

Abstract
Photosystem I (PS I) contains two symmetric branches of electron transfer cofactors. In both the A- and B-branches, the phylloquinone in the A1 site is π-stacked with a tryptophan residue and is H-bonded to the backbone nitrogen of a leucine residue. In this work, we use optical and electron paramagnetic resonance (EPR) spectroscopies to investigate cyanobacterial PS I complexes, where these tryptophan residues are changed to phenylalanine. The time-resolved optical data show that backward electron transfer from the terminal electron acceptors to P·+700 is affected in the A- and B-branch mutants, both at ambient and cryogenic temperatures. These results suggest that the quinones in both branches take part in electron transport at all temperatures. The electron-nuclear double resonance (ENDOR) spectra of the spin-correlated radical pair P·+700A·−1 and the photoaccumulated radical anion A·−1, recorded at cryogenic temperature, allowed the identification of characteristic resonances belonging to protons of the methyl group, some of the ring protons and the proton hydrogen-bonded to phylloquinone in the wild type and both mutants. Significant changes in PS I isolated from the A-branch mutant are detected, while PS I isolated from the B-branch mutant shows the spectral characteristics of wild-type PS I. A possible short-lived B-branch radical pair cannot be detected by EPR due to the available time resolution; therefore, only the A-branch quinone is observed under conditions typically employed for EPR and ENDOR spectroscopies.
Introduction
Photosystem I (PS I) is a membrane-bound multi-subunit protein complex found in plants, algae and cyanobacteria, which is an indispensible part of the photosynthetic electron transfer chain (for a collection of recent reviews on PS I, see Ref. [1]). PS I mediates light-induced translocation of electrons across the membrane, from the lumenal side to the stromal side [2].
Research in PS I in the past few years has been greatly aided by the availability of a 2.5 Å resolution crystal structure of cyanobacterial PS I [3] (Protein Data Bank [PDB] entry 1JB0) and, more recently, by a 3.4 Å resolution crystal structure of plant PS I [4] (PDB entry 2O01). These structures, together with structural information obtained for the individual subunits [5–10], have allowed for rapid progress in PS I research. In particular, the availability of detailed structural information has benefited site-directed mutagenesis studies, thereby allowing precise targeting of important amino acids in the environment of particular cofactors and on the protein–protein interface. This information has been successfully coupled with modeling of the properties of a particular cofactor or of protein–protein interactions. A combination of mutagenesis work and modeling is expected to continue to provide insights into the function of membrane-bound, energy transforming protein complexes, with PS I as one of the best-studied models.
Cyanobacterial PS I is composed of 12 protein subunits, which bind a large number of pigments involved in capturing light energy and cofactors involved in carrying out electron transfer [3]. The subunits PsaA, PsaB and PsaC bind all of the cofactors that participate in electron transfer. The core of the PS I complex is formed by the PsaA/PsaB heterodimer, which consists of two large, homologous subunits, each containing 11 transmembrane helixes. Within this hydrophobic milieu, the electron transfer cofactors P700 (Chl a/Chl a′ heterodimer), A0 (Chl a), A1 (phylloquinone), and the [4Fe–4S] cluster FX are found. The two terminal [4Fe–4S] clusters FA and FB are bound to the PsaC subunit on the stromal side of a membrane (reviewed in Refs. [11, 12]).
Two branches of electron transfer cofactors exist in PS I that are related by a pseudo-C 2 symmetry axis. The cofactors of the A-branch are bound to the PsaA subunit and the cofactors of the B-branch are bound by the PsaB subunit (Fig. 1a), with the exception of the accessory Chls. Within the resolution of the crystal structure [3], the cofactors and their binding sites are very similar on the A- and B-branches. The presence of two structurally equivalent branches of cofactors gives rise to the question of the directionality of electron transfer in PS I, i.e., whether electron transfer occurs through the cofactors of the A-branch, or through the cofactors of the B-branch or through the cofactors bound to both branches. Ever since the issue of directionality was re-invigorated by Joliot and Joliot [13], it has been a hot topic in the literature (reviewed, e.g., in Refs. [14–16]).
Fig. 1.
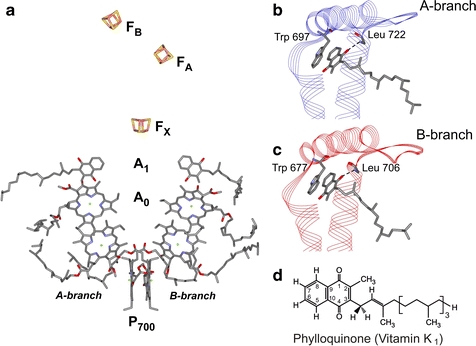
a Arrangement of the cofactors in the electron transfer chain of PS I from T. elongatus taken from the 2.5 Å resolution X-ray crystal structure (PDB entry 1JB0) [3]. The two branches are denoted as A- and B-branches. The spectroscopic names are indicated for each cofactor. Both branches of cofactors are highly similar. Moreover, the A1 binding sites in the A- and B-branches are nearly identical except for the conformation of the quinone tail. The color coding of the amino acids is as follows: carbon, gray; oxygen, red; nitrogen, blue; sulfur, yellow. b Structure of the A1 binding site in the A-branch of PS I taken from the 2.5 Å resolution crystal structure. The backbone amide proton of L722PsaA is in H-bonding distance to one of the oxygens of the quinone (dashed line), while no H-bonding donor is present to the other oxygen of the quinone. W697PsaA is π-stacked to the quinone, with a plane-to-plane distance of 3.0–3.5 Å. c Structure of the A1 binding site in the B-branch of PS I taken from the 2.5 Å resolution crystal structure. The backbone amide proton of L706PsaB is in H-bonding distance to one of the oxygens of the quinone (dashed line), while no H-bonding donor is present to the other oxygen of the quinone. W677PsaB is π-stacked to the quinone, with a plane-to-plane distance of 3.0–3.5 Å. d Chemical structure of phylloquinone (vitamin K1) with numbering scheme (color figure online)
The study of PS I complexes with site-directed mutations in the A1 binding sites by optical and electron paramagnetic resonance (EPR) spectroscopies has been central to the directionality debate. In Fig. 1b and c, the environment of the phylloquinone (vitamin K1) within the A1 binding site in the A- and B-branches is compared. The near-perfect similarity between the two branches is apparent; the only notable difference is the conformation of the quinone tail (see Fig. 1d for numbering scheme). Within the A1 binding site, phylloquinone is hydrogen bonded to the amide nitrogen of a leucine residue (L722PsaA and L706PsaB), and is π-stacked to a conserved tryptophan (W697PsaA and W677PsaB). The latter is hydrogen bonded to a conserved serine (S692PsaA and S672PsaB) [3]. These interactions are part of a wider intricate network of hydrogen bonds and other interactions extending from A0 through A1 to the FX binding site [3].
The conserved tryptophan (Trp) residues that are π-stacked to A1 were one of the first targets for studies by site-directed mutagenesis in both the eukaryote Chlamydomonas reinhardtii [17–20] and the prokaryote Synechocystis sp. PCC 6803 [21, 22]. Note that the nomenclature is different in C. reinhardtii (W693PsaA and W673PsaB are π-stacked to A1 in the A- and B-branches, respectively).
In the first study of its kind, the π-stacking tryptophan on the A-branch was changed to leucine (W693LPsaA) or histidine (W693HPsaA) in C. reinhardtii [19]. The results obtained in these mutants were nearly identical. Both resulted in a lower amount of PS I in the cells, and both mutants were unable to grow on minimal media with oxygen. The rate of forward electron transfer between A1 and FX, when measured by transient EPR at 260 K, was decreased in PS I isolated from both mutants. On photoaccumulation in the presence of sodium dithionite, a much larger contribution of A0 was detected by EPR in these mutants, as compared to the wild type. X-band continuous-wave (CW) electron-nuclear double resonance (ENDOR) studies of the same samples revealed reduced hyperfine coupling (hfc) of the characteristic methyl group protons of the quinone radical trapped by photoaccumulation. It was concluded that the A-branch quinone is involved in electron transfer in PS I. The complimentary B-branch mutants W673LPsaB and W673HPsaB were studied together with mutants of the methionine, which ligates the A0 acceptor in C. reinhardtii [20], with the emphasis on the latter one. It was concluded that the W673LPsaB and W673HPsaB substitutions also affect the electronic structure of A·−1. Thus, substitution of π-stacking tryptophan by leucine or histidine in both A- and B-branches affects electron transfer in C. reinhardtii [19, 20].
Guergova-Kuras et al. [18] replaced the π-stacked tryptophans in the A- and B-branches by phenylalanine in C. reinhardtii, generating W693FPsaA and W673FPsaB mutants. A double mutant, in which both A- and B-branch tryptophans were replaced by phenylalanine, was also generated (W693FPsaA/W673FPsaB). In contrast to Ref. [19], all of their mutants were capable of photoautotrophic growth in the presence and in the absence of oxygen, although the double mutant was unable to grow under high light intensity [17]. Both the wild type and mutants were thoroughly investigated by time-resolved optical spectroscopy. The kinetics of A·−1 oxidation, monitored by transient absorption at 380 nm in whole cells of wild-type C. reinhardtii, is characterized by fast (t 1/2 = 13 ns) and slow (t 1/2 = 150 ns) kinetic decay phases. When the electrochromic band shift (from 480 to 457 nm) was used as a reporter of A·−1 re-oxidation, similar results were obtained. Analogous measurements on the mutant cells showed that in the A-branch mutant (W693FPsaA), the decay rate of the slow kinetic phase decreased (ca. 3 times) and its decay-associated spectrum was changed. Conversely, in the corresponding B-branch mutant (W673FPsaB), the decay rate of the fast kinetic phase decreased (ca. 5 times) and its decay-associated spectrum was changed. In the double mutant (W693FPsaA/W673FPsaB), the decay rates of both the slow and fast kinetic phases decreased by ca. 3 and 5 times, respectively. Photoaccumulated samples of thylakoid membranes isolated from wild-type C. reinhardtii and all three mutants were probed by CW EPR at Q-band [17]. Under the experimental conditions employed, a mixture of A·−1 and A·−0 was detected in the wild type and double mutant (W693FPsaA/W673FPsaB) samples. In thylakoid membranes isolated from W673FPsaB, a nearly pure A·−1 signal was detected, with very little A·−0 contamination. In the W693FPsaA sample, only A·−0 was detected. This differs from the finding of Purton et al. [19], in which photoaccumulated PS I samples of W693LPsaA and W693HPsaA showed a mixture of A·−1 and A·−0 (although with a larger A·−0 contribution than in the wild type). Study of these mutants by transient EPR spectroscopy at X-band showed that in PS I from the A-branch mutant and from the double mutant, the hfc of the methyl protons of the quinone is partially resolved, and is more pronounced than in spectra of either wild-type PS I or the B-branch mutant. The kinetics of P·+700 re-reduction was also studied in thylakoid membranes isolated from the wild type and mutant strains. The appearance of major phases with lifetimes in the millisecond time range indicated that in all samples, P·+700(FA/FB)− had been formed. The data in Refs. [17, 18] have shown that in PS I from eukaryotes, both the A- and B-branches are active in photosynthetic electron transfer. It was estimated that there is a slight preference for electron transfer via the A-branch (55–66% of the total flux) [18].
Xu et al. [21, 22] replaced the π-stacked tryptophans in the A- and B-branches by phenylalanine in cyanobacterium Synechocystis sp. PCC 6803, generating the W697PsaA and W677PsaB mutants. Both mutants were able to grow photoautotrophically; however, both had reduced growth rates, especially under high light intensities [21]. The PS I content of cells of both mutants was only 80% of the wild-type level. The PS I-mediated electron transfer activity was significantly lower in both mutants [21]. Time-resolved optical difference spectroscopy on wild-type Synechocystis sp. PCC 6803 whole cells and on isolated PS I complexes showed well-resolved fast and slow kinetic phases [22].1 Analogous measurements on the mutant cells and PS I complexes showed that in the A-branch mutant (W697FPsaA) the decay rate of the slow kinetic phase decreased (ca. 3 times), in the B-branch mutant (W677FPsaB) the decay rate of the fast kinetic phase similarly decreased (ca. 4 times). This agrees very well with the results obtained by optical spectroscopy on the π-stacked tryptophan mutants in C. reinhardtii [18]. The kinetics of A·−1 oxidation was also measured by X-band EPR at 260 K and room temperature in PS I isolated from the W697FPsaA and W677FPsaB mutants [22]. EPR results showed that the rate of forward electron transfer is decreased in the A-branch mutant (ca. 3 times) as compared to wild type, while no effect is found in the B-branch mutant. In summary, the discrepancy between the kinetic measurements by EPR and optical spectroscopies was reported.
Photoaccumulated samples of PS I isolated from the W697FPsaA and the W677FPsaB mutants were studied by low-temperature CW EPR at Q-band [21]. A mixture of A·−1 and A·−0 was found in the wild type and both mutant samples. In the B-branch mutant, the contribution of A0 was found to be comparable to the wild type. However, a much larger contribution of A0 was found in A-branch mutant as compared to the wild type and B-branch mutant. This is in agreement with results obtained on PS I isolated from the C. reinhardtii W693H/LPsaA mutants, where an increase in the amount of A·−0 was found [19]. However, this does not agree with results obtained on the W693FPsaA mutant in C. reinhardtii, for which no A·−1 was observed [17]. Studies of PS I isolated from the W697FPsaA and the W677FPsaB mutants by transient EPR at 80 and 260 K showed that the hfcs of the methyl group protons are more pronounced in the A-branch mutant (W697FPsaA) as compared to the wild type or the B-branch mutant (W677FPsaB) [21, 22]. This agrees well with results obtained on thylakoid membranes isolated from the W693FPsaA mutant of C. reinhardtii [17]. Pulsed ENDOR studies of the spin-polarized radical pair in the PS I mutant samples at X-band revealed that the methyl proton hfc was increased by ca. 5% in the W697FPsaA mutant as compared to the W677FPsaB mutant [21], and to the Thermosynechococcus elongatus wild-type PS I complex reported elsewhere [23].
In summary, only changes in the A-branch mutants are detected by EPR-based methods, while physiological and biochemical characterizations and time-resolved optical spectroscopy detect changes in PS I of both A- and B-branch mutants of cyanobacteria [21, 22]. The transient radical pair spectrum involving B-branch quinone was observed previously only under strongly reducing conditions [24].
The interpretation of the data on π-stacked tryptophan mutants differs for C. reinhardtii and Synechocystis sp. PCC 6803. The electron transfer chain in PS I and the environment of the cofactors involved in electron transfer, in particular, the A1 binding site, are conserved in these organisms. Hence, the differences in the interpretation of the data have to be reconciled.
We present here a further investigation of PS I from wild type and the W697FPsaA and W677FPsaB mutants of Synechocystis by time-resolved optical spectroscopy and by advanced EPR techniques. The advantage of this work is that the kinetics of P·+700 re-reduction in PS I isolated from the wild type and both mutant samples was studied by optical spectroscopy at room temperature as well at low temperature, thereby allowing comparison of optical and EPR data. Spin-polarized transient X- and Q-band EPR spectra of PS I isolated from all three samples were recorded and compared to those of previous studies. For the first time samples of PS I from Synechocystis sp. PCC 6803 wild type, W697FPsaA and W677FPsaB were studied simultaneously by ENDOR, investigating both the transient radical pair and also the stationary photoaccumulated radical. Moreover, our study was carried out at Q-band, thereby benefiting from an increased spectral resolution at higher frequency [25]. Unlike the previous work [21], ENDOR spectra were recorded for wild type and mutant samples from the same strain of cyanobacteria, studied in parallel. The analysis of our data obtained by both optical and EPR spectroscopies, which were carried out under similar experimental conditions, allows conclusions to be made with regard to the effect of substitution of the π-stacking Trp residues in the A- and B-branches on the electron transfer chain in cyanobacterial PS I.
Methods
EPR Sample Preparation
The concentration of Chl a in PS I samples varied between 5 and 10 mM. For radical pair measurements, samples were reduced by adding a solution of sodium ascorbate (final concentration, 5 mM). After 5 min dark incubation at 4°C, the sample was dark frozen in liquid nitrogen. The photoaccumulated quinone radical anion A·−1 was generated using a procedure identical to that previously described in Ref. [25]. Briefly, a 20 μl solution of trimeric PS I was mixed with 2.5 μl 1 M glycine buffer (pH 10), after which 2.5 μl of fresh sodium dithionite solution in 1 M glycine buffer (pH 10; final sodium dithionite concentration, 30 mM) was added. This was followed by 30 min of dark adaptation at 4°C and subsequent freezing. All buffers were degassed and purged with argon prior to use and all steps were carried out under anaerobic conditions. The sample was illuminated at 200 K by halogen lamps (150 W each). A water filter, cold glass filter and a concentrated CuSO4 solution were used to minimize infrared and ultraviolet irradiation.
EPR Spectroscopy
Transient X-band EPR measurements of the spin-polarized radical pair P·+700A·−1 were carried out using a Bruker ER200E spectrometer equipped with a Bruker ER042 MRH E microwave (MW) bridge and a dielectric ring resonator (Bruker ER 4118X-MD5), using the direct detection method without field modulation [26–28]. The data acquisition was performed by an external personal computer (PC) equipped with the SpecMan program [29] and a high-speed digitizer (Acqiris AP235). Transient Q-band EPR measurements were carried out on the same spectrometer using a Bruker ER051QG MW bridge. A homebuilt Q-band resonator, similar to that described previously in Ref. [32], was used. The resonator contained 12 horizontal slits of 0.3 mm width to allow in situ light excitation of the sample (>65% light transmission) [25]. Light excitation at 532 nm was achieved with the Vibrant 355 II Laser system from OPOTEK. It consists of an optical parametric oscillator, pumped by short (~ 8 ns) light pulses at 355 nm provided by a Nd:YAG Laser at a repetition rate of 10 Hz. The light was coupled into the cavity by an optical fiber with output energy of ~7 mJ.
Pulse Q-band EPR and 1H ENDOR experiments were carried out on a Bruker ELEXSYS E580 Q-band spectrometer with a Super Q-FT MW bridge equipped with a homebuilt ENDOR resonator [30, 31], similar to that described previously in Ref. [32]. The resonator used for measurements of the light-induced radical pair P·+700A·−1 contained 12 horizontal slits of 0.3 mm width to allow in situ light excitation of the sample (>65% light transmission) [25]. Field-swept echo-detected (FSE) EPR spectra were recorded using a two-pulse echo sequence (π/2–τ–π–τ–echo), in which the echo intensity was registered as a function of the external magnetic field. MW pulses of 40 ns (π/2) and 80 ns (π-pulse), and τ = 400 ns were used. The 1H ENDOR spectra of photoaccumulated A·−1 were recorded using the Davies ENDOR sequence [33] (π–t–π/2–τ–π–τ–echo) with an inversion π-pulse of 200 ns, t = 21 μs, and a radio-frequency (RF) π-pulse of 17 μs and the detection sequence similar to the FSE EPR experiment. An ENI 3200L (300 W) RF amplifier was used.
Light excitation at 532 nm was achieved using a frequency-doubled GCR-130 Nd:YAG Laser system from Spectra Physics (pulse width, ca. 8 ns). The repetition rate was 10 Hz. The light was coupled into the cavity using prisms with an energy of ~10 mJ in front of the resonator. FSE EPR spectra were recorded as described above. The delay after laser flash (DAF) was 1 μs. The 1H ENDOR spectra of the radical pair P·+700A·−1 were recorded using the Davies ENDOR sequence with an inversion π-pulse of 200 ns, t = 13 μs, and RF π-pulse of 9 μs and the detection sequence similar to the FSE EPR experiment. The DAF was 1 μs. An AR2500L RF amplifier (Amplifier Research) was used for 1H ENDOR measurements on the radical pair. The generation of RF pulses and the detection were performed by an external PC equipped with the SpecMan program [29], the digitizer mentioned above and a SMT 02 Rhode and Schwarz synthesizer. The pulse sequence was repeated twice 10 and 20 ms after the laser flash to allow subtraction of stationary background signals.
Time-Resolved Optical Spectroscopy at 820 nm
The PS I samples for kinetic measurements were prepared anaerobically in 50 mM Tris buffer (pH 8.3), containing 4 mM sodium ascorbate and 0.04% n-dodecyl-β-d-maltoside, at a Chl a concentration of 70–100 μg/ml. Flash-induced absorbance changes were measured in the microsecond to second time range at 820 nm on a homebuilt double-beam spectrometer as described previously [34]. The actinic flash was provided by a frequency-doubled, Nd-YAG Laser (λ = 532 nm, 7 ns pulse duration, flash energy of ~1 mJ/cm2, Quanta-Ray DCR-11, Spectra Physics, CA). Typically, 12–16 transients were recorded and averaged. Multiexponential fits of the kinetic data were performed using the PLUK software [35]. The interval between flashes was in the 30–90 s range depending on the temperature.
Results and Discussion
Optical Spectroscopy
Charge recombination kinetics between P·+700 and the terminal electron acceptors was measured at 820 nm on isolated PS I complexes from Synechocystis sp. PCC 6803 wild type and π-stacking tryptophan mutants on the A-branch (W697FPsaA) and B-branch (W677FPsaB) at temperatures of 73 and 292 K. The measurements were performed under anaerobic conditions, thereby ensuring multiple turnovers. Figure 2a shows the flash-induced absorbance changes of P·+700 recorded at room temperature. In wild-type PS I complexes, the typical [2, 36, 37] recombination kinetics in the millisecond time range is observed with lifetimes (τ) of the main component of 30 ms (66% contribution) and 76 ms (28% contribution). In PS I complexes from both Trp mutants, the charge recombination kinetics was significantly faster than in the wild type, with the main kinetic phases of 5.7 and 16.4 ms (in a 1:1 ratio) in the W697FPsaA mutant, and 8.2 and 38.5 ms (in a 4:1 ratio) in the W677FPsaB mutant.2 Thus, kinetics of the P·+700 re-reduction is influenced by changes in both A- and B-branches. A similar acceleration of P·+700 decay was also observed in the millisecond time range in thylakoid membranes of C. reinhardtii mutants W693FPsaA and W673FPsaB [17] (note that W693FPsaA and W673FPsaB are π-stacked to A1 in the A- and B-branches of this organism). The effect was even stronger in the double mutant W693FPsaA/W673FPsaB. The conclusion derived from analysis of the optical data is consistent with that derived from the physiological data [18, 19, 21], indicating that mutations on both A- and B-branches significantly reduce the photoautotrophic growth rate as well as PS I-mediated electron transfer activity and the amount of PS I.
Fig. 2.
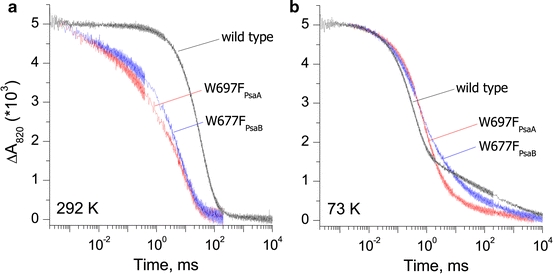
Comparison of the charge recombination kinetics of P·+700 reduction measured at 820 nm in PS I isolated from the wild type, and the W697FPsaA and W677FPsaB mutants of Synechocystis sp. PCC 6803 at 292 K (a) and 73 K (b)
At cryogenic temperatures, the rate of the forward electron transfer to the terminal iron–sulfur clusters becomes slower than the rate of charge recombination from phylloquinone [38], thus re-reduction of P·+700 occurs mostly from the electron acceptor A1 rather than from the terminal iron–sulfur clusters. In wild-type PS I, the main kinetic components with lifetimes of 285 and 954 μs are observed with relative contributions of 47 and 20%, respectively. The differences in kinetics between the Trp mutants and wild-type PS I at 73 K are less apparent than at 292 K (Fig. 2b). In comparison to the wild type, the charge recombination kinetics is slowed down in both mutants, with the main phases of 530 μs and 1.7 ms (37 and 33%, respectively) in PS I isolated from the W697FPsaA mutant, and 440 μs and 2.8 ms (37 and 23%, respectively) in PS I isolated from the W677FPsaB mutant. Slower recombination from [FA/FB]− occurs in ca. 30% of wild-type PS I and in ca. 25% of both A- and B-branch mutant PS I.
According to the latest experimental investigations and theoretical estimation of redox properties of A1, the electron transfer between A1 and FX is viewed slightly thermodynamically unfavorable in the A-branch (occurs uphill in free energy) and thermodynamically favorable in the B-branch (occurs downhill in free energy) (reviewed in Ref. [39]). The P·+700 charge recombination kinetics was modeled by assuming that both A- and B-branches are active, with the above-described thermodynamics for the A1 to FX electron transfer step. The experimentally observed four- to fivefold increase in the recombination rate (Fig. 2) can be modeled by shifting the redox potential of A1/A1 − 40–60 mV in the positive direction in both mutants. The change in the W697FPsaA mutant will increase the uphill free energy gap between A1 and FX; conversely, the change in the W677FPsaB mutant will decrease the downhill free energy gap between A1 and FX. A change in the redox potential of A1/A1 − explains why the difference between both mutants and wild type is much smaller at cryogenic temperatures, during which recombination occurs mostly from A1 (reviewed in Ref. [2]). Our estimated change in redox potential of phylloquinone agrees with that previously made by Boudreaux et al. [17]. The change in the redox potential of A1/A1 − is in agreement with a slowing down of the forward electron transfer from A1 to FX, as reported previously in both mutants [18, 22].
In summary, our optical data clearly show that mutations of π-stacking tryptophan in both A- and B-branches affect backward electron transfer in PS I. This can be explained by the effect of the mutation on the redox potential of A1/A1 −. As such, the change in redox potential of the A-side and B-side quinones influences backward as well as forward electron transfer, in agreement with previous data [17–19, 21, 22].
EPR and ENDOR Studies
To gain detailed insight into changes of the electronic structure of A·−1 caused by replacing the π-stacked tryptophan, we studied the quinone radical anion in both mutants using EPR and ENDOR spectroscopies at X- and Q-band frequencies. In Fig. 3, the radical pair EPR spectra of wild-type PS I are compared with those of the W697FPsaA and W677FPsaB mutants at X- and Q-band. Similar spectra were reported previously [21]. The polarization patterns of the radical pair EPR spectra are sensitive to the orientation of the quinone with respect to the P·+700 [40, 41]. Since the overall patterns of either mutant are very similar to the wild type, we can conclude that the quinone in the P·+700A·−1 spin-correlated radical pair is oriented identically in wild type and both mutants. The g-values of the quinone in PS I from the W697FPsaA and W677FPsaB mutants are also similar to those observed in wild-type PS I, indicating that the protein environment is not strongly distorted by the replacement of tryptophan by phenylalanine. The X-band EPR spectra show the characteristic splitting due to partially resolved hyperfine structure that originates from the rather large methyl group proton hfc of the quinone in the A1 binding site [23, 42]. In the radical pair EPR spectrum of the A-branch mutant, this pattern is more pronounced than in the wild type and B-branch mutant, which is an indication that the hfc of the methyl group protons is increased in the A-branch mutant. This finding is in agreement with previously reported data for cyanobacteria [21] and C. reinhardtii [17].
Fig. 3.
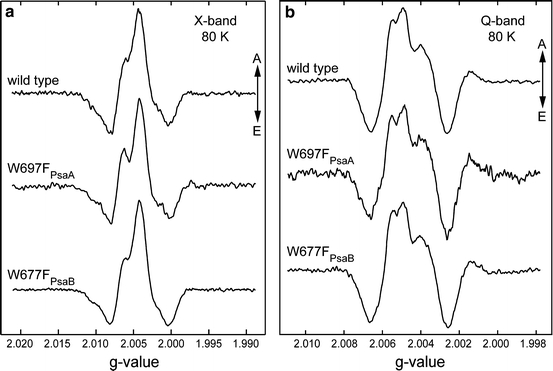
Spin-polarized transient EPR spectra of PS I isolated from the wild type, and the W697FPsaA and W677FPsaB mutants of Synechocystis sp. PCC 6803 at X-band (a) and Q-band (b). Positive signals correspond to absorption (A) and negative signals correspond to emission (E). Measurements were performed at 80 K
To resolve the methyl proton hfcs fully and to determine the hfcs of other weaker coupled protons, it is necessary to significantly increase the spectral resolution. This is achieved by use of ENDOR spectroscopy. Previously, Xu et al. [21] recorded X-band radical pair ENDOR spectra of the W697FPsaA and W677FPsaB mutants, however, only at a single field position, where powder-type ENDOR spectra were obtained. Furthermore, Synechocystis sp. PCC 6803 wild-type PS I was not measured, and both mutants were compared with T. elongatus wild-type PS I published elsewhere [23]. The radical pair X-band ENDOR spectrum recorded on thylakoid membranes isolated from the W693FPsaA mutant of C. reinhardtii [17] was reported to resemble the wild-type spectrum; however, the spectra were not shown. Thus, the previous investigation of the wild type and π-stacking mutants by radical pair ENDOR needs to be completed. In our recently published study of the phylloquinone radical anion in solution [43] and in the A1 site of PS I [25], ENDOR spectroscopy at Q-band was used to resolve hfcs and to assign them to specific protons. The 3.5 times higher microwave frequency of Q-band as compared to X-band allows orientation selection, which results in superior resolution of the ENDOR spectra [44–47].
In this work, we report the first Q-band radical pair ENDOR spectra of PS I from wild-type Synechocystis sp. PCC 6803 PS I (Fig. 4). The spectra are very similar to those reported previously for PS I from T. elongatus [25]; therefore, assignments made in the previous work can be transferred. The radical pair 1H ENDOR spectra of PS I isolated from the wild type and from the W697FPsaA and W677FPsaB mutants are compared in Fig. 4.
Fig. 4.
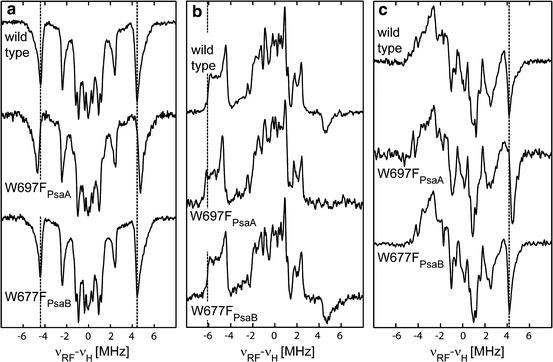
Comparison of pulse Q-band radical pair 1H ENDOR spectra of PS I isolated from wild type, and the W697FPsaA and W677FPsaB mutants of Synechocystis sp. PCC 6803. Spectra were recorded at different external magnetic field positions: a close to g x (g = 2.0070), b close to g y (g = 2.0055) and c close to g z (g = 2.0018) of the quinone radical anion. The dotted line indicates the ENDOR signals assigned to the methyl protons in wild type. Measurements were performed at 80 K
The ENDOR spectra recorded at the low-field wing of the EPR spectrum (g = 2.0070), close to the g
x orientation of the quinone, are almost single crystal-like (Fig. 4a). At this spectral position, mostly emissive EPR transitions of the quinone are excited [25, 48]. Consequently, the ENDOR spectrum is dominated by emissive signals of protons belonging to A·−1. In these ENDOR spectra, the signals of methyl group protons are the most prominent; they are well separated from the other signals. This makes them an ideal probe for changes in the spin density and thus in the electronic structure of the quinone. The hfcs of the methyl group protons are identical in the wild type and W677FPsaB mutant, while in the W697FPsaA mutant they are increased by 0.5 MHz (Table 1). Based on our recently published assignment of the ENDOR signals of A·−1 [25], the resonances of several ring protons, the hydrogen bond proton, as well as protons with smaller hfcs can be identified in the wild type and both mutants [see Fig. 1d for numbering of protons in phylloquinone (vitamin K1)]. The resonances are similar in the wild type and the W677FPsaB mutant, while some differences can be observed in the W697FPsaA mutant. Note that the 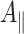 component of the H-bond proton is the same in wild type and both mutants, which allows us to conclude that this important interaction of phylloquinone with the protein environment is not changed.
component of the H-bond proton is the same in wild type and both mutants, which allows us to conclude that this important interaction of phylloquinone with the protein environment is not changed.
Table 1.
1H hfcs of A·−1 of PS I from Synechocystis sp. PCC 6803 wild type (wt) and W697FPsaA (WFA) and W677FPsaB (WFB) mutants (MHz)
| Pos. 2 (CH3)a | Pos. 5 (α) | Pos. 6 (α) | Pos. 7 (α) | Pos. 8 (α) | H-bond | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| wt | WFA | WFB | wt | WFA | WFB | wt | WFA | WFB | wt | WFA | WFB | wt | WFA | WFB | wt | WFA | WFB | |
| A x |
+8.8 (+9.2)b |
+9.3 (+9.8) |
+8.8 (+9.2) |
−1.9 (−1.9) |
−2.0 | −1.9 |
−2.1 (−2.0) |
−2.1 |
−5.0 (−5.2) |
−5.0 | −4.9 |
−0.8 (−0.8) |
−0.8 | −0.8 |
+7.4 (+7.4) |
+7.4 | +7.4 | |
| A y |
+12.4 (+12.7) |
+12.9 (+13.2) |
+12.4 (+12.6) |
(−0.3) | (+0.5) | (−0.7) | (+3.5) | (−3.5) | ||||||||||
| A z |
+8.4 (+8.7) |
+8.9 (+9.3) |
+8.4 +8.7) |
(−3.6) | (−1.9) |
−3.50 (−3.40) |
(−1.3) | (−3.5) | ||||||||||
| a iso |
+9.9 (+10.2) |
+10.4 (+10.8) |
+9.9 (+10.2) |
(−1.9) | (−1.1) | (−3.1) | (+0.5) | (+0.1) | ||||||||||
| φ c | +30 | +30 | +30 | +12 | +10 | −12 | −5 | −20 | ||||||||||
| θ c | 0 | 0 | 0 | 0 | 0 | 0 | 0 | +30 | ||||||||||
aNumbering of molecular positions is according to Fig. 1d
bValues that are given in parentheses refer to the photoaccumulated radical anion A·−1
cAngles (φ, θ) of the hfc tensor principal axes with respect to the principal axes of the g-tensor are given in degrees. Angles are assumed to be identical in the radical pair and the photoaccumulated radical anion. The error in determination of the hfcs is typically in the order of 100 kHz, but may be larger in some cases of broad or overlapping lines (e.g., the H-bond proton)
Figure 4b shows ENDOR spectra recorded around g = 2.0055, close to the g y orientation of the quinone. The contribution of P·+700 to the ENDOR spectra at this position is still rather small [48]. At this spectral position, both emissive and absorptive EPR transitions are excited. In the ENDOR spectrum, emissive and absorptive lines overlap, which results in a partial cancellation. Consequently, only the A y component of the methyl group proton can be identified with confidence. As described above (Fig. 4a), the wild type and the W677FPsaB mutant have the same hfcs, while in the W697FPsaA mutant the hfcs are increased by 0.5 MHz. Overall, the spectra of the wild type and B-branch mutant are very similar, while the spectrum of the A-branch mutant is different.
Figure 4c shows ENDOR spectra recorded around g = 2.0018, close to g z orientation of the quinone, which is similar to a single crystal-like orientation for the A·−1 radical anion. At this magnetic field position, the contribution of P·+700 to the EPR and ENDOR spectra is significant. As in Fig. 4b, at this spectral position, both emissive and absorptive EPR transitions are excited, leading to overlap of emissive and absorptive lines in ENDOR, which results in their partial cancellation. Hence, in the radical pair ENDOR spectra at this position, only the hfc of the methyl group protons can be unambiguously determined. Similar to the radical pair ENDOR spectra shown in Fig. 4a and b, the methyl group hfc of the W697FPsaA is increased by 0.5 MHz as compared to the wild type and the W677FPsaB mutant.
In summary, only the mutation in the A-branch changes the radical pair ENDOR spectra, while the corresponding mutation in the B-branch shows radical pair ENDOR spectra in good agreement with those of the wild type. The A
x and A
y hfcs determined here for the W697FPsaA and W677FPsaB mutants are in agreement with the 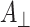 and
and 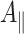 values measured in Ref. [21] at X-band.
values measured in Ref. [21] at X-band.
In order to study the pure A·−1 radical anion without a contribution from P·+700, a photoaccumulation procedure has been employed. However, this procedure typically used to generate the stationary A·−1 radical anion often leads to the co-accumulation of other radicals. In particular, contamination with the Chl a radical anion A·−0 frequently occurs [17, 19, 21, 49]. In Synechocystis sp. PCC 6803 wild-type PS I, a contamination with the A0 − radical was found previously [21]. This is in contrast to T. elongatus PS I, where almost pure A·−1 could be photoaccumulated [25]. It was also found previously that in PS I isolated from the W697FPsaA and W677FPsaB mutants, the amount of A·−0 is larger than in wild-type Synechocystis sp. PCC 6803 PS I [21]. Purton et al. [19] also detected an increased amount of A·−1 in the W693LPsaA and W693HPsaA mutants of C. reinhardtii. Boudreaux et al. [17] reported that in thylakoid membranes isolated from the W693FPsaA mutant, only A·−0 was found upon photoaccumulation, and in the W673FPsaB mutant, nearly pure A·−1 was found.
Using the photoaccumulation protocol described in Ref. [25], PS I samples from wild type and the W697FPsaA and W677FPsaB mutants were prepared. Figure 5 shows the orientation-selected 1H ENDOR spectra of these samples. The spectral orientations reported here are similar to those used in the more comprehensive analysis of T. elongatus carried out previously [25]. Broad resonances from protons with hfcs at about 12 MHz indicate the presence of photoaccumulated A·−0 in all three samples. In both mutant samples, the A·−0 contamination is much larger than in wild-type samples. Despite this contamination, the signals of the methyl group of the A·−1 are clearly visible in the ENDOR spectra of photoaccumulated PS I. In agreement with the radical pair ENDOR data, only the hfcs of the methyl group in the phylloquinone of the W697FPsaA mutant change when compared with wild-type PS I, while no changes of the hfcs were in the W677FPsaB mutant. Note that A·−0 and A·−1 can be detected in both mutants of cyanobacteria, in contrast to the results reported for C. reinhardtii [17].
Fig. 5.
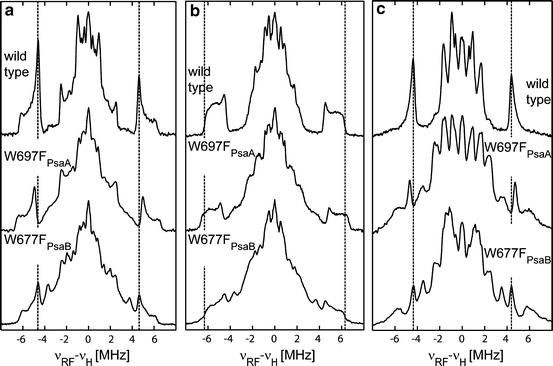
Comparison of pulse Q-band 1H ENDOR spectra of photoaccumulated A·−1 stationary radical anion in PS I isolated from wild type, and the W697FPsaA and W677FPsaB mutants of Synechocystis sp. PCC 6803. Spectra were recorded at different external magnetic field positions: a close to g x (g = 2.0065), b close to g y (g = 2.0046–2.0049) and c close to g z (g = 2.0026) spectral orientations of the quinone. The dotted line indicates the ENDOR signals assigned to the methyl protons in the wild type. Measurements were performed at 80 K
In summary, PS I from wild type, the A-branch mutant (W697FPsaA) and the B-branch (W677FPsaB) mutant were studied by transient EPR at X- and Q-band frequencies, while pulsed 1H ENDOR was applied to study A·−1 in radical pair and photoaccumulated samples. Changes in the electronic structure of A·−1 are clearly recognizable in the W697FPsaA mutant, while results obtained for the W677FPsaB mutant are in agreement with wild type.
Conclusions
PS I isolated from Synechocystis sp. PCC 6803 wild type and from π-stacked tryptophan mutants (W697FPsaA and W677FPsaB) was studied by time-resolved optical absorption and advanced EPR spectroscopies. The optical data show that the kinetics of the re-reduction of P·+700 is altered at room and at cryogenic temperatures in both A-branch and B-branch mutants. The transient EPR spectra recorded at X- and Q-band frequencies on wild type, W697FPsaA and W677FPsaB samples exhibit the same polarization pattern, indicating that the quinone in the spin-correlated radical pair does not change orientation upon mutation. The pulsed Q-band ENDOR spectra of the radical pair and the stationary photoaccumulated A·−1 were recorded at three field positions, corresponding approximately to the principal orientations of the g-tensor of the quinone. The spectra of A·−1 from Synechocystis sp. PCC 6803 wild-type PS I closely resemble those of T. elongatus, which allowed us to transfer the assignment made in our previous work [25]. Comparison of the ENDOR spectra of the wild type with those of the W697FPsaA and W677FPsaB mutants revealed changes only in the A-branch mutant, while ENDOR spectra of the B-branch mutant are very similar to wild-type PS I. A possible short-lived B-branch radical pair could not be detected by EPR due to the insufficient time resolution. In summary, our EPR and ENDOR results clearly show that only A-branch quinone is observed by EPR spectroscopy at cryogenic temperature.
Acknowledgments
We are grateful to Prof. John H. Golbeck from the Pennsylvania State University for providing the samples of PS I isolated from wild type, W697FPsaA and W677FPsaB mutants as well as for the possibility to use the optical setup in his laboratory. We are also indebted to John H. Golbeck for comprehensive discussion of our results. The technical help of Gudrun Klihm (Max-Planck Institut, Mülheim) is gratefully acknowledged. The Max Planck Society, Sfb 498 (TP A3) and Sfb 663 (TP A7) are acknowledged for generous funding of this work.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
Note that in isolated PS I complexes the amplitude of the fast phase is smaller.
Note that in both mutants ~30% contribution in charge recombination kinetics comes from the faster μs-kinetic phase that is attributed to PS I complexes that have no PsaC subunit.
References
- 1.J.H. Golbeck (ed.), Photosystem I. The Light-driven Plastocyanin:Ferredoxin Oxidoreductase (Springer, The Netherlands, 2006)
- 2.Brettel K. Biochim. Biophys. Acta Bioenerg. 1997;1318:322–373. doi: 10.1016/S0005-2728(96)00112-0. [DOI] [Google Scholar]
- 3.Jordan P, Fromme P, Witt HT, Klukas O, Saenger W, Krauss N. Nature. 2001;411:909–917. doi: 10.1038/35082000. [DOI] [PubMed] [Google Scholar]
- 4.Amunts A, Drory O, Nelson N. Nature. 2007;447:58–63. doi: 10.1038/nature05687. [DOI] [PubMed] [Google Scholar]
- 5.Falzone CJ, Kao YH, Zhao JD, Bryant DA, Lecomte JTJ. Biochemistry. 1994;33:6052–6062. doi: 10.1021/bi00186a004. [DOI] [PubMed] [Google Scholar]
- 6.Xia ZC, Broadhurst RW, Laue ED, Bryant DA, Golbeck JH, Bendall DS. Eur. J. Biochem. 1998;255:309–316. doi: 10.1046/j.1432-1327.1998.2550309.x. [DOI] [PubMed] [Google Scholar]
- 7.Mayer KL, Shen GZ, Bryant DA, Lecomte JTJ, Falzone CJ. Biochemistry. 1999;38:13736–13746. doi: 10.1021/bi9910373. [DOI] [PubMed] [Google Scholar]
- 8.Barth P, Savarin P, Gilquin B, Lagoutte B, Ochsenbein F. Biochemistry. 2002;41:13902–13914. doi: 10.1021/bi0259599. [DOI] [PubMed] [Google Scholar]
- 9.Antonkine ML, Bentrop D, Bertini I, Luchinat C, Shen G, Bryant DA, Stehlik D, Golbeck JH. J. Biol. Inorg. Chem. 2000;5:381–392. doi: 10.1007/PL00010667. [DOI] [PubMed] [Google Scholar]
- 10.Antonkine ML, Liu GH, Bentrop D, Bryant DA, Bertini I, Luchinat C, Golbeck JH, Stehlik D. J. Biol. Inorg. Chem. 2002;7:461–472. doi: 10.1007/s00775-001-0321-3. [DOI] [PubMed] [Google Scholar]
- 11.Vassiliev IR, Antonkine ML, Golbeck JH. Biochim. Biophys. Acta Bioenerg. 2001;1507:139–160. doi: 10.1016/S0005-2728(01)00197-9. [DOI] [PubMed] [Google Scholar]
- 12.M.L. Antonkine, J.H. Golbeck, in Photosystem I. The Light-driven Plastocyanin:Ferredoxin Oxidoreductase: Advances in Photosynthesis and Respiration, ed. by J.H. Golbeck, vol. 24 (Springer, The Netherlands, 2006), pp. 79–98
- 13.Joliot P, Joliot A. Biochemistry. 1999;38:11130–11136. doi: 10.1021/bi990857c. [DOI] [PubMed] [Google Scholar]
- 14.F. Rappaport, B.A. Diner, K. Redding, in Photosystem I. The Light-driven Plastocyanin:Ferredoxin Oxidoreductase: Advances in Photosynthesis and Respiration, ed. by J.H. Golbeck, vol. 24 (Springer, The Netherlands, 2006), pp. 223–244
- 15.K. Redding, A. van der Est, in Photosystem I. The Light-driven Plastocyanin:Ferredoxin Oxidoreductase: Advances in Photosynthesis and Respiration, ed. by J.H. Golbeck, vol. 24 (Springer, The Netherlands, 2006), pp. 413–437
- 16.A. van der Est, in Photosystem I. The Light-driven Plastocyanin:Ferredoxin Oxidoreductase: Advances in Photosynthesis and Respiration, ed. by J.H. Golbeck, vol. 24 (Springer, The Netherlands, 2006), pp. 387–411
- 17.Boudreaux B, MacMillan F, Teutloff C, Agalarov R, Gu FF, Grimaldi S, Bittl R, Brettel K, Redding K. J. Biol. Chem. 2001;276:37299–37306. doi: 10.1074/jbc.M102327200. [DOI] [PubMed] [Google Scholar]
- 18.Guergova-Kuras M, Boudreaux B, Joliot A, Joliot P, Redding K. Proc. Natl. Acad. Sci. USA. 2001;98:4437–4442. doi: 10.1073/pnas.081078898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Purton S, Stevens DR, Muhiuddin IP, Evans MCW, Carter S, Rigby SEJ, Heathcote P. Biochemistry. 2001;40:2167–2175. doi: 10.1021/bi0019489. [DOI] [PubMed] [Google Scholar]
- 20.Fairclough WV, Forsyth A, Evans MCW, Rigby SEJ, Purton S, Heathcote P. Biochim. Biophys. Acta-Bioenerg. 2003;1606:43–55. doi: 10.1016/S0005-2728(03)00083-5. [DOI] [PubMed] [Google Scholar]
- 21.Xu W, Chitnis P, Valieva A, van der Est A, Pushkar YN, Krzystyniak M, Teutloff C, Zech SG, Bittl R, Stehlik D, Zybailov B, Shen GZ, Golbeck JH. J. Biol. Chem. 2003;278:27864–27875. doi: 10.1074/jbc.M302962200. [DOI] [PubMed] [Google Scholar]
- 22.Xu W, Chitnis PR, Valieva A, van der Est A, Brettel K, Guergova-Kuras M, Pushkar YN, Zech SG, Stehlik D, Shen GZ, Zybailov B, Golbeck JH. J. Biol. Chem. 2003;278:27876–27887. doi: 10.1074/jbc.M302965200. [DOI] [PubMed] [Google Scholar]
- 23.Fursman CE, Teutloff C, Bittl R. J. Phys. Chem. B. 2002;106:9679–9686. [Google Scholar]
- 24.Poluektov OG, Paschenko SV, Utschig LM, Lakshmi KV, Thurnauer MC. J. Am. Chem. Soc. 2005;127:11910–11911. doi: 10.1021/ja053315t. [DOI] [PubMed] [Google Scholar]
- 25.Niklas J, Epel B, Antonkine ML, Sinnecker S, Pandelia ME, Lubitz W. J. Phys. Chem. B. 2009;113:10367–10379. doi: 10.1021/jp901890z. [DOI] [PubMed] [Google Scholar]
- 26.D. Stehlik, C.H. Bock, M.C. Thurnauer, in Advanced EPR—Applications in Biology and Biochemistry, ed. by A.J. Hoff, (Elsevier, Amsterdam, 1989), pp. 371–403
- 27.K.A. McLauchlan, in Advanced EPR—Applications in Biology and Biochemistry, ed. by A.J. Hoff (Elsevier, Amsterdam, 1989), pp. 345–369
- 28.H. Levanon, in Biophysical Techniques in Photosynthesis, ed. by J. Amesz, A.J. Hoff (Kluwer Academic Publishers, Dordrecht, 1996), pp. 211–234
- 29.Epel B, Gromov I, Stoll S, Schweiger A, Goldfarb D. Concepts Magn. Reson. B. 2005;26:36–45. doi: 10.1002/cmr.b.20037. [DOI] [Google Scholar]
- 30.Sinnecker S, Reijerse E, Neese F, Lubitz W. J. Am. Chem. Soc. 2004;126:3280–3290. doi: 10.1021/ja0392014. [DOI] [PubMed] [Google Scholar]
- 31.Silakov A, Reijerse EJ, Albracht SPJ, Hatchikian EC, Lubitz W. J. Am. Chem. Soc. 2007;129:11447–11458. doi: 10.1021/ja072592s. [DOI] [PubMed] [Google Scholar]
- 32.Sienkiewicz A, Smith BG, Veselov A, Scholes CP. Rev. Sci. Instrum. 1996;67:2134–2138. doi: 10.1063/1.1147027. [DOI] [Google Scholar]
- 33.Davies ER. Phys. Lett. A. 1974;47:1–2. doi: 10.1016/0375-9601(74)90078-4. [DOI] [Google Scholar]
- 34.Vassiliev IR, Jung YS, Smart LB, Schulz R, Mcintosh L, Golbeck JH. Biophys. J. 1995;69:1544–1553. doi: 10.1016/S0006-3495(95)80026-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kalaidzidis YL, Gavrilov AV, Zaitsev PV, Kalaidzidis AL, Korolev EV. Program. Comput. Softw. 1997;23:206–211. [Google Scholar]
- 36.Hiyama T, Ke B. Arch. Biochem. Biophys. 1971;147:99–108. doi: 10.1016/0003-9861(71)90314-6. [DOI] [PubMed] [Google Scholar]
- 37.Hiyama T, Ke B. Proc. Natl. Acad. Sci. USA. 1971;68:1010–1013. doi: 10.1073/pnas.68.5.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schlodder E, Falkenberg K, Gergeleit M, Brettel K. Biochemistry. 1998;37:9466–9476. doi: 10.1021/bi973182r. [DOI] [PubMed] [Google Scholar]
- 39.Srinivasan N, Golbeck JH. Biochim. Biophys. Acta Bioenerg. 2009;1787:1057–1088. doi: 10.1016/j.bbabio.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 40.van der Est A, Prisner T, Bittl R, Fromme P, Lubitz W, Mobius K, Stehlik D. J. Phys. Chem. B. 1997;101:1437–1443. doi: 10.1021/jp9622086. [DOI] [Google Scholar]
- 41.Stehlik D, Bock CH, Petersen J. J. Phys. Chem. 1989;93:1612–1619. doi: 10.1021/j100341a084. [DOI] [Google Scholar]
- 42.Rigby SEJ, Evans MCW, Heathcote P. Biochemistry. 1996;35:6651–6656. doi: 10.1021/bi952619x. [DOI] [PubMed] [Google Scholar]
- 43.Epel B, Niklas J, Sinnecker S, Zimmermann H, Lubitz W. J. Phys. Chem. B. 2006;110:11549–11560. doi: 10.1021/jp060548d. [DOI] [PubMed] [Google Scholar]
- 44.Flores M, Isaacson R, Abresch E, Calvo R, Lubitz W, Feher G. Biophys. J. 2007;92:671–682. doi: 10.1529/biophysj.106.092460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Flores M, Isaacson R, Abresch E, Calvo R, Lubitz W, Feher G. Biophys. J. 2006;90:3356–3362. doi: 10.1529/biophysj.105.077883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Flores M, Isaacson RA, Calvo R, Feher G, Lubitz W. Chem. Phys. 2003;294:401–413. doi: 10.1016/S0301-0104(03)00321-5. [DOI] [Google Scholar]
- 47.Lubitz W, Feher G. Appl. Magn. Reson. 1999;17:1–48. doi: 10.1007/BF03162067. [DOI] [Google Scholar]
- 48.Epel B, Niklas J, Antonkine ML, Lubitz W. Appl. Magn. Reson. 2006;30:311–327. doi: 10.1007/BF03166203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang F, Shen GZ, Schluchter WM, Zybailov BL, Ganago AO, Vassiliev IR, Bryant DA, Golbeck JH. J. Phys. Chem. B. 1998;102:8288–8299. doi: 10.1021/jp981952i. [DOI] [Google Scholar]