

Abstract
Cardiac transplantation is an effective treatment for multiple types of heart failure refractive to therapy. Although immunosuppressive therapeutics have increased survival rates within the first year post-transplant, chronic rejection (CR) remains a significant barrier to long term graft survival. Indicators of CR include patchy interstitial fibrosis, vascular occlusion, and progressive loss of graft function. Multiple factors have been implicated in the onset and progression of CR, including TGFβ, IL-6, and connective tissue growth factor (CTGF). While associated with CR, the role of CTGF in CR and the factors necessary for CTGF induction in vivo are not understood. To this end, we utilized forced expression and neutralizing antibody approaches. Transduction of allografts with CTGF significantly increased fibrotic tissue development, though not to levels observed with TGFβ transduction. Further, intragraft CTGF expression was inhibited by IL-6 neutralization while TGFβ expression remained unchanged, indicating that IL-6 effects may potentiate TGFβ-mediated induction of CTGF. Finally, neutralizing CTGF significantly reduced graft fibrosis without reducing TGFβ and IL-6 expression levels. These findings indicate that CTGF functions as a downstream mediator of fibrosis in CR, and that CTGF neutralization may ameliorate fibrosis and hypertrophy associated with CR.
Keywords: Chronic rejection, CTGF, TGFβ, IL-6, fibrosis
Introduction
Chronic rejection (CR) is a significant barrier to long term graft acceptance. Manifestations of CR include interstitial fibrosis, occlusion of luminal structures, and progressive loss of graft function (1–7). The etiology of CR is not fully understood. However, multiple factors have been associated with its onset and progression, especially TGFβ. TGFβ overexpression is linked with chronic rejection (8, 9), and may negatively impact graft survival through chemotactic and pro-fibrotic effects (10). However, in addition to its deleterious fibrotic effects on the graft, TGFβ’s immunosuppressive and anti-proliferative functions may be indispensable for graft and host survival (11). For example, TGFβ plays a critical role in the induction and function of T regulatory cells (Treg), which are believed to contribute to graft acceptance (12–14). Further, TGFβ inhibits T and B cell proliferation (10) and represses cancers of epithelial cell origin (15). These opposing effects make TGFβ a suboptimal target for CR treatments and have prompted investigation into the downstream mediators of TGFβ in CR pathology. Identifying downstream mediators of CR may facilitate the development of therapeutics that negate the fibrosis-inducing activity of TGFβ while sparing its anti-inflammatory and anti-proliferative effects.
One such downstream mediator, known to be induced by TGFβ in multiple cell types (16), including cardiac myocytes and fibroblasts (17), is CTGF. CTGF plays an important role in the development of connective tissue as well as the formation of scar tissue (18, 19), and is upregulated in multiple fibrotic disorders, including CR of cardiac and kidney grafts (8, 20–22). CTGF mediates multiple pro-fibrotic effects ascribed to TGFβ including increased extra cellular matrix production, fibroblast proliferation, and enhancement of adhesive responses (22). Thus, as CTGF is induced by TGFβ and because CTGF mediates pro-fibrotic effects, CTGF has been proposed as a therapeutic target for limiting the deleterious fibrotic effects of TGFβ while sparing its immune-modulatory functions (8, 23, 24).
CTGF induction by TGFβ has been observed in settings of cardiac fibrosis (22). However, we have previously reported that transduction of syngeneic grafts with TGFβ is insufficient to induce CTGF or CR (8). Hence, TGFβ-mediated induction of CTGF in vivo is contextually dependent. One such contextual difference between allogeneic and syngeneic grafts is the development of alloimmune responses which may provide factors that crosstalk with TGFβ signaling (25). This prompted further investigation into immune parameters that potentiated TGFβ-induced fibrosis and led to the identification of a critical role in the initiation and progression of CR for IL-6 (26), a cytokine that modulates the effects of TGFβ in multiple cell types (27–29).
Since TGFβ, CTGF, and IL-6 have established associations with CR (8, 26), we investigated the relationships between these cytokines utilizing overexpression and neutralization approaches. These findings support the role of CTGF as a promoter of cardiac graft fibrosis and indicate that it functions downstream of TGFβ and IL-6. Further, these findings indicate that CTGF neutralization holds promise as a therapeutic approach for limiting the fibrosis associated with CR.
Materials and Methods
Mice
Female C57BL/6 (H-2b) and BALB/c (H-2d) mice were obtained from Charles River Laboratories (Raleigh, NC) and were kept under micro-isolator conditions. The use of mice for these studies was reviewed and approved by the University of Michigan’s Committee On The Use And Care Of Animals.
Vascularized cardiac transplantation
Heterotopic cardiac transplantation was performed as described (30). Briefly, the aorta and pulmonary artery of the donor heart were anastomosed end-to-side to the recipient’s abdominal aorta and inferior vena cava, respectively. Upon perfusion with the recipient’s blood, the transplanted heart resumes contraction. Graft function is monitored by abdominal palpation.
In vivo mAb therapy
Anti-CD4 (hybridoma GK1.5, obtained from American Type Culture Collection, Manassas, VA), anti-CD40L (hybridoma MR1, kindly provided by Dr. Randy Noelle, Dartmouth College), and anti-IL-6 mAb (hybridoma MP5-20F3, obtained from American Type Culture Collection, Manassas VA, with permission of DNAX) mAbs were prepared by Bio X Cell (West Lebanon, NH). Allograft recipients were transiently depleted of CD4+ cells by i.p. injection of 1 mg of anti-CD4 mAb on days -1, 0, and 7 post transplant (8, 26). For inductive anti-CD40L therapy, allograft recipients were injected i.p. with 1 mg of anti-CD40L on days 0, 1, and 2 post transplant (8, 26). Anti-IL-6 mAb or control rat IgG (Sigma, St. Louis, MO) was administered by i.p. injection of 1 mg on days -1, 1, and 3 and weekly thereafter (26, 31). Allograft recipients treated with anti-CTGF mAb (FG-3019, kindly provided by FibroGen, Inc., San Francisco, CA (32, 33)) or control human IgG (Sigma, St. Louis, MO) received 0.5 mg i.p. twice weekly beginning on day 7 postransplant.
Adenoviral-mediated transduction of cardiac grafts
Transduction was performed as previously described (8, 34, 35). Briefly, cardiac grafts were perfused via the aorta with 5 × 108 pfu of E1/E3 deleted adenoviral vectors encoding the active form of human TGFβ1 (AdTGFβ) (8, 34), human CTGF (AdCTGF) (36), or beta-galactosidase (Adβgal) (8, 34, 35). Following perfusion, donor grafts were placed in iced Ringer’s solution for 1 hour prior to transplantation. Previous studies with Adβgal have revealed a patchy distribution of transgene expression by both cardiac and vascular cells that persists for at least 8 weeks post transplant (35).
Morphometric analysis of cardiac graft fibrosis and hypertrophy
Graft fibrosis was quantified by morphometric analysis of Masson’s trichrome stained sections using iPLab software (Scanalytics Inc., Fairfax, VA). Mean fibrotic area was calculated from 10 to 12 areas per heart section analyzed at 200X magnification (26, 37). To quantify cardiomyocyte area as a measure of hypertrophy, digital outlines were drawn around at least 80 cardiomyocytes from views of H&E stained sections at 200X magnification. Areas within outlines were quantified using SCION IMAGE Beta4.0.2 software (Scion Corporation, Frederick, MD) to measure cardiomyocyte cell size (38). A minimum of 8 hearts were analyzed per group for both analysis techniques.
Quantitative real time PCR
Graft RNA was isolated by homogenizing tissues in TRIzol reagent (Invitrogen, Carlsbad, CA) as per manufacturer’s protocol. Five μg of total RNA were reverse transcribed using Oligo dT, dNTPs, MMLV-RT (Invitrogen, Carlsbad, CA), RNAsin (Promega, Madison, WI) in PCR Buffer (Roche, Indianapolis, IN). Resulting cDNA was purified by a 1:1 extraction with phenol/chloroform/isoamyl (25:24:1) then precipitated in one volume 3M NaOAc and two volumes absolute ethanol. Levels of atrial natriuretic peptide (ANP), CTGF, IL-6, TGFβ, IL-17, and T cell receptor β constant region (TCRβ) message were determined by quantitative real time PCR using iQ SYBR master mix (Bio-Rad, Hercules, CA) in a Rotor-Gene 3000 thermocycler (Corbett Life Science, San Francisco, CA). Expression levels were determined relative to GAPDH using the Rotor-Gene Comparative Concentration utility.
Primer sequences were as follows:
ANP (Nppa) forward 5′ GGAGGTCAACCCACCTCTG 3′
ANP (Nppa) reverse 5′ GCTCCAATCCTGTCAATCCTAC 3′
CTGF (Ctgf) forward 5′ GGAAAACATTAAGAAGGGCAAAA 3′
CTGF (Ctgf) reverse 5′ CCGCAGAACTTAGCCCTGTA 3′
GAPDH (Gapdh) forward 5′ CTGGTGCTGAGTATGTCGTG 3′
GAPDH (Gapdh) reverse 5′ CAGTCTTCTGAGTGGCAGTG 3′
IL-6 (Il6) forward 5′ CGTGGAAATGAGAAAAGAGTTGT 3′
IL-6 (Il6) reverse 5′ TCCAGTTTGGTAGCATCCATC 3′
TGFβ (Tgfb 1) forward 5′ CCTGAGTGGCTGTCTTTTGAC 3′
TGFβ (Tgfb 1) reverse 5′ CCTGTATTCCGTCTCCTTGGT 3′
IL-17 (Il17a) forward 5′ GGACTCTCCACCGCAATGA 3′
IL-17 (Il17a) reverse 5′ GACCAGGATCTCTTGCTGGA 3′
TCRβ (Tcrb-C) forward 5′ CTGCCAAGTGCAGTTCCAT 3′
TCRβ (Tcrb-C) reverse 5′ GGCCTCTGCACTGATGTTCT 3′
Flow cytometry
Splenocytes were labeled with FITC-conjugated anti-CD3, PE-conjugated anti-CD4, and CY5-conjugated anti-CD8 (PharMingen San Jose, CA). Cell analyses were performed on lymphocytes gated using forward vs. side scatter using a Becton Dickinson FACSCalibur (San Jose, CA).
Statistical analysis
Statistical significance was calculated using an unpaired t-test with Welch’s correction. p values ≤0.05 were considered statistically significant.
Results
Experimental system
BALB/c cardiac allografts in C57BL/6 recipients receiving anti-CD40L mAb continue to function for >60 days and do not develop CR, unless transduced with TGFβ (8). In contrast, allografts in recipients transiently depleted of CD4+ cells develop CR as CD4+ cells begin to repopulate the periphery between 3 and 4 weeks following initial depletion (8, 39–41). Echocardiographic and histologic analysis revealed that day 30 post transplant represents a critical point in this CR model as extensive graft hypertrophy and fibrosis are present at this time and are followed by degradation of cardiac contractility (26). Therefore, grafts were assessed at day 30 post transplant in these studies. We have used these models to better understand the roles of TGFβ, IL-6, and CTGF in CR.
Elevated intragraft TGFβ, IL-6, and CTGF expression correlate with CR
Transduction of allografts, but not syngeneic grafts, with TGFβ is sufficient to induce CTGF and CR (8), indicating the involvement of an immune component in TGFβ-mediated fibrosis. This is further supported by our recent identification of IL-6 as a critical inducer of CR (26). Hence, the in vivo interactions of TGFβ, CTGF, and IL-6 in CR were the focus of this study. TGFβ, CTGF, and IL-6 transcripts were measured in grafts whose recipients were transiently depleted of CD4+ cells, which develop CR, and compared to allografts whose recipients were treated with anti-CD40L, which do not develop CR, or untreated syngeneic grafts. Intragraft levels of TGFβ, IL-6, and CTGF were significantly increased (p=0.0476, p=0.0254, and p=0.0079 respectively) in cardiac allografts whose recipients were transiently depleted of CD4+ cells than in grafts whose recipients were treated with anti-CD40L or syngeneic controls (Figure 1). Thus, the upregulation of all three cytokines was observed in grafts undergoing CR.
Figure 1. Elevated intragraft expression of TGFβ, IL-6, and CTGF in cardiac allografts undergoing CR.

TGFβ, IL-6, and CTGF message levels were determined at day 30 post transplant using quantitative real time PCR in syngeneic cardiac grafts, cardiac allografts from recipients treated with anti-CD40L mAb therapy (Anti-CD40L), or cardiac allografts whose recipients were transiently depleted of CD4+ cells (Anti-CD4). Bars represent mean + S.E.M. of 4–9 grafts with expression relative to GAPDH normalized to the syngeneic group.
Forced expression of CTGF or TGFβ promotes allograft fibrosis
To determine whether exogenous expression of CTGF promotes cardiac fibrosis, allografts and syngeneic grafts were transduced with AdCTGF. AdCTGF transduction of allografts in recipients treated with anti-CD40L caused a significant increase in fibrotic area by day 30 post transplant compared to allografts transduced with control virus (Figure 2A). In contrast, syngeneic grafts transduced with AdCTGF had similar levels of fibrosis to controls. It should be noted that the mean fibrotic area for AdCTGF-transduced allografts was less than in hearts transduced with AdTGFβ, consistent with previous descriptions in lung transductions (42). This difference could not be accounted for by differences in transgene expression levels, as AdTGFβ and AdCTGF expression were comparable in these studies as determined by real time PCR (data not shown). Thus, while forced expression of either TGFβ or CTGF promoted cardiac allograft fibrosis, they did so to different extents (Figure 2). This could in part be due to TGFβ induction of endogenous CTGF expression (8, 17, 43), thereby producing an additive effect.
Figure 2. Forced expression of TGFβ or CTGF promotes allograft fibrosis.

(A) Morphometric analysis of Masson’s trichrome staining at day 30 post transplant in cardiac grafts that were left untransduced or transduced with adenoviral vectors encoding βgal (Adβgal), CTGF (AdCTGF), or TGFβ (AdTGFβ) prior to grafting into syngeneic recipients or allogeneic recipients treated with anti-CD40L. Bars represent the combined mean + S.E.M. of fibrotic (blue) area of 10–12 frames of view per heart taken from 5 to 12 different cardiac grafts per group. (B) Intragraft IL-6 message levels were determined at day 30 post transplant using quantitative real time PCR in groups from (A). Bars represent mean + S.E.M. of at least 4 hearts per group with expression relative to GAPDH normalized to naïve, untransplanted BALB/c hearts. (C) Intragraft IL-17 message levels were determined using quantitative real time PCR in syngeneic grafts transduced with AdTGFβ or allogeneic grafts transduced with AdTGFβ whose recipients received anti-CD40L treatment. Bars represent mean + S.E.M. of at least 5 independent hearts per group with expression relative to GAPDH normalized to the naïve BALB/c group.
It has been observed that TGFβ and CTGF are potently fibrotic in tandem while less fibrotic individually (44, 45). Therefore, we asked whether co-transduction of both TGFβ and CTGF vectors would induce fibrosis and CR in syngeneic grafts. No increases in fibrosis were observed upon co-transduction of syngeneic grafts compared to single virus transduction (data not shown). Thus, while injection of TGFβ and CTGF synergize to cause fibrotic responses in the skin (45), forced expression of both was insufficient to induce fibrosis or CR in syngeneic cardiac grafts, further supporting the requirement of an immune component.
We next considered whether the greater fibrotic activity of AdTGFβ relative to AdCTGF could be due to immunologic effects. TGFβ is chemotactic for multiple immune cell types (10) that are able to produce IL-6, which we have recently reported to play a critical role in CR (26). Therefore we asked whether differences in intragraft IL-6 expression might account for these disparate outcomes. IL-6 transcript levels exhibited a suggested increase in AdTGFβ, but not AdCTGF transduced allografts whose recipients received anti-CD40L therapy. No increases in IL-6 expression were observed in AdTGFβ or AdCTGF-transduced syngeneic grafts (Figure 2B).
TGFβ and IL-6 have been implicated in the development of Th17 responses (27), which have recently been linked to CR (46, 47). Hence, we assessed the expression of IL-17 in allogeneic and syngeneic grafts transduced with AdTGFβ. IL-17 expression was significantly greater (p=0.0107) in allografts than in syngeneic grafts (Figure 2C) while IL-17 expression was similar in allogeneic and syngeneic grafts transduced with AdCTGF (data not shown). Thus, increased IL-17 and CTGF transcript levels may promote fibrosis associated with AdTGFβ-transduced allografts, but not AdTGFβ-transduced syngeneic grafts that do not develop fibrosis.
IL-6 neutralization reduces intragraft CTGF and IL-17 transcripts
The association between TGFβ, IL-6, and CTGF (Figure 1) may be strengthened by previous reports that IL-6 enhances TGFβ signaling by altering receptor localization in the cell membrane (29) and that IL-6 can alter the outcome of TGFβ signaling (27, 28). Indeed, we have previously reported that IL-6 neutralization prevents CR of cardiac allografts (26). We therefore asked whether IL-6 neutralization would inhibit CTGF or IL-17 expression (Figure 3). In allografts whose recipients were transiently depleted of CD4+ cells, treatment with anti-IL-6 mAb significantly reduced intragraft IL-6, IL-17, and CTGF expression (p=0.0216, p=0.0044, and p=0.0180 respectively) compared to control antibody treatment. In contrast, TGFβ expression levels remained unchanged (Figure 3). Thus, IL-6 promotes intragraft IL-6, IL-17, and CTGF expression.
Figure 3. IL-6 neutralization reduces expression of IL-6, IL-17, and CTGF but not TGFβ in cardiac allografts undergoing CR.

Intragraft IL-6, IL-17, CTGF, and TGFβ message levels were determined at day 30 post transplant using quantitative real time PCR in cardiac allograft recipients that were transiently depleted of CD4+ cells and received either neutralizing anti-IL-6 (Anti-IL-6) or control rat IgG (rIgG). Bars represent mean + S.E.M. of 6–8 grafts per group with expression relative to GAPDH normalized against rIgG-treated contols.
CTGF neutralization ameliorates allograft fibrosis
We next asked whether CTGF neutralization would inhibit the fibrosis associated with CR. To this end, we treated allograft recipients that were transiently depleted of CD4+ cells with neutralizing anti-CTGF mAb or control antibody. Treatment with anti-CTGF mAb resulted in significant reduction of fibrotic area (p<0.0001, Figures 4A and B), but was not accompanied by reduction of intragraft TGFβ, CTGF, or IL-6 transcripts (Figure 4C). These observations support a role for CTGF as a downstream mediator of fibrosis associated with CR.
Figure 4. CTGF neutralization ameliorates fibrosis.

(A) Representative sections of Masson’s trichrome stains, in which fibrotic tissue stains blue, of cardiac allografts from recipients transiently depleted of CD4+ cells (Anti-CD4) at day 30 post transplant in recipients treated with control IgG or neutralizing anti-CTGF mAb (200X magnification). (B) Morphometric analysis of trichrome staining of groups in (A). Bars represent mean + S.E.M. of 10–12 frames of view from each of 6 to 9 hearts. (C) TGFβ, IL-6, and CTGF message levels were determined at day 30 post transplant using quantitative real time PCR in cardiac allografts described in (A). Bars represent mean + S.E.M. of samples taken from 8–12 different cardiac grafts with expression relative to GAPDH normalized against hIgG-treated controls.
CTGF neutralization decreases cardiomyocyte hypertrophy associated with CR
CTGF can induce cardiomyocyte hypertrophy (48, 49), a function it shares with IL-6 (26). Since IL-6 neutralization inhibited CTGF expression (Figure 3), we assessed the effect of neutralizing CTGF on cardiomyocyte hypertrophy. Anti-CTGF treatment resulted in a significant decrease (p<0.0001) in cardiomyocyte hypertrophy (Figure 5A) and significantly reduced (p=0.0102) the intragraft expression of ANP (Figure 5B), a molecular marker of cardiac hypertrophy (50, 51). For reference, cardiomyocyte area and ANP expression levels for naïve, untransplanted BALB/c hearts and allografts transplanted into recipients receiving anti-CD40L therapy are depicted.
Figure 5. CTGF neutralization ameliorates cardiac hypertrophy in CR grafts.

(A) Cardiomyocyte area was quantified from H&E stains of day 30 post transplant cardiac allografts taken from recipients transiently depleted of CD4+ cells (Anti-CD4) and receiving CTGF neutralizing mAb (Anti-CTGF) or control antibodies (hIgG), recipients treated with Anti-CD40L mAb, or naïve, untransplanted BALB/c hearts. Bars represent mean + S.E.M. of area measurements taken from ≥100 cardiomyocytes per heart from 5 (naïve BALB/c and Anti-CD40L), 8 (Anti-CD4+hIgG), or 10 (Anti-CD4+Anti-CTGF) different hearts per group. (B) Intragraft message levels of atrial natriuretic peptide (ANP), a marker of cardiac hypertrophy, were quantified with real time PCR in cardiac grafts from groups in (A) at day 30 post transplant. Bars represent mean + S.E.M. of 8–12 grafts per experimental group (Anti-CD4+hIgG or Anti-CTGF) and 4 grafts per control group (Anti-CD40L and naïve BALB/c) with expression relative to GAPDH normalized against the naïve BALB/c hearts.
CTGF neutralization inhibits T cell infiltration of grafts
CTGF promotes integrin-mediated adhesive responses in multiple cell types (52–61) and induces the production of chemokines (62). We therefore asked whether CTGF neutralization might also alter the infiltration of immune cells into grafts undergoing CR. Histologic analysis indicated reduced cellular infiltrate in grafts receiving anti-CTGF (Figure 6A). Indeed, a significant decrease (p=0.0238) in TCRβ constant region expression, a marker of graft infiltrating T cells (63), was observed (Figure 6B). To verify that this difference wasn’t due to CTGF neutralization preventing peripheral repopulation of CD4+ cells, we compared the percentage of CD4+ cells in anti-CTGF and control treated graft recipients. No significant differences were observed between these groups (Figure 6C).
Figure 6. CTGF neutralization limits graft infiltration by T cells in CR grafts.

(A) Representative H&E stains of day 30 post transplant cardiac allografts taken from recipients transiently depleted of CD4+ cells (Anti-CD4) and receiving CTGF neutralizing mAb (Anti-CTGF) or control antibodies (hIgG). Stains suggest a reduction in perivascular infiltrate density in grafts treated with neutralizing Anti-CTGF. (B) Intragraft message levels of T cell receptor β constant region (TCRβ) were quantified at day 30 post transplant with real time PCR as a measure of T cell infiltration of allografts in recipients transiently depleted of CD4+ cells (Anti-CD4) and receiving anti-CTGF mAb or control hIgG antibodies, recipients treated with Anti-CD40L mAb, or naïve BALB/c hearts. Bars represent mean + S.E.M. of 8–12 grafts per group with expression relative to GAPDH normalized against the hIgG group. (C) Repopulation of CD4+ cells in the periphery at day 30 post transplant was determined by flow cytometric analysis of splenocytes isolated from graft recipients. Bars represent mean + S.E.M. of the percentage CD4+ cells of the gated cell population in 5 to 7 recipients tested.
Discussion
CR has been associated with multiple factors, perhaps most frequently with TGFβ (9). However, the role of TGFβ in CR is complicated by its pleiotropic activity encompassing immunosuppressive and anti-proliferative effects in immune (10, 64–66) and non-immune (15, 67) cells as well as the induction of Treg (68–70), which are associated with graft acceptance (12, 13, 24). Thus, TGFβ may promote graft survival and global immune tolerance while suppressing malignancy, making it ill-suited as a therapeutic target in the treatment of CR. This has prompted investigation into the downstream mediators of fibrotic TGFβ function (8, 23).
Multiple reports indicate that TGFβ requires additional factors to drive fibrosis (8, 44, 45). Indeed, syngeneic grafts do not develop fibrosis in response to TGFβ, while allografts whose recipients receive anti-CD40L mAb develop marked fibrosis in response to TGFβ (Figure 2, (8)). Hence, alloimmune responses potentiate the pro-fibrotic effects of TGFβ. We have reported a critical role for IL-6 in CR (26), whose elevated expression correlated with TGFβ and CTGF (Figure 1). Correlations of TGFβ with CTGF (8) and IL-6 (26) have previously been described. Further, we have previously observed CTGF expression associated with areas of graft-infiltrating mononuclear cells (8), whose recruitment during inflammatory responses has been linked to IL-6 (71, 72). Therefore, we considered that there may be connectivity between all three cytokines.
To ascertain the sufficiency of TGFβ and CTGF to induce CR, allogeneic and syngeneic cardiac grafts were transduced with AdTGFβ or AdCTGF and transplanted into recipients receiving anti-CD40L mAb or syngeneic recipients. AdTGFβ and AdCTGF significantly increased mean fibrotic area compared to untransduced or control vector treated allografts (Figure 2A). Consistent with a previous report of adenoviral transduction of lungs (42), the fibrotic response to TGFβ transduction in the heart was significantly greater than the response to CTGF transduction (Figure 2A). Greater fibrotic responses to AdTGFβ could be from synergy of TGFβ-induced immune factors and CTGF in cardiac allografts, an effect which is not observed in syngeneic grafts (8). Further, in cardiac allografts, TGFβ induction of endogenous CTGF may synergize with TGFβ-mediated chemotactic effects on multiple immune lineage cells (10), which may explain the suggested upregulation of IL-6 and significant upregulation of IL-17 (Figure 2).
Given the differences in AdTGFβ responses between allografts and syngeneic grafts and the correlation of TGFβ and CTGF with IL-6 in CR (Figure 1), we asked whether the presence of IL-6 was required for CTGF upregulation. In cardiac allograft recipients transiently depleted of CD4+ cells, IL-6 neutralization reduced the expression of IL-6 and CTGF without altering TGFβ transcript levels (Figure 3). This suggests that TGFβ transcript regulation lies upstream of IL-6 and CTGF in CR. It should be noted that IL-6 neutralization does not prevent repopulation of CD4+ cells in the periphery (26). This indicates that the ability of IL-6 neutralization to prevent CR (26) could function in part through reduction of intragraft CTGF. Further, IL-6 neutralization significantly inhibited IL-17 expression (Figure 3), indicating that IL-17 might play a role in CTGF induction, as IL-17 has been reported to induce collagen production in cardiac fibroblasts (73). Another explanation for this effect might be decreased recruitment of graft infiltrating cells which may express or induce local cells to express CTGF (8), IL-6 and IL-17. Indeed, IL-6 induces chemotaxis and migration of immune cells (74, 75).
Since IL-6 neutralization ameliorated CR (26) and decreased intragraft CTGF expression (Figure 3), we treated cardiac allograft recipients with neutralizing CTGF mAb. CTGF neutralization significantly reduced allograft fibrosis (Figures 4A, B) without significantly reducing intragraft TGFβ, IL-6, or CTGF expression (Figure 4C). These findings are consistent with CTGF being a downstream mediator of fibrosis in CR (16, 23, 42, 76).
The significant but incomplete reduction in fibrotic area in response to CTGF neutralization may be explained by multiple factors. Our neutralization protocol, though effective, may not be optimal. Another possibility is the presence of CTGF-independent pro-fibrotic effects of TGFβ and/or IL-6 (77). A further consideration is whether the mAb FG-3019, which recognizes CTGF module 2 in humans and rodents (33), might inhibit some but not all pro-fibrotic effects of CTGF. However, this possibility seems unlikely in light of a recent report evaluating the anti-fibrotic efficacy of anti-CTGF antibodies directed against each of the four CTGF modules. In this report, only mAb directed against the von Willebrand factor type C domain (module 2) was able to inhibit TGFβ-induced fibrosis (78). Indeed, this is the same domain that the anti-CTGF mAb utilized in our study binds (32, 33).
Beyond its roles in fibrosis, CTGF can exert other effects relevant to CR. Recent studies have described a concomitance of cardiomyocyte hypertrophy with CR (26, 79, 80). CTGF is produced by hypertrophic chondrocytes during development (81), and is produced by cardiac myocytes in response to hypertrophic stimuli (49). In addition, CTGF itself can induce cardiomyocyte hypertrophy (48). Treatment with neutralizing anti-CTGF mAb significantly reduced mean cardiomyocyte area (Figure 5A) and intragraft levels of ANP (Figure 5B), a marker of cardiac hypertrophy in multiple settings (26, 50, 51). However, it should be noted that anti-CTGF mAb did not inhibit cardiac hypertrophy to the extent previously observed with anti-IL-6 (26). This finding indicates that in addition to driving cardiac fibrosis, CTGF may augment cardiomyocyte hypertrophy associated with CR. Interestingly, hypertrophy is associated with downregulation of two recently discovered CTGF-inhibiting micro RNAs in cardiac myocytes (82). Thus, CTGF may be linked to cardiac hypertrophy on multiple levels.
Finally, as CTGF is known to play an important role in fibroblast adhesion in response to TGFβ (55, 76), we asked whether CTGF might similarly influence recruitment of lymphocytes to the graft. Histologic assessment of infiltrating cells was indicative of reduced numbers of graft infiltrating lymphocytes (Figure 6A). This observation was further supported by significant reduction of intragraft TCRβ constant region expression (Figure 6B) in response to CTGF neutralization.
Based on these observations and others in the literature, we propose a model representing the interactions of TGFβ, IL-6, and CTGF and their induction of hypertrophy and fibrosis associated with CR (Figure 7A). In cardiac allografts that undergo CR, TGFβ (Figure 1, (8)) and IL-6 (Figures 1, 2B, (26)) are induced. In syngeneic grafts, forced expression of TGFβ is insufficient to upregulate CTGF and fibrosis (8), and IL-6 remains at basal levels (Figure 2B). IL-6 neutralization inhibits hypertrophy and fibrosis associated with CR (26), which may be in part through inhibition of CTGF and IL-17 expression while TGFβ expression remains unchanged (Figure 3). Thus, TGFβ and IL-6 appear to be cooperative upstream factors promoting CTGF expression and CR. CTGF neutralization limits fibrosis (Figures 4A, B) and cardiomyocyte hypertrophy (Figure 5), without altering intragraft TGFβ, IL-6, or CTGF transcripts (Figure 4C). These effects of CTGF neutralization coincide with reduction in graft infiltrating T cells (Figure 6). Together, these observations support a downstream role for CTGF in fibrosis and hypertrophy.
Figure 7. Proposed model of cytokine interactions in chronic rejection.
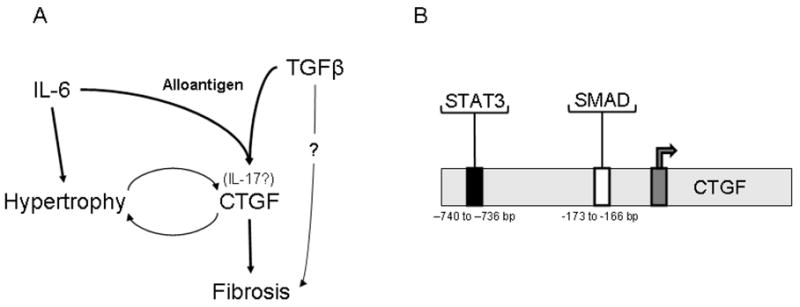
(A) In cardiac allografts, TGFβ and IL-6 contribute to CTGF production. IL-6 and CTGF are both known to promote hypertrophy in cardiac myocytes, which in turn can produce CTGF. CTGF functions as a downstream mediator of fibrosis. (B) Induction of CTGF downstream of TGFβ and IL-6 could be explained by the respective presence of a consensus SMAD binding element and a STAT3 response element in 5′ region upstream of the CTGF promoter. For expanded explanations, please see text.
Contexts in which TGFβ and IL-6 are present coincide with intragraft IL-17 expression, which has been implicated in promoting cardiac remodeling (83), fibrosis (73), bronchiolitis obliterans syndrome in lung transplant patients (46), and cardiac allograft vasculopathy (47). However, the effects of IL-17 on hypertrophy and CTGF expression are unclear and merit further investigation. Our proposed model of CTGF induction downstream of IL-6 and TGFβ (and perhaps IL-17) might be explained by previous identification of both a STAT3 responsive element (740 to 736 bp) (84) and a consensus SMAD binding element (−173 and −166) (85) upstream of the CTGF promoter (Figure 7B). Hence, optimal induction of CTGF in CR may require that CTGF producing cells receive both SMAD and STAT3 signals, likely provided by TGFβ (10) and IL-6 (86) respectively.
This study supports a role for CTGF as a downstream mediator of fibrosis and highlights the essential contributions of immune elements to CR and fibrosis of cardiac grafts while elucidating relationships between TGFβ, IL-6, and CTGF. Further, these studies indicate for the first time that mAb neutralizing CTGF can ameliorate fibrosis and hypertrophy associated with CR. These findings further implicate IL-6 as a critical immune factor in CR that may potentiate TGFβ-mediated CTGF induction. Finally, TGFβ-mediated induction of fibrosis in allogeneic but not syngeneic grafts was associated with a suggested increase in intragraft IL-6 expression and a significant increase in IL-17 expression, supporting the notion that TGFβ induction of fibrosis and CR requires interaction with immune parameters.
Acknowledgments
Supported by NIH grants R01 HL070613 (DKB) and R01 AI061469 (DKB) and by an American Heart Association Predoctoral Fellowship (AJB).
References
- 1.Orosz CG, Pelletier RP. Chronic remodeling pathology in grafts. Curr Opin Immunol. 1997;9(5):676–680. doi: 10.1016/s0952-7915(97)80048-9. [DOI] [PubMed] [Google Scholar]
- 2.Paul LC. Current knowledge of the pathogenesis of chronic allograft dysfunction. Transplant Proc. 1999;31(4):1793–1795. doi: 10.1016/s0041-1345(99)00171-2. [DOI] [PubMed] [Google Scholar]
- 3.Waaga AM, Gasser M, Laskowski I, Tilney NL. Mechanisms of chronic rejection. Curr Opin Immunol. 2000;12(5):517–521. doi: 10.1016/s0952-7915(00)00132-1. [DOI] [PubMed] [Google Scholar]
- 4.Womer KL, Vella JP, Sayegh MH. Chronic allograft dysfunction: mechanisms and new approaches to therapy. Semin Nephrol. 2000;20(2):126–147. [PubMed] [Google Scholar]
- 5.Weiss MJ, Madsen JC, Rosengard BR, Allan JS. Mechanisms of chronic rejection in cardiothoracic transplantation. Front Biosci. 2008;13:2980–2988. doi: 10.2741/2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mehra MR. Contemporary concepts in prevention and treatment of cardiac allograft vasculopathy. Am J Transplant. 2006;6(6):1248–1256. doi: 10.1111/j.1600-6143.2006.01314.x. [DOI] [PubMed] [Google Scholar]
- 7.Valantine H. Cardiac allograft vasculopathy after heart transplantation: risk factors and management. J Heart Lung Transplant. 2004;23(5 Suppl):S187–193. doi: 10.1016/j.healun.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 8.Csencsits K, Wood SC, Lu G, Faust SM, Brigstock D, Eichwald EJ, et al. Transforming growth factor beta-induced connective tissue growth factor and chronic allograft rejection. Am J Transplant. 2006;6(5 Pt 1):959–966. doi: 10.1111/j.1600-6143.2006.01292.x. [DOI] [PubMed] [Google Scholar]
- 9.Jain S, Furness PN, Nicholson ML. The role of transforming growth factor beta in chronic renal allograft nephropathy. Transplantation. 2000;69(9):1759–1766. doi: 10.1097/00007890-200005150-00001. [DOI] [PubMed] [Google Scholar]
- 10.Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- 11.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342(18):1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 12.Wood KJ, Sakaguchi S. Regulatory T cells in transplantation tolerance. Nat Rev Immunol. 2003;3(3):199–210. doi: 10.1038/nri1027. [DOI] [PubMed] [Google Scholar]
- 13.Yong Z, Chang L, Mei YX, Yi L. Role and mechanisms of CD4+CD25+ regulatory T cells in the induction and maintenance of transplantation tolerance. Transpl Immunol. 2007;17(2):120–129. doi: 10.1016/j.trim.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 14.Walsh PT, Taylor DK, Turka LA. Tregs and transplantation tolerance. J Clin Invest. 2004;114(10):1398–1403. doi: 10.1172/JCI23238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brattain MG, Markowitz SD, Willson JK. The type II transforming growth factor- beta receptor as a tumor-suppressor gene. Curr Opin Oncol. 1996;8(1):49–53. doi: 10.1097/00001622-199601000-00009. [DOI] [PubMed] [Google Scholar]
- 16.Leask A, Abraham DJ. The role of connective tissue growth factor, a multifunctional matricellular protein, in fibroblast biology. Biochem Cell Biol. 2003;81(6):355–363. doi: 10.1139/o03-069. [DOI] [PubMed] [Google Scholar]
- 17.Chen MM, Lam A, Abraham JA, Schreiner GF, Joly AH. CTGF expression is induced by TGF- beta in cardiac fibroblasts and cardiac myocytes: a potential role in heart fibrosis. J Mol Cell Cardiol. 2000;32(10):1805–1819. doi: 10.1006/jmcc.2000.1215. [DOI] [PubMed] [Google Scholar]
- 18.de Winter P, Leoni P, Abraham D. Connective tissue growth factor: structure-function relationships of a mosaic, multifunctional protein. Growth Factors. 2008;26(2):80–91. doi: 10.1080/08977190802025602. [DOI] [PubMed] [Google Scholar]
- 19.Bonniaud P, Martin G, Margetts PJ, Ask K, Robertson J, Gauldie J, et al. Connective tissue growth factor is crucial to inducing a profibrotic environment in “fibrosis-resistant” BALB/c mouse lungs. Am J Respir Cell Mol Biol. 2004;31(5):510–516. doi: 10.1165/rcmb.2004-0158OC. [DOI] [PubMed] [Google Scholar]
- 20.Cheng O, Thuillier R, Sampson E, Schultz G, Ruiz P, Zhang X, et al. Connective tissue growth factor is a biomarker and mediator of kidney allograft fibrosis. Am J Transplant. 2006;6(10):2292–2306. doi: 10.1111/j.1600-6143.2006.01493.x. [DOI] [PubMed] [Google Scholar]
- 21.Yuan YC, Xia ZK, Mu JJ, Zhang QC, Yin BL. Increased connective tissue growth factor expression in a rat model of chronic heart allograft rejection. J Formos Med Assoc. 2009;108(3):240–246. doi: 10.1016/S0929-6646(09)60058-9. [DOI] [PubMed] [Google Scholar]
- 22.Daniels A, van Bilsen M, Goldschmeding R, van der Vusse GJ, van Nieuwenhoven FA. Connective tissue growth factor and cardiac fibrosis. Acta Physiol (Oxf) 2009;195(3):321–338. doi: 10.1111/j.1748-1716.2008.01936.x. [DOI] [PubMed] [Google Scholar]
- 23.Mannon RB. Therapeutic targets in the treatment of allograft fibrosis. Am J Transplant. 2006;6(5 Pt 1):867–875. doi: 10.1111/j.1600-6143.2006.01261.x. [DOI] [PubMed] [Google Scholar]
- 24.Csencsits K, Wood SC, Lu G, Bishop DK. Transforming growth factor-beta1 gene transfer is associated with the development of regulatory cells. Am J Transplant. 2005;5(10):2378–2384. doi: 10.1111/j.1600-6143.2005.01042.x. [DOI] [PubMed] [Google Scholar]
- 25.Guo X, Wang XF. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 2009;19(1):71–88. doi: 10.1038/cr.2008.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diaz JA, Booth AJ, Lu G, Wood SC, Pinsky DJ, Bishop DK. Critical role for IL-6 in hypertrophy and fibrosis in chronic cardiac allograft rejection. Am J Transplant. 2009;9(8):1773–1783. doi: 10.1111/j.1600-6143.2009.02706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441(7090):235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 28.Chen RH, Chang MC, Su YH, Tsai YT, Kuo ML. Interleukin-6 inhibits transforming growth factor-beta-induced apoptosis through the phosphatidylinositol 3-kinase/Akt and signal transducers and activators of transcription 3 pathways. J Biol Chem. 1999;274(33):23013–23019. doi: 10.1074/jbc.274.33.23013. [DOI] [PubMed] [Google Scholar]
- 29.Zhang XL, Topley N, Ito T, Phillips A. Interleukin-6 regulation of transforming growth factor (TGF)-beta receptor compartmentalization and turnover enhances TGF-beta1 signaling. J Biol Chem. 2005;280(13):12239–12245. doi: 10.1074/jbc.M413284200. [DOI] [PubMed] [Google Scholar]
- 30.Corry RJ, Winn HJ, Russell PS. Primarily vascularized allografts of hearts in mice. The role of H-2D, H-2K, and non-H-2 antigens in rejection. Transplantation. 1973;16(4):343–350. doi: 10.1097/00007890-197310000-00010. [DOI] [PubMed] [Google Scholar]
- 31.Burrell BE, Csencsits K, Lu G, Grabauskiene S, Bishop DK. CD8+ Th17 mediate costimulation blockade-resistant allograft rejection in T-bet-deficient mice. J Immunol. 2008;181(6):3906–3914. doi: 10.4049/jimmunol.181.6.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aikawa T, Gunn J, Spong SM, Klaus SJ, Korc M. Connective tissue growth factor-specific antibody attenuates tumor growth, metastasis, and angiogenesis in an orthotopic mouse model of pancreatic cancer. Mol Cancer Ther. 2006;5(5):1108–1116. doi: 10.1158/1535-7163.MCT-05-0516. [DOI] [PubMed] [Google Scholar]
- 33.Dornhofer N, Spong S, Bennewith K, Salim A, Klaus S, Kambham N, et al. Connective tissue growth factor-specific monoclonal antibody therapy inhibits pancreatic tumor growth and metastasis. Cancer Res. 2006;66(11):5816–5827. doi: 10.1158/0008-5472.CAN-06-0081. [DOI] [PubMed] [Google Scholar]
- 34.Chan SY, Goodman RE, Szmuszkovicz JR, Roessler B, Eichwald EJ, Bishop DK. DNA-liposome versus adenoviral mediated gene transfer of transforming growth factor beta1 in vascularized cardiac allografts: differential sensitivity of CD4+ and CD8+ T cells to transforming growth factor beta1. Transplantation. 2000;70(9):1292–1301. doi: 10.1097/00007890-200011150-00006. [DOI] [PubMed] [Google Scholar]
- 35.Chan SY, Li K, Piccotti JR, Louie MC, Judge TA, Turka LA, et al. Tissue-specific consequences of the anti-adenoviral immune response: implications for cardiac transplants. Nat Med. 1999;5(10):1143–1149. doi: 10.1038/13467. [DOI] [PubMed] [Google Scholar]
- 36.Haberberger TC, Kupfer K, Murphy JE. Profiling of genes which are differentially expressed in mouse liver in response to adenoviral vectors and delivered genes. Gene Ther. 2000;7(11):903–909. doi: 10.1038/sj.gt.3301181. [DOI] [PubMed] [Google Scholar]
- 37.Burrell BE, Lu G, Li XC, Bishop DK. OX40 costimulation prevents allograft acceptance induced by CD40-CD40L blockade. J Immunol. 2009;182(1):379–390. doi: 10.4049/jimmunol.182.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nozato T, Ito H, Tamamori M, Adachi S, Abe S, Marumo F, et al. G1 cyclins are involved in the mechanism of cardiac myocyte hypertrophy induced by angiotensin II. Jpn Circ J. 2000;64(8):595–601. doi: 10.1253/jcj.64.595. [DOI] [PubMed] [Google Scholar]
- 39.Bishop DK, Li W, Chan SY, Ensley RD, Shelby J, Eichwald EJ. Helper T lymphocyte unresponsiveness to cardiac allografts following transient depletion of CD4-positive cells. Implications for cellular and humoral responses. Transplantation. 1994;58(5):576–584. doi: 10.1097/00007890-199409150-00009. [DOI] [PubMed] [Google Scholar]
- 40.Piccotti JR, Li K, Chan SY, Eichwald EJ, Bishop DK. Cytokine regulation of chronic cardiac allograft rejection: evidence against a role for Th1 in the disease process. Transplantation. 1999;67(12):1548–1555. doi: 10.1097/00007890-199906270-00008. [DOI] [PubMed] [Google Scholar]
- 41.Csencsits K, Burrell BE, Lu G, Eichwald EJ, Stahl GL, Bishop DK. The classical complement pathway in transplantation: unanticipated protective effects of C1q and role in inductive antibody therapy. Am J Transplant. 2008;8(8):1622–1630. doi: 10.1111/j.1600-6143.2008.02295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bonniaud P, Margetts PJ, Kolb M, Haberberger T, Kelly M, Robertson J, et al. Adenoviral gene transfer of connective tissue growth factor in the lung induces transient fibrosis. Am J Respir Crit Care Med. 2003;168(7):770–778. doi: 10.1164/rccm.200210-1254OC. [DOI] [PubMed] [Google Scholar]
- 43.Grotendorst GR, Okochi H, Hayashi N. A novel transforming growth factor beta response element controls the expression of the connective tissue growth factor gene. Cell Growth Differ. 1996;7(4):469–480. [PubMed] [Google Scholar]
- 44.Frazier K, Williams S, Kothapalli D, Klapper H, Grotendorst GR. Stimulation of fibroblast cell growth, matrix production, and granulation tissue formation by connective tissue growth factor. J Invest Dermatol. 1996;107(3):404–411. doi: 10.1111/1523-1747.ep12363389. [DOI] [PubMed] [Google Scholar]
- 45.Mori T, Kawara S, Shinozaki M, Hayashi N, Kakinuma T, Igarashi A, et al. Role and interaction of connective tissue growth factor with transforming growth factor-beta in persistent fibrosis: A mouse fibrosis model. J Cell Physiol. 1999;181(1):153–159. doi: 10.1002/(SICI)1097-4652(199910)181:1<153::AID-JCP16>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 46.Burlingham WJ, Love RB, Jankowska-Gan E, Haynes LD, Xu Q, Bobadilla JL, et al. IL-17-dependent cellular immunity to collagen type V predisposes to obliterative bronchiolitis in human lung transplants. J Clin Invest. 2007;117(11):3498–3506. doi: 10.1172/JCI28031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yuan X, Paez-Cortez J, Schmitt-Knosalla I, D’Addio F, Mfarrej B, Donnarumma M, et al. A novel role of CD4 Th17 cells in mediating cardiac allograft rejection and vasculopathy. J Exp Med. 2008;205(13):3133–3144. doi: 10.1084/jem.20081937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hayata N, Fujio Y, Yamamoto Y, Iwakura T, Obana M, Takai M, et al. Connective tissue growth factor induces cardiac hypertrophy through Akt signaling. Biochem Biophys Res Commun. 2008;370(2):274–278. doi: 10.1016/j.bbrc.2008.03.100. [DOI] [PubMed] [Google Scholar]
- 49.Matsui Y, Sadoshima J. Rapid upregulation of CTGF in cardiac myocytes by hypertrophic stimuli: implication for cardiac fibrosis and hypertrophy. J Mol Cell Cardiol. 2004;37(2):477–481. doi: 10.1016/j.yjmcc.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 50.Caron KM, James LR, Kim HS, Knowles J, Uhlir R, Mao L, et al. Cardiac hypertrophy and sudden death in mice with a genetically clamped renin transgene. Proc Natl Acad Sci U S A. 2004;101(9):3106–3111. doi: 10.1073/pnas.0307333101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fredj S, Bescond J, Louault C, Potreau D. Interactions between cardiac cells enhance cardiomyocyte hypertrophy and increase fibroblast proliferation. J Cell Physiol. 2005;202(3):891–899. doi: 10.1002/jcp.20197. [DOI] [PubMed] [Google Scholar]
- 52.Babic AM, Chen CC, Lau LF. Fisp12/mouse connective tissue growth factor mediates endothelial cell adhesion and migration through integrin alphavbeta3, promotes endothelial cell survival, and induces angiogenesis in vivo. Mol Cell Biol. 1999;19(4):2958–2966. doi: 10.1128/mcb.19.4.2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ball DK, Rachfal AW, Kemper SA, Brigstock DR. The heparin-binding 10 kDa fragment of connective tissue growth factor (CTGF) containing module 4 alone stimulates cell adhesion. J Endocrinol. 2003;176(2):R1–7. doi: 10.1677/joe.0.176r001. [DOI] [PubMed] [Google Scholar]
- 54.Chen CC, Chen N, Lau LF. The angiogenic factors Cyr61 and connective tissue growth factor induce adhesive signaling in primary human skin fibroblasts. J Biol Chem. 2001;276(13):10443–10452. doi: 10.1074/jbc.M008087200. [DOI] [PubMed] [Google Scholar]
- 55.Chen Y, Abraham DJ, Shi-Wen X, Pearson JD, Black CM, Lyons KM, et al. CCN2 (connective tissue growth factor) promotes fibroblast adhesion to fibronectin. Mol Biol Cell. 2004;15(12):5635–5646. doi: 10.1091/mbc.E04-06-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gao R, Brigstock DR. Connective tissue growth factor (CCN2) induces adhesion of rat activated hepatic stellate cells by binding of its C-terminal domain to integrin alpha(v)beta(3) and heparan sulfate proteoglycan. J Biol Chem. 2004;279(10):8848–8855. doi: 10.1074/jbc.M313204200. [DOI] [PubMed] [Google Scholar]
- 57.Hoshijima M, Hattori T, Inoue M, Araki D, Hanagata H, Miyauchi A, et al. CT domain of CCN2/CTGF directly interacts with fibronectin and enhances cell adhesion of chondrocytes through integrin alpha5beta1. FEBS Lett. 2006;580(5):1376–1382. doi: 10.1016/j.febslet.2006.01.061. [DOI] [PubMed] [Google Scholar]
- 58.Nishida T, Kawaki H, Baxter RM, Deyoung RA, Takigawa M, Lyons KM. CCN2 (Connective Tissue Growth Factor) is essential for extracellular matrix production and integrin signaling in chondrocytes. J Cell Commun Signal. 2007;1(1):45–58. doi: 10.1007/s12079-007-0005-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gao R, Brigstock DR. A novel integrin alpha5beta1 binding domain in module 4 of connective tissue growth factor (CCN2/CTGF) promotes adhesion and migration of activated pancreatic stellate cells. Gut. 2006;55(6):856–862. doi: 10.1136/gut.2005.079178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heng EC, Huang Y, Black SA, Jr, Trackman PC. CCN2, connective tissue growth factor, stimulates collagen deposition by gingival fibroblasts via module 3 and alpha6- and beta1 integrins. J Cell Biochem. 2006;98(2):409–420. doi: 10.1002/jcb.20810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jedsadayanmata A, Chen CC, Kireeva ML, Lau LF, Lam SC. Activation-dependent adhesion of human platelets to Cyr61 and Fisp12/mouse connective tissue growth factor is mediated through integrin alpha(IIb)beta(3) J Biol Chem. 1999;274(34):24321–24327. doi: 10.1074/jbc.274.34.24321. [DOI] [PubMed] [Google Scholar]
- 62.Wu SH, Lu C, Dong L, Chen ZQ. Signal transduction involved in CTGF-induced production of chemokines in mesangial cells. Growth Factors. 2008:1. doi: 10.1080/08977190802227828. [DOI] [PubMed] [Google Scholar]
- 63.Zhao XM, Frist WH, Yeoh TK, Miller GG. Expression of cytokine genes in human cardiac allografts: correlation of IL-6 and transforming growth factor-beta (TGF-beta) with histological rejection. Clin Exp Immunol. 1993;93(3):448–451. doi: 10.1111/j.1365-2249.1993.tb08199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Letterio JJ, Roberts AB. Regulation of immune responses by TGF-beta. Annu Rev Immunol. 1998;16:137–161. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- 65.Prud’homme GJ, Piccirillo CA. The inhibitory effects of transforming growth factor-beta-1 (TGF-beta1) in autoimmune diseases. J Autoimmun. 2000;14(1):23–42. doi: 10.1006/jaut.1999.0339. [DOI] [PubMed] [Google Scholar]
- 66.Wahl SM. Transforming growth factor beta: the good, the bad, and the ugly. J Exp Med. 1994;180(5):1587–1590. doi: 10.1084/jem.180.5.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103(2):295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 68.Fu S, Zhang N, Yopp AC, Chen D, Mao M, Zhang H, et al. TGF-beta induces Foxp3 + T-regulatory cells from CD4 + CD25− precursors. Am J Transplant. 2004;4(10):1614–1627. doi: 10.1111/j.1600-6143.2004.00566.x. [DOI] [PubMed] [Google Scholar]
- 69.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198(12):1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zheng SG, Gray JD, Ohtsuka K, Yamagiwa S, Horwitz DA. Generation ex vivo of TGF-beta-producing regulatory T cells from CD4+CD25− precursors. J Immunol. 2002;169(8):4183–4189. doi: 10.4049/jimmunol.169.8.4183. [DOI] [PubMed] [Google Scholar]
- 71.Marin V, Montero-Julian FA, Gres S, Boulay V, Bongrand P, Farnarier C, et al. The IL-6-soluble IL-6Ralpha autocrine loop of endothelial activation as an intermediate between acute and chronic inflammation: an experimental model involving thrombin. J Immunol. 2001;167(6):3435–3442. doi: 10.4049/jimmunol.167.6.3435. [DOI] [PubMed] [Google Scholar]
- 72.Hurst SM, Wilkinson TS, McLoughlin RM, Jones S, Horiuchi S, Yamamoto N, et al. Il-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity. 2001;14(6):705–714. doi: 10.1016/s1074-7613(01)00151-0. [DOI] [PubMed] [Google Scholar]
- 73.Venkatachalam K, Mummidi S, Cortez DM, Prabhu SD, Valente AJ, Chandrasekar B. Resveratrol inhibits high glucose-induced PI3K/Akt/ERK-dependent interleukin-17 expression in primary mouse cardiac fibroblasts. Am J Physiol Heart Circ Physiol. 2008;294(5):H2078–2087. doi: 10.1152/ajpheart.01363.2007. [DOI] [PubMed] [Google Scholar]
- 74.Jones SA. Directing transition from innate to acquired immunity: defining a role for IL-6. J Immunol. 2005;175(6):3463–3468. doi: 10.4049/jimmunol.175.6.3463. [DOI] [PubMed] [Google Scholar]
- 75.Weissenbach M, Clahsen T, Weber C, Spitzer D, Wirth D, Vestweber D, et al. Interleukin-6 is a direct mediator of T cell migration. Eur J Immunol. 2004;34(10):2895–2906. doi: 10.1002/eji.200425237. [DOI] [PubMed] [Google Scholar]
- 76.Shi-wen X, Stanton LA, Kennedy L, Pala D, Chen Y, Howat SL, et al. CCN2 is necessary for adhesive responses to transforming growth factor-beta1 in embryonic fibroblasts. J Biol Chem. 2006;281(16):10715–10726. doi: 10.1074/jbc.M511343200. [DOI] [PubMed] [Google Scholar]
- 77.Qi W, Chen X, Polhill TS, Sumual S, Twigg S, Gilbert RE, et al. TGF-beta1 induces IL-8 and MCP-1 through a connective tissue growth factor-independent pathway. Am J Physiol Renal Physiol. 2006;290(3):F703–709. doi: 10.1152/ajprenal.00254.2005. [DOI] [PubMed] [Google Scholar]
- 78.Ikawa Y, Ng PS, Endo K, Kondo M, Chujo S, Ishida W, et al. Neutralizing monoclonal antibody to human connective tissue growth factor ameliorates transforming growth factor-beta-induced mouse fibrosis. J Cell Physiol. 2008;216(3):680–687. doi: 10.1002/jcp.21449. [DOI] [PubMed] [Google Scholar]
- 79.Raichlin E, Villarraga HR, Chandrasekaran K, Clavell AL, Frantz RP, Kushwaha SS, et al. Cardiac allograft remodeling after heart transplantation is associated with increased graft vasculopathy and mortality. Am J Transplant. 2009;9(1):132–139. doi: 10.1111/j.1600-6143.2008.02474.x. [DOI] [PubMed] [Google Scholar]
- 80.Torre-Amione G. Cardiac allograft hypertrophy: a new target for therapy, a surrogate marker for survival? Am J Transplant. 2009;9(1):7–8. doi: 10.1111/j.1600-6143.2008.02503.x. [DOI] [PubMed] [Google Scholar]
- 81.Nishida T, Kubota S, Fukunaga T, Kondo S, Yosimichi G, Nakanishi T, et al. CTGF/Hcs24, hypertrophic chondrocyte-specific gene product, interacts with perlecan in regulating the proliferation and differentiation of chondrocytes. J Cell Physiol. 2003;196(2):265–275. doi: 10.1002/jcp.10277. [DOI] [PubMed] [Google Scholar]
- 82.Duisters RF, Tijsen AJ, Schroen B, Leenders JJ, Lentink V, van der Made I, et al. miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res. 2009;104(2):170–178. doi: 10.1161/CIRCRESAHA.108.182535. 176p following 178. [DOI] [PubMed] [Google Scholar]
- 83.Liu W, Feng W, Wang F, Li W, Gao C, Zhou B, et al. Osteoprotegerin/RANK/RANKL axis in cardiac remodeling due to immuno-inflammatory myocardial disease. Exp Mol Pathol. 2008;84(3):213–217. doi: 10.1016/j.yexmp.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 84.Okada H, Kikuta T, Inoue T, Kanno Y, Ban S, Sugaya T, et al. Dexamethasone induces connective tissue growth factor expression in renal tubular epithelial cells in a mouse strain-specific manner. Am J Pathol. 2006;168(3):737–747. doi: 10.2353/ajpath.2006.050656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Holmes A, Abraham DJ, Sa S, Shiwen X, Black CM, Leask A. CTGF and SMADs, maintenance of scleroderma phenotype is independent of SMAD signaling. J Biol Chem. 2001;276(14):10594–10601. doi: 10.1074/jbc.M010149200. [DOI] [PubMed] [Google Scholar]
- 86.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7(6):454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]