

Abstract
Phosphoinositide 3-kinases (PI3Ks) have diverse and profound roles in health and disease. The primordial PI3K, Vps34, is present in all eukaryotes and has essential roles in autophagy, endosomal sorting, phagocytosis and signalling upstream of mTOR in nutrient sensing. The crystal structure of Vps34 reveals a constricted adenine-binding pocket, shedding light on why specific inhibitors of this class of PI3K have proven elusive. Both the phosphoinositide-binding loop and the C-terminal helix of Vps34 have dual roles: they are essential for catalysis on membranes and they suppress futile ATPase cycles. Vps34 appears to alternate between a closed form in the cytosol and an open form on the membrane. Structures of Vps34 complexes with a series of inhibitors show why the autophagy inhibitor 3-methyladenine preferentially inhibits Vps34 and lay a foundation for generating new potent and specific Vps34 inhibitors.
The class III PI3K, Vps34, is the most ancient paralog of the three classes of phosphoinositide 3-kinases in mammals (1). It engages in a wide range of intracellular transport activities, including transport to lysosomes via multivesicular bodies (2), endosome to trans-Golgi transport via retromers (3), phagosome maturation (4, 5) and autophagy (6). More recently, signalling roles of Vps34 have been described in nutrient sensing in the mTOR pathway (7, 8) and signalling downstream of G-protein-coupled receptors (9). Given the role of Vps34 in activating mTOR signalling, Vps34 inhibitors could have application in treatment of obesity or insulin resistance (10). One of the obstacles to understanding the cellular roles of Vps34 is that currently there is no inhibitor capable of specifically inhibiting class III PI3K.
Vps34 phosphorylates the D-3 hydroxyl of PtdIns to produce PtdIns3P. Proteins containing binding modules such as FYVE or PX domains that specifically recognise PtdIns3P, initiate the assembly of complexes on endosomes, phagosomes or autophagosomes. Vps34 associates with the N-terminally myristoylated, putative Ser/Thr protein kinase Vps15 (hVps15/p150 in humans), which leads to activation of Vps34 (11, 12). Regulatory proteins such as Rab5 and Rab7 bind to Vps15 and enable activation of the Vps34/Vps15 complex at membranes (6, 13, 14).
The Vps34/Vps15 heterodimer is found in multiple complexes in eukaryotes (10), and some of these complexes have a fundamental role in autophagy (15). Autophagy has diverse intracellular roles including degradation of long-lived proteins and organelles, and in maintaining a balance between cell growth and death during development (16, 17). In yeast, Vps15/Vps34/Vps30 form the core of complexes I and II, while Atg14 and Vps38 recruit this core for autophagy and endosome-to-TGN sorting, respectively (18). The mammalian ortholog of Vps30 is Beclin1, which in autophagy associates with hAtg14/Barkor (19, 20), and, in a separate complex, UV irradiation resistance-associated gene (UVRAG) (21) and Bax-interacting factor-1 (Bif-1) (22). UVRAG has also been proposed to function in endosomal sorting (23).
We have determined the structure of Vps34, and complexes of it with inhibitors. We have produced an initial Vps34-selective inhibitor and the structures will aid in further development of these inhibitors, with potential applications both in the clinic and as tools for understanding intracellular signalling. A construct of Drosophila melanogaster Vps34 (DmVps34) lacking the C2 domain (Δ1-257), referred to as HELCAT (helical and catalytic domains), was used for the 2.9 Å resolution structure determination (Fig. 1A). The C2 domain has no influence on catalytic activity in vitro (Fig. S1, S2) but its role may be to bind Beclin1 (21). The overall fold of the enzyme shows a solenoid helical domain packed against a catalytic domain, forming a compact unit with extensive inter-domain contacts (Fig. 1B). The asymmetric unit of the crystals contains a dimer of Vps34 with 1800 Å2 of the solvent-accessible surface buried in the interface. The C-terminal helix of one subunit inserts into a prominent slot on the surface of the other subunit (Fig. S3). However, light scattering analyses indicate that Vps34 is a monomer in solution (Fig. S4).
Fig. 1.
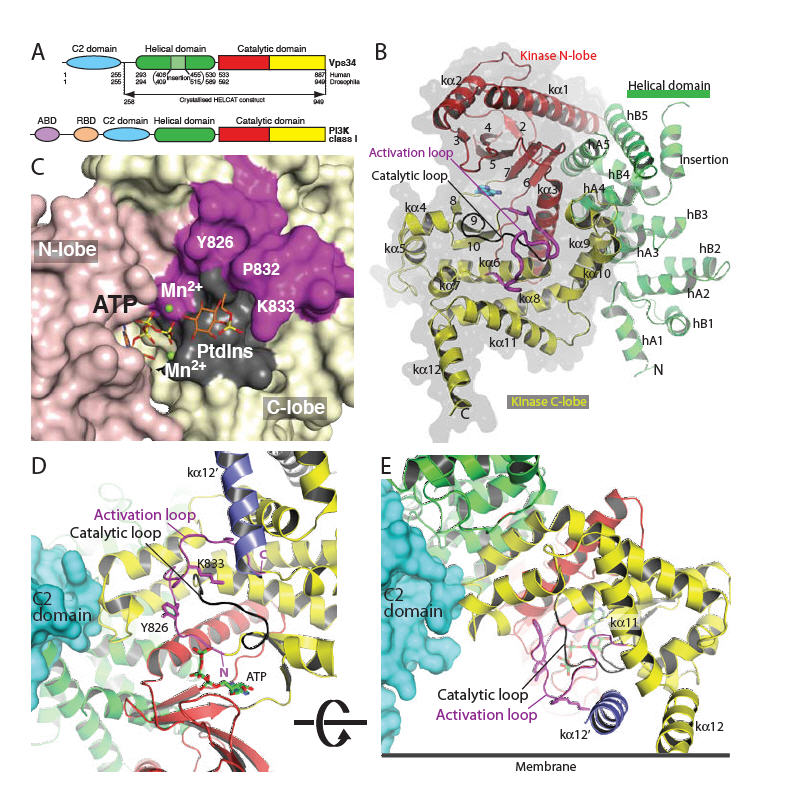
Structure of Vps34 catalytic core (HELCAT). (A) Domain organisation of Vps34 and class I PI3Ks. (B) Overall fold of the DmVps34 HELCAT. (C) A view of the hook-shaped activation loop (magenta) encircling the catalytic loop (black). The C2 domain (cyan) is that of p110γ after superimposing DmVps34 residues 291-949 onto p110γ. The kα12′ helix (slate) is the C-terminal helix from the adjacent molecule in the crystal dimer. (D) The 2mFo-DFc electron density, contoured at 1.1σ, for the activation loop. (E) A model for PtdIns headgroup binding to Vps34, suggesting that Lys833-Dm (K771-Hs) interacts with the 1-phosphate. (F) The putative orientation of Vps34 on a membrane.
One of the most striking features of the Vps34 structure is the completely ordered phosphoinositide-binding or “activation” loop (Fig. 1B-D). This loop is critical for the characteristic lipid substrate preferences of the PI3K catalytic subunits (24), but in other PI3K structures, it has been largely disordered (25, 26). The proximal (N-terminal) end of the Vps34 activation loop forms an essential part of the phosphotransferase reaction center (Fig. 1C,E,F). The intermediate section forms a vertical wall reaching the membrane surface (Fig. 1F). The distal (C-terminal) end of the loop cradles the C-terminal helix from the other molecule in the crystal dimer. Although we have been unable to obtain a Vps34/PtdIns complex structure, it is possible to model phosphoinositide headgroup binding that would facilitate direct transfer of the ATP γ-phosphate to the 3-OH of the inositol ring (Fig. 1E). The 1-phosphate of the substrate is likely to be adjacent to the ε-amino group of Lys771-Hs (Lys833-Dm) at the apex of the loop, which is consistent with our observation that the K771A mutant dramatically impairs activity (Fig. 2A). The inositol ring would stack on the hydrophobic surface created by Pro770-Hs and Tyr764-Hs (832-Dm and 826-Dm) (Fig. 1E). Consistent with this, the Y764A mutation inactivates the enzyme (Fig. 2A). The D-3 hydroxyl would be in a small pocket lined with catalytic loop residues Asp743-Hs, Arg744-Hs, His745-Hs and Asn748-Hs (Dm 805-DRHxxN-810). The guanidinium group of Arg744-Hs interacts with, and potentially stabilises, the backbone of the DFG motif in the activation loop, and the positive charge may also help neutralise negative charge in the transition state of γ-phosphate transfer.
Fig. 2.
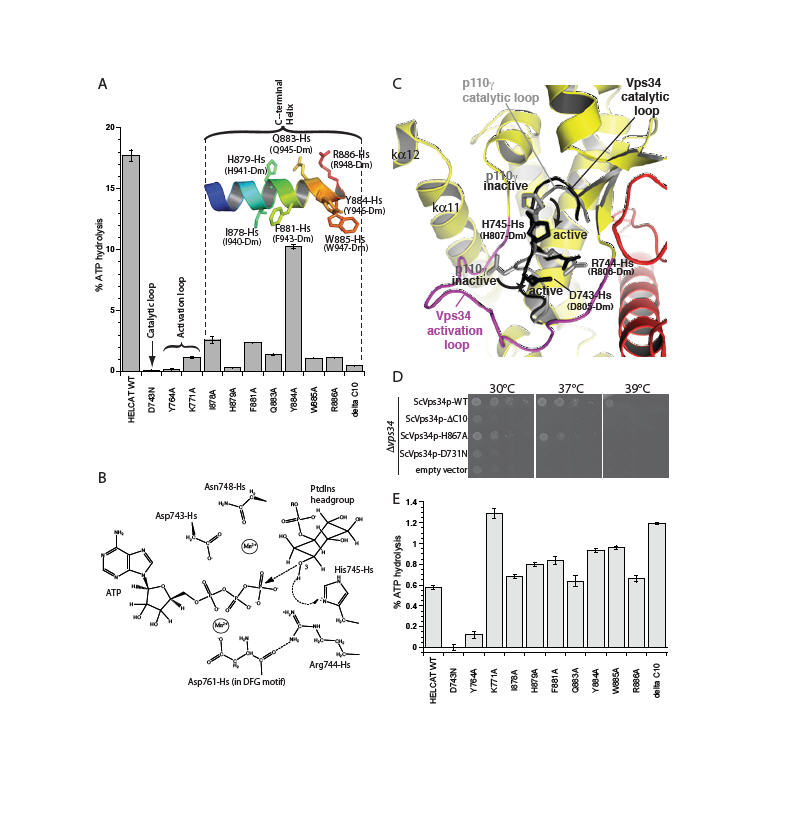
Essential structural elements for Vps34 catalysis. (A) The catalytic loop, the activation loop and the amphipathic C-terminal helix are critical for catalysis on PtdIns:PS vesicles. (B) Proposed catalytic mechanism of Vps34. (C) A close-up view of the proposed movements of catalytic loop residues His745-Hs and Asp743-Hs between the inactive and active conformations represented by the p110γ (grey) and DmVps34 crystal structure (black), respectively. (D) The ability of a yeast Vps34p-expressing plasmid to complement the growth defect of a Δvps34 yeast strain at elevated temperatures is impaired by deletion of the C-terminal helix (Vps34p ΔC10), a point mutation in this helix (ScH867A) or a mutation in the catalytic loop (ScD731N). (E) Basal ATPase activities in the absence of vesicles.
Vps34 residues within the conserved catalytic loop DRH motif (Hs 743-DRH-745 and Dm 805-807) have a conformation that suggests a mechanism whereby His745-Hs could act as the catalytic base, abstracting a proton from the substrate 3-OH to facilitate nucleophilic attack on the γ-phosphate of ATP. Two acidic residues, Asp743-Hs and Asp761-Hs (Asp805-Dm and Asp823-Dm) are well positioned to act as metal ligands that could help neutralize negative charge in the transition state (Fig. 2B). The p110γ/ATP structure appears to have captured the catalytic loop in an inactive state in which neither the histidine nor the aspartate of the DRH is properly oriented for catalysis. The difference between the p110γ and Vps34 catalytic loops may reflect an inactive-to-active transition that is possible for all PI3Ks (Fig. 2C).
An earlier study noted the importance of a C-terminal element for Vps34 activity in vivo (27). The structure shows that this element is part of the C-terminal helix (kα12). This helix has a critical role in catalysis both in vitro (Fig. 2A) and in vivo (Fig. 2D). Truncation of the C-terminal 10 residues of human and yeast Vps34 almost completely abrogates catalytic activity. Even single point mutations in the conserved C-terminal motif ΦHxΦxQYWRx greatly reduce enzymatic activity on PtdIns-containing vesicles (Fig. 2A) and in vivo (Fig. 2D). Surprisingly, truncation of the ten C-terminal residues enhances basal ATPase activity in the absence of lipid substrate (Fig. 2E). The HsVps34 W885A and Y884A mutations in the C-terminus also increase the basal ATPase activity (Fig. 2E). This suggests that in the closed form, the C-terminal helix would fold over the catalytic loop locking the catalytic His745-Hs (His807-Dm) in its inactive conformation (Fig. 3A). In this arrangement, the C-terminal helix would be cradled by the activation loop (Fig. S3). Consistent with this, the activation loop mutant K771A increases basal ATPase activity like the C-terminal helix mutations. The loop between the last two helices would act as a hinge that enables an open-to-closed form transition (Fig. 3B). Consequently, the C-terminal tail appears to have a dual role: auto-inhibitory off the membrane and activating on the membrane. FRET and lipid sedimentation analyses also show that the C-terminal helix has a role in membrane binding (Fig. S5).
Fig. 3.
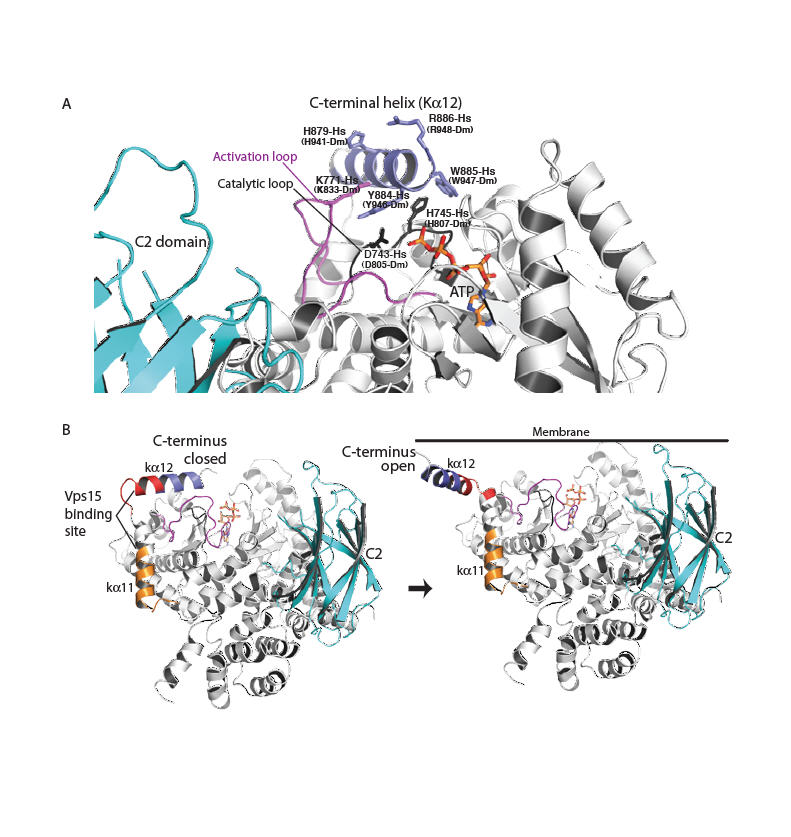
A model for Vps34 activation on membranes. (A) A close-up for the closed form of the enzyme in the cytosol. The C-terminal helix protects the phosphotransferase center from water. (B) The transition of the enzyme from a closed form in the cytosol (C-terminal helix in) to an open form with the C-terminal helix interacting with the membrane. The C2 domain (cyan) modelled as in Fig. 1. Vps15-interacting regions are classified as strong (red) or weak (orange) (27).
The Vps34 ATP-binding pocket (Fig. 4A) has a smaller volume than the corresponding pocket of the class I p110γ pocket (800 Å3 versus 1200 Å3). In Vps34, the P-loop (known to bind the phosphates of ATP (25)) curls inward toward the ATP binding pocket, and this is coincident with a parallel inward bending of the kα1/kα2 loop (Fig. S6). Furthermore, the hinge between the N- and C-lobes is one residue shorter in Vps34 than class I PI3Ks, and therefore lacks the bulged-out space at the adenine-binding pocket hinge, which is characteristic of class I PI3Ks (Fig. S7).
Fig. 4.
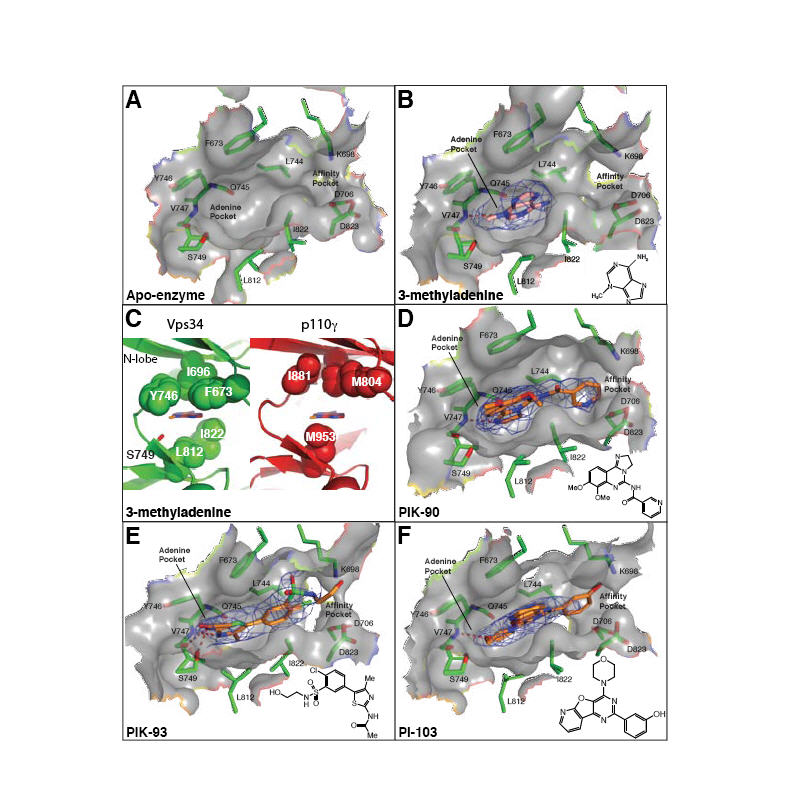
Inhibitor binding in the ATP pocket. (A-B,D-F) 2mFo-DFc electron densities are shown (contoured at 1.0σ). (C) A comparison of the ATP-binding pocket of the Vps34/3-MA complex (left, green) with p110γ (right, red). A ring of hydrophobic residues encircles 3-MA, and may provide specificity for Vps34. The p110γ structure is of the ATP/enzyme complex (PDB ID 1E8X), but a 3-MA has been modelled in the pocket as a reference point for comparison with the Vps34/3-MA complex. (G) Substitution of a cyclopentanecarboxamide in PT210 for the acetamide moiety of PIK93 reverses the specificity of the compound to selectively inhibit Vps34.
Class I PI3Ks can form an allosteric or “specificity” pocket (adjacent to the adenine pocket) only in the presence of propeller-like inhibitors (28). The IC50s for the propeller-like PI3K inhibitors (e.g. PIK-39, Fig. S8) are generally much worse for Vps34 than other PI3Ks. This is probably due to increased rigidity of the Vps34 pocket arising from a bulky residue substituted in the P-loop (Phe612-Hs, Phe673-Dm) that packs against the aromatic hinge residue unique to Vps34 (Phe684-Hs, Tyr746-Dm). These differences effectively close off a corner of the adenine-binding pocket, giving it a more constrained appearance.
Currently there is no high-affinity, specific inhibitor of Vps34. We have determined the structure of a complex of Vps34 with 3-methyladenine (3-MA)(Fig. 4B,C), which is often used as a specific inhibitor of autophagy. We also have determined the structures of Vps34 in complexes with three multi-targeted inhibitors: PIK-90, PIK-93 and PI-103 (Fig. 4D-F). These complexes provide insight into developing more potent and specific Vps34 inhibitors.
Although at the 10 mM concentration typically used for inhibiting autophagy in cells, 3-MA inhibits both class I and III PI3Ks, in vitro assays show that 3-MA has a preference for Vps34 (Fig. S8). There is a hydrophobic ring consisting of Phe673-Dm, Tyr746-Dm and Leu812-Dm, which encircles the 3-methyl group of 3-MA and is unique to and conserved in Vps34. The corresponding residues in class I PI3Ks are not in close proximity of the 3-methyl group, and the methionine equivalent of Leu812-Dm may cause steric hindrance (Fig. 4B,C). 3-MA appears to bind to the hinge, as does the adenine moiety of ATP in p110γ, hydrogen bonding to the Val747-Dm amide and the Gln745 carbonyl.
All PI3K inhibitors have at least one canonical bond to the hinge. PIK-90 and PI-103 form a single H-bond, while PIK-93 forms two H-bonds to Val747-Dm, coinciding with a lower IC50 of PIK-93 relative to PIK-90 (Fig. S8). The affinity pocket of PI3Ks is lined with several hydrophobic and polar residues with which inhibitors can interact to greatly augment their potency, these include in DmVps34: Lys698 (modified by wortmannin), Asp823 (from the DFG) and Asp706 (in helix kα3, equivalent to helix αC of protein kinases). The pyridine ring of PIK-90, the chlorophenyl group of PIK-93 and the m-phenol group of PI-103 are within hydrogen bonding distance of these residues. In addition, the pyridinylfuranopyrimidine group of PI-103 extends out of the pocket over the surface analogous to hydrophobic region II in protein kinases.
Our initial attempts to synthesize new Vps34 inhibitors, based on the structure of Vps34, indicate that there are ample opportunities to improve their chemical properties and significantly increase specificity for class III PI3Ks. Elaborating the ethanolamine moiety of the PIK-93 sulphonamide (compound 3-94B) that extends out of the affinity pocket, and simultaneous elaborations of the sulphonamide and the amide (compound 3-94C) have little impact on IC50 values (Fig. S9).
In order to exploit potential differences within the affinity pocket between Vps34 and Class I PI3Ks, we increased the steric bulk of the Chloro-substituent of the central phenyl ring of PIK-93. Addition of the methoxy group (compound PT21) showed little change in IC50 for Vps34 (88nM), but over a 10-fold increase in IC50 for the most potently inhibited Class I PI3K (p110γ, 61 nM), compared to PIK93 (Fig. S9). To further improve specificity for Vps34, we synthesized an analog of PT21 with additional modifications oriented toward the hinge region differences between Vps34 and PI3Kγ (Fig. S7). Compound PT210 (Fig. 4G) contains a cyclopentanecarboxamide substitution for the acetamide moiety of PIK93 and exhibits a modest 13-fold loss in potency for Vps34 (IC50 450 nM) compared to PIK93, yet has an 1100-fold higher IC50 for PI3Kγ (IC50 ~4μM) compared to PIK93, resulting in a compound with reversed kinase specificity compared to PIK-93.
In summary, the structure of Vps34, with a completely ordered phosphoinositide-recognition loop, has enabled us to model substrate binding and the catalytic mechanism. The C-terminal helix plays a critical role in catalysis on membranes. In addition, it also has an auto-inhibitory role that prevents ATP hydrolysis when it is not at the membrane. The structures of Vps34 in complexes with PI3K inhibitors have provided clues as to how 3-MA can preferentially inhibit Vps34, and they have illustrated how additional moieties can be incorporated into inhibitors without affecting affinity for the enzyme, while greatly increasing their specificity for Vps34. This can be crucial in the design of new generations of Vps34 inhibitors with improved specificity, solubility and cellular availability.
Supplementary Material
Acknowledgments
We thank ESRF beamline scientists on ID14-4, ID23-1 and ID29. We are grateful to Yohei Ohashi and Alison Gillingham for help with yeast experiments, Carsten Sachse for electron microscopy, Mark Allen for help with X-ray data collection, Alex Berndt for advice and discussions and Sean Munro for critically reading the manuscript. We thank Beatríz González for a clone of HsVps34, and Greg Ducker for help in optimizing Vps34 inhibitor assays. B.T.H is supported by the Mount Zion Health Fund, B.T. is supported by the Achievement Rewards for College Scientists (ARCS) Fellowship at UCSF, and K.M.S thanks the Waxman Foundation for support. The coordinates have been deposited in the Protein Data Bank, with identification numbers (PDB Ids): 2X6H (apo), 2X6I (PIK-90), 2X6J (PIK-93), 2X6K (PI-103) and 2X6F (3-MA).
Footnotes
Supporting Online Material
Materials and Methods
Figs. S1, S2, S3, S4, S5, S6, S7, S8, S9, S10
Table S1
References and Notes
- 1.Engelman JA, Luo J, Cantley LC. Nat Rev Genet. 2006;7:606. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 2.Schu PV, et al. Science. 1993;260:88. doi: 10.1126/science.8385367. [DOI] [PubMed] [Google Scholar]
- 3.Burda P, Padilla SM, Sarkar S, Emr SD. J Cell Sci. 2002;115:3889. doi: 10.1242/jcs.00090. [DOI] [PubMed] [Google Scholar]
- 4.Vieira OV, et al. J Cell Biol. 2001;155:19. doi: 10.1083/jcb.200107069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vergne I, et al. Proc Natl Acad Sci U S A. 2005;102:4033. doi: 10.1073/pnas.0409716102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simonsen A, Tooze SA. J Cell Biol. 2009;186:773. doi: 10.1083/jcb.200907014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Byfield MP, Murray JT, Backer JM. J Biol Chem. 2005;280:33076. doi: 10.1074/jbc.M507201200. [DOI] [PubMed] [Google Scholar]
- 8.Nobukuni T, et al. Proc Natl Acad Sci U S A. 2005;102:14238. doi: 10.1073/pnas.0506925102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slessareva JE, Routt SM, Temple B, Bankaitis VA, Dohlman HG. Cell. 2006;126:191. doi: 10.1016/j.cell.2006.04.045. [DOI] [PubMed] [Google Scholar]
- 10.Backer JM. Biochem J. 2008;410:1. doi: 10.1042/BJ20071427. [DOI] [PubMed] [Google Scholar]
- 11.Panaretou C, Domin J, Cockcroft S, Waterfield MD. J Biol Chem. 1997;272:2477. doi: 10.1074/jbc.272.4.2477. [DOI] [PubMed] [Google Scholar]
- 12.Yan Y, Flinn RJ, Wu H, Schnur RS, Backer JM. Biochem J. 2009;417:747. doi: 10.1042/BJ20081865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shin HW, et al. J Cell Biol. 2005;170:607. doi: 10.1083/jcb.200505128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murray JT, Backer JM. Methods Enzymol. 2005;403:789. doi: 10.1016/S0076-6879(05)03068-5. [DOI] [PubMed] [Google Scholar]
- 15.Obara K, Sekito T, Ohsumi Y. Mol Biol Cell. 2006;17:1527. doi: 10.1091/mbc.E05-09-0841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Axe EL, et al. J Cell Biol. 2008;182:685. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fimia GM, et al. Nature. 2007;447:1121. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- 18.Kihara A, Noda T, Ishihara N, Ohsumi Y. J Cell Biol. 2001;152:519. doi: 10.1083/jcb.152.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Itakura E, Kishi C, Inoue K, Mizushima N. Mol Biol Cell. 2008;19:5360. doi: 10.1091/mbc.E08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun Q, et al. Proc Natl Acad Sci U S A. 2008;105:19211. doi: 10.1073/pnas.0810452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang C, et al. Nat Cell Biol. 2006;8:688. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- 22.Takahashi Y, et al. Nat Cell Biol. 2007;9:1142. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang C, et al. Nat Cell Biol. 2008;10:776. doi: 10.1038/ncb1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bondeva T, et al. Science. 1998;282:293. doi: 10.1126/science.282.5387.293. [DOI] [PubMed] [Google Scholar]
- 25.Walker EH, Perisic O, Ried C, Stephens L, Williams RL. Nature. 1999;402:313. doi: 10.1038/46319. [DOI] [PubMed] [Google Scholar]
- 26.Huang CH, et al. Science. 2007;318:1744. doi: 10.1126/science.1150799. [DOI] [PubMed] [Google Scholar]
- 27.Budovskaya YV, Hama H, DeWald DB, Herman PK. J Biol Chem. 2002;277:287. doi: 10.1074/jbc.M109263200. [DOI] [PubMed] [Google Scholar]
- 28.Berndt A, et al. Nat Chem Biol. 2010;6:117. doi: 10.1038/nchembio.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.