

Abstract
Translocations are known to affect the expression of genes at the breakpoints and, in the case of unbalanced translocations, alter the gene copy number. However, a comprehensive understanding of the functional impact of this class of variation is lacking. Here, we have studied the effect of balanced chromosomal rearrangements on gene expression by comparing the transcriptomes of cell lines from controls and individuals with the t(11;22)(q23;q11) translocation. The number of differentially expressed transcripts between translocation-carrying and control cohorts is significantly higher than that observed between control samples alone, suggesting that balanced rearrangements have a greater effect on gene expression than normal variation. Many of the affected genes are located along the length of the derived chromosome 11. We show that this chromosome is concomitantly altered in its spatial organization, occupying a more central position in the nucleus than its nonrearranged counterpart. Derivative 22-mapping chromosome 22 genes, on the other hand, remain in their usual environment. Our results are consistent with recent studies that experimentally altered nuclear organization, and indicated that nuclear position plays a functional role in regulating the expression of some genes in mammalian cells. Our study suggests that chromosomal translocations can result in hitherto unforeseen, large-scale changes in gene expression that are the consequence of alterations in normal chromosome territory positioning. This has consequences for the patterns of gene expression change seen during tumorigenesis-associated genome instability and during the karyotype changes that lead to speciation.
In addition to the millions of individual base pair changes that distinguish any two unrelated copies of our genome (The International HapMap Consortium 2003), recent reports have also described large numbers of copy number variable regions (CNVRs) (Redon et al. 2006; Korbel et al. 2007; Kidd et al. 2008; Conrad et al. 2009).
Nucleotide changes and copy number variation contribute to functional variation by modifying gene expression both in cis and in trans (Cheung et al. 2005; Merla et al. 2006; Stranger et al. 2007a,b; Guryev et al. 2008; Henrichsen et al. 2009b). Interestingly, CNVRs contribute to functional variation not only by modifying the expression levels of genes within the aneuploid segments in a somewhat dosage-dependent manner (Stranger et al. 2007a; Henrichsen et al. 2009b), but also by inducing altered expression of genes lying near to the breakpoints, even though these do not vary in copy number (Merla et al. 2006; Reymond et al. 2007; Stranger et al. 2007a; Guryev et al. 2008; Molina et al. 2008; Cahan et al. 2009; Henrichsen et al. 2009b). Consistently, these segmental aneuploidies are often associated with human phenotypes and diseases (for review, see Henrichsen et al. 2009a; Ionita-Laza et al. 2009; Zhang et al. 2009).
A third type of variation comprises balanced chromosomal rearrangements, such as reciprocal translocations and inversions, which elicit no gain or loss of genetic material. Balanced rearrangements occur in approximately one in 500 individuals in the general population (Ogilvie et al. 2001), and recent studies have identified hundreds of polymorphic inversions (Tuzun et al. 2005; Korbel et al. 2007; Kidd et al. 2008). Approximately 6% of reciprocal translocations are associated with some sort of phenotype (Warburton 1991). This may result from the direct disruption of a gene, or genes, at the breakpoints or by dissociating genes from their long-range regulatory elements (Kleinjan and van Heyningen 2005). At the transcriptome level, however, the functional impact of balanced translocations remains poorly studied.
Here, we report a comprehensive genome-wide analysis into the effect of the t(11;22)(q23;q11) reciprocal translocation on gene expression. This translocation between chromosome 11 (HSA11) and 22 (HSA22) is a recurrent constitutional, non-Robertsonian translocation (Emanuel and Saitta 2007) (see Supplemental Fig. S1), with the vast majority of carrier families not sharing common haplotypes (N Simon Thomas, pers. comm.). Carriers of the translocation are phenotypically normal, although some studies have shown they have up to a 10-fold greater risk of developing breast cancer than cytogenetically normal individuals (Lindblom et al. 1994; Jobanputra et al. 2005; Wieland et al. 2006; Carter et al. 2010). Carriers are also at risk of having progeny with Emanuel syndrome (OMIM no. 609029) (Fraccaro et al. 1980; Zackai and Emanuel 1980), due to malsegregation of the translocation products through meiosis. These children inherit an unbalanced complement of the balanced translocation and are partially trisomic for chromosomes 11 and 22 [+der(22)t(11;22)(q23;q11); see Supplemental Figure S1].
At the transcriptome level, the functional impact of balanced translocations is yet to be elucidated. However, it is known that these translocations can impact on the spatial organization of the derived chromosomes within the nucleus (Croft et al. 1999; Taslerova et al. 2003, 2006). Gene regulation is dependent not only on regulatory sequences and chromatin context located in cis, but also on nuclear position (for review, see Cremer et al. 2006; Fraser and Bickmore 2007; Heard and Bickmore 2007; Meaburn and Misteli 2007). This positioning is nonrandom with gene-rich and gene-poor chromosomes usually located toward the nuclear interior and nuclear periphery, respectively (Croft et al. 1999; Boyle et al. 2001). Consequently, HSA22 is normally more centrally positioned than HSA11 (Boyle et al. 2001). Since the radial organization of chromosomes in the nucleus impacts upon the nuclear positioning of translocation chromosomes (Croft et al. 1999), and proximity to the nuclear edge can directly affect gene expression in mammalian cells (Finlan et al. 2008; Reddy et al. 2008; Deniaud and Bickmore 2009), we reasoned that a possible repositioning of the derivative chromosomes 11 and 22 in translocation cell lines could have an effect on gene expression along the length of the derivative chromosomes.
Results and Discussion
To assess the effect of a balanced chromosomal rearrangement on the transcriptome, we individually analyzed genome-wide expression levels, using Affymetrix expression arrays, in lymphoblastoid cell lines from 26 individuals: 13 with cytogenetically normal karyotypes, nine with the t(11;22)(q23;q11)-balanced translocation, and four with the +der(22)t(11;22)(q23;q11) karyotype (Emanuel syndrome patients) (see Supplemental Table S1 for the complete list of cell lines; see Methods for details).
Emanuel syndrome patients show significant changes in gene expression as compared with controls
Comparison of unbalanced individuals with controls revealed significantly more transcript expression changes than the balanced versus control comparison (312 and 116 transcripts, respectively, when considering only the most variable transcripts; 1313 and 985 transcripts, respectively, when considering all transcripts; see Methods). This was anticipated, since Emanuel syndrome patients are partially trisomic for HSA11 and HSA22 and are phenotypically affected (Fraccaro et al. 1980; Zackai and Emanuel 1980) (see OMIM no. 609029 for phenotype details; see Supplemental Fig. S1 for karyotype). Indeed, a statistically significant fraction of the differentially expressed transcripts mapped to these two chromosomes (Fig. 1A–D) (e.g., 10 transcripts, half mapping to HSA11 and half to HSA22 among the top 25-ranked compared with the expected 1.2 [P < 0.01] and 0.5 [P < 0.0003], respectively, Fisher's exact test; see Supplemental Table S2 for details). Consistently, all differentially expressed genes that map to the trisomic regions of chromosome 11 and 22 and are ranked in the top 100 most differentially expressed transcripts show an increase in expression levels in cells of Emanuel syndrome patients (Fig. 1D). The comparison between unbalanced and controls was performed as an internal control and possible links between dysregulated genes and the Emanuel syndrome phenotype will be discussed elsewhere.
Figure 1.

Mapping of t(11;22) and Emanuel syndrome differentially expressed genes. Ensembl Karyoview ideograms showing the chromosomal location of the top 100 differentially expressed genes between Emanuel patients (unbalanced) and controls (A–D) and between carriers of the balanced t(11;22) translocation and controls (E–G). Complete karyograms are shown in A and E, while detailed views of chromosomes 11 and 22 are shown in B and F, and C and G, respectively. The derivative chromosome 22, which corresponds to the trisomic regions in Emanuel syndrome patients, is shown in D. The blue lines indicate the translocation breakpoints. Green arrowheads mark the genes showing an increase in expression in cell lines from Emanuel syndrome patients (A–D) and translocation carriers (E–G), while red arrowheads depict those showing a decrease.
To confirm our results, we performed quantitative PCR (Merla et al. 2006; Molina et al. 2008) on a total of 14 different cell lines from the three different genotypes (four Emanuel syndrome, five t(11;22) carriers, and five control cell lines) to determine the relative expression levels of 10 genes that appeared to show modified expression in the cell lines of Emanuel syndrome patients, but not of translocation carriers. Eight of these genes (four on HSA11 and four on HSA22) are trisomic in the patients (genes and assays used are in Supplemental Table S3). The results are consistent with the data obtained using the expression microarrays (correlation coefficient, R2 = 0.96). For example, the trisomic transcripts are expressed on average 1.64-fold (SD = 0.30) more in cells of Emanuel syndrome patients as compared with normal controls (Supplemental Fig. S2).
Balanced individuals show gene expression changes as compared with controls
To assess whether the number of differentially expressed transcripts between controls and balanced t(11;22) individuals was significant, or whether it could be reached by sampling the control population alone, we randomly permuted the labels of the control and balanced samples 1000 times (results were comparable using as few as 10 permutations), and each time computed the number of differentially expressed transcripts using a standard t-test, thus providing an empirical distribution of this metric (Fig. 2A). To prevent bias due to an arbitrary choice of significance threshold, several different P-value cutoffs were used (19 thresholds between 10−6 and 1). The number of differentially expressed transcripts between unbalanced versus full-genome complement cohorts (unbalanced vs. others), and male versus female or young versus old cohorts were also computed for reference (Fig. 2A). As expected (see above), we found a significant number of differentially expressed transcripts when comparing transcriptomes of Emanuel syndrome patients with those of balanced and controls together (unbalanced vs. others comparison). Similarly, the male versus female comparison revealed a significant number of transcripts showing differential expression between the genders. Reasoning that a substantial proportion of these would map to the sex chromosomes, we replicated our analyses, removing this bias by considering only autosome-mapping transcripts (Fig. 2B; Supplemental Figs, S3, S4, S5). This showed that the total number of autosomal transcripts differentially expressed between male and female samples was not significant and could be achieved by chance alone. On the other hand, comparison of individuals with and without the t(11;22) translocation (controls vs. balanced) showed numbers of differentially expressed transcripts that ran along the upper limit of the area containing 95% of the permutation distribution corresponding to the conventional P-value threshold of 0.05 (Fig. 2), and thereby closely bordering significance (e.g., at threshold 0.01 to call a gene differentially expressed, we observe 16 differentially expressed genes, corresponding to the top 3% of the permutation results). The number of differentially expressed transcripts between the two groups with complete genome complements is therefore large enough to place t(11;22) carriers at one tail of the normal transcriptome profile distribution (Fig. 2). This indicates that balanced rearrangements have a greater effect on gene expression than normal variation, and that these individuals show a greater degree of gene expression changes than would be expected given their apparently normal phenotype.
Figure 2.

Numbers of differentially expressed transcripts between t(11;22) carriers and control individuals. Graphs showing the number of differentially expressed transcripts between the studied cohorts identified with variable P-value thresholds and mapping on all (A) or autosomal only (B) chromosomes. Axes are in log scale. The following comparisons are shown: balanced [i.e., t(11;22) carriers] versus control cohorts (red line; nine vs. 13 samples), male versus female cohorts (blue line; six vs. 16 samples), young versus old cohorts (green line; nine vs. 13), and unbalanced (Emanuel syndrome patients, with partial trisomy 11 and 22) versus full-genome complement cohorts (unbalanced [four] vs. others [22]; orange line). Note that the sex and aneuploid/euploid comparisons are provided for information only. They involve different numbers of samples and therefore cannot be directly compared with the “balanced versus control” statistics or to the permutation distribution (see below). These two comparisons and the age comparison are compared with their own permutation results in Supplemental Figures S3, S4, and S5. The gray lines and shaded area represent 50%, 70%, 90%, and 95% of the permutation distribution (nine vs. 13; see main text for details). We observe that the number of transcripts differentially expressed between the control and the balanced cohorts follows the line demarcating 95% of the permutations, suggesting that t(11;22) carriers are at one tail of the distribution of transcriptome profiles.
Examination of the fold changes for the most differentially expressed genes between controls and balanced translocation carriers showed that they are for the greater part up-regulated (e.g., 73 up-regulated genes out of the top 100) and are distributed throughout the genome, with genes being located on almost every chromosome (Fig. 1E). Interestingly, some of these genes map to contiguous chromosomal regions, indicating that they may be under a common mechanism of expression modulation (Fig. 1F). For example, the group of genes located in the region 11q23.1-q25 show more differential expression than would be expected by chance (P = 0.03; software developed at the Swiss Institute of Bioinformatics and implementing the GSEA algorithm) (Subramanian et al. 2005). If changes in gene expression between controls and translocation carrying cells were caused solely by physical separation of cis-acting regulatory elements, we would expect an enrichment of affected genes close to the breakpoints on HSA11 and HSA22. This was not the case. On the contrary, we find genes with modified expression mapping tens of megabases from the breakpoints, on the opposite arm of the chromosome or on different chromosomes altogether (Fig. 1E–G; Supplemental Fig. S6). Furthermore, this separation, termed “position effect” or “cis-ruption” (Kleinjan and van Heyningen 2005; Kleinjan and Coutinho 2009), cannot explain why transcripts mapping to HSA11, but not HSA22, are over-represented amongst affected transcripts (e.g., four transcripts mapping to HSA11 among top 25-ranked compared with the expected 1.2 [P < 0.04, Fisher's exact test] and no HSA22 transcripts [expected 0.5, P = 1; see Supplemental Table S4 for details]).
The derivative chromosomes occupy different nuclear positions than their normal counterparts
Since chromosome position within the nucleus is nonrandom and a previous study has shown that a different balanced translocation, also involving HSA11 and HSA22, and present in over 85% of Ewing sarcomas, modified the position of the derivative chromosomes within the interphase nucleus (Taslerova et al. 2003, 2006), we reasoned that repositioning of the derivative chromosomes 11 and 22 could be the basis of the perturbed gene expression that we observed. We therefore used interphase fluorescence in situ hybridization (FISH) to measure the relative position of HSA11, HSA22, derivative 11, and derivative 22 chromosomes in lymphoblastoid nuclei from multiple controls and translocation carriers. To determine the positions of the normal and derivative chromosome 11, we used a chromosome 11–specific paint and BAC clone RP11-422P18 that maps to 22q12, just distal to the translocation breakpoint (Table 1; Fig. 3A). In nuclei from seven different control lymphoblastoid cell lines, HSA11 occupied, on average, a position midway between the center and the edge of the nucleus, as previously described (Fig. 3C; Boyle et al. 2001). In nuclei from four balanced t(11;22) cell lines, the normal HSA11 had a nuclear distribution similar to that found in control cell lines. However, the distribution of the derivative 11 territory was shifted significantly toward the center of the nucleus as compared with the normal HSA11 (P = 0.00073, permutation test with Kolmogorov-Smirnov statistics; Fig. 3C). In contrast, the 22q BAC signal was located toward the center of the nucleus in both control and t(11;22) cells when marking the normal HSA22, consistent with the known central location of chromosome 22 (Boyle et al. 2001), but shifted to a significantly more peripheral position when on the derivative 11 (P = 0.002; Fig. 3D).
Table 1.
List of BAC and fosmid clones used for FISH analysis

aFrom Ensembl release 55, July 2009.
bFrom the UCSC Genome Browser, Human Mar 2006 Assembly (hg18).
Figure 3.

Nuclear position of normal and derivative chromosome territories in t(11;22) cells and control cell lines. BAC coordinates are shown in Table 1. (A) Chromosome ideograms and schematic representation of the regions recognized by FISH probes used to distinguish the normal and derivative chromosome 11s, and an example of FISH on lymphoblastoid cells from a t(11;22) carrier. (Green) Chromosome 11 paint; (red) BAC RP11-422P18. DNA is counterstained with DAPI (blue). In nuclei of cells carrying the balanced t(11;22), the normal HSA11 is marked solely by the green chromosome paint signal, whereas the green painted territory for the derivative chromosome 11 is also associated with a red BAC hybridization signal. The normal HSA22 is indicated by a lone red BAC signal. (B) As in A, but chromosome 11 paint is in red and BAC RP11-93E4 is in green. In nuclei of cells carrying the balanced t(11;22), the normal HSA11 is marked by the red chromosome paint signal associated with a green BAC hybridization signal, whereas the derivative chromosome 11 is marked solely by the red painted territory. The derivative chromosome 22 is indicated by a lone green BAC signal. (C) Mean (+ SEM) percent of chromosome 11 paint hybridization signal present in five shells of equal area eroded from the edge (shell 1) to the center (shell 5) of the nucleus in seven control cell lines of normal karyotype (left) and four t(11;22) cell lines (right), showing the relative positions of the normal (blue) and derivative chromosome 11 (green). n = 40–50 nuclei each for each cell lines. The derivative 11 territory is shifted to a more central position in the nucleus compared with the normal 11. (D) As in C, but using BAC RP11-422P18 on four normal cell lines (left) and four t(11;22) cell lines (right) to compare positioning of the normal chromosome 22 (red) from that of the derivative 11 (green). The BAC signal on the derivative 11 is situated in a more peripheral position than that on the normal chromosome 22. (E) As in C, but using BAC RP11-93E4 on four normal cell lines (left) and five t(11;22) cell lines (right) to compare positioning of the normal chromosome 11 (blue) from that of the derivative 22 (yellow). The BAC signal on the derivative 22 is situated in a more central position than that on the normal chromosome 11.
To examine the part of 11q that is distal to the translocation breakpoint at 11q23, we used BAC RP11-93E4 that maps to 11q24 (Table 1; Fig. 3B). This portion of the genome is relocated to a more internal nuclear position when on the derivative 22, relative to its position on normal HSA11 (Fig. 3E). Because of the central position of chromosomes 22 within the nuclei of lymphoblastoid cells (Boyle et al. 2001) and their close association with the nucleolus, we were unable to use a chromosome 22–specific paint to distinguish the normal and derivative 22 in the same nucleus. However, we could not detect a significant difference in the nuclear position of a BAC on the derivative 22 (Fig. 3E) and the BAC that detects the normal HSA22 (Fig. 3D). Therefore, we conclude that the normal and derivative chromosome 22 do not significantly differ in their nuclear position. This may be because the derivative 22 retains the ribosomal DNA (rDNA) arrays on the p arm, thereby anchoring this chromosome in the nucleolus.
This (re)positioning of the derivative chromosomes is consistent with the change in gene density within a 2-Mb window around the breakpoints, which has been shown to be a good predictor of radial chromosome positioning within lymphoblastoid nuclei (Boyle et al. 2001; Murmann et al. 2005; Federico et al. 2008). The transposition of the derivative chromosome 11 to a more central position within the nucleus results in HSA11 genes retained on this chromosome being placed into an anomalous chromatin environment, potentially causing changes in their level of expression. Similarly, HSA11 genes located distal to the breakpoint, which therefore map to the derivative 22, are also relocated to a more central position. Conversely, HSA22 genes that map to the derivative 11 show a shift to a more peripheral position than those on the normal HSA22. The fact that only a limited number of genes on the rearranged chromosomes are detectably misexpressed is consistent with previous studies in which chromosome position was experimentally manipulated (Finlan et al. 2008; Reddy et al. 2008). Both these “tethering” experiments and our analyses suggest that not all genes in the genome are sensitive to their nuclear environment. However, we note that the observed changes in expression are effective even with a single erroneously positioned allele, suggesting that nuclear chromosome organization has a substantial influence on the regulation of gene expression.
Chromosomes not involved in the translocation also show nuclear position changes
To determine whether the position of other chromosomes within the nucleus is affected in cells from balanced translocation carriers, we repeated our FISH analysis using a chromosome 17-specific paint. The results showed that HSA17, a gene-dense chromosome, is shifted to a significantly more peripheral position in balanced cell lines as compared with controls (Fig. 4A; Kolmogorov-Smirnov statistics, P = 0.03). This suggests that movement of the large derivative 11 to a more central position has a knock on effect that results in displacement of other chromosomes from their usual nuclear positions.
Figure 4.

Nuclear position of chromosome 17 territories and fosmids covering differentially expressed genes in normal and t(11;22) cells. The fosmid coordinates and mapping positions are shown in Table 1 and Supplemental Figure S6, respectively. (A) Mean (+SEM) percent of chromosome 17 paint hybridization signal present in five shells of equal area eroded from the edge (shell 1) to the center (shell 5) of the nucleus in four control cell lines of normal karyotype (dark purple) and five t(11;22) cell lines (light purple) showing the relative positions of the chromosome 17s. n = 40–50 nuclei each for each cell lines. The chromosome 17 territory is shifted to a more peripheral position in nuclei from balanced translocation carriers as compared to controls. (B) Mean (+SEM) percent of the hybridization signal of chromosome 9q21.32 fosmid G248P81402D5 (D5) in four control cell lines of normal karyotype (dark blue) and four t(11;22) cell lines (pink) showing the relative positions of the fosmid. The fosmid signal is shifted to a slightly more peripheral position in nuclei from balanced cell lines as compared with controls. (C) As in B, but using fosmid G248P8942D11 (D11) on three control cell lines of normal karyotype (right) and four t(11;22) cell lines (left), showing the relative positions of the fosmid on the normal 11 (blue) and on the derivative chromosome 11 (green). The fosmid signal on the derivative 11 is shifted to a slightly more peripheral position than that on the normal 11. (D) As in B, but using fosmid G248P84992F11 (F11) on three control cell lines (left) and four t(11;22) cell lines (right), showing the relative positions of the normal 22 (red) and the derivative 11 (green). The fosmid signal on the derivative 11 is shifted to a slightly more central position than that on the normal 22. (E) As in B, but using fosmid G248P87917B7 (B7) on four control cell lines (left) and five t(11;22) cell lines (right), showing the relative positions of the normal 22 (red) and the derivative 11 (green). This fosmid covers a gene that does not show any differential expression between balanced translocation carriers and controls and the fosmid signal on the derivative 11 shows no difference in position compared with that on the normal 22.
The repositioning of the derivative and other chromosomes within the nucleus and associated changes in gene expression may have many causes. The most active chromosomes are situated at the nuclear center (Boyle et al. 2001) and active and inactive copies of monoallelically expressed genes occupy different positions within the nucleus (Takizawa et al. 2008). The placement of genes into anomalous chromatin environments or their relocation relative to the center or periphery of the nucleus can affect their expression, and gene expression levels have been shown to be reduced or increased in correlation with movement toward the nuclear periphery or interior, respectively (Williams et al. 2006; Finlan et al. 2008; Reddy et al. 2008; Szczerbal et al. 2009). Consistently, we observed that all HSA11-mapping genes that belong to the 100 most differentially expressed genes in t(11;22) cells showed increased expression levels concomitant to their movement to a more central nuclear location (see Fig. 1E,F). Although it appears that HSA22 genes that map to the derivative 22 chromosome do not show a significant change in their nuclear location as compared with those on the normal HSA22 (although given that chromosome 22 paint could not be used in this study, we have not mapped the full extent of the derivative chromosome 22 territory), those genes that show differential expression may still have undergone a shift in their position, as genes can loop out from their chromosome territory, and the repositioning of whole chromosome territories has been shown to be less dramatic than that of individual gene loci (Szczerbal et al. 2009). Differentially expressed HSA22 genes that map to the derivative 22 may, therefore, have undergone an alteration in their position that coincides with their observed expression changes.
Differentially expressed genes show position changes within the nucleus
To determine whether individual differentially expressed genes were repositioned within the nucleus, we repeated the FISH analyses using four different fosmid probes that cover three genes differentially expressed in cell lines from balanced individuals as compared with controls, and one control gene that did not show any expression differences between these cohorts (fosmid coordinates and mapping positions are shown in Table 1 and Supplemental Fig. S6, respectively). The fosmids mapping to differentially expressed genes showed slight, yet not significant (Kolmogorov-Smirnov statistics) nuclear position changes (Fig. 4B–D), whereas the fosmid covering the control gene showed no alteration in position (Fig. 4E). This adds to the growing body of evidence suggesting that changes in nuclear position are linked with gene expression changes. Our results only partially agree with the view that increases in gene expression correlate with a shift toward the center of the nucleus, and vice versa (Williams et al. 2006; Finlan et al. 2008; Reddy et al. 2008; Szczerbal et al. 2009), as fosmids F11 and D5, which map to genes showing an increase and decrease in expression, respectively, are concomitantly shifted more centrally and more peripherally. On the contrary, fosmid D11 moves to a more peripheral position within the nucleus, despite it mapping to a gene showing an increase in expression.
The reorganization of chromosomes within the nucleus may result in disruption of cis interactions. A recent comprehensive mapping of long-range interactions revealed that intrachromosomal contacts decreased as a function of genomic distance reaching a plateau at 90 Mb in lymphoblastoid cells (Lieberman-Aiden et al. 2009). Co-associations of genes situated 25-Mb apart on the same chromosome (Osborne et al. 2004; Simonis et al. 2006; Mitchell and Fraser 2008) have also been shown to occur at “transcription factories”—discrete foci within the nucleus that contain high concentrations of active RNA polymerase II, at which actively transcribed alleles have been shown to be positioned (Iborra et al. 1996; Osborne et al. 2004)—or around aggregations of splicing-related factors, termed “Nuclear Speckles” (Brown et al. 2008; Sutherland and Bickmore 2009). Interactions are not restricted to those genes situated on the same chromosome—trans interactions have also been reported (Lieberman-Aiden et al. 2009) and may be supposed to have a functional basis (Misteli 2004; Osborne et al. 2007). Thus, relocation of not only the derivative chromosomes, but also other chromosomes within the nuclei of cells from balanced translocation carriers, may disrupt some of these interactions. The cis and trans interactions that occur at these specific foci could also explain why genes differentially expressed between balanced translocation carriers and controls mapped not only to the derivative chromosomes but to nearly every chromosome. These genes are then likely to modify the expression of other downstream targets, further extending the number of affected genes and their respective mapping positions.
While we speculate that many of the gene expression changes we observe are a direct consequence of nuclear reorganization caused by the altered positioning of the translocation chromosomes within the nucleus, we cannot exclude that they are caused by other facets of genome structure and function, such as replication timing, which also correlate with nuclear position and repositioning (Hiratani et al. 2008).
Here, we have shown large-scale changes in gene expression concurrent with translocation-induced nuclear repositioning. Although carriers of the balanced t(11;22)(q23;q11) are generally phenotypically normal, we note that there are reports of increased cancer incidence in these carriers (Lindblom et al. 1994; Jobanputra et al. 2005; Wieland et al. 2006; Carter et al. 2010), and speculate that the altered gene expression we observe may underlie this. Furthermore, our observations raise the possibility that translocations in cancer cells may contribute to oncogenesis not only by disrupting the gene, or genes mapping around the breakpoints, but also through changes in expression of genes positioned elsewhere on the affected chromosomes and on other displaced chromosomes, a hypothesis that warrants detailed investigation in the future. Our results could also have implications on reproductive separation. Major genome rearrangements may play important roles in sympatric and parapatric speciation, a phenomenon known as chromosome speciation (for review, see Ayala and Coluzzi 2005; Noor and Feder 2006) and balanced translocations have been shown to reinforce the reproductive isolation of Saccharomyces species (Delneri et al. 2003). It was postulated that natural selection can fix such changes because they confer adaptive significance either through changes in expression of specific genes or possibly by modifying the structure of the implicated chromosomes themselves (Perez-Ortin et al. 2002; Colson et al. 2004). Consistently, Marguès-Bonet et al. (2004) found that genes mapping on chromosomes rearranged between human and chimpanzee showed more varied relative expression levels between the two species than genes situated on colinear chromosomes. Here, we show that the human t(11;22) balanced translocation modifies the relative expression levels of a significant number (2%–3%) of expressed transcripts. Our results, therefore, suggest that in vertebrates a single major genome rearrangement (e.g., a translocation) may provide both a selective advantage and a genetic barrier without the requirement of secondary changes.
Methods
Cell culture
Age and sex matched lymphoblastoid cell lines were obtained from either the Coriell Institute for Medical Research (http://www.coriell.org/), the European Collection of Cell Cultures (ECACC; http://www.ecacc.org.uk/), or from Elisabeth Blennow at the Karolinska Institute, Stockholm. A complete list of cells is shown in Supplemental Table S1. Appropriate informed consent was obtained for each sample by the physician in charge. All cells were cultured in RPMI-1640 Medium with GlutaMAX (GIBCO, Invitrogen), supplemented with 10% (v/v) Fetal calf serum, and 1% (v/v) penicillin and streptomycin at 37°C in 5% CO2. DNA was extracted from each cell line of the sample population using the Nucleon BACC genomic DNA extraction kit (GE Healthcare) according to the manufacturer's instructions. The absence of DNA sequence loss, as well as the presence/absence of derivative 11 and derivative 22 chromosomes were assessed with PCR assays using the following pairs of primers that map near to the HSA11 and HSA22 breakpoints: primer pair c14irev GGAAGTTAGAGAAAACTGAGAA and JF22.2 CCTCCAACGGATCCATACT; and primer pair JF22.2 CCTCCAACGGATCCATACT and c14hfor AACACTCCCACTGACAGCTA (see Kurahashi et al. 2000a,b; Kurahashi and Emanuel 2001). The amplification reactions were performed as follows: 95°C for 5 min; five cycles of 94°C for 30 sec, 58°C for 30 sec, and 72°C for 1 min; followed by 35 cycles of 94°C for 30 sec, 56°C for 30 sec, and 72°C for 1 min; and 72°C for 5 min.
RNA isolation and expression array
Total RNA was extracted using TRIzol reagent (Invitrogen), cleaned on RNeasy columns (Qiagen) according to the manufacturers' instructions and used as a template for complementary DNA (cDNA) synthesis and biotinylated antisense cRNA preparation. The synthesis of cDNA and cRNA, labeling, hybridization, and scanning of the samples were performed as described by Affymetrix (http://www.affymetrix.com). GeneChip Human Genome U133 Plus 2.0 arrays, each interrogating over 47,000 target sequences with appropriate probesets (Affymetrix) were used to hybridize the labeled cRNA. Lymphoblastoid cell lines established from blood samples of individuals with normal (13 samples), balanced (nine), or unbalanced (four) karyotypes were processed for a total of 26 expression arrays. These data have been deposited in the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO series accession number GSE13122.
Expression data analyses were performed using R (version 2.7.1) (The R Development Core Team 2004) and bioconductor (Gentleman et al. 2004). Low-level analysis and normalization were done using RMA (bioconductor package affy, version 1.16.0) (Irizarry et al. 2003). Two datasets were analyzed: In addition to the complete set of target sequences (transcripts), a filtered data set that included only target sequences with hybridization signals with standard deviation over all samples greater than 0.5 was also created. For differential expression analysis, the following two pairwise comparisons were performed: control versus balanced, and as an internal control, control versus unbalanced. The number of differentially expressed transcripts in each comparison was determined using the moderated t-statistic provided by the limma package (version 2.14.5) (Smyth 2004, 2005). A threshold of P < 0.01 was used; multiple testing was accounted for by correcting the P-values using the Benjamini and Hochberg (1995) method to control the false discovery rate (FDR). Genome mapping of the expressed transcripts was determined following version na22 (March 9, 2007) of the annotation provided by Affymetrix. To assess whether the number of differentially expressed transcripts between the two groups with full-genome complements was significant and could not be reached by chance alone (controls vs. balanced comparison, Fig. 2), we performed a permutation test. We randomly permuted the labels of the control and balanced samples 1000 times, and each time computed the number of differentially expressed transcripts using a standard t-test, thus providing an empirical distribution of this metric (Fig. 2). To prevent bias due to an arbitrary choice of significance threshold, several different P-value cutoffs were used (19 thresholds between 10−6 and 1; Fig. 2). No correction for multiple testing was performed, since the permutation distribution already provides an assessment of the expected number of significant results obtained by chance. We note that tests performed with only 10 and 100 permutations gave similar results.
Quantitative PCR
Quantitative PCR was performed using SYBRGREEN PCR Master Mix (Applied Biosystems) following the manufacturer's specifications. Oligonucleotides were designed using the Primer3 program with default parameters (Supplemental Table S3). The efficiency of each assay was tested in a cDNA dilution series as described (Livak and Schmittgen 2001). All RT-PCR reactions were performed in a 10-μL final volume and three replicates per sample and set up in a 384-well plate format using a Freedom EVO robot (TECAN) and run in an ABI 7900 Sequence Detection System (Applied Biosystems) with the following amplification conditions: 50°C for 2 min, 95°C for 10 min, and 50 cycles of 95°C 15 sec/60°C for 1 min. Each plate included the appropriate normalization genes to control for any variability between the different plate runs. Raw threshold cycle (CT) values were obtained using SDS2.2 (Applied Biosystems). To calculate the normalized relative expression ratio, we followed the method described in Merla et al. (2006) and Molina et al. (2008), and exploited the geNorm method to select GUSB, SDHA, and TBP as normalization genes (Vandesompele et al. 2002). Relative expressions were measured in four cell lines from Emanuel syndrome patients, five from t(11;22) carriers, and five from controls.
Fluorescence in situ hybridization
A complete list of the BACs and fosmid clones used for this report is shown in Table 1. They were obtained from BACPAC Resource Center (BPRC) at the Children's Hospital Oakland Research Institute. Clones were grown and DNA extracted according to BPRC protocols. Chromosome paints were a kind gift from J. Trent (TGen, Pheonix, AZ). FISH probes were produced and FISH performed on fixed cell suspensions according to standard methods (Guan et al. 1996; Chong et al. 1997). Image capture and analyses were performed using in-house scripts written for IP Lab Spectrum (Scanalytics Corp.).
The radial positions of the chromosome territories and BAC and fosmid clones were determined using an adaptation of a method previously described (Boyle et al. 2001; Finlan et al. 2008). A total of 40–50 nuclei for each of three to seven different cell lines were analyzed per experiment and five shells (annuli) of equal area were calculated from the periphery (annulus 1) to the center (annulus 5) of the nucleus based on the segmented DAPI signal. Signals were treated independently. An area of interest was drawn around the relevant signal and the mean proportion (%) of hybridization signal in each annulus calculated and normalized to the proportion of hybridization signal for that fluorochrome in the whole nucleus. The difference between the two distributions of positions (averaged over the replicates) was assessed using the cumulative proportions of hybridization signal and a Kolmogorov-Smirnov statistics. Significance was determined by performing a permutation test over all possible permutations.
Acknowledgments
We thank Otto Hagenbüchle, Keith Harshman, and Alexandra Paillusson, from the Lausanne DNA Array Facility, and Jacqueline Chrast for technical help; Elisabeth Blennow and Jeff Trent for reagents; N. Simon Thomas for sharing unpublished data; and Henrik Kaessmann and Nicolas Salamin for comments on the manuscript. This work was supported by grants from the Jérôme Lejeune Foundation, the Telethon Action Suisse Foundation, the Swiss National Science Foundation, and the European Commission anEUploidy Integrated Project (grant no. 037627) to A.R.; from the UK Medical Research Council to W.A.B.; and from the NCCR Molecular Oncology, a program of the Swiss National Science Foundation, to M.D.
Footnotes
[Supplemental material is available online at http://www.genome.org. The gene expression data from this study have been submitted to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under series accession no. GSE13122.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.103622.109.
References
- Ayala FJ, Coluzzi M 2005. Chromosome speciation: Humans, Drosophila, and mosquitoes. Proc Natl Acad Sci 102 (Suppl 1): 6535–6542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y 1995. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57: 289–300 [Google Scholar]
- Boyle S, Gilchrist S, Bridger JM, Mahy NL, Ellis JA, Bickmore WA 2001. The spatial organization of human chromosomes within the nuclei of normal and emerin-mutant cells. Hum Mol Genet 10: 211–219 [DOI] [PubMed] [Google Scholar]
- Brown JM, Green J, Pires das Neves R, Wallace HA, Smith AJ, Hughes J, Gray N, Taylor S, Wood WG, Higgs DR, et al. 2008. Association between active genes occurs at nuclear speckles and is modulated by chromatin environment. J Cell Biol 182: 1083–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahan P, Li Y, Izumi M, Graubert TA 2009. The impact of copy number variation on local gene expression in mouse hematopoietic stem and progenitor cells. Nat Genet 41: 430–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter MT, Barrowman NJ, St Pierre SA, Emanuel BS, Boycott KM 2010. Risk of breast cancer not increased in translocation 11;22 carriers: Analysis of 80 pedigrees. Am J Med Genet A 152A: 212–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung VG, Spielman RS, Ewens KG, Weber TM, Morley M, Burdick JT 2005. Mapping determinants of human gene expression by regional and genome-wide association. Nature 437: 1365–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong SS, Pack SD, Roschke AV, Tanigami A, Carrozzo R, Smith AC, Dobyns WB, Ledbetter DH 1997. A revision of the lissencephaly and Miller-Dieker syndrome critical regions in chromosome 17p13.3. Hum Mol Genet 6: 147–155 [DOI] [PubMed] [Google Scholar]
- Colson I, Delneri D, Oliver SG 2004. Effects of reciprocal chromosomal translocations on the fitness of Saccharomyces cerevisiae. EMBO Rep 5: 392–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, et al. 2009. Origins and functional impact of copy number variation in the human genome. Nature 10.1028/nature08516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer T, Cremer M, Dietzel S, Muller S, Solovei I, Fakan S 2006. Chromosome territories––a functional nuclear landscape. Curr Opin Cell Biol 18: 307–316 [DOI] [PubMed] [Google Scholar]
- Croft JA, Bridger JM, Boyle S, Perry P, Teague P, Bickmore WA 1999. Differences in the localization and morphology of chromosomes in the human nucleus. J Cell Biol 145: 1119–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delneri D, Colson I, Grammenoudi S, Roberts IN, Louis EJ, Oliver SG 2003. Engineering evolution to study speciation in yeasts. Nature 422: 68–72 [DOI] [PubMed] [Google Scholar]
- Deniaud E, Bickmore WA 2009. Transcription and the nuclear periphery: Edge of darkness? Curr Opin Genet Dev 19: 187–191 [DOI] [PubMed] [Google Scholar]
- Emanuel BS, Saitta SC 2007. From microscopes to microarrays: Dissecting recurrent chromosomal rearrangements. Nat Rev Genet 8: 869–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federico C, Cantarella CD, Di Mare P, Tosi S, Saccone S 2008. The radial arrangement of the human chromosome 7 in the lymphocyte cell nucleus is associated with chromosomal band gene density. Chromosoma 117: 399–410 [DOI] [PubMed] [Google Scholar]
- Finlan LE, Sproul D, Thomson I, Boyle S, Kerr E, Perry P, Ylstra B, Chubb JR, Bickmore WA 2008. Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet 4: e1000039 doi: 10.1371/journal.pgen.1000039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraccaro M, Lindsten J, Ford CE, Iselius L 1980. The 11q;22q translocation: A European collaborative analysis of 43 cases. Hum Genet 56: 21–51 [DOI] [PubMed] [Google Scholar]
- Fraser P, Bickmore W 2007. Nuclear organization of the genome and the potential for gene regulation. Nature 447: 413–417 [DOI] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. 2004. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol 5: R80 doi: 10.1186/gb-2004-5-10-r80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan XY, Zhang H, Bittner M, Jiang Y, Meltzer P, Trent J 1996. Chromosome arm painting probes. Nat Genet 12: 10–11 [DOI] [PubMed] [Google Scholar]
- Guryev V, Saar K, Adamovic T, Verheul M, van Heesch SA, Cook S, Pravenec M, Aitman T, Jacob H, Shull JD, et al. 2008. Distribution and functional impact of DNA copy number variation in the rat. Nat Genet 40: 538–545 [DOI] [PubMed] [Google Scholar]
- Heard E, Bickmore W 2007. The ins and outs of gene regulation and chromosome territory organisation. Curr Opin Cell Biol 19: 311–316 [DOI] [PubMed] [Google Scholar]
- Henrichsen CN, Chaignat E, Reymond A 2009a. Copy number variants, diseases and gene expression. Hum Mol Genet 18: R1–R8 [DOI] [PubMed] [Google Scholar]
- Henrichsen CN, Vinckenbosch N, Zollner S, Chaignat E, Pradervand S, Schutz F, Ruedi M, Kaessmann H, Reymond A 2009b. Segmental copy number variation shapes tissue transcriptomes. Nat Genet 41: 424–429 [DOI] [PubMed] [Google Scholar]
- Hiratani I, Ryba T, Itoh M, Yokochi T, Schwaiger M, Chang CW, Lyou Y, Townes TM, Schubeler D, Gilbert DM 2008. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol 6: e245 doi: 10.1371/journal.pbio.0060245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iborra FJ, Pombo A, Jackson DA, Cook PR 1996. Active RNA polymerases are localized within discrete transcription “factories” in human nuclei. J Cell Sci 109: 1427–1436 [DOI] [PubMed] [Google Scholar]
- The International HapMap Consortium 2003. The International HapMap Project. Nature 426: 789–796 [DOI] [PubMed] [Google Scholar]
- Ionita-Laza I, Rogers AJ, Lange C, Raby BA, Lee C 2009. Genetic association analysis of copy-number variation (CNV) in human disease pathogenesis. Genomics 93: 22–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP 2003. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4: 249–264 [DOI] [PubMed] [Google Scholar]
- Jobanputra V, Chung WK, Hacker AM, Emanuel BS, Warburton D 2005. A unique case of der(11)t(11;22),-22 arising from 3:1 segregation of a maternal t(11;22) in a family with co-segregation of the translocation and breast cancer. Prenat Diagn 25: 683–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, Hansen N, Teague B, Alkan C, Antonacci F, et al. 2008. Mapping and sequencing of structural variation from eight human genomes. Nature 453: 56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinjan DJ, Coutinho P 2009. Cis-ruption mechanisms: Disruption of cis-regulatory control as a cause of human genetic disease. Brief Funct Genomics Proteomics 8: 317–332 [DOI] [PubMed] [Google Scholar]
- Kleinjan DA, van Heyningen V 2005. Long-range control of gene expression: Emerging mechanisms and disruption in disease. Am J Hum Genet 76: 8–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, Kim PM, Palejev D, Carriero NJ, Du L, et al. 2007. Paired-end mapping reveals extensive structural variation in the human genome. Science 318: 420–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurahashi H, Emanuel BS 2001. Long AT-rich palindromes and the constitutional t(11;22) breakpoint. Hum Mol Genet 10: 2605–2617 [DOI] [PubMed] [Google Scholar]
- Kurahashi H, Shaikh TH, Emanuel BS 2000a. Alu-mediated PCR artifacts and the constitutional t(11;22) breakpoint. Hum Mol Genet 9: 2727–2732 [DOI] [PubMed] [Google Scholar]
- Kurahashi H, Shaikh TH, Zackai EH, Celle L, Driscoll DA, Budarf ML, Emanuel BS 2000b. Tightly clustered 11q23 and 22q11 breakpoints permit PCR-based detection of the recurrent constitutional t(11;22). Am J Hum Genet 67: 763–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. 2009. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326: 289–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblom A, Sandelin K, Iselius L, Dumanski J, White I, Nordenskjold M, Larsson C 1994. Predisposition for breast cancer in carriers of constitutional translocation 11q;22q. Am J Hum Genet 54: 871–876 [PMC free article] [PubMed] [Google Scholar]
-
Livak KJ, Schmittgen TD 2001. Analysis of relative gene expression data using real-time quantitative PCR and the
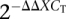 method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar] - Marques-Bonet T, Caceres M, Bertranpetit J, Preuss TM, Thomas JW, Navarro A 2004. Chromosomal rearrangements and the genomic distribution of gene-expression divergence in humans and chimpanzees. Trends Genet 20: 524–529 [DOI] [PubMed] [Google Scholar]
- Meaburn KJ, Misteli T 2007. Cell biology: Chromosome territories. Nature 445: 379–381 [DOI] [PubMed] [Google Scholar]
- Merla G, Howald C, Henrichsen CN, Lyle R, Wyss C, Zabot MT, Antonarakis SE, Reymond A 2006. Submicroscopic deletion in patients with Williams-Beuren syndrome influences expression levels of the nonhemizygous flanking genes. Am J Hum Genet 79: 332–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T 2004. Spatial positioning; a new dimension in genome function. Cell 119: 153–156 [DOI] [PubMed] [Google Scholar]
- Mitchell JA, Fraser P 2008. Transcription factories are nuclear subcompartments that remain in the absence of transcription. Genes & Dev 22: 20–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina J, Carmona-Mora P, Chrast J, Krall PM, Canales CP, Lupski JR, Reymond A, Walz K 2008. Abnormal social behaviors and altered gene expression rates in a mouse model for Potocki-Lupski syndrome. Hum Mol Genet 17: 2486–2495 [DOI] [PubMed] [Google Scholar]
- Murmann AE, Gao J, Encinosa M, Gautier M, Peter ME, Eils R, Lichter P, Rowley JD 2005. Local gene density predicts the spatial position of genetic loci in the interphase nucleus. Exp Cell Res 311: 14–26 [DOI] [PubMed] [Google Scholar]
- Noor MA, Feder JL 2006. Speciation genetics: Evolving approaches. Nat Rev Genet 7: 851–861 [DOI] [PubMed] [Google Scholar]
- Ogilvie CM, Braude P, Scriven PN 2001. Successful pregnancy outcomes after preimplantation genetic diagnosis (PGD) for carriers of chromosome translocations. Hum Fertil 4: 168–171 [DOI] [PubMed] [Google Scholar]
- Osborne CS, Chakalova L, Brown KE, Carter D, Horton A, Debrand E, Goyenechea B, Mitchell JA, Lopes S, Reik W, et al. 2004. Active genes dynamically colocalize to shared sites of ongoing transcription. Nat Genet 36: 1065–1071 [DOI] [PubMed] [Google Scholar]
- Osborne CS, Chakalova L, Mitchell JA, Horton A, Wood AL, Bolland DJ, Corcoran AE, Fraser P 2007. Myc dynamically and preferentially relocates to a transcription factory occupied by Igh. PLoS Biol 5: e192 doi: 10.1371/journal.pbio.0050192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Ortin JE, Querol A, Puig S, Barrio E 2002. Molecular characterization of a chromosomal rearrangement involved in the adaptive evolution of yeast strains. Genome Res 12: 1533–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The R Development Core Team 2004. R: A language and environment for statistical computing. R Foundation for Statistical Computing R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- Reddy KL, Zullo JM, Bertolino E, Singh H 2008. Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature 452: 243–247 [DOI] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, et al. 2006. Global variation in copy number in the human genome. Nature 444: 444–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymond A, Henrichsen CN, Harewood L, Merla G 2007. Side effects of genome structural changes. Curr Opin Genet Dev 17: 381–386 [DOI] [PubMed] [Google Scholar]
- Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B, de Laat W 2006. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat Genet 38: 1348–1354 [DOI] [PubMed] [Google Scholar]
- Smyth GK 2004. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3: Article3. doi: 10.2202/1544-6115.1027 [DOI] [PubMed] [Google Scholar]
- Smyth G 2005. Linear models for microarray data. In Bioinformatics and Computational Biology Solutions using R and Bioconductor, (ed. Gentleman RC et al. ), pp. 397–420 Springer, New York [Google Scholar]
- Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, Redon R, Bird CP, de Grassi A, Lee C, et al. 2007a. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 315: 848–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Nica AC, Forrest MS, Dimas A, Bird CP, Beazley C, Ingle CE, Dunning M, Flicek P, Koller D, et al. 2007b. Population genomics of human gene expression. Nat Genet 39: 1217–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland H, Bickmore WA 2009. Transcription factories: Gene expression in unions? Nat Rev Genet 10: 457–466 [DOI] [PubMed] [Google Scholar]
- Szczerbal I, Foster HA, Bridger JM 2009. The spatial repositioning of adipogenesis genes is correlated with their expression status in a porcine mesenchymal stem cell adipogenesis model system. Chromosoma 118: 647–663 [DOI] [PubMed] [Google Scholar]
- Takizawa T, Gudla PR, Guo L, Lockett S, Misteli T 2008. Allele-specific nuclear positioning of the monoallelically expressed astrocyte marker GFAP. Genes & Dev 22: 489–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taslerova R, Kozubek S, Lukasova E, Jirsova P, Bartova E, Kozubek M 2003. Arrangement of chromosome 11 and 22 territories, EWSR1 and FLI1 genes, and other genetic elements of these chromosomes in human lymphocytes and Ewing sarcoma cells. Hum Genet 112: 143–155 [DOI] [PubMed] [Google Scholar]
- Taslerova R, Kozubek S, Bartova E, Gajduskova P, Kodet R, Kozubek M 2006. Localization of genetic elements of intact and derivative chromosome 11 and 22 territories in nuclei of Ewing sarcoma cells. J Struct Biol 155: 493–504 [DOI] [PubMed] [Google Scholar]
- Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, Pertz LM, Haugen E, Hayden H, Albertson D, Pinkel D, et al. 2005. Fine-scale structural variation of the human genome. Nat Genet 37: 727–732 [DOI] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F 2002. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3: research0034.1 doi: 10.1186/gb-2002-3-7-research0034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburton D 1991. De novo balanced chromosome rearrangements and extra marker chromosomes identified at prenatal diagnosis: Clinical significance and distribution of breakpoints. Am J Hum Genet 49: 995–1013 [PMC free article] [PubMed] [Google Scholar]
- Wieland I, Muschke P, Volleth M, Ropke A, Pelz AF, Stumm M, Wieacker P 2006. High incidence of familial breast cancer segregates with constitutional t(11;22)(q23;q11). Genes Chromosomes Cancer 45: 945–949 [DOI] [PubMed] [Google Scholar]
- Williams RR, Azuara V, Perry P, Sauer S, Dvorkina M, Jorgensen H, Roix J, McQueen P, Misteli T, Merkenschlager M, et al. 2006. Neural induction promotes large-scale chromatin reorganisation of the Mash1 locus. J Cell Sci 119: 132–140 [DOI] [PubMed] [Google Scholar]
- Zackai EH, Emanuel BS 1980. Site-specific reciprocal translocation, t(11;22) (q23;q11), in several unrelated families with 3:1 meiotic disjunction. Am J Med Genet 7: 507–521 [DOI] [PubMed] [Google Scholar]
- Zhang F, Gu W, Hurles ME, Lupski JR 2009. Copy number variation in human health, disease, and evolution. Annu Rev Genomics Hum Genet 10: 451–481 [DOI] [PMC free article] [PubMed] [Google Scholar]