

Abstract
The C-terminal domain of the fibrinogen γ chain (γC) has been shown to bind to the integrins αIIbβ3, αMβ2 and αVβ3. It has also been reported that a peptide derived from the αMβ2-binding site of γC can suppress an animal model of multiple sclerosis, experimental autoimmune encephalomyelitis (EAE). Here we have truncated γC at position 399 to remove the prothrombotic αIIbβ3-binding site. We show that this truncated version of γC, termed γC399tr, can bind to activated T cells. In addition, T cells incubated with γC399tr secreted less IFN-γ when stimulated with antigen and APC; however, cytokine secretion was unaltered when T cells were stimulated non-specifically with a mixture of anti-CD3 and anti-CD28 antibodies. Thus, only antigen-dependent T cell activation is inhibited by γC399tr. When administered intraperitoneally, γC399tr potently inhibited actively induced EAE and reversed ongoing disease. We hypothesize that the ability of γC399tr to inhibit autoreactive immune responses is a result of its ability to bind integrins. This activity was not solely dependent on the αMβ2 integrin-binding site. When polyalanine was substituted for the αMβ2-binding site, the resulting γC390polyA was still able to inhibit EAE. To our knowledge, this is the first demonstration that T cells can bind to fibrin(ogen), an important extracellular matrix protein that is deposited at sites of inflammation. Our results also identify γC399tr as a novel therapeutic molecule.
Keywords: autoimmunity, experimental autoimmune encephalomyelitis (EAE), MOG, multiple sclerosis
Introduction
The fibrinogen γ chain has a conserved globular domain at its C-terminus, designated γC. This region encompasses fibrinogen residues 151-411. According to the crystal structure of the human γC domain [1], γC has a C-terminal fibrin-polymerization domain, a single calcium-binding site, and a deep binding pocket. Although this structure lacks a classical integrin-binding motif, γC has the ability to bind a variety of integrins including, αIIbβ3, αMβ2 and αVβ3. Elimination of residues 400-411 effectively removes the prothrombotic αIIbβ3-binding site. This truncated version of γC is designated γC399tr and corresponds to fibrinogen residues 151-399.
Integrins are a family of cell adhesion receptors that recognize extracellular matrix ligands, including fibrin(ogen) as well as many cell surface ligands [2]. Integrins are transmembrane α β heterodimers and at least 18 α and 8 β subunits are known. It has been well-established that, after binding to its ligand, an integrin can transduce signals from the outside to the inside of a cell. The activation state of an integrin molecule depends on its conformation as well as the clustering of individual integrin subunits. The activation state can be modulated by signals from inside the cell, a process called “inside-out” signaling [2]. For example, when a T cell is activated integrins on its surface may undergo a conformational change, assuming a more “on” position, extending out from the cell surface. The known integrins that bind to γC: αIIbβ3 on platelets, αVβ3 on endothelial cells, and αMβ2 on leukocytes, play an important role in thrombus formation, angiogenesis, and inflammation, respectively.
Many integrins are important for normal immune function. Integrin α4β1 binds to the vascular cell-adhesion molecule-1 (VCAM-1) and facilitates extravasation of lymphocytes from the peripheral circulation to sites of inflammation. Integrin αLβ2 is important for the formation of the immunologic synapse. It localizes to the pSMAC portion of the synapse, playing an important role in the ability of a T cell to recognize MHC-bound peptide antigens.
Biologics are a new class of medications that target key components of the immune response and, in some cases, target integrins. For example, natalizumab is directed against the integrin α4β1, present on the surface of lymphocytes. This medication is FDA approved and used to treat patients with multiple sclerosis and inflammatory bowel disease [3, 4]. Efalizumab is a biologic directed against the integrin LFA-1 (αLβ2) that until recently was used to treat psoriasis [5]. Given the proven therapeutic potential of integrin-binding biologics, we sought to identify an endogenous integrin-binding molecule that possesses anti-inflammatory properties. Specifically, we focused on γC because of its ability to bind several integrins, including some important for immune function. Takada and colleagues identified the αVβ3-binding site within γC [6]. Although αVβ3 is an arginine-glycine-aspartic acid (RGD)-binding integrin, it can also recognize γC, which lacks a classic integrin-binding motif. The ability of γC to bind this integrin was of interest because osteopontin also binds to β3 integrins and this interaction is important in inducing Th1 immune responses [7]. In addition to the αVβ3-binding site, an αMβ2-binding site has also been described within γC [8]. This integrin is expressed on the surface of microglia and macrophages and it plays a role in their activation and migration.
We hypothesized that the truncated version of γC, γC399tr, would be able to bind to leukocytes via their surface integrin molecules and alter the leukocytes' effector function. Here we characterize the cell-binding properties of γC399tr and demonstrate its ability to inhibit autoimmunity. Specifically, we found that γC399tr was able to: bind to T cells, inhibit EAE and reverse ongoing autoimmunity.
Methods
Peptides
The encephalitogenic myelin oligodendrocyte glycoprotein (MOG) peptide p:35-55 (MEVGWYRSPFSRVVHLYRNGK) and the ovalbumin (OVA) peptide p:323-339 (ISQAVHAAHAEINEAGR) were purchased from A @ A (San Diego, CA). Peptides were purified by HPLC and purity was confirmed by mass spectroscopy.
Mice
C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and housed in the UC Davis AAALAC-approved vivarium. OVA-specific TCR Tg OTII mice were also obtained from the Jackson Laboratory. Mice used in all experiments were age-matched female mice housed in laminar flow units and fed autoclaved chow. The UC Davis Institutional Animal Care and Use Committee approved all experiments.
Experimental autoimmune encephalomyelitis (EAE)
Experimental autoimmune encephalomyelitis was actively induced in C57BL/6 mice by subcutaneous immunization with 150 μl of a CFA emulsion containing 150 μg of Mycobacterium tuberculosis H37Ra (Difco) and 150 μg of MOG p:35-55. On days 0 and 2 mice were injected i.p. with 150 ng of purified pertussis toxin (PTX, List Biologicals) dissolved in 0.5 ml PBS. This protocol yields consistent results. EAE severity was scored as follows: 0, no clinical signs of disease or mild tail weakness; 1, complete tail limpness; 2, limp tail and moderate hind limb weakness or unsteady gait; 3, complete hind limb paralysis; 4, hind limb paralysis and some forelimb paralysis; 5, moribund [9]. Mice treated with γC399tr or γC390polyA received 100 μg daily via intraperitoneal injections starting on day four after MOG p:35-55 immunization. Representative mice were sacrificed and CNS samples were sent for H&E analysis to determine the extent of the CNS pathology.
Cell culture
The medium employed in all cell culture was RPMI 1640 (Invitrogen), supplemented with 5 × 10-5 M 2-mercaptoethanol (Sigma, St. Louis, MO), 4mM L-glutamine (Invitrogen), 100 U/ml benzylpenicillin (Invitrogen), 100 μg/ml streptomycin sulfate (Invitrogen) and 10% heat-inactivated fetal bovine serum (Hyclone). All cultures were incubated at 37°C in a humidified atmosphere of 5% CO2. For T cell stimulation assays splenocytes isolated from OT II mice (Jackson Labs) were incubated at 4 × 105 cells per well in a 96-well flat bottom plate with indicated amount of peptide antigen. ELISA antibodies were purchased from BD biosciences and the manufacturer's protocol was followed.
Flow cytometry analysis
All samples were pretreated with Fc-block. PE conjugated antibodies directed against CD3, CD19, and CD11c (BD Biosciences) were used in conjunction with a PE-specific selection kit (Stem Cell Technologies) to isolate T cells, B cells, and dendritic cells from spleen cell suspensions. Alexa 488-labeled γC399tr was then used to stain purified leukocyte populations for flow cytometry analysis. Peritoneal derived macrophages; a mast cell line, RBL (ATCC); and a human cutaneous T cell lymphoma line, CRL2105, were also stained with Alexa 488-labeled γC399tr for flow cytometry analysis. Finally, freshly isolated splenocytes were depleted of red blood cells and stained with Alexa 488-labeled γC399tr. These cells were also stained with antibodies to CD11c, NK1.1, and PDCA-1 conjugated to PE, APC and Alexa 647, respectively. All samples were run on a BD FACSCalibur flow cytometer and acquired data was analyzed using Flowjo 8.7 software.
Mutant γC399tr
Individual amino acid residues in the NRLSIGE sequence (residues 390-396 of γC) were simultaneously substituted to alanine as previously described [8].
Results
γC399tr binds to leukocytes
In order to determine which cells of the immune system have the ability to bind γC399tr, PE antibodies directed against CD19 and CD11c were used in conjunction with PE-specific magnetic selection beads to isolate B cells and dendritic cells respectively from spleen cell suspensions. These isolated populations were then stained with Alexa 488-labeled γC399tr. Figure 1 reveals that γC399tr-Alexa 488 effectively bound to B cells (Figure 1A). As expected Alexa 488-labeled γC399tr also bound to peritoneal derived macrophages, which express αMβ2 (Figure 1B). Binding of γC399tr-Alexa 488 to CD11c-isolated cells revealed two distinct γC399tr-binding populations, one with brighter staining than the other (Figure 1C). We therefore set out to better characterize the CD11c positive population. Staining with Alexa647-conjugated PDCA-1-specific antibody revealed that the CD11c positive population did not include significant numbers of plasmacytoid dendritic cells (Figure 1D). However, staining red blood cell-depleted splenocytes with APC-conjugated NK1.1-specific antibody and PE-conjugated CD11c specific antibody revealed a CD11clow, NK1.1 positive population. This population clearly bound to γC399tr. Thus, CD11c positive splenocytes are comprised of both myeloid dendritic cells and NK1.1 positive cells, both of which bind to γC399tr, albeit with different affinities.
Figure 1. γC399tr binds to B cells, macrophages, and dendritic cells.
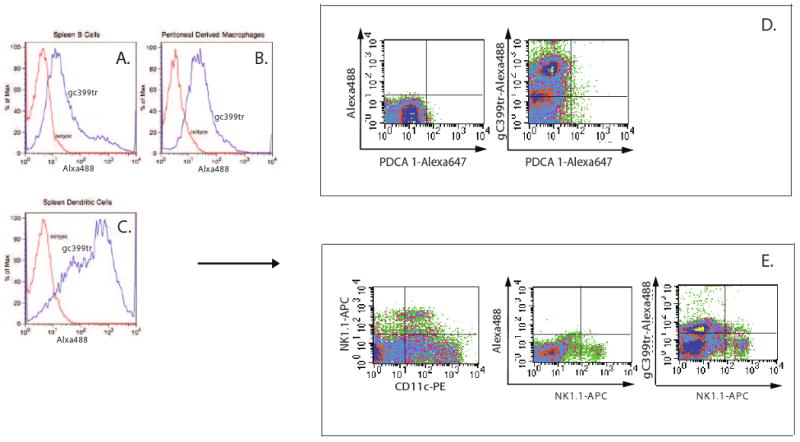
A) B cells were purified from splenocytes using a PE-labeled anti-CD19 antibody and a PE selection kit. Staining with Alexa 488-labeled γC399tr revealed that this population effectively bound to γC399tr. B) Peritoneal derived macrophages were further purified with a PE-labeled F4/80 antibody and a PE selection kit. Staining with Alexa 488-labeled γC399tr revealed that this population also effectively bound to γC399tr. C) CD11c+ cells were purified from splenocytes using a PE-labeled anti-CD11c antibody and a PE selection kit. Staining with Alexa 488-labeled γC399tr revealed that this population effectively bound to γC399tr with a bimodal distribution. D) To further characterize the two CD11c+ populations, splenocytes were depleted of red blood cells and then stained with PE-conjugated anti-CD11c, Alexa 488-conjugated γC399tr and Alexa 647-conjugated anti-PDCA-1. This revealed that there were almost no contaminating PDCA-1+ cells in the CD11c+ population. Again, CD11c+ cells bound well to Alexa 488-conjugated γC399tr. E) Splenocytes were then double stained with PE-conjugated anti-CD11c and APC-conjugated anti-NK1.1. Results show that a population of CD11clow NK1.1+ cells exists in the spleen. These cells bound poorly to γC399tr.
Next we stained immortalized cell lines with Alexa 488-labeled γC399tr. When compared to control, Alexa 488-labeled γC399tr effectively bound to a macrophage cell line JM774A-1 and poorly to a cutaneous T cell lymphoma cell line (Figure 2). In addition, Alexa 488-labeled γC399tr did not bind to the mast cell line, RBL (Figure 2).
Figure 2. A macrophage cell line binds to γC399tr.
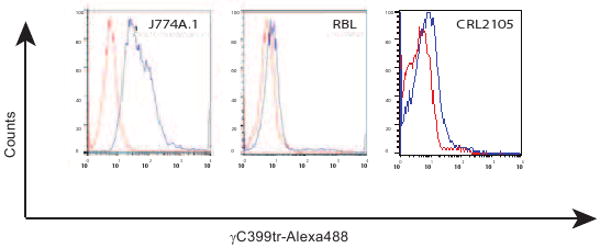
Alexa 488-labeled γC399tr effectively bound to a macrophage cell line JM774A.1 and bound weakly to a cutaneous T cell lymphoma, CRL2105. However, Alexa 488-labeled γC399tr did not bind to a mast cell line, RBL.
γC399tr an Inhibitor of Autoimmunity
After discovering γC399tr's ability to bind to leukocytes, the next logical step was to determine whether or not this molecule possessed any therapeutic potential. We therefore set out to characterize its effect on the induction of autoimmunity. We hypothesized that γC399tr would be able to bind to leukocytes via their surface integrin molecules and alter the immune response. C57BL/6 animals were immunized subcutaneously with MOG p:35-55 in a fashion as to induce EAE. Animals were then mixed randomly and divided into two groups, a control group to receive refolding buffer alone and a group to receive γC399tr treatments. Treatments started on day four after immunization. Animals were treated daily until the end of the experimentation on day 25. Table 1 reveals that none of the γC399tr-treated animals developed EAE (0/6). Four of the six control animals treated with refolding buffer developed severe and chronic EAE (maximum disease scores: 5,4,4,2,0,0). All animals were scored blindly.
Table 1.
| Treatment | Maximum Disease score | Mean Day of Onset |
|---|---|---|
| γC399tr 100 μg | (0,0,0,0,0,0) | NA |
| Refolding Buffer | (5,4,4,2,0,0) | 12.25 |
(*Mice were scored blindly.)
Central nervous system tissue samples with coded labels were also sent to a pathologist for Hematoxylin and Eosin (H&E) staining and analysis (Figure 3). The pathologist was blinded, unaware of the treatment groups. All samples from the γC399tr-treated group were read as having no CNS inflammation. CNS samples isolated from the control mice treated with refolding buffer were read as positive for CNS inflammation. The pathologist photographed the H&E slides and representative slides are shown in Figure 3A-3F.
Figure 3. γC399tr protects animals from EAE induction.
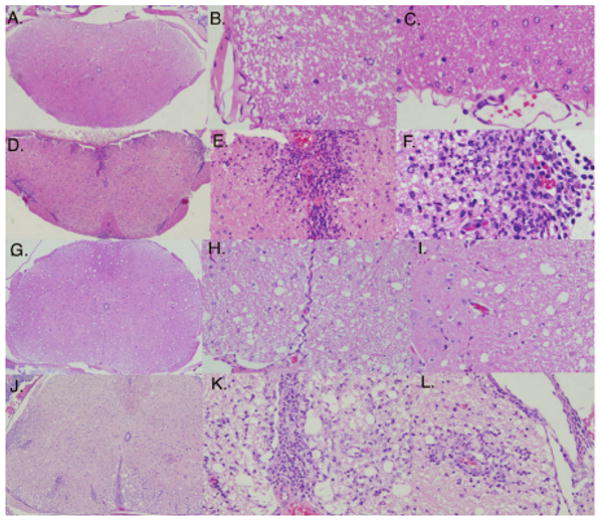
Coded CNS samples from mice depicted in Table 1 were paraffin-embedded and stained with H&E. A representative from each group is shown in images 3A-3F. All mice treated with γC399tr (3A-3C) were free of CNS inflammatory cells. In contrast, marked inflammation was seen in mice treated with refolding buffer alone (3D-3F). In 3D scanning magnification reveals multiple sites of perivascular inflammation. At closer magnification (3E and 3F) the CNS inflammation can be identified as mixed cellular (macrophages and lymphocytes). G-L) A crossover experiment was then performed in which control mice receiving refolding buffer were switched to γC399tr and γC399tr-treated animals were switched to refolding buffer. CNS samples were obtained from mice in both groups at the end of the experiment, day 25. CNS samples obtained from mice in the refolding buffer to γC399tr treatment group completely lacked CNS inflammation [3G (scanning), 3H and 3I (closer magnification)]. CNS samples from the γC399tr to refolding buffer group 3J-3L had marked CNS perivascular inflammatory cells [3J (scanning), 3K and 3L (closer magnification) ]. A pathologist blinded to treatment groups read all H&E samples.
To further characterize the anti-inflammatory properties of γC399tr we then designed a crossover study in which MOG p:35-55 immunized animals were again divided randomly into two groups, a control group receiving refolding buffer alone and a γC399tr-treated group. Then, on day 16 after MOG p:35-55 immunization, treatments were switched; the control group receiving refolding buffer was switched to γC399tr treatments and the γC399tr-treated group began to receive refolding buffer alone. Figure 4 shows the results of our crossover study. Prior to day 16 none (0/8) of the γC399tr-treated mice developed EAE, while seven out of eight animals in the control group developed severe disease (maximum disease scores: 4,4,4,4,4,4,3,0). In contrast, after switching therapies on day 16, four of the eight mice initially receiving γC399tr developed EAE (maximum disease scores: 4,3,2,2,0,0,0,0); and all of the animals that had initially developed EAE recovered once they began receiving γC399tr-treatment (Figure 4). Coded CNS samples were again sent to a pathologist for H&E analysis. The results confirmed our clinical scores. On day 25 no CNS inflammation was detected in any of the mice receiving γC399tr since crossover day 16 (Figure 3). Thus, γC399tr possesses strong anti-inflammatory properties and may be a potential therapeutic agent for the treatment of states of chronic inflammation.
Figure 4. γC399tr protects mice from actively induced EAE.

Sixteen C57BL/6 mice were immunized subcutaneously with MOG p:35-55 in an encephalitogenic fashion. On day 4, mice were randomly divided into two groups of eight mice each. One group received daily injections of 100 μg of γC399tr while the other group received an equal volume of refolding buffer alone. The degree of paralysis was then scored daily as described in the materials and methods. On day 16, treatments were crossed; animals initially receiving refolding buffer started to receive γC399tr and vice versa. Treatment injections were given in the morning and animals scored blindly in the evening. Averages for each group are graphed as days post injection versus EAE disease score. 4A) Upper left- seven of eight mice receiving control treatments with buffer alone developed EAE. 4B) Upper right- mice receiving γC399tr treatment were protected from EAE induction (0/8) (P<0.00001, student T test). 4C) Lower left- once animals developed EAE i.p. treatments with γC399tr could reverse their active disease. 4D) Lower right- four of the eight mice that were initially protected by γC399tr treatments developed EAE once this therapy was switched to refolding buffer.
γC399tr inhibits activated T cell
Since EAE is known to be a CD4+ T cell mediated autoimmune disease we sought to further characterize the specific effects that γC399tr has on activated T cells. The first step was to determine if γC399tr had a differential binding capacity for naïve and activated T cells. To do this spleen cells obtained from C57BL/6 mice were incubated with anti-CD28 and anti-CD3 antibodies for 48 hours to activate the resident T cells. Following activation PE conjugated anti-TCR antibodies were used in conjunction with a PE selection kit (StemCell Technologies) to isolate activated T cells. Naïve T cells were also isolated from fresh spleen using the same PE-selection kit. The cells were then stained with γC399tr-Alexa 488 and analyzed by flow cytometry. Figure 5 shows that γC399tr binding is increased on activated T cells. With this in mind we then sought to identify the physiologic effect that γC399tr binding had on T cells, using the OT II TCR transgenic mouse model [11]. These mice have T cells specific to the OVA peptide, p:323-339. Splenocytes isolated from OT II mice were incubated with increasing concentration of γC399tr in the presence or absence of OVA peptide at a concentration of 5 μg/ml. Results shown in Figure 6 reveal that γC399tr effectively inhibits IL-4, IFN-γ, and IL-17 produced by OT II T cells in response to antigen (Figures 6A, 6B and 6C, respectively). To determine if this inhibition was due to a nonspecific effect of γC399tr, we conducted a similar experiment in which splenocytes were incubated with increasing concentrations of γC399tr in the presence or absence of anti-CD3 and anti-CD28 antibodies. Figure 6D reveals that γC399tr was unable to inhibit T cell cytokine secretion when cells were stimulated non-specifically in an antigen independent fashion. This rules out a non-specific toxic effect of γC399tr on cultured cells. From this data we hypothesize that the ability of γC399tr to inhibit autoimmunity is partly due to its ability to inhibit activated T cells.
Figure 5. γC399tr binds with increased affinity to activated T cells.
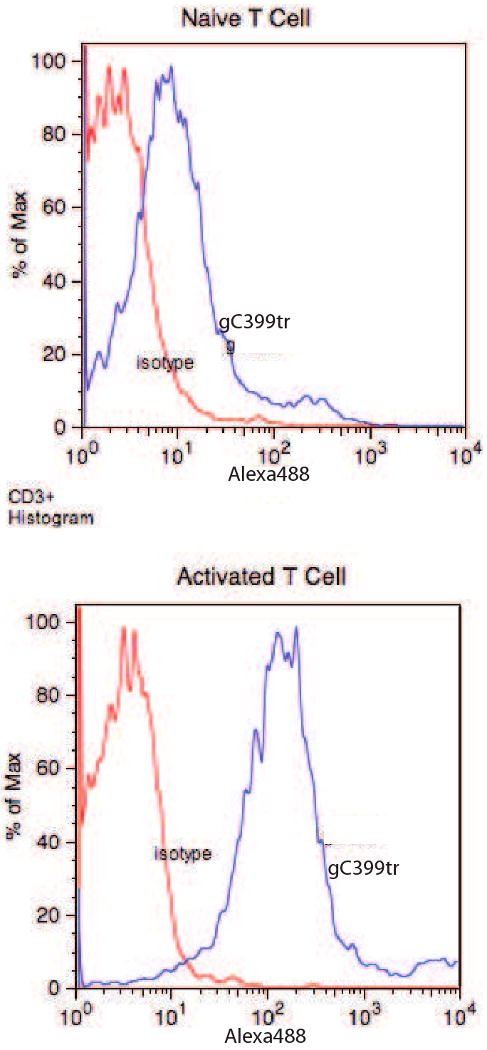
Naïve T cells and T cells activated with a mixture of CD3 and CD28 antibodies were stained with Alexa 488-labeled γC399tr. Upper histogram reveals that naïve T cells bound poorly to γC399tr. In contrast, activated T cells bound very strongly γC399tr (lower histogram).
Figure 6. γC399tr can inhibit cytokine production by activated T cells.

Splenocytes isolated from OTII Tg mice were in vitro cultured with either OVA peptide 5 μg/ml (6A-6C) or anti-CD3/anti-CD28 antibodies (6D) in the presence of increasing concentrations of γC399tr. 6A) Upper left- As the concentration of γC399tr is increased OVA-specific T cells secrete less IL-4 (circles). OTII cells incubated with control peptide did not secrete IL-4 (squares). 6B) Upper right- As the concentration of γC399tr is increased OVA-specific T cells secrete less IFNγ (circles). OTII cells incubated with control peptide did not secrete IFNγ (diamonds). 6C Bottom left- As the concentration of γC399tr is increased OVA-specific T cells secrete less IL-17 (circles). OTII cells incubated with control peptide did not secrete IL-17 (squares). 6D) Bottom right- γC399tr DID NOT inhibit IFNγ production by OTII T cells stimulated nonspecifically with anti-CD3 and anti-CD28 antibodies (circles). As the concentration of γC399tr is increased secretion of IFNγ is unchanged (triangles). OTII cells incubated with control peptide did not secrete IFNγ (squares).
Mutating the αMβ2 -binding site within γC399tr does not eliminate its therapeutic potential
Importantly, the Mac-1 binding site within γC399tr has been well-characterized and appears to be distinct from the αvβ3-binding site [12]. Alanine substitutions within the Mac-1 binding site have been shown to abolish Mac-1 binding [8]. To determine if the protective effect of γC399tr is dependent on this Mac-1-binding site we mutated this site within γC399tr to alanine. The resulting γC390PolyA was then administered to mice once daily beginning on day four after EAE induction with MOG p:35-55. Similar to the results obtained with wild-type γC399tr, the γC390PolyA molecule was effective at inhibiting EAE. As shown in Figure 7 only 2/6 mice developed EAE. Thus, even in the absence of an αMβ2-binding site γC399tr possessed some inhibitory properties.
Figure 7. The αMβ2-binding defective mutant γC390polyA effectively inhibits EAE.

EAE was induced using the same protocol as in Figure 4. γC399tr and γC390polyA were able to effectively inhibit EAE (7A upper left and 7B upper right, respectively. Asterix represents P<0.05, student T test). 7C) Lower left- mice treated with buffer alone as a control developed severe EAE. 7D) Lower right- animals that had initially developed EAE were able to recover after administration of γC399tr. This figure better highlights the chronic nature of the untreated animals.
Discussion
Fibrinogen is deposited at sites of tissue injury, including sites of inflammation. The ability of cells to migrate through the deposited fibrin scaffold is key to a variety of processes. Integrins bind to extracellular matrix proteins, including fibrin(ogen). Given the importance of this interaction one might predict that the integrin-binding motifs within fibrinogen would be conserved evolutionarily. However, the classic RGD-integrin-binding motif is not conserved within fibrinogen across different species. Thus, it was hypothesized that fibrin(ogen) likely contains other integrin-binding sequences. The γC region of fibrinogen is conserved evolutionarily and has been found to bind a variety of integrins. We have sought to determine the leukocyte populations capable of binding to γC399tr and to characterize the effect that binding of soluble γC399tr has on the immune response. Although we were unable to prove that the γC399tr-leukocyte interactions were mediated through cell surface integrins, a variety of circumstantial evidence was found in support of this possibility. For one, the fibrinogen fragment γC399tr has been already been shown to bind to a variety of integrins, some of which are present on immune cells. For example, an αMβ2-binding site within γC399tr has been well-characterized [8]. Not surprisingly, γC399tr bound well to macrophages, which express αMβ2 (Figure 1B). However, interactions between fibrin fragments and integrins on the surface of T cells, B cells, and dendritic cells have not been reported. Given that fibrin and its degradation products are deposited perivascularly at sites of inflammation [10], one would expect that such an interaction would enhance T cell and B cell migration and navigation through inflammatory tissue. It would also facilitate cellular interactions between these cells and growth factors bound to the deposited fibrin scaffold. Activated integrins extend out from the surface of the cell, and are more accessible for ligand binding. When T cells are activated or are exposed to chemokines their integrins also assume a more active state; γC399tr bound with a higher affinity to activated T cells. Integrins also participate in antigen recognition by helping the T cell bind to the antigen presenting cell (APC). They form part of the immunologic synapse, which is required for antigen-dependent T cell activation. The formation of this synapse is not required when T cells are stimulated with anti-CD3 and anti-CD28; γC399tr was only able to inhibit antigen dependent T cell activation. Lastly, it appeared that the integrin expression level correlated with the binding capacity of γC399tr. Cells that expressed higher concentrations of integrins bound more strongly to γC399tr. Together this data identifies γC399tr-integrin interactions as one possible mechanism for γC399tr binding to leukocytes.
This manuscript demonstrates that γC399tr is a potent inhibitor of EAE, the animal model of MS. It is possible that some of this protection is mediated through the αMβ2 binding site within γC399tr. However, this site is not solely responsible for all of the protective effects of γC399tr. When mutated to polyalanine the resulting γC390polyA was still able to inhibit EAE induction (Figure 7). Thus, other properties of γC399tr are likely contributing to its role in EAE inhibition. One possibility is that γC399tr-has an inhibitory effect on T cells. The secretion of T cell cytokines IFN-γ and IL-17, known to be important for EAE induction [13-15], were inhibited by soluble γC399tr (Figure 6).
Since fibrinogen degradation products are deposited at sites of inflammation, the ability to bind to fibrino(gen) would allow leukocytes to better migrate through inflammatory sites. Why then was soluble γC399tr able to inhibit autoimmunity? In contrast to a fibrin scaffold, binding of soluble γC399tr would not help leukocytes orient themselves for migration. In addition, γC399tr was able to inhibit antigen-dependent T cell activation, something a fibrin scaffold would not be expected to do.
Importantly, our results reveal a novel approach to identify integrin-binding mediators of autoimmunity. We have focused on identifying “self” molecules rather than identifying monoclonal integrin-blocking antibodies. Integrins have relatively low affinities for their ligands (Kd=10-6 to 10-7 M). Thus, any interaction between γC399tr and its integrin ligands is likely quite different than that of a high affinity specific monoclonal antibody. Although the broad binding specificity of this ligand might limit its potential for drug development, we are hopeful that this study will aid in the design of future biologic medications.
Acknowledgments
EM is a recipient of a Career Award for Medical Scientists from the Burroughs Wellcome Fund and a Howard Hughes Medical Institute Physician Scientist Early Career Award. This work was supported by the NIH (A1075029 awarded to EM and CA131015 to YT).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yee VC, Pratt KP, Cote HC, Trong IL, Chung DW, Davie EW, Stenkamp RE, Teller DC. Crystal structure of a 30 kDa C-terminal fragment from the gamma chain of human fibrinogen. Structure. 1997;5:125–138. doi: 10.1016/s0969-2126(97)00171-8. [DOI] [PubMed] [Google Scholar]
- 2.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 3.Miller DH, Khan OA, Sheremata WA, Blumhardt LD, Rice GP, Libonati MA, Willmer-Hulme AJ, Dalton CM, Miszkiel KA, O'Connor PW. A controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2003;348:15–23. doi: 10.1056/NEJMoa020696. [DOI] [PubMed] [Google Scholar]
- 4.Gordon FH, Lai CW, Hamilton MI, Allison MC, Srivastava ED, Fouweather MG, Donoghue S, Greenlees C, Subhani J, Amlot PL, Pounder RE. A randomized placebo-controlled trial of a humanized monoclonal antibody to alpha4 integrin in active Crohn's disease. Gastroenterology. 2001;121:268–274. doi: 10.1053/gast.2001.26260. [DOI] [PubMed] [Google Scholar]
- 5.Gottlieb AB, Krueger JG, Wittkowski K, Dedrick R, Walicke PA, Garovoy M. Psoriasis as a model for T-cell-mediated disease: immunobiologic and clinical effects of treatment with multiple doses of efalizumab, an anti-CD11a antibody. Arch Dermatol. 2002;138:591–600. doi: 10.1001/archderm.138.5.591. [DOI] [PubMed] [Google Scholar]
- 6.Yokoyama K, Erickson HP, Ikeda Y, Takada Y. Identification of amino acid sequences in fibrinogen gamma -chain and tenascin C C-terminal domains critical for binding to integrin alpha vbeta 3. J Biol Chem. 2000;275:16891–16898. doi: 10.1074/jbc.M000610200. [DOI] [PubMed] [Google Scholar]
- 7.Ashkar S, Weber GF, Panoutsakopoulou V, Sanchirico ME, Jansson M, Zawaideh S, Rittling SR, Denhardt DT, Glimcher MJ, Cantor H. Eta-1 (osteopontin): an early component of type-1 (cell-mediated) immunity. Science. 2000;287:860–864. doi: 10.1126/science.287.5454.860. [DOI] [PubMed] [Google Scholar]
- 8.Flick MJ, Du X, Witte DP, Jirouskova M, Soloviev DA, Busuttil SJ, Plow EF, Degen JL. Leukocyte engagement of fibrin(ogen) via the integrin receptor alphaMbeta2/Mac-1 is critical for host inflammatory response in vivo. J Clin Invest. 2004;113:1596–1606. doi: 10.1172/JCI20741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Menezes JS, van den Elzen P, Thornes J, Huffman D, Droin NM, Maverakis E, Sercarz EE. A public T cell clonotype within a heterogeneous autoreactive repertoire is dominant in driving EAE. J Clin Invest. 2007;117:2176–2185. doi: 10.1172/JCI28277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adams RA, Bauer J, Flick MJ, Sikorski SL, Nuriel T, Lassmann H, Degen JL, Akassoglou K. The fibrin-derived gamma377-395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease. J Exp Med. 2007;204:571–582. doi: 10.1084/jem.20061931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998;76:34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- 12.Yakubenko VP, Solovjov DA, Zhang L, Yee VC, Plow EF, Ugarova TP. Identification of the binding site for fibrinogen recognition peptide gamma 383-395 within the alpha(M)I-domain of integrin alpha(M)beta2. J Biol Chem. 2001;276:13995–14003. doi: 10.1074/jbc.M010174200. [DOI] [PubMed] [Google Scholar]
- 13.Ando DG, Clayton J, Kono D, Urban JL, Sercarz EE. Encephalitogenic T cells in the B10.PL model of experimental allergic encephalomyelitis (EAE) are of the Th-1 lymphokine subtype. Cell Immunol. 1989;124:132–143. doi: 10.1016/0008-8749(89)90117-2. [DOI] [PubMed] [Google Scholar]
- 14.Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med. 2006;203:1685–1691. doi: 10.1084/jem.20060285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]