


Abstract
We developed orthogonal ribosome−mRNA pairs in which the orthogonal ribosome (O-ribosome) specifically translates the orthogonal mRNA and the orthogonal mRNA is not a substrate for cellular ribosomes. O-ribosomes have been used to create new cellular circuits to control gene expression in new ways, they have been used to provide new information about the ribosome, and they form a crucial part of foundational technologies for genetic code expansion and encoded and evolvable polymer synthesis in cells. The production of O-ribosomes in the cell makes it challenging to study the properties of O-ribosomes in vitro, because no method exists to purify functional O-ribosomes from cellular ribosomes and other cellular components. Here we present a method for the affinity purification of O-ribosomes, via tagging of the orthogonal 16S ribosomal RNA. We demonstrate that the purified O-ribosomes are pure by primer extension assays, and structurally homogenous by gel electrophoresis and sucrose gradients. We demonstrate the utility of this purification method by providing a preliminary in vitro characterization of Ribo-X, an O-ribosome previously evolved for enhanced unnatural amino acid incorporation in response to amber codons. Our data suggest that the basis of Ribo-X’s in vivo activity is a decreased affinity for RF1.
INTRODUCTION
Orthogonal, parallel and independent systems are a key foundation for synthetic biology. The synthesis of orthogonal systems that are uncoupled from evolutionary constraints, and selectively abstracted from cellular regulation, is an emerging approach to making biology more amenable to engineering (1,2).
We have described the creation of orthogonal ribosomes (O-ribosomes) that efficiently and specifically direct translation from orthogonal mRNAs, (O-mRNA) which are not substrates for natural ribosomes, in Escherichia coli (3). Unlike natural ribosomes, O-ribosomes are non-essential for cellular survival and their activity can be measured, independently of that of natural ribosomes, by the translation of orthogonal messages. We have shown that O-ribosomes allow the construction of translational AND and OR functions (4) and the invention of transcription−translation feed forward loops in the cell (5). These genetic circuits have allowed the temporal control of gene expression and the introduction of information processing delays in ways that are impossible using natural gene expression.
O-ribosomes have also been used to provide information on natural ribosome function that would be difficult to obtain by using the natural ribosome alone. For example, we have shown that the rrnB operon (from which 16S rRNA and 23S rRNA are transcribed) can be re-factored to allow the independent production of each rRNA, thereby defining the boundaries for the minimal 16S RNA transcript that can be processed for functional ribosome production (5). Moreover, we have used O-ribosomes to perform large-scale mutagenesis and selection experiments on nucleotides in the structurally defined 6,000Å2 interface between the two subunits of the ribosome (6). These experiments revealed that a subset of the structurally defined nucleotides in the ribosomal subunit interface form ‘functional hotspots’ that are disproportionately important.
Finally, O-ribosomes provide a crucial component in orthogonal translation, an approach to writing orthogonal genetic codes for synthesizing proteins containing unnatural amino acids and ultimately the cellular synthesis of entirely unnatural polymers. There are two major challenges in encoding the incorporation of unnatural amino acids into proteins. First, orthogonal aminoacyl−tRNA synthetase/tRNA pairs are required that specifically use an unnatural amino acid. Second, since each of the 64 triplet codons is used up encoding natural protein synthesis, new codons are required with which to encode unnatural amino acids. In several cases orthogonal aminoacyl−tRNA synthetases that use unnatural amino acids have been developed allowing incorporation of unnatural amino acids with low efficiency in response to the amber stop codon (7−9). However, competition with release factor at the amber codon limits the efficiency of amino acid incorporation on cellular ribosomes to ∼20%. We showed that, while the natural ribosome is responsible for synthesizing the proteome and is intolerant to mutation, the O-ribosome can be synthetically evolved in the laboratory (creating Ribo-X) to allow near quantitative unnatural amino acid incorporation in response to amber codons on the orthogonal message (10). This work showed that it is possible to differentiate the genetic code that is read by two different messages in the cell using orthogonal translational components. In recent work, we have shown that it is possible to evolve O-ribosomes (Ribo-Q) that decode quadruplet codons using extended anticodon tRNAs, that are poorly decoded by natural ribosomes. This provides a series of blank codons on the orthogonal message. We have combined Ribo-Q with mutually orthogonal aminoacyl−tRNA synthetase/tRNA pairs that use distinct unnatural amino acids, to create a first generation orthogonal genetic code (11).
While much characterization of O-ribosome function can be done in vivo, we have been interested in purifying O-ribosomes from endogenous ribosomes and other cellular components- to allow us to investigate the properties of O-ribosomes and their mutants in vitro. To address this challenge we have developed a system for the affinity purification of highly active in vivo assembled O-ribosomes, which is based on methods reported for the purification of tagged wild-type ribosomes (12). Here, we describe the method for purifying O-ribosomes by affinity tagging the orthogonal 16S ribosomal RNA. We demonstrate that the tagging does not affect O-ribosome function in vivo and that the purified ribosomes are pure and structurally homogenous, by primer extension, gel electrophoresis and sucrose gradients. Moreover, we demonstrate that this system allows the properties of O-ribosomes to be characterized in vitro, by providing an initial characterization of the effects of mutations in ribo-X on ribosome:RF1 interactions.
MATERIALS AND METHODS
Bacterial strains, media and growth conditions
All cloning steps were performed in the E. coli strain DH10B (F- mcrA Δ (mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara, leu)7697 galU galK λ- rpsL nupG). Subsequent expression experiments were conducted in E.coli strain BL21-DE3 (Invitrogen) [F− ompT hsdSB(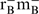 ) gal dcm (DE3)]. Co-expression of O-mRNA and O-ribosome constructs for the in vivo O-ribosome activity assay used the Genehog strain (Invitrogen) [F- mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(ara- leu)7697 galU galK rpsL (StrR) endA1 nupG fhuA::IS2 (confers phage T1 resistance)]. Ribosome purifications were performed from the MRE 600 strain (a natural RNase 1-deficient strain) (13). Cultures were grown in LB or 2XTY liquid media as described in text. The media was supplemented with ampicillin at 100 μg/ml and/or kanamycin at 50 μg/ml where necessary unless described in text.
) gal dcm (DE3)]. Co-expression of O-mRNA and O-ribosome constructs for the in vivo O-ribosome activity assay used the Genehog strain (Invitrogen) [F- mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(ara- leu)7697 galU galK rpsL (StrR) endA1 nupG fhuA::IS2 (confers phage T1 resistance)]. Ribosome purifications were performed from the MRE 600 strain (a natural RNase 1-deficient strain) (13). Cultures were grown in LB or 2XTY liquid media as described in text. The media was supplemented with ampicillin at 100 μg/ml and/or kanamycin at 50 μg/ml where necessary unless described in text.
MS2-GST expression system
A mutated MS2 coat protein coding sequence containing the FG mutation (amino acid residues 68−80 deleted, WT numbering) to prevent oligomerization (14,15) and the V29I mutation (wild-type numbering) to increase tag binding affinity (16,17) was amplified by PCR using the primers MS2fwd (gcgcaGGATCCgcttctaactttactcagttcgttctcgtc) and MS2rev (gcgcaCTCGAGttagtagatgccggagtttgctgcgattg) bearing the XhoI (fwd), BamHI (rev) restriction sites and subcloned into the expression vector pGex 4T1 (GE Healthcare) (pBR322 origin, the ampicillin resistance and the GST gene expression given by the PTrc promoter) using these sites resulting in a GST-MS2 construct expressed under the control of the strong inducible PTrc promoter.
To express and purify GST-MS2, cultures were grown overnight in non-inducing 2XTY liquid media and then diluted to an OD600 of 0.1 in 2 l of fresh 2XTY and grown to an OD600 of 0.6, induced with IPTG (final concentration 1 mg/ml) and incubated (16 h, 20°C, 220 rpm). Cultures were then pelleted, the pellets kept on ice and resuspended in 50 ml of ice-cold GST-MS2 lysis buffer (20 mM Tris–HCl pH 7.5, 150 mM KCl, 2 mM DTT, 5% glycerol, 6 mM BME, 100 µM Benzamidine, 100 µM PMSF) and lysed using an emulsiflux at 4°C. The lysates were kept on ice and clarified by centrifugation (1 h, 20 000 rpm 4°C), in SS34 tubes (Beckman) and the supernatant (40−45 ml) applied to phosphate buffered solution (PBS) (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM NaHPO4) pre-incubated 10 ml column of 4b glutathione−sepharose resin (GE Healthcare) (at RT with a flow rate of 0.5 ml/min, washed with 100 ml of PBS (1 ml/min) and GST-MS2 eluted with 40 ml GST-MS2 elution buffer (50 mM Tris–HCl pH 8, 10 mM reduced glutathione). The elution was then dialyzed (snakeskin dialysis tubing, Roche) in 3 l of PBS containing 20% glycerol (16 h, 4°C).The dialysis was repeated in the same volume (2 h, 4°C), and the protein separated into aliquots and flash frozen. The protein concentration was measured and the protein purity determined by SDS-PAGE gel.
Tag-ribosome expression system
The tag sequence was introduced into the O-16S by enzymatic inverse PCR. The template was pJC73-O-16S(3) and the primers were Tagfwd (gcgcaGGTCTCAgattacccatctttactagtCTTCTTTGCTGACGAGTGGCGGACGGGTGAG), Tagrev (gcgcaGGTCTCAaatcctcatcaaaactagtCTTCTTCCTGTTACCGTTCGACTTGCATGTG), that contain BsaI sites. The PCR reaction mix was digested with BsaI and DpnI, circularized by ligation and transformed into E. coli DH10B. pSC101*-O-ribosome-tag was created by subcloning the entire tag-O-16S sequence into pSC101*-O-ribosome (10) (which contains the O-1623S operon under the control of the ribosomal P1P2 promoter) using the XhoI XbaI sites flanking the O-16S gene. The Ribo-X mutations (10) were subcloned into the O-ribosome-tag construct using the unique Sca1 and Xba1 sites creating pSC101*-Ribo-X-tag.
To express and purify actively translating tagged O-ribosomes the pSC101*-O-ribosome-tag or pSC101*-Ribo-X-tag constructs were co-transformed with O-MBP-GST (10), plated onto LB-KA (containing 50 μg/ml kanamycin and 50 μg/ml ampicillin). The cultures were grown overnight (16 h, 20°C, 220 rpm) in 2XTY-KA, diluted to an OD600 of 0.1 in 4 L of fresh 2XTY-KA, grown to an OD600 of 0.9 (16 h, 20°C, 220 rpm). The cultures were then cooled at 4°C for 2 h, pelleted, and resuspended in 30 ml of ribosome lysis buffer (20 mM Tris–HCl pH 7.5, 100 mM NH4Cl, 10 mM MgCl2, 0.5 mM EDTA, 6 mM BME, 100 µM Benzamidine, 100 µM PMSF). Cells were lysed using an emulsiflux at 4°C. The lysate were kept on ice and clarified by centrifugation (1 h, 20 000 rpm 4°C, in SS34 tubes (Beckman) and the supernatant (25 ml) were purified for the ribosome fraction by applying it to an ice-cold 35 ml sucrose cushion (20 mM Tris–HCl pH 7.5, 500 mM NH4Cl, 10 mM MgCl2, 0.5 mM EDTA, 1.1 M sucrose, 6 mM BME, 100 µM Benzamidine) in a Ti45 ultracentrifuge tube (Beckman) and the ribosome fraction sedimented by centrifugation (18 h, 37 000 rpm 4°C) to reveal a glassy ribosome pellet. This pellet was used for affinity purification of tagged ribosomes or further processed for storage as described below. The glassy pellet was rinsed with ice-cold ribosome lysis buffer and then resuspended in 2 ml ribosome lysis buffer by application of the buffer and allowing the glassy pellet to dissolve on ice. The ribosomes were diluted in ribosome binding buffer (20 mM Tris–HCl pH 7.5, 100 mM NH4Cl and 10 mM MgCl2, 6 mM BME, 100 µM Benzamidine, 100 µM PMSF) to a total volume of 10 ml, giving a ribosome concentration in 7−14 mg/ml range (determined using the conversion 1 A260 = 62 mg). The ribosomes were split into aliquots and flash frozen for affinity purification.
RF1 expression system
To express RF1, the RF1expression plasmid from the PURE expression system (18) was transformed into BL21-DE3 and the cultures grown overnight (16 h, 20°C, 220 rpm) in 2XTY liquid media. The culture was diluted to an OD600 of 0.1 in 2 L of fresh 2XTY, grown to an OD600 of 0.9 (16h, 20°C, 220 rpm) and cooled at 4°C for 2 h. Cultures were pelleted and resuspended in 50 ml RF1 lysis buffer (50 mM Na2HPO4 pH 8, 300 mM NaCl), and lysed at 4°C using an emulsiflux. The supernatant was further diluted with RF1-lysis buffer to a total volume of 150 ml and applied (4°C, 5 ml/min) to a 50 ml Ni-chelating fast flow sepharose column (GE Healthcare). The column was washed with 150 ml of RF1-wash buffer (50 mM Na2HPO4, pH 8 300 mM NaCl, 10 mM imidazole) and eluted using a gradient of imidazole (upto 250 mM imidazole) using RF1-elution buffer (50 mM Na2HPO4 pH 8, 300 mM NaCl, 250 mM imidazole), the elution was collected in 5 ml fractions. RF1 containing fractions were pooled and further purified using a 300S gel filtration column: the pooled fractions were diluted into 300 ml of RF1 buffer (5 mM HEPES pH 7.5, 50 mM KCl, 100 mM NH4Cl, 10 mM MgCl2) and separated into 5 ml fractions using a 5 ml/min flow rate. RF1 was precipitated by slowly stirring in ammonium sulphate to a final concentration of 50% and incubating (16 h, 4°C). The precipitated proteins were resuspended in 500 µl of RF1 buffer containing 50% glycerol.
Tag MS2-GST affinity measurements
The activity of MS2-GST for binding the tag was assayed by fluorescence anisotropy using the chemically synthesized, fluorescently labelled tag RNA (5′ fluorescein GCGAGGATTACCCGC). The change in fluorescence anisotropy was measured (excitation wavelength 494 nm, emission 521 nm) with the incremental addition of 10 µM Tag RNA to a 200 µM solution of MS2-GST in ribosome buffer (supplemented with 0.1 mg/ml bovine serine albumen). The measurements were performed in a quartz cuvette with mixing. The change in fluorescence anisotropy (binding) with increasing RNA tag concentration was plotted and the Kd value extracted from a best-fit.
Tag-O-ribosome in vivo translational activity
To determine the effect of the tag on O-ribosome activity we transformed pSC101*-O-ribosome or pSC101*-O-ribosome-tag into Genehog E. coli containing the O-Cat reporter (10). The cultures were grown overnight in LB liquid media supplemented with 50 μg/ml kanamycin and 25 μg/ml tetracycline, diluted 10-fold into LB supplemented with 12.5 μg/ml kanamycin and 6.25 μg/ml tetracycline, grown to mid log phase (3 h, 37°C, 220 rpm) and then plated onto LB-agar supplemented with 12.5 μg/ml kanamycin and 6.25 μg/ml tetracycline and a range of chloramphenicol concentrations (0–1000 μg/ml) and incubated (overnight, 37°C).
Tagged O-ribosome affinity purification
Four milligrams of purified GST-MS2 was diluted in 40 ml of PBS and bound to 10 ml of glutathione sepharose beads (in PBS) by incubating with gentle mixing for 30 min at RT. The beads were pelleted by centrifugation (5 min, 500 rpm, 4°C) and washed; twice with 40 ml of PBS and twice with 40 ml of ribosome binding buffer (20 mM Tris–HCl pH 7.5, 100 mM NH4Cl and 10 mM MgCl2, 6 mM BME, 100 µM benzamidine, 100 µM PMSF).
The sucrose cushion purified glassy ribosome pellet was resuspended in 40 ml of ribosome binding buffer, added to the pre-chilled beads and incubated (rocked gently for at 4°C for 1 h). The beads were then washed three times with 40 ml of ribosome binding buffer at 4°C; the wash steps separated by 45 min. The final bead pellet was resuspended in 10 ml of ribosome buffer (20 mM Tris–HCl pH 7.5, 100 mM NH4Cl, 10 mM MgCl2, 6 mM BME, 100 µM Benzamidine, 100 µM PMSF) + 3 mM RNA tag and incubated (18 h, gently rolling, 4°C).
The synthesized RNA Tag sequence [5′PGCGAGGAUCACCCGC, a known competitive ligand for MS2 (16)] was dissolved in Tris–HCl pH 7.5. The tag was folded by heating to 98°C and slow cooling to RT, it was then diluted in ribosome binding buffer to a concentration 3 mM and used to elute the bound ribosomes 10 ml of the 3 mM RNA tag solution (4°C) was added to the washed beads and incubated (18 h, gentle rolling, 4°C). The eluted ribosomes were separated from the beads by pelleting the beads (20 min, 4000 rpm, 4°C). Any remaining beads were removed from the supernatant by filtering through a 0.2 µM filter.
The tagged O-ribosomes were concentrated using a 30 000 Da molecular weight cut-off Amicon concentrator (Millipore), pre-equilibrated with ribosome buffer + 5% Tween, by centrifugation (4°C, 2000 rpm) to a final volume of ∼500 µl, split into aliquots and flash frozen.
Characterization of affinity purified tag-ribosomes
The purity of the affinity purified tagged O-ribosome was assayed by reverse transcription (12). The template rRNA was prepared by taking a 50–100µg aliquot of concentrated eluted ribosomes or ribosomal RNA extracted using Trizol (Invitrogen), according to the manufactures instructions. The RT primer (GGCCATGATACTTGAC) used was gel purified and subsequently 32P labelled (5 µl reaction mixture consisting of 5 µM of RT primer, 3.36 pmols γ-32P ATP, 1× PNK buffer (NEB) and 3 U polynucleotide kinase (NEB) and incubated at 37°C for 45 min followed by 65°C for 15 min). One microlitre of this labelled primer mixture and 1 µg of template was then added to a 50 µl reaction mixture consisting of 1× superscript 3 buffer (Invitrogen), 5 mM DTT, 100 mM ddATP, dGTP, dTTP, dCTP. Annealing was performed by incubating the mixture at 94°C for 2 min followed by a temperature gradient down to 25°C at 0.1°C/s, 200 U Superscript 3 (Invitrogen) was then added and extension preformed by incubating at 50°C 16 h. The completed RT reaction was mixed with a equal volume of formamide running buffer (5% sucrose, 0.1% bromophenol blue, formamide) incubated at 85°C for 15 min and 2–5 µl loaded onto a denaturing gel [10% formamide, 20% acylamide, 1× TBE (90 mM Tris–Borate 2 mM EDTA)] run at 37 W for 2.5 h using 1× TBE running buffer. The gel was imaged using a Typhoon scanner and the intensity of the bands analyzed using the Image Quant TL program (GE Health Care).
The integrity of the eluted ribosomes was determined by non-denaturing electrophoresis [gel: 0.5% agarose, 3% acylamide, 1× TBM (89 mM tris base, 89 mM boric acid 1 mM MgCl2)]. The gel was pre-run (1 h, 4°C, 100 V) in 1× TBM running buffer, the running buffer changed for fresh running buffer, and 0.1–2 OD260 of the eluted ribosomes [diluted in a equal volume of non-denaturing loading buffer (20% sucrose, 1× TBM, 0.1% bromophenol blue)] loaded. Gels were run for 2 h, (4°C, 100 V) before visualizing the nucleic acids by incubating in 10 µg/ml ethidium bromide for 10 min and then imaging under UV.
Analytical sucrose gradients were also used to look at the integrity of purified ribosomes. An analytical sucrose gradient was prepared with 15% and 30% (w/v) sucrose in ribosome buffer. These two solutions were added to a gradient pourer to pour even 13 ml 15–30% sucrose gradients in 40Ti tubes. The gradients were cooled to 4°C and then 2 OD260 of the ribosome preparations were carefully added to the very top layer of the gradients. The gradients were spun (20 h, 20 500 rpm, 4°C) and slowly removed from the bottom of the tubes through a fine needle. The OD254 was measured along the gradient to give the gradient profile.
Preparation of tRNAfMet-[3H]fMet for the RF1 termination assay (19)
To prepare tRNAfMet-[3H]fMet the tRNAfmet was first aminoacylated with [3H]Met. A 200 µl reaction mix consisting of 1× fMet reaction buffer (20 mM Tris–HCl pH 7.5, 7 mM MgCl2, 150 mM KCl), 12 µM L-[3H]Met, 4 mM ATP (pH 7), 80 µM tRNAfmet and 1 µM MetRS was incubated (15 min, 37°C). The reaction was driven to completion by quenching with a high concentration of cold fMet; so the reaction mix was then supplemented with 15 µmol of fMet, a volume of reaction buffer to keep the buffer concentration constant and 0.4 µMoles of ATP and incubated (15 min, 37°C).
The charged tRNAfmet-Met was formylated by the addition of 4 µl of formyl donor (preparation is described below) (N5H10 methenyl-tetrahydrofolic acid) and 5 µM of formylase. This reaction mix was then incubated (15 min, 37°C).
The formyl donor was prepared by dissolving 100 mg folinic acid calcium salt in 8 ml of 50 mM β-mercaptoethanol, adding 880 µl of 1 M HCl and incubating for 3 h. This is frozen and before use, 100 µl thawed on ice and 10 µl of 1 M Tris–HCl pH 8 and 10 µl 1 M KOH added, vortexed and incubated at RT for 20 min and any precipitation removed by centrifugation.
The tRNAfMet-[3H]fMet mixture was then phenol/chloroform (pH 4.3) extracted, ethanol precipitated and resuspended in 50 µl of tRNA storage buffer (10 mM Ammonium acetate pH 5, 50 mM KCl). Finally the aminoacylated tRNA was desalted using a Sephadex G25 column (GE), pre-equilibrated with tRNA storage buffer, by centrifugation at 7000g for 2 min. The specific activity was calculated by measuring the RNA concentration (OD260, Mr 25 000 Da) and the scintillation counts (1 µl of purified tRNAfMet-[3H]fMet was added to 3.8 ml of scintillation fluid [Fluoran-safer 2 (BDH)] in triplicate and 3H counted (Beckham LS6000SC).
In vitro RF1 termination assay
The initiation complex/pre-termination complex was assembled as described (19), by incubation (37°C, 20 min) of a reaction mix of 1 µM purified ribosome, 2 µM O-mRNA amber (5′P GCGGCCGCUUUCAUAUCCCUCCGCAAAUGUAGUUU) or mRNA amber (5′P AGGAGGUGAGGUAUGUAGUUU), 2 µM tRNAfMet-[3H]fMet in ribosome buffer (20 mM Tris–HCl pH 7.5, 100 mM NH4Cl, 10 mM MgCl2, 6 mM BME, 100 µM Benzamidine, 100 µM PMSF) (either 50 or 100 µl). 0.1 µg/ml BSA was added to the ribosome buffer for the reactions and RF1 dilutions.
Termination was then induced by the addition of RF1 (19), initial experiments were performed with saturating concentration of RF1 and [3H]fMet release measured by filter binding. RF1 was added to the 50 or 100 μl pre-termination reaction mix to a final concentration of 5 µM, a control was also prepared where no RF1 was added (incubated in parallel to control for spontaneous tRNAfMet-[3H]fMet hydrolysis). The incubation was continued at 37°C, 15 μl aliquots were then removed at 1, 5 and 20 min, added to 1 ml of ice-cold ribosome buffer, separated into three replicates and added to a nitrocellulose filter pre-soaked in ribosome buffer, washed three times with 900 μl of ribosome buffer, air dried and added to 3.8 ml of scintillation fluid and the bound 3H counted. This measures the amount of ribosome bound [3H]fMet.
Subsequent experiments were performed with a range of RF1 concentrations and measured [3H]fMet release. The pre-termination complex was assembled as described above and split into 18 μl aliquots in a row of a 96-well plate, with the next row containing 2 μl of RF1 at various concentrations in ribosome buffer. A no RF1 control containing only ribosome buffer was also prepared. The plate was incubated at 37°C for 2 min to equilibrate the solution to the reaction temperature. To start the reaction the pre-termination complex was added to the RF1 samples using a multi-channel pipette and mixed rapidly. After 10 s, 200 μl of ice-cold 5% TCA (trichloroacetic acid) was added to quench the reaction and precipitate the complex and unhydrolyzed tRNAfMet-[3H]fMet leaving hydrolyzed [3H]fMet in solution. 200 µl of the quenched reaction was removed and added to 800 μl ice-cold 5% TCA and mixed thoroughly. The sample was then spun (20 min, 25 000g, 4°C) and the top 650 µl removed split into three replicates, added to 3.8 ml scintillation fluid and 3H counted.
RESULTS
Tagged O-ribosomes are active in vivo and can be affinity purified
To provide a molecular handle with which to purify the O-ribosome from cells we introduced the MS2 stem loop tag into the O-16S rRNA in place of nucleotides 83–86 (Figure 1). Previous work has shown that introducing this sequence into wild-type 16S rRNA does not interfere with the activity of endogenous ribosomes (12).
Figure 1.

Design of the ribosome RNA affinity tag. The structure of the 70S ribosome (21) showing the position of the tag (shown in red) with the 30S shown in yellow and the 50S in grey. The position and sequence of the tag in the 16S secondary sequence is shown below. The blue nucleotides are the linker sequence and the affinity tag is shown in green (12).
To demonstrate that the tag does not affect the activity of O-ribosomes we measured the chloramphenicol resistance of cells containing the O-cat reporter (which contains the chloramphenicol acetyl-transferase gene on an O-ribosome binding site) and the tagged and untagged O-ribosome (produced from pSC101*-O-ribosome and pSC101*-O-ribosome-tag). Both tagged and untagged O-ribosomes support growth to 600 μg/ml (data not shown) demonstrating that the tag has no measurable effect on translational efficiency, within the limits of this assay.
To produce tagged O-ribosomes for purification MRE600 E. coli containing pO-GST-MBP(10) (a vector containing GST-MBP expressed from an O-ribosome binding site) and the relevant ribosome construct (10) were grown at 20°C, the growth cooled to 4°C, lysed using an emulsiflux and the lysate clarified by centrifugation. To prepare crude ribosomes the lysates were applied to a sucrose cushion, the resulting glassy ribosome pellet resuspended and used in the affinity purifications.
To purify the tagged O-ribosomes from cellular ribosomes (Figure 2) we bound total ribosomes to glutathione sepharose coated with MS2-GST (we confirmed the activity of our MS2-GST preparations by measuring its affinity for fluorescently labelled MS2-tag RNA by fluorescence anisotropy), and washed away endogenous ribosomes. To specifically elute the tagged O-ribosomes we first tried glutathione, as previously reported for wild-type ribosomes (12). This method worked, leading to the elution of stable GST-MS2:ribosome-tag complexes [eluted ribosomes were tagged as measured by allele specific primer extension (12) (data not shown)]. However, since the GST-MS2 fusion is large the GST-MS2:30S and GST-MS2:70S complexes eluted are shifted in sucrose gradient profiles, relative to the normal 30S and 70S peak positions. This leads to the GST-MS2:30S peak overlapping with the 50S peak, making sucrose gradient profiles difficult to interpret. Moreover, we found that aggregates formed in the elution, presumably as a result of multimerization via GST, MS2 or both, making it difficult to quantify the soluble ribosome concentration accurately for downstream assays. These issues made the method challenging for purifying tagged ribosomes, whether orthogonal or wild-type.
Figure 2.

O-ribosome affinity purification strategy. Initial binding of the tagged O-ribosome to glutathione bound GST-MS2 is followed by elution using the RNA tag sequence.
Ideally, tagged ribosomes could be specifically eluted without the large GST-MS2 tag. This would give normal sucrose gradient profiles for the tagged ribosome, avoid multimerization and aggregation and avoid attaching a large protein fusion to the ribosome for further assays. To address this challenge, we eluted bound ribosomes with a chemically synthesized MS2-RNA hairpin. Moreover, in our final protocol we optimized several critical parameters to preserve the structural integrity of tagged ribosomes during purification: a batch affinity purification was used and the entire purification was done at 4°C to further stabilize ribosome structure.
Initially, the effectiveness of the modified protocol in producing a pure pool of tagged O-ribosome from cellular mixture was tested using an allele specific primer extension assay. Subsequently to ensure the presence of the tag and the modified purification process does not interfere with ribosome function in vitro we assayed the structural integrity and termination activity of eluted tagged-O-ribosomes, using untagged wild-type ribosomes as a control.
O-ribosomes purified using the MS2 system are 95% pure
To assess the purity of eluted tagged O-ribosomes, we took advantage of the C1192U spectinomycin resistance mutation in the tagged O-ribosome constructs for allele-specific primer extension (20). An oligonucleotide probe complementary to nucleotides 1193–1209 was extended in the presence of ddATP giving rise to a +1 species for tagged O-ribosomes and a +4 species for endogenous ribosomes (Figure 3a). RNA was extracted from ribosomes and primer extension performed using a 5′-[32P] labelled primer. The primer extension products were resolved on a denaturing polyacrylamide gel and the relative intensities of the bands resulting from tagged and untagged ribosomes quantified. The ratio of extension product demonstrates that, following the MS2 purification method, the tagged O-ribosome pool is ∼95 % pure.
Figure 3.

Characterization of affinity purified tagged O-ribosomes. (a) Purity of eluted O-ribosome measured by allele-specific primer extension. (b) Analytical sucrose gradient profiles of untagged wild-type and purified tagged O-ribosome. (c) Non-denaturing gel separation the indicated ribosomes.
MS2-tag does not affect the integrity of O-ribosomes
To further investigate the integrity of MS2 purified ribosomes, we compared the eluted tagged O-ribosomes with untagged wild-type control ribosomes using analytical sucrose gradient separation (Figure 3b) and non-denaturing gel electrophoresis (Figure 3c).
The wild-type ribosome sucrose gradient profile, separated at a high magnesium concentration reveals a large 70S peak, a 50S peak and a small 30S peak (Figure 3b). In comparison, the purified orthogonal tagged O-ribosome reveals a profile with a sharp 70S peak, increased 30S and no 50S peak. This further confirms the success of the purification, since the tag is in the 16S rRNA and should only facilitate purification of 30S and 70S, while free 50S should be washed away. Non-denaturing gel electrophoresis (Figure 3c) of the purified tagged O-ribosome (which dissociates the subunits, but keeps the majority of proteins associated with the ribosome) reveals strong 30S and 50S bands and a similar pattern of additional bands to untagged wild-type ribosome control. Taken together these data suggest that we have been able to purify soluble, intact O-ribosomes.
Tagged O-ribosomes show comparable in vitro termination activity to wild-type ribosomes
Since we wanted to use purified tagged O-ribosomes to investigate the molecular basis for enhanced efficiency unnatural amino acid incorporation in response to the amber stop codon by Ribo-X(10) we were interested in benchmarking the release factor mediated termination activity of the tagged O-ribosome against wild-type ribosomes.
We compared the eluted tagged O-ribosome and untagged wild-type ribosomes using an in vitro termination assay (19) in which an mRNA (containing a ribosome binding site followed by a start codon immediately followed by an amber stop codon) is assembled with a ribosome and [3H]fMet-tRNAfMet in the P-site, creating the pre-termination complex. RF1 is added to this complex to initiate termination. RF1 binds to the A site amber stop codon and catalyzes the hydrolysis of P-site [3H]fMet from tRNAfMet (Figure 4a). The maximum amount of [3H]fMet that can be released is determined in a filter binding assay, using saturating concentration of RF1 (Figure 4a). This is a measure of the size of the ribosome pool that is competent to bind [3H]fMet-tRNAfMet, RF1 and catalyze the release of [3H]fMet. We observed similar activity for untagged wild-type and purified tagged O-ribosomes in this assay. This indicates that the purified tagged O-ribosomes have comparable termination activity to wild-type ribosomes (Figure 4b).
Figure 4.

In vitro termination assays reveal that Ribo-X has a decreased affinity for RF1. (a) Schematic of termination assays. A complex of the O-ribosome with an initiator tRNA aminoacylated with 35S formyl-methionine (red circle) is assembled (Step 1), in subsequent rounds this requires dissociation of deacetylated tRNA. RF1 is added (Step 2) directing hydrolysis of 35S formyl-methionine from the tRNA (Step 3). RF1 dissociates (Step 4). The filter-binding assay measures the ribosome associated 35S, while the TCA precipitation measures the 35S released (b) RF1 termination activity measured in vitro with untagged wild-type and purified tagged O-ribosome, using filter-binding assays. (c) O-ribosome and Ribo-X termination activity with RF1. Measurement of initial rate of tRNA-fMet hydrolysis over a range of RF1 concentrations is shown for O-ribosome and Ribo-X. RF1 concentration was increased till there was a measurable release in fMet and this release plotted. tRNA-fMet hydrolysis (37°C, 10 s) is measured by TCA precipitation.
Ribo-X has a decreased affinity for RF1
Finally, we used our method to prepare O-ribosomes and Ribo-X for functional comparison using the RF1 termination assay. The assay was performed over a range of RF1 concentrations and initial rates measured using TCA precipitation (Figure 4a). In our preliminary experiments we observe a substantial difference in RF1 activity with the O-ribosome and Ribo-X (Figure 4c). With O-ribosomes half-maximal termination is observed at an RF1 concentration between 0.125 and 0.25 nM. In contrast, under identical conditions, the same amount of fMet release with Ribo-X requires a concentration of RF1 of between 2 and 4 nM. The maximal catalytic activity of Ribo-X at saturating RF1 concentrations, however, does not appear to change. Our data demonstrate that the mutations in Ribo-X reduce the affinity of the ribosome for RF1 at the amber codon by a factor of ∼8-fold. This may provide a mechanistic explanation for the increased amber suppression activity observed in vivo.
DISCUSSION
In this work we have developed a purification scheme to isolate pure, homogenous and active tagged O-ribosomes. We introduced a number of technical advances that allow useful ribosomes to be purified following tagging. We demonstrated the utility of this approach for making in vitro measurements on O-ribosomes. Moreover, we were able to make in vitro measurements on an evolved O-ribosome and demonstrate that Ribo-X is likely to enhance unnatural amino acid incorporation in response to the amber codon in vivo by decreasing binding of RF1 to the amber codon/ribosome complex. In the future the approach presented here may contribute to the detailed molecular characterization of Ribo-X and O-ribosomes that have been evolved to decode quadruplet codons for genetic code expansion.
FUNDING
Funding for open access charge: Medical Research Council.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors are very grateful to Ann Kelley and Sabine Petry for providing reagents and for their expert advice and help.
REFERENCES
- 1.Chin JW. Modular approaches to expanding the functions of living matter. Nat. Chem. Biol. 2006;2:304–311. doi: 10.1038/nchembio789. [DOI] [PubMed] [Google Scholar]
- 2.Lu TK, Khalil AS, Collins JJ. Next-generation synthetic gene networks. Nat. Biotechnol. 2009;27:1139–1150. doi: 10.1038/nbt.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rackham O, Chin JW. A network of orthogonal ribosome x mRNA pairs. Nat. Chem. Biol. 2005;1:159–166. doi: 10.1038/nchembio719. [DOI] [PubMed] [Google Scholar]
- 4.Rackham O, Chin JW. Cellular logic with orthogonal ribosomes. J. Am. Chem. Soc. 2005;127:17584–17585. doi: 10.1021/ja055338d. [DOI] [PubMed] [Google Scholar]
- 5.An W, Chin JW. Synthesis of orthogonal transcription-translation networks. Proc. Natl Acad. Sci. USA. 2009;106:8477–8482. doi: 10.1073/pnas.0900267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rackham O, Wang K, Chin JW. Functional epitopes at the ribosome subunit interface. Nat. Chem. Biol. 2006;2:254–258. doi: 10.1038/nchembio783. [DOI] [PubMed] [Google Scholar]
- 7.Xie J, Schultz PG. A chemical toolkit for proteins – an expanded genetic code. Nat. Rev. Mol. Cell Biol. 2006;7:775–782. doi: 10.1038/nrm2005. [DOI] [PubMed] [Google Scholar]
- 8.Neumann H, Peak-Chew SY, Chin JW. Genetically encoding N(epsilon)-acetyllysine in recombinant proteins. Nat. Chem. Biol. 2008;4:232–234. doi: 10.1038/nchembio.73. [DOI] [PubMed] [Google Scholar]
- 9.Chin JW, Cropp TA, Anderson JC, Mukherji M, Zhang Z, Schultz PG. An expanded eukaryotic genetic code. Science. 2003;301:964–967. doi: 10.1126/science.1084772. [DOI] [PubMed] [Google Scholar]
- 10.Wang K, Neumann H, Peak-Chew SY, Chin JW. Evolved orthogonal ribosomes enhance the efficiency of synthetic genetic code expansion. Nat. Biotechnol. 2007;25:770–777. doi: 10.1038/nbt1314. [DOI] [PubMed] [Google Scholar]
- 11.Neumann H, Wang K, Davis L, Garcia-Alai M, Chin JW. Encoding multiple unnatural amino acids via a quadruplet decoding ribosome. Nature. 2010 doi: 10.1038/nature08817. [Epub ahead of print, doi:10.1038/nature08817] [DOI] [PubMed] [Google Scholar]
- 12.Youngman EM, Green R. Affinity purification of in vivo-assembled ribosomes for in vitro biochemical analysis. Methods. 2005;36:305–312. doi: 10.1016/j.ymeth.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 13.Wimberly BT, Brodersen DE, Clemons WM, Jr, Morgan-Warren RJ, Carter AP, Vonrhein C, Hartsch T, Ramakrishnan V. Structure of the 30S ribosomal subunit. Nature. 2000;407:327–339. doi: 10.1038/35030006. [DOI] [PubMed] [Google Scholar]
- 14.Peabody DS. The RNA binding site of bacteriophage MS2 coat protein. EMBO J. 1993;12:595–600. doi: 10.1002/j.1460-2075.1993.tb05691.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peabody DS, Ely KR. Control of translational repression by protein-protein interactions. Nucleic Acids Res. 1992;20:1649–1655. doi: 10.1093/nar/20.7.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johansson HE, Dertinger D, LeCuyer KA, Behlen LS, Greef CH, Uhlenbeck OC. A thermodynamic analysis of the sequence-specific binding of RNA by bacteriophage MS2 coat protein. Proc. Natl Acad. Sci. USA. 1998;95:9244–9249. doi: 10.1073/pnas.95.16.9244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shtatland T, Gill SC, Javornik BE, Johansson HE, Singer BS, Uhlenbeck OC, Zichi DA, Gold L. Interactions of Escherichia coli RNA with bacteriophage MS2 coat protein: genomic SELEX. Nucleic Acids Res. 2000;28:E93. doi: 10.1093/nar/28.21.e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shimizu Y, Inoue A, Tomari Y, Suzuki T, Yokogawa T, Nishikawa K, Ueda T. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 2001;19:751–755. doi: 10.1038/90802. [DOI] [PubMed] [Google Scholar]
- 19.Caskey CT, Beaudet AL, Scolnick EM, Rosman M. Hydrolysis of fMet-tRNA by peptidyl transferase. Proc. Natl Acad. Sci. USA. 1971;68:3163–3167. doi: 10.1073/pnas.68.12.3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sigmund CD, Ettayebi M, Borden A, Morgan EA. Antibiotic resistance mutations in ribosomal RNA genes of Escherichia coli. Methods Enzymol. 1988;164:673–690. doi: 10.1016/s0076-6879(88)64077-8. [DOI] [PubMed] [Google Scholar]
- 21.Selmer M, Dunham CM, Murphy FVt, Weixlbaumer A, Petry S, Kelley AC, Weir JR, Ramakrishnan V. Structure of the 70S ribosome complexed with mRNA and tRNA. Science. 2006;313:1935–1942. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]