only, the m/z value of 2575.1 corresponded to the peptide 365DAQTNSSSNNNNNNNNNNLGIEGR388 in the known pJV2 amino acid sequence (Figure 5). However, because trypsin cleaves peptide chains at the carboxyl side of the amino acids lysine and arginine, the only conclusion is that peptide 365DAQTNSSSNNNNNNNNNNLGIEGR388 is present in pJV2 (MBP-FXa-FKBP12-myc-His6) product (Table 1). Therefore, we were not able to exclude other amino acids downstream from IEGR in the fusion protein.
Figure 5. MALDI-TOF analysis of trypsin digestion of pJV2 (MBP-FXa-FKBP12-myc-His6) product.

Table 1. C-terminal residues of the proteolyzed pJV2, pHL-ΔIEGR, and pAW2 proteins as determined by mass spectrometry.
Superscript numbers refer to the location of amino acids in the mature fusion peptides. The C-terminus of the MBP region contains a glutamic acid alanine sequence (deleted in pHL-ΔEA), a string of ten asparagines (N10, deleted in pHL-ΔpolyN), and the Factor Xa recognition site, IEGR (deleted in pHL-ΔIEGR). The R represents the arginine which was changed to a valine for pHL-IEGV. The FKBP12 region and the histidine tag are italicized. The IEGRGIP sequence of pJV2 was replaced in pAW2 with DDDDKVPNS, which contains the enterokinase recognition site. Amino acids determined to be the C-termini by mass spectrometry are underlined.
| pJV2 (MBP-FXa-FKBP12-myc-His6) |
| pHL-ΔIEGR (MBP-FKBP12-myc-His6) |
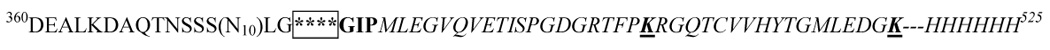 |
| pAW2 (MBP-EK-FKBP12-myc-His6) |
To determine if IEGR was truly the C-terminal sequence of the pJV2 proteolytic fragment, further mass spectrometric analysis was performed. Figure 6 shows the MALDI-TOF mass spectrum of endoproteinase Asp-N digestion of the pJV2 product. The peak at m/z 2574.9 corresponds to peptide 365DAQTNSSSNNNNNNNNNNLGIEGR388. The identity of the peptide was supported by MS/MS analysis shown in Figure 7. According to the known amino acid sequence from cDNA, the amino acid after IEGR is glycine, which will not be cleaved by Asp-N. Taken together, the mass spectrometric data analyses indicated that the C-terminus of processed pJV2 (MBP-FXa-FKBP12-myc-His6) protein is IEGR, the Factor Xa recognition site (Table 1).
Figure 6. (A) MALDI-TOF analysis of endoproteinase Asp-N digestion of pJV2 (MBP-FXa-FKBP12-myc-His6) product.

Figure 7. MS/MS of selected ion 365DAQTNSSSNNNNNNNNNNLGIEGR388 in MALDI-QIT-TOF.

Mutagenesis of cleavage site
Proteolysis can be caused by the actual primary sequence of the peptide or a structural feature, such as hydrophobicity, associated with the peptide. To confirm our hypothesis that the amino acids in the Factor Xa recognition site IEGR triggered proteolysis, a series of mutations were introduced in the spacer region between MBP and the C-terminal cargo proteins in pJV2 (MBP-FXa-FKBP12-myc-His6). The C-terminus of MBP contains some intriguing regions: a glutamic acid alanine (EA) sequence which is part of the recognition site (EAEA) of the yeast protease Ste13 as well as a stretch of ten consecutive asparagines (Table 1) [3, 31]. Therefore, the following constructs were made: pHL-IEGV construct had a missense mutation in the Factor Xa site from IEGR to IEGV, pHL-ΔIEGR did not have the IEGR region at all (Figure 1), pHL-ΔEA lacked the glutamic acid alanine sequence, and pHL-ΔpolyN lacked the ten asparagines upstream of the FXa site (Table 1). Furthermore, pAW1 (MBP-EK-myc-His6) was created by the replacement of the Factor Xa IEGR with the protease enterokinase (EK) site DDDDK. In analogy to pAW1, pAW2 (MBP-EK-FKBP12-myc-His6) and pAW3 (MBP-EK-EGFP-myc-His6) were also created with the in frame fusion of FKBP12 and EGFP, respectively (Figure 1). The strains harboring these constructs were induced as usual. However, none of the new constructs showed resistance to proteolysis (Figures 8 and 9), as westerns with anti-MBP antibody indicated that the processed proteins accumulated as truncated 45 kDa proteins (pHL-ΔEA samples not shown). Furthermore, anti-myc antibodies were unable to identify any protein species (data not shown). The only exception was the pAW3 strain, which showed a minor species of around 75 kDa corresponding to the expected full length of the MBP-EK-EGFP-myc-His6 fusion protein in the cell culture supernatant (Figure 9, lane 6). As seen previously in Figure 3, the presence of EGFP as the cargo protein partially inhibits this proteolytic phenomenon.
Figure 8. Western of pJV2, pHL-IEGV, pHL-ΔIEGR and pHL-ΔpolyN with anti-MBP antibody.

Lane 1: MBP-2 standard; lane 2: pJV2 supernatant; lane 3: pPICZαB supernatant; lane 4: pHL-IEGV supernatant; lane 5: pHL-ΔIEGR supernatant; lane 6: pHL-ΔpolyN supernatant.
Figure 9. SDS-PAGE with (A) silver staining and (B) western analysis with anti-MBP antibody of pAW1 (MBP-EK-myc-His6), pAW2 (MBP-EK-FKBP12-myc-His6) and pAW3 (MBP-EK-EGFP-myc-His6).

Lane M: Marker; lane 1: MBP-2; lane 2: pPICZαB supernatant; lane 3: empty; lane 4: pAW1 supernatant; lane 5: pAW2 supernatant; lane 6: pAW3 supernatant.
Taken together, these observations are consistent with three scenarios. It is possible that one non-specific protease cleaves in close proximity at or around the IEGR region regardless of modifications. It is also possible that multiple proteases are operating. Finally, one specific protease (e.g. for basic amino acids R or K) possibly cleaves preferentially at IEGR when present but reverts to other basic sites in close proximity if IEGR is modified.
In order to further elucidate details of the proteolytic event, the real C-termini of the other fusion proteins needed to be identified. Here, we used the fusion protein pHL-ΔIEGR (MBP-FKBP12-myc-His6) and pAW2 (MBP-EK-FKBP12-myc-His6) as the model proteins because the linker region IEGR was deleted in the pHL-ΔIEGR while pAW2 had a completely different linker region between the MBP and the FKBP12 cargo protein. The proteins were purified by amylose resin followed by subsequent SDS-PAGE. The gel bands were excised, digested with trypsin or Glu-C, and the peptide pool was loaded onto a C18 column for nano-reverse phase HPLC-MS analysis (LCQ Deca XP+). 407RGQTCVVHYTGMLEDGK423 and 395TISPGDGRTFPK406 were identified as the possible C-termini for the pHL-ΔIEGR (MBP-FKBP12-myc-His6) product, while 369NSSSNNNNNNNNNNLGDDDDKVPN392 and 402TISPGDGRTFPKRGQTC418 were identified as the two possible C-termini for the pAW2 (MBP-EK-FKBP12-myc-His6) strain (data not shown). We could not conclude which was the major species. The cleavage sites were found either in the spacer region or the cargo protein (Table 1). However, there appeared no similarity or common theme to these residues. Thus, proteolysis occurred at different target amino acids in the pJV2 (MBP-FXa-FKBP12-myc-His6), pHL-ΔIEGR (MBP-FKBP12-myc-His6) and pAW2 (MBP-EK-FKBP12-myc-His6) as a result of residue changes in an upstream spacer region (Table 1).
Use of MBP as a secretion enhancer
Although proteolysis occurred during the secretory process, we were interested in exploring the fates of the cargo proteins after they were cleaved from MBP. Specifically we wished to determine if the temporary association with MBP enhanced the secretion of the cargo proteins. Therefore, we compared the export of the cargo protein expressed by themselves to cargo proteins fused to MBP (Figure 1). In the case of FKBP12, the peptide by itself was secreted efficiently from a strain expressing the plasmid pCA2 (FKBP12-myc-His6); the anti-FKBP12 antibody detected high levels of antigen in the extracellular supernatant (Figure 10B, lane 1). We hypothesize that recombinant FKBP12 dimerized with itself, producing the 30 kDa band along with the expected 15 kDa species. However, when it was fused to MBP and subsequently cleaved, FKBP12 was undetectable in western analysis of the extracellular medium of pJV2 (Figure 10B, lane 2) although the MBP portion was secreted efficiently (Figure 10A, lane 2). Thus, MBP actually decreased FKBP12’s export from the cell, presumably by increasing its degradation. However, fusion to MBP enhanced the secretion of EGFP. EGFP, when expressed as a singular moiety from pKS1 (EGFP-myc-His6), was undetectable in westerns with anti-EGFP antibodies (Figure 10D, lane 4). When EGFP was fused to an N-terminal MBP, as in pJV4 (MBP-FXa-EGFP-myc-His6), the majority was proteolyzed (Figure 10C, lane 3). However the released EGFP (Figure 10D, lane 3) was secreted at a much higher rate than the EGFP that was not fused to MBP(Figure 10D, lane 4).
Figure 10. Western analysis of supernatants to determine fates of cargo proteins.

Parts A and B. Lane 1:pCA2 (FKBP-myc-His6) supernatant and lane 2: pJV2 (MBP-FXa-FKBP12-myc-His6) supernatant probed with anti-MBP (A) and anti-FKBP12 (B) antibodies. Part C. Lane 1: MBP-2 standard; lane 2: pPICZαB supernatant; lane 3: pJV4 (MBP-FXa-EGFP-myc-His6) supernatant; lane 4: pKS1 (EGFP-myc-His6) supernatant probed with anti-MBP antibody. Part D. Lane 1: E.coli expressing pGLO plasmid (EGFP standard); lane 2: pPICZαB supernatant; lane 3: pJV4 (MBP-FXa-EGFP-myc-His6) supernatant; lane 4: pKS1 (EGFP-myc-His6) supernatant probed with anti-EGFP antibody. Part E. Lane 1: MBP-2 standard; lane 2: pCH1 (HRP) supernatant; lane 3: pHL2 (MBP-FXa-HRP-myc-His6) supernatant probed with anti-MBP antibody. Part F. Lane 1: native horseradish peroxidase; lane 2: pCH1 (HRP) supernatant; lane 3: pHL2 (MBP-FXa-HRP-myc-His6) supernatant probed with anti-HRP antibody.
Because of the opposite effects of MBP on FKBP12 and EGFP, we tested MBP’s effect on a third cargo protein, horseradish peroxidase (HRP). HRP was shown previously to be exported with relatively moderate efficiency from P. pastoris [41]. Western analysis indicated that in our studies, the MBP-HRP fusion was proteolyzed as expected (Figure 10E, lane 3). The HRP released from this MBP fusion, a product of pHL2 (MBP-FXa-HRP-myc-His6), was secreted at much higher levels (Figure 10F, lane 3) than the HRP produced by itself from pCH1 (HRP) (Figure 10F, lane 2). The P.pastoris-produced HRP is a larger molecular weight than the purchased HRP because of hyperglycosylation. This 80 kDa peptide does not react with anti-MBP antibody (Figure 10E, lane 3), demonstrating that it does not contain the escort protein. Our attempts to purify the proteolyzed cargo protein utilizing the 6His tag were unsuccessful. Furthermore enzymatic assays of extracellular media from both strains indicated higher levels of HRP activity in the strain expressing pHL2 (MBP-FXa-HRP-myc-His6) compared to the strain expressing pCH1 (HRP) (data not shown). Thus, in two of three cases, MBP can serve as a secretion enhancer even though it is cleaved from its cargo protein.
DISCUSSION
In this study, several constructs were generated to test whether or not maltose binding protein (MBP) was able to improve the production and purification of recombinant proteins in P. pastoris. Although many investigators have exploited MBP for this purpose in the E. coli, the mechanism by which MBP increases the solubility and export of its cargo proteins is not fully understood in bacteria, where it has been utilized for protein expression for almost 20 years. The most popular model is that MBP possesses chaperonin-like characteristics [42]. MBP contains hydrophobic residues in its ligand bonding cleft, which appear to play a central role guiding the cargo proteins to their native structures [42]. These properties are believed to allow MBP to bind reversibly to folding intermediates of its fusion partners and temporarily keep them in a conformation that prevents their self-association and aggregation [19, 43, 44].
We expected the N-terminal MBP fusions expressed in P.pastoris to be secreted into the extracellular medium. Although all the constructs were expressed at high levels and could be purified with an amylose chromatography (suggesting proper folding), our westerns indicated that the fusion proteins were truncated to a polypeptide approximately the same molecular weight as MBP. We varied expression conditions by altering the growth temperature and components of the culturing media; however, this cleavage could not be inhibited.
In our constructs, there is a protease recognition site for the human blood Factor Xa in spacer region between MBP and the cargo protein. Normally, the Factor Xa site is used after purification to separate the cargo protein from MBP. This protease recognition site (Ile-Glu-Gly-Arg) should not be recognized in P. pastoris [36, 37]. Because of our initial findings, we hypothesized that the Factor Xa site was being recognized by an uncharacterized protease inside the P. pastoris cell and, as a result, the Factor Xa site was replaced with an enterokinase cleavage site. However, all fusion proteins were truncated regardless of whether: a) the protease site in between the fusion partners was the cleavage recognition site for Factor Xa (Ile-Glu-Gly-Arg), a mutated Factor Xa (Ile-Glu-Gly-Val), enterokinase (Asp-Asp-Asp-Asp-Lys), or completely absent or, b) the cargo protein was FKBP12, Spe-9, HRP or EGFP. Because all the secreted proteins, which differed in the spacer area harboring the protease cleavage site or in the cargo peptide regions, appeared the same size on westerns, we hypothesize that a P. pastoris protease possibly recognized a three-dimensional structure in MBP, not a specific amino acid sequence, to cause cleavage further downstream of the MBP region. When sequences in the MBP or spacer region were deleted, mass spectrometry indicated that the site of proteolysis changed to amino acids in the cargo protein, partially confirming our hypothesis. Taken together, the results suggest that some property other than a specific amino acid sequence in the MBP fusion triggers proteolytic attack in regions C-terminal of the MBP moiety and that the cleavage sites change with variations in the linker regions.
The existence of this type of protease in P. pastoris has been proposed by other investigators [45, 46]. Gustavsson and colleagues studied a stretch of amino acids called a linker situated between 2 domains of a fusion protein. Specifically, the authors found that a CBD (cellulose binding domain) –CALB (Candida antarctica lipase B) fusion protein was unstable due to a proteolytic cleavage at the linker, which resulted in a mixture of CBD-CALB and a degradation product equal in size to the wild type CALB produced in P. pastoris. Substitution of different amino acids in the linker in some cases decreased the rate of proteolysis; however even with completely different residues occupying the linker, this region was still the major site of cleavage [46]. In fact two linkers with the same approximate length but different amino acid composition were cleaved at an equal extent. Whether the same or different proteases were required for the proteolysis of the different linker regions was not discussed [46].
Several proteases have been characterized in P. pastoris, including carboxypeptidase Y, Kex2, proteinase A (encoded by PEP4) and proteinase B (encoded by PRB1) [14]. Proteases garner great interest because protein degradation is a major obstacle when expressing recombinant proteins in large scale. As P. pastoris cells die in culture, they release intracellular proteases, which in turn cause degradation of the valuable heterologous peptides in the extracellular culture [40]. Identifying, characterizing and neutralizing these enzymes would improve the yeast’s application as a host for protein production. We have initiated efforts to identify the MBP-associated protease, including a screen to isolate novel mutant strains that secrete nonproteolyzed MBP fusions. In addition, we expressed our pJV2 product MBP-FXa-FBP12-myc-His6 in a double mutant strain, SMD1163, which lacked both proteinase A and B activities [3]; however proteolysis still occurred as usual, suggesting that these two major proteases are not involved in this mechanism (data not shown). Very little is known about proteases that recognize structural features other than specific amino acid residues; therefore, identifying this gene may provide novel insights.
Although proteolysis occurred between MBP and its cargo proteins, in two of three cases, MBP still acted to enhance secretion. The export of EGFP and HRP were aided by temporary association with MBP. MBP’s ability to facilitate export is dependent on the specific cargo protein’s structure; some peptides, such as FKBP12, are not aided by the escort’s presence [47]. Therefore, at this time, it is difficult to predict whether MBP can improve the secretion of a particular cargo protein. The MBP fusion must be constructed and tested on an individual basis. However, the potential increase in yield, as seen with EGFP and HRP, makes this option worth pursuing. In these cases, an affinity tag needs to be present on the N- or C-terminus of the cargo protein to assist in purification since MBP is separated from the cargo protein due to cleavage. Nevertheless, MBP’s ability to enhance export may be extremely helpful to scientists who struggle with important recombinant proteins that are poorly secreted from the P. pastoris cell.
Acknowledgements
This work was supported by NIH-AREA grant GM65882-02 to J. L.-C. and G. P. L.-C. Z. L. was supported with intramural research funds from the Department of Chemistry, University of the Pacific. We thank Dr. Chris Adams (Stanford University Mass Spectrometry) for his assistance with mass spectrometry.
Abbreviations
- MBP
maltose binding protein
- EGFP
enhanced green fluorescent protein
- FXa
Factor Xa
- EK
enterokinase
- HRP
horseradish peroxidase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cregg JM, Madden KR. Development of the methylotrophic yeast,Pichia pastoris, as a host system for the production of foreign proteins. Dev. Ind. Microbiol. 1988;29:33–41. [Google Scholar]
- 2.Higgins DR, Cregg JM. Introduction to Pichia pastoris. In: Higgins DR, Cregg JM, editors. Pichia Protocols. Totowa, New Jersey: Humana Press; 1998. pp. 1–15. [Google Scholar]
- 3.Cereghino JL, Cregg JM. Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol Rev. 2000;24:45–66. doi: 10.1111/j.1574-6976.2000.tb00532.x. [DOI] [PubMed] [Google Scholar]
- 4.Cregg JM, Shen S, Johnson M, Waterham HR. Classical genetic manipulation. In: Higgins DR, Cregg JM, editors. Pichia protocols. Totowa, New Jersey: Humana Press; 1998. pp. 17–26. [Google Scholar]
- 5.Lin-Cereghino GP, Leung W, Lin-Cereghino J. Expression of protein in Pichia pastoris. In: Dyson M, Durocher Y, editors. Expression Systems: Methods Express. Oxfordshire: Scion Publishing Limited; 2007. pp. 123–145. [Google Scholar]
- 6.Jahic M, Veide A, Charoenrat T, Teeri T, Enfors SO. Process technology for production and recovery of heterologous proteins with Pichia pastoris. Biotechnol Prog. 2006;22:1465–1473. doi: 10.1021/bp060171t. [DOI] [PubMed] [Google Scholar]
- 7.Stratton J, Chiruvolu V, Meagher M. High cell-density fermentation. Methods Mol Biol. 1998;103:107–120. doi: 10.1385/0-89603-421-6:107. [DOI] [PubMed] [Google Scholar]
- 8.Brake AJ. Alpha-factor leader-directed secretion of heterologous proteins from yeast. Methods Enzymol. 1990;185:408–421. doi: 10.1016/0076-6879(90)85036-n. [DOI] [PubMed] [Google Scholar]
- 9.Caplan S, Green R, Rocco J, Kurjan J. Glycosylation and structure of the yeast MF alpha 1 alpha-factor precursor is important for efficient transport through the secretory pathway. J Bacteriol. 1991;173:627–635. doi: 10.1128/jb.173.2.627-635.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kjeldsen T. Yeast secretory expression of insulin precursors. Appl Microbiol Biotechnol. 2000;54:277–286. doi: 10.1007/s002530000402. [DOI] [PubMed] [Google Scholar]
- 11.Payne WE, Kaiser CA, Bevis BJ, Soderholm J, Fu D, Sears IB, Glick BS. Isolation of Pichia pastoris genes involved in ER-to-Golgi transport. Yeast. 2000;16:979–993. doi: 10.1002/1097-0061(200008)16:11<979::AID-YEA594>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 12.Rothblatt JA, Webb JR, Ammerer G, Meyer DI. Secretion in yeast: structural features influencing the post-translational translocation of prepro-alpha-factor in vitro. Embo J. 1987;6:3455–3463. doi: 10.1002/j.1460-2075.1987.tb02669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inan M, Fanders SA, Zhang W, Hotez PJ, Zhan B, Meagher MM. Saturation of the secretory pathway by overexpression of a hookworm (Necator americanus) Protein (Na-ASP1) Methods Mol Biol. 2007;389:65–76. doi: 10.1007/978-1-59745-456-8_5. [DOI] [PubMed] [Google Scholar]
- 14.Macauley-Patrick S, Fazenda ML, McNeil B, Harvey LM. Heterologous protein production using the Pichia pastoris expression system. Yeast. 2005;22:249–270. doi: 10.1002/yea.1208. [DOI] [PubMed] [Google Scholar]
- 15.Tsai CW, Duggan PF, Shimp RL, Jr, Miller LH, Narum DL. Overproduction of Pichia pastoris or Plasmodium falciparum protein disulfide isomerase affects expression, folding and O-linked glycosylation of a malaria vaccine candidate expressed in P. pastoris. J Biotechnol. 2006;121:458–470. doi: 10.1016/j.jbiotec.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 16.Sakaguchi M. Eukaryotic protein secretion. Curr Opin Biotechnol. 1997;8:595–601. doi: 10.1016/s0958-1669(97)80035-3. [DOI] [PubMed] [Google Scholar]
- 17.Lee C, Lee S-G, Takahashi S, Kim B-G. The soluble expression of the human renin binding protein using fusion partners: a comparison of ubiquitin, thioredoxin, maltose binding protein and NusA. Biotechnol Bioproc Eng. 2003;8:89–93. [Google Scholar]
- 18.Terpe K. Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol. 2003;60:523–533. doi: 10.1007/s00253-002-1158-6. [DOI] [PubMed] [Google Scholar]
- 19.Kapust RB, Waugh DS. Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 1999;8:1668–1674. doi: 10.1110/ps.8.8.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riggs P. Expression and purification of recombinant proteins by fusion to maltose-binding protein. Mol Biotechnol. 2000;15:51–63. doi: 10.1385/MB:15:1:51. [DOI] [PubMed] [Google Scholar]
- 21.Lichty JJ, Malecki JL, Agnew HD, Michelson-Horowitz DJ, Tan S. Comparison of affinity tags for protein purification. Protein Expr Purif. 2005;41:98–105. doi: 10.1016/j.pep.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 22.Arnau J, Lauritzen C, Petersen GE, Pedersen J. Current strategies for the use of affinity tags and tag removal for the purification of recombinant proteins. Protein Expr Purif. 2006;48:1–13. doi: 10.1016/j.pep.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 23.Daly R, Hearn MT. Expression of heterologous proteins in Pichia pastoris: a useful experimental tool in protein engineering and production. J Mol Recognit. 2005;18:119–138. doi: 10.1002/jmr.687. [DOI] [PubMed] [Google Scholar]
- 24.Lin SC, Lin IP, Chou WI, Hsieh CA, Liu SH, Huang RY, Sheu CC, Chang MD. CBM21 starch-binding domain: a new purification tag for recombinant protein engineering. Protein Expr Purif. 2009;65:261–266. doi: 10.1016/j.pep.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 25.Ahn JO, Choi ES, Lee HW, Hwang SH, Kim CS, Jang HW, Haam SJ, Jung JK. Enhanced secretion of Bacillus stearothermophilus L1 lipase in Saccharomyces cerevisiae by translational fusion to cellulose-binding domain. Appl Microbiol Biotechnol. 2004;64:833–839. doi: 10.1007/s00253-003-1547-5. [DOI] [PubMed] [Google Scholar]
- 26.Rotticci-Mulder JC, Gustavsson M, Holmquist M, Hult K, Martinelle M. Expression in Pichia pastoris of Candida antarctica lipase B and lipase B fused to a cellulose-binding domain. Protein Expr Purif. 2001;21:386–392. doi: 10.1006/prep.2000.1387. [DOI] [PubMed] [Google Scholar]
- 27.Yang D, Peng M, Yang H, Yang Q, Xu J. Expression, purification and characterization of Gloydius shedaoensis venom gloshedobin as Hsp70 fusion protein in Pichia pastoris. Protein Expr Purif. 2009;66:138–142. doi: 10.1016/j.pep.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 28.Piskur J, Kielland-Brandt MC. Folding and secretion of Saccharomyces cerevisiae carboxypeptidase Y are influenced by fusion with short heterologous peptides. Biotechnol. Appl. Biochem. 1993;18:239–257. [PubMed] [Google Scholar]
- 29.Balbas P. Understanding the art of producing protein and nonprotein molecules in Escherichia coli. Mol Biotechnol. 2001;19:251–267. doi: 10.1385/MB:19:3:251. [DOI] [PubMed] [Google Scholar]
- 30.Tropea JE, Cherry S, Nallamsetty S, Bignon C, Waugh DS. A generic method for the production of recombinant proteins in Escherichia coli using a dual hexahistidine-maltose-binding protein affinity tag. Methods Mol Biol. 2007;363:1–19. doi: 10.1007/978-1-59745-209-0_1. [DOI] [PubMed] [Google Scholar]
- 31.DiGuan C, Li P, Riggs PD, Inouye H. Vectors that facilitate the expression and purification of foreign proteins in E. coli by fusion to the maltose binding protein. Gene. 1988;67:21–30. doi: 10.1016/0378-1119(88)90004-2. [DOI] [PubMed] [Google Scholar]
- 32.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A Laboratory Handbook. New York: Cold Spring Harbor Laboratory Press, Cold Spring Harbor; 1989. [Google Scholar]
- 33.Cregg JM, Russell KA. Transformation. In: Higgins DR, Cregg JM, editors. Pichia Protocols. Totowa, New Jersey: Humana Press; 1998. pp. 27–39. [Google Scholar]
- 34.Thor D, Xiong S, Orazem CC, Kwan AC, Cregg JM, Lin-Cereghino J, Lin-Cereghino GP , Cloning and characterization of the Pichia pastoris MET2 gene as a selectable marker. FEMS Yeast Res. 2005;5:935–942. doi: 10.1016/j.femsyr.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 35.Kang CB, Hong Y, Dhe-Paganon S, Yoon HS. FKBP family proteins: immunophilins with versatile biological functions. Neurosignals. 2008;16:318–325. doi: 10.1159/000123041. [DOI] [PubMed] [Google Scholar]
- 36.Calik P, Orman MA, Celik E, Halloran SM, Calik G, Ozdamar TH. Expression system for synthesis and purification of recombinant human growth hormone in Pichia pastoris and structural analysis by MALDI-ToF Mass Spectrometry. Biotechnol Prog. 2008;24:221–226. doi: 10.1021/bp070294t. [DOI] [PubMed] [Google Scholar]
- 37.Celik E, Calik P, Halloran SM, Oliver SG. Production of recombinant human erythropoietin from Pichia pastoris and its structural analysis. J Appl Microbiol 103. 2007:2084–2094. doi: 10.1111/j.1365-2672.2007.03448.x. [DOI] [PubMed] [Google Scholar]
- 38.Singson A, Mercer KB, L'Hernault SW. The C. elegans spe-9 gene encodes a sperm transmembrane protein that contains EGF-like repeats and is required for fertilization. Cell. 1998;93:71–79. doi: 10.1016/s0092-8674(00)81147-2. [DOI] [PubMed] [Google Scholar]
- 39.Cormack B. Green fluorescent protein as a reporter of transcription and protein localization in fungi. Curr Opin Microbiol. 1998;1:406–410. doi: 10.1016/s1369-5274(98)80057-x. [DOI] [PubMed] [Google Scholar]
- 40.Sinha J, Plantz BA, Inan M, Meagher MM. Causes of proteolytic degradation of secreted recombinant proteins produced in methylotrophic yeast Pichia pastoris: case study with recombinant ovine interferon-tau. Biotechnol Bioeng. 2005;89:102–112. doi: 10.1002/bit.20318. [DOI] [PubMed] [Google Scholar]
- 41.Morawski B, Lin Z, Cirino P, Joo H, Bandara G, Arnold FH. Functional expression of horseradish peroxidase in Saccharomyces cerevisiae and Pichia pastoris. Protein Eng. 2000;13:377–384. doi: 10.1093/protein/13.5.377. [DOI] [PubMed] [Google Scholar]
- 42.Nallamsetty S, Waugh DS. Mutations that alter the equilibrium between open and closed conformations of Escherichia coli maltose-binding protein impede its ability to enhance the solubility of passenger proteins. Biochem Biophys Res Commun. 2007;364:639–644. doi: 10.1016/j.bbrc.2007.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fox JD, Kapust RB, Waugh DS. Single amino acid substitutions on the surface of Escherichia coli maltose-binding protein can have a profound impact on the solubility of fusion proteins. Protein Sci. 2001;10:622–630. doi: 10.1110/ps.45201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fox JD, Routzahn KM, Bucher MH, Waugh DS. Maltodextrin-binding proteins from diverse bacteria and archaea are potent solubility enhancers. FEBS Lett. 2003;537:53–57. doi: 10.1016/s0014-5793(03)00070-x. [DOI] [PubMed] [Google Scholar]
- 45.Fitches E, Wiles D, Douglas AE, Hinchliffe G, Audsley N, Gatehouse JA. The insecticidal activity of recombinant garlic lectins towards aphids. Insect Biochem Mol Biol. 2008;38:905–915. doi: 10.1016/j.ibmb.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 46.Gustavsson M, Lehtio J, Denman S, Teeri TT, Hult K, Martinelle M. Stable linker peptides for a cellulose-binding domain-lipase fusion protein expressed in Pichia pastoris. Protein Eng. 2001;14:711–715. doi: 10.1093/protein/14.9.711. [DOI] [PubMed] [Google Scholar]
- 47.Nallamsetty S, Waugh DS. Solubility-enhancing proteins MBP and NusA play a passive role in the folding of their fusion partners. Protein Expr Purif. 2006;45:175–182. doi: 10.1016/j.pep.2005.06.012. [DOI] [PubMed] [Google Scholar]