

Abstract
Separation of milligram amounts of heparin oligosaccharides ranging in degree of polymerization from 4 to 32 is achieved within 6 hours using continuous-elution polyacrylamide gel electrophoresis (CE-PAGE) on commercially available equipment. The purity and structural integrity of CE-PAGE-separated oligosaccharides are confirmed by strong-anion exchange high-pressure liquid chromatography, electrospray ionization Fourier transform mass spectrometry and two-dimensional nuclear magnetic resonance spectroscopy. The described method is straightforward and time-efficient, affording size-homogeneous oligosaccharides that can be used in sequencing, protein binding, and other structure-function relationship studies.
Keywords: ESI-FTMS, heparin oligosaccharides, preparative PAGE
Introduction
Heparin is a linear, sulfated glycosaminoglycan (GAG) consisting of 1→4 linked iduronic or glucuronic acid (IdoA or GlcA) and glucosamine (GlcN) disaccharide building blocks with an average of 2.7 sulfo groups per disaccharide repeat (Figure 1). Heparin mediates a number of biological pathways through its interactions with various regulatory proteins, such as growth factors, chemokines, cytokines, and protease inhibitors[1; 2]. Structure-function relationship studies involving heparin-protein interactions often require purified heparin oligosaccharides of various sizes and sulfation states obtained through enzymatic depolymerization of heparin polysaccharide. The ability to prepare high-purity oligosaccharides in sufficient amounts for the x-ray crystallography, protein-GAG binding studies, and domain sequencing is critical for the development of new drug targets and potential GAG-based therapeutics.
Figure 1.
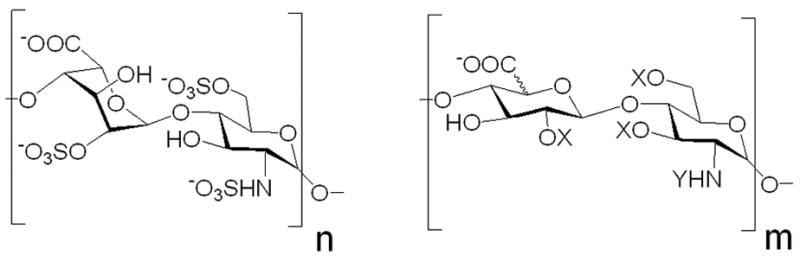
Structure of bovine lung heparin. An average chain contains major trisulfated disaccharide repeating units (n ~ 27) and minor disaccharide repeating units (m ~ 3) where X = H or SO3− and Y = SO3− and occasionally COCH3 or H.
Fractionation and purification of heparin oligosaccharides can be achieved in several ways using strong-anion exchange (SAX) chromatography [3; 4; 5], ion-pairing reverse-phase high-pressure liquid chromatography (IP-RP HPLC) [6; 7; 8], size-exclusion chromatography (SEC) [9], and micro-preparative polyacrylamide gel electrophoresis (PAGE) [10; 11; 12] in combination or alone. Under low-pH conditions, SAX chromatography separates GAG-derived oligosaccharides according to the number of sulfo groups. Bound oligosaccharides are eluted with an increasing salt gradient, and must be desalted for most downstream applications. During the IP-RP HPLC separation, an alkyl ammonium mobile phase modifier interacts with the negatively charged carboxy and sulfo groups in heparin through the positively charged ammonium moiety. The alkyl chain of the modifier interacts with the alkyl chains of the reverse-phase support imparting longer retention times to the more highly sulfated oligosaccharides, which are eluted with an increasing organic solvent gradient. SEC separates the oligosaccharides by size and may be used with a high ionic strength, volatile buffer, such as 30–50 mM ammonium formate [13]. Native, discontinuous PAGE affords unmatched resolution for separation of GAG-derived oligosaccharides and provides an additional analytical dimension in the process of GAG characterization [11; 12]. On a micro-preparative scale, native PAGE in a slab format has been used in our laboratory for the separation of heparin-derived oligosaccharides [10] and intact chondroitin sulfate polysaccharides from a proteoglycan bikunin [14]. Recovery of sample by soaking a cut portion of a fixed and stained gel containing a band is difficult. Instead, a narrow strip of the gel is stained to serve as a guide for the localization of bands in the unstained portion of the gel from which a sample is recovered. This approach results in a reduction of both the purity and amount of sample recovered. Alternatively, following PAGE, oligosaccharide bands can be electro-transferred onto positively charged membranes, excised, and eluted from the membrane strips with 2 M sodium chloride. Electro-transfer from an unstained membrane, is similarly guided only by a narrow vertical strip cut from the membrane and visualized with a dye. In both soaking and electro-transfer band curving during PAGE (smiling effect) can result in the contamination of a band of interest by neighboring bands. The recovered oligosaccharide also contains oligo- and poly-acrylamide and these contaminating substances are difficult to remove, making it hard to obtain sample of sufficient purity to afford clean NMR and mass spectral data required for structural assignment. Native, continuous-elution (CE) PAGE has been used by a number of workers for the micro-preparative separation of proteins, N-glycans, lipopolysaccharides, and nucleic acids [15]. CE-PAGE combines the convenience of an LC system with many advantages of PAGE, especially the possibility of customizing the experimental conditions for a particular sample mixture without the high additional cost.
Here, we report a high-resolution separation of milligram amounts of oligosaccharide mixture obtained by enzymatic depolymerization of bovine lung heparin (BLH) using native discontinuous CE-PAGE. The method described here permits simultaneous purification of a broad range of heparin oligosaccharides with MW 1,155 Da–8,660 Da within 6 hours using a commercial electrophoresis cell. Purity and structural integrity of PAGE-separated oligosaccharides were verified using proton nuclear magnetic resonance (1H NMR) spectroscopy, strong-anion exchange high-performance liquid chromatography (SAX-HPLC) and electrospray ionization mass spectrometry (ESI MS).
Materials and methods
Chemicals
Electrophoresis grade acrylamide, N,N′-methylene-bis-acrylamide, sucrose, glycine, ammonium persulfate (APS), N,N,N′,N′-tetramethylenediamine (TEMED), and bromophenol blue were from Bio-Rad (Hercules, CA). Boric acid, disodium salt of ethylenediaminetetraacetic acid (Na2EDTA), phenol red, Azure A and Alcian blue were from Fisher (Pittsburg, PA). Heparin lyases I, II, and III (E.C4.2.2.7, EC4.2.2.8 ) were from Cape Cod Associates (Seikagaku America, East Falmouth, MA). All solvents were HPLC grade and all other chemicals were molecular biology grade.
Preparation of heparin oligosaccharides
Bovine lung heparin (Celsus Laboratories, Cincinnati, OH) was partially depolymerized using a mixture of heparinases I, II, and III. The enzymes were denatured by boiling and removed by centrifugation. The supernatant containing the oligosaccharide mixture was collected, and the disaccharides and buffer salts were removed using size-exclusion resin (Bio-Gel P-4, Bio-Rad, Hercules, CA) and double-distilled water mobile phase. The resulting BLH oligosaccharide ladder was lyophilized and stored at −80° C.
PAGE
Heparin oligosaccharides were separated by the native CE-PAGE using Mini Prep Cell with 7 mm ID gel tube or Model 491 Prep Cell with 28 mm ID gel tube (Bio-Rad, Hercules, CA) and discontinuous buffer system [11]. The electrode running buffer was 1 M glycine, 0.2 M Tris, pH 8.8 (achieved upon dissolution), and the resolving gel buffer was 0.1 M boric acid, 0.1 M Tris, 0.01 M disodium EDTA, pH 8.3. A 15% total acrylamide (T) monomer solution contained 14.08% w/v acrylamide, 0.92% w/v N,N′-methylene-bis-acrylamide, and 5% w/v sucrose in the resolving gel buffer. A 10 cm × 7 mm diameter resolving gel column was cast from 4 mL 15% T monomer solution, to which 4 μL TEMED and 12 μL 10% APS were added to catalyze polymerization. A 10 cm × 28 mm diameter resolving gel column was cast from 40 mL 15% T monomer solution with 20 μL TEMED and 100 μL 10% APS. Resolving gel was allowed to polymerize overnight, after which a 5% T stacking gel was cast. The stacking gel monomer solution was prepared in the resolving gel buffer with pH adjusted to 6.3 using HCl and contained 4.75% w/v acrylamide and 0.25% w/v N,N′-methylene-bis-acrylamide. The volume of the stacking gel was twice that of the sample and contained 1 μL TEMED and 30 μL 10% APS per 1 mL monomer solution.
The BLH oligosaccharide mixture was dissolved in the running buffer, mixed with equal volume of 50% w/v sucrose containing 10 μg/mL phenol red, a tracking dye. Upper buffer chamber was filled with the electrode running buffer; the elution buffer reservoir and the lower buffer chamber were filled with the resolving gel buffer. A peristaltic pump (Econo-Pump, Bio-Rad, Hercules, CA) was set to a flow rate of 80 μL/min (Mini Prep Cell) or 500 μL/min (Model 491 Prep Cell) and connected to a fraction collector (Model 2110, Bio-Rad, Hercules, CA) set to collect 3-min fractions. Electrophoresis was carried out at a constant power of 1 W (Mini Prep Cell) or 5 W (Model 491 Prep Cell) for a total of 6 hrs; fractions were collected after approximately 2 hrs when the tracking dye was 1 cm above the bottom of the gel column. Collected fractions were analyzed by analytical PAGE on 0.75 mm × 6.8 cm × 8.6 cm mini-gels cast in-house using 15% T or 22% T resolving gel monomer solution and 5% T stacking gel monomer solution. The mini-gels were subjected to electrophoresis at constant 200 V using the discontinuous buffer system described above, stained in 0.5% w/v Alcian blue containing 2% v/v aqueous acetic acid solution for 30 min, and de-stained in water.
Sample purification
Prior to MS and NMR analyses, Tris-HCl and disodium EDTA were removed from the fractions collected during CE-PAGE using SAX spin-columns (High-capacity Mini-Q or Maxi-Q, Sartorius, Goettingen, Germany). To each fraction, a one-half volume of 225 mM NaCl solution was added, and the resulting sample was loaded onto the spin column, pre-equilibrated with 150 mM NaCl. The sample was washed twice with the 150 mM NaCl, eluted with 300 μL (Mini-Q) or 1 mL (Maxi-Q) 2 M NaCl, and desalted using a centrifugal filter (Microcon YM-3, for dp4 or YM-10, for dp6 and higher, or Amicon Ultra, 3,000 MWCO Millipore, Billerica, MA).
Strong-anion exchange chromatography
SAX-HPLC analysis of gel-eluted fractions was performed on a Shimadzu LC-10Ai liquid chromatography system equipped with an SPD-20A UV-VIS detector using a 4.6×250 mm Waters Spherisorb S5 SAX column. A two-segment gradient elution was achieved using mobile phases A, water, pH 3.4 (adjusted with HCl) and B, 2 M aqueous NaCl, pH 3.4 (adjusted with HCl) at a flow rate of 1 mL/min. The bound sample was washed with 0 % B–40 % B over 5 min, and eluted with 40 % B–75 % B over 125 min. The elution was monitored at 232 nm, the λmax for the 4,5-unsaturated uronic acid residue generated on the oligosaccharide non-reducing end by the heparin lyases treatment. For semi-preparative separation, a 20×250 mm Waters Spherisorb S5 SAX column was used with the same gradient but at a flow-rate of 4 mL/min.
Mass Spectrometry
CE-PAGE fractions containing dp4-dp14 were analyzed by ESI-FTMS on an LTQ XL Orbitrap mass spectrometer (Thermo Fisher Scientific, San-Jose, CA). Mobile phase consisting of 0.1% formic acid in 50% aqueous methanol [16] was delivered by an Agilent 1200 nano-LC pump at a flow rate of 50 μL/min. Desalted and lyophilized samples were dissolved in the mobile phase and 2–8 μL/injection were introduced by direct infusion through an Agilent 1200 autosampler. Mass spectra were acquired at a resolution of 30,000 in the negative-ion mode under the following conditions: spray voltage 3 kV, capillary temperature 200°C, capillary voltage −15 V, tube lens −100 V, the sheath and auxiliary gas flow rates were set to 20 and 5 units respectively.
Proton NMR analysis of gel-eluted heparin
Purity and structural integrity of gel-eluted heparin was assessed using 1H-NMR. Heparin oligosaccharides mixture and oligosaccharides eluted from gel were purified by SAX spin chromatography as described above, desalted, and freeze-dried. Dry samples were dissolved in 0.5 mL 99.996% deuterium oxide (2H2O, Sigma, St. Louis, MO) and freeze-dried. The 2H2O dissolution/freeze-drying was repeated three times before NMR analysis. NMR spectra were acquired in 99.996% 2H2O on a Bruker Ultrashield 600 MHz (14.1 Tesla) NMR instrument equipped with an ultrasensitive HCN cryoprobe with a z-axis gradient. The spectra were acquired at a probe temperature of 298 K and acquisition time of 2.6 s.
Azure A metachromatic assay
Recovery of oligosaccharides from CE-PAGE was estimated by measuring their metachromatic activity in an Azure A solution [17]. An assay working solution, 0.02 mg/mL Azure A was prepared from a 1 mg/mL stock immediately before the assay. BLH ladder solutions for the standard curve were prepared in the lower chamber buffer (elution buffer), and the buffer was also used for the blank. The Azure A working solution, 190 mL was thoroughly mixed with 10 mL of heparin solution, and the absorbance of this mixture at 620 nm was measured against the elution buffer blank. The decrease in absorbance at 620 nm was plotted as a function of heparin oligosaccharide concentration, and the amounts of recovered oligosaccharides were calculated using the resulting linear equation (Supplementary Figure 1). The assay standards and samples were prepared in triplicate in a 96-well plate, and their absorbance was measured on a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA).
Results and discussion
BLH oligosaccharide mixture used in the present study contained approximately 15 components with dp ranging from 4 to 32 monosaccharides. The difference in size between individual chains is mainly due to a disaccharide repeating unit since BLH has fairly uniform disaccharide composition (Figure 1). As the chain length increases, the percent difference in size between the chains decreases, making it difficult to separate large heparin oligosaccharides by SEC. Native, discontinuous PAGE, affords high-resolution separation over a wider range of MW compared with SEC. The goal of this work was to assess the utility of the continuous elution native PAGE for the preparative separation of heparin oligosaccharides in the amounts and purity sufficient for downstream structure-function relationship studies and in a time-efficient way.
PAGE conditions
Taking into account the number and MW distribution of the BLH ladder components, the following PAGE parameters were optimized: (1) total acrylamide monomer concentration (%T), (2) gel dimensions, and (3) running conditions. During the %T optimization, 10 μg of BLH oligosaccharide mixture was loaded on 10%T, 15%T, and 22%T slab mini-gels and subjected to electrophoresis for 30 min at constant 200 V. In the 22%T gel, a tetrasaccharide band migrated only half of the gel length during the 30-minute run, making the separation time-inefficient. In the 10%T gel, a portion of the sample migrated off of the gel, causing the loss of smaller oligosaccharides. The 15%T afforded the best sample resolution and was selected for the preparative separation.
Next, the preparative gel column length was selected guided by the basic idea that while a longer gel column would improve separation, it would also require a longer running time than a shorter column. Using Mini Prep cell with 7-mm diameter gel tube, 5-cm, 7-cm, 9-cm, and 10-cm gel columns were compared in terms of separation achieved under the same experimental conditions: 1 mg sample loading, 1 W constant power electrophoresis, 80 μL/min elution buffer flow rate, 240 μL fraction volume. Predictably, there was clear advantage in using 10-cm gel length, since the increase in the total running time was compensated by improved resolution.
Another aspect of method optimization was the maximum amount of sample that could be separated without compromising the resolution. In the mixture of 15 or more BLH oligosaccharides, the amounts of individual components decrease with the increasing size, as evidenced from the Alcian blue staining intensity of oligosaccharide bands (Supplementary Figure 2, Figure 2). Thus, increasing the sample loading negatively impacts resolution of the smaller oligosaccharides to a greater extent than the larger ones. In this study, the maximum amount of the BLH oligosaccharide mixture that could be separated into its individual components on a 7-mm diameter, 10-cm 15%T gel was 2.3 mg, which afforded 20–40 μg of purified oligosaccharide per fraction as estimated by Azure A assay. (Supplementary Figure 1)
Figure 2.

PAGE and ESI-FTMS analysis of CE-PAGE purified heparin oligosaccharides (insets): A. tetrasaccharide, dp4; and B. tetradecasaccharide, dp14.
ESI-FTMS analysis
Fractions collected from the 7-mm diameter gel were analyzed by PAGE and the like fractions were pooled. Oligosaccharides with dp4 through dp14 were analyzed by ESI-FTMS in the negative ion mode. Based on the MS data, the major component in each gel band consisted of trisulfated disaccharide building blocks (Figures 1 and 2). Peaks corresponding to lower sulfation states are probably artifacts of MS analysis caused by the analyte fragmentation through a loss of SO3 as their intensity is higher than expected based on the known composition of BLH. Major contaminant in the negative-ion ESI-FTMS analyses of PAGE-eluted, desalted oligosaccharides was EDTA, which formed partially sodiated clusters at m/z 605.1615, [2(EDTA)+Na-2H]−; m/z 627.1433, [2(EDTA)+2Na-3H]−; and m/z 649.1252, [2(EDTA)+3Na-4H]−. Apparently, EDTA interacts with heparin oligosaccharides and remains in the sample even after extensive desalting. We compared two methods for removing this contaminant: disrupting electrostatic interactions with 2 M NaCl and SAX spin-column purification. The contaminant peaks were completely absent in the mass spectra acquired from samples purified by either method. Thus, for MS-based studies, addition of 2 M NaCl followed by desalting is a fast and cost-efficient method for the EDTA removal.
SAX-HPLC analysis
The detection of peaks corresponding to the loss of SO3 in the mass spectra of purified oligosaccharides could reflect the sulfation heterogeneity within the sample and/or be a result of fragmentation during the MS analysis. To determine the sulfation states of major components in the gel-eluted oligosaccharide fractions, several purified oligosaccharide samples were analyzed by SAX-HPLC. Fractions eluted from two 7-mm diameter gels and containing dp8 were pooled, divided, and one portion was separated on an analytical SAX column with UV detection at 232 nm (4,5-unsaturated uronic acid residue). Two major chromatographic peaks were collected, desalted, and their mass spectra compared to that of PAGE-eluted dp8 before SAX-HPLC. The two mass spectra appeared virtually identical including the abundance of ions corresponding to lower sulfation states. (Supplementary Figure 3)
Fractions eluted from a 28-mm diameter gel and containing dp8, dp10, dp12, and dp14 were separated on a semi-preparative SAX column, and in each case, the largest chromatographic peak was collected, desalted, and analyzed by ESI-FTMS. As was the case with the dp8 fraction, resulting mass spectra contained peaks corresponding to lower sulfation states of each oligosaccharide, which can be attributed to the analyte fragmentation through the loss of SO3 during the MS analysis (Supplementary Figure 4).
NMR analysis
For NMR characterization, BLH oligosaccharides were separated on a larger scale, using 28-mm gel tube (Model 491 Cell) which accommodates a 40-mL, 10-cm gel. Increasing the gel volume 10-fold permitted a 10-fold increase in sample loading. Following the PAGE analysis of fractions, like fractions were pooled, 1 vol. of 4 M NaCl was added to each fraction, and the resulting solutions were desalted using 3,000 MWCO centrifugal filters (Amicon Ultra, 3000 MWCO, Millipore). The 1H NMR spectra of gel-eluted fractions purified by this method indicated that both Tris and EDTA were still present in the oligosaccharide sample. EDTA is an undesirable contaminant because it chelates Ca2+ and other cations, interfering with heparin-protein interactions and particularly Ca2+-dependent protein interactions [18; 19], and rendering the sample unusable for the protein interactions studies. A simple SAX spin-column purification procedure of the gel-eluted oligosaccharides permitted complete removal of both EDTA and Tris according to the NMR data (Figure 3). Structural integrity of gel-eluted oligosaccharides was assessed by two-dimensional (2D) correlation spectroscopy (COSY) and 2D heteronuclear multiple quantum coherence (HMQC) spectroscopy [20]. Comparison of the 2D COSY and HMQC spectra of the BLH oligosaccharide mixture and gel-eluted oligosaccharides confirmed that the saccharide backbone structure remains unchanged after the CE-PAGE (Figure 4).
Figure 3.

1H-NMR analysis of A. BLH oligosaccharide mixture; B. gel-eluted dp6 + dp8 fraction after desalting using 3,000 MWCO spin-column; and C. gel-eluted dp6 + dp8 fraction after purification using SAX spin-column.
Figure 4.

Two-dimensional NMR analysis of BLH oligosaccharide mixture (A, B) and CE-PAGE purified BLH decasaccharide, dp10 (C, D). The NMR peaks corresponding to H-H (COSY, panels A,C) or H-C (HMQC, panels B, D) correlations are labeled N for GlcN and I for IdoA residues, and A for non-reducing end unsaturated uronic acid residue.
In summary, the native CE-PAGE method for separation of BLH oligosaccharide mixture described here affords high resolution of the mixture components over broad MW range. After a simple buffer removal procedure, gel-eluted oligosaccharides are amenable for structural characterization and/or protein binding studies. The amount of a purified oligosaccharide eluted from a 7-mm diameter gel was sufficient for SAX-HPLC and FTMS analysis; however, the NMR experiments required a 10-fold increase in sample amounts which was achieved using a 28-mm diameter gel tube and a larger electrophoresis cell. The results of PAGE, SAX-HPLC, ESI-FTMS, and NMR characterization of the gel-eluted oligosaccharides demonstrate that this method of separation does not introduce chemical artifacts or alter the oligosaccharide structure. To our knowledge, this is the first time that GAG oligosaccharides have been recovered by PAGE that are of sufficient purity for the spectral analysis of their structure.
Supplementary Material
Acknowledgments
Funding
This work was supported by the National Institutes of Health grants GM38060, GM090127, and HL096972.
The authors thank Dennis Pu and Samantha Cheong for their help in collecting SAX-HPLC fractions and preparation of Azure A assay.
Abbreviations
- %T
percent total acrylamide
- 2D COSY
two-dimensional correlation spectroscopy
- 2D HMQC
two-dimensional heteronuclear multiple quantum coherence spectroscopy
- BLH
bovine lung heparin
- CE-PAGE
continuous-elution polyacrylamide gel electrophoresis
- dp
degree of polymerization
- ESI-FTMS
electrospray ionization Fourier transform mass spectrometry
- GAG
glycosaminoglycan
- GlcA
D-glucuronic acid
- GlcN
glucosamine
- HPLC
high-pressure liquid chromatography
- IdoA
L-iduronic acid
- IP-RP HPLC
ion-pairing reverse-phase high-pressure liquid chromatography
- LC
liquid chromatography
- MW
molecular weight
- MWCO
molecular weight cut-off
- NMR
nuclear magnetic resonance
- PAGE
polyacrylamide gel electrophoresis
- SAX
strong anion exchange chromatography
- SEC
size-exclusion chromatography
- TEMED
N,N,N′,N′-tetramethylene diamine
- UV
ultraviolet
Footnotes
Conflict of interest
None declared
Financial support from the National Institutes of Health grants GM38060, GM090127, HL096972 is gratefully acknowledged
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Capila I, Linhardt RJ. Heparin-protein interactions. Angew Chem Int Ed Engl. 2002;41:391–412. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 2.Kreuger J, Spillmann D, Li J-p, Lindahl U. Interactions between heparan sulfate and proteins: the concept of specificity. J Cell Biol. 2006;174:323–327. doi: 10.1083/jcb.200604035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Korir AK, Larive CK. Advances in the separation, sensitive detection, and characterization of heparin and heparan sulfate. Anal Bioanal Chem. 2009;393:155–69. doi: 10.1007/s00216-008-2412-2. [DOI] [PubMed] [Google Scholar]
- 4.Rice KG, Kim YS, Grant AC, Merchant ZM, Linhardt RJ. High-performance liquid chromatographic separation of heparin-derived oligosaccharides. Anal Biochem. 1985;150:325–31. doi: 10.1016/0003-2697(85)90518-4. [DOI] [PubMed] [Google Scholar]
- 5.Skidmore M, Atrih A, Yates E, Turnbull JE. Labelling heparan sulphate saccharides with chromophore, fluorescence and mass tags for HPLC and MS separations. In: Packer NH, Karlsson NG, editors. Methods in molecular biology. Humana Press; 2009. pp. 157–69. [DOI] [PubMed] [Google Scholar]
- 6.Doneanu CE, Chen W, Gebler JC. Analysis of oligosaccharides derived from heparin by ion-pair reversed-phase chromatography/mass spectrometry. Anal Chem. 2009;81:3485–99. doi: 10.1021/ac802770r. [DOI] [PubMed] [Google Scholar]
- 7.Thanawiroon C, Linhardt RJ. Separation of a complex mixture of heparin-derived oligosaccharides using reversed-phase high-performance liquid chromatography. J Chromatogr A. 2003;1014:215–23. doi: 10.1016/s0021-9673(03)00779-9. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Z, Xie J, Liu H, Liu J, Linhardt RJ. Quantification of heparan sulfate disaccharides using ion-pairing reversed-phase microflow high-performance liquid chromatography with electrospray ionization trap mass spectrometry. Anal Chem. 2009;81:4349–55. doi: 10.1021/ac9001707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ziegler A, Zaia J. Size-exclusion chromatography of heparin oligosaccharides at high and low pressure. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;837:76–86. doi: 10.1016/j.jchromb.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 10.al-Hakim A, Linhardt RJ. Isolation and recovery of acidic oligosaccharides from polyacrylamide gels by semi-dry electrotransfer. Electrophoresis. 1990;11:23–8. doi: 10.1002/elps.1150110106. [DOI] [PubMed] [Google Scholar]
- 11.Rice KG, Rottink MK, Linhardt RJ. Fractionation of heparin-derived oligosaccharides by gradient polyacrylamide-gel electrophoresis. Biochem J. 1987;244:515–22. doi: 10.1042/bj2440515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Volpi N, Maccari F. Electrophoretic approaches to the analysis of complex polysaccharides. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;834:1–13. doi: 10.1016/j.jchromb.2006.02.049. [DOI] [PubMed] [Google Scholar]
- 13.Zaia J. On-line separations combined with MS for analysis of glycosaminoglycans. Mass Spectrom Rev. 2009;28:254–72. doi: 10.1002/mas.20200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chi L, Wolff JJ, Laremore TN, Restaino OF, Xie J, Schiraldi C, Toida T, Amster IJ, Linhardt RJ. Structural analysis of bikunin glycosaminoglycan. J Am Chem Soc. 2008;130:2617–25. doi: 10.1021/ja0778500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seelert H, Krause F. Preparative isolation of protein complexes and other bioparticles by elution from polyacrylamide gels. Electrophoresis. 2008;29:2617–2636. doi: 10.1002/elps.200800061. [DOI] [PubMed] [Google Scholar]
- 16.Wolff JJ, Laremore TN, Busch AM, Linhardt RJ, Amster IJ. Influence of Charge State and Sodium Cationization on the Electron Detachment Dissociation and Infrared Multiphoton Dissociation of Glycosaminoglycan Oligosaccharides. J Am Soc Mass Spectrom. 2008;19:790–798. doi: 10.1016/j.jasms.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grant AC, Linhardt RJ, Fitzgerald GL, Park JJ, Langer R. Metachromatic activity of heparin and heparin fragments. Anal Biochem. 1984;137:25–32. doi: 10.1016/0003-2697(84)90341-5. [DOI] [PubMed] [Google Scholar]
- 18.Coyne E, Messmore HL, Walenga JM, Fareed J, Wehrmacher WH. Ethylenediaminetetraacetic Acid (EDTA) in Heparin. Thrombosis Research. 1998;90:245–246. doi: 10.1016/s0049-3848(98)00063-2. [DOI] [PubMed] [Google Scholar]
- 19.Fiore MM, Mackie IM. Mechanism of low-molecular-weight heparin reversal by platelet factor 4. Thrombosis Research. 2009;124:149–155. doi: 10.1016/j.thromres.2008.12.047. [DOI] [PubMed] [Google Scholar]
- 20.Guerrini M, Zhang Z, Shriver Z, Naggi A, Masuko S, Langer R, Casu B, Linhardt RJ, Torri G, Sasisekharan R. Orthogonal analytical approaches to detect potential contaminants in heparin. Proc Natl Acad Sci USA. 2009;106:16956–61. doi: 10.1073/pnas.0906861106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.