

Abstract
Structural variation (SV) is a rich source of genetic diversity in mammals, but due to the challenges associated with mapping SV in complex genomes, basic questions regarding their genomic distribution and mechanistic origins remain unanswered. We have developed an algorithm (HYDRA) to localize SV breakpoints by paired-end mapping, and a general approach for the genome-wide assembly and interpretation of breakpoint sequences. We applied these methods to two inbred mouse strains: C57BL/6J and DBA/2J. We demonstrate that HYDRA accurately maps diverse classes of SV, including those involving repetitive elements such as transposons and segmental duplications; however, our analysis of the C57BL/6J reference strain shows that incomplete reference genome assemblies are a major source of noise. We report 7196 SVs between the two strains, more than two-thirds of which are due to transposon insertions. Of the remainder, 59% are deletions (relative to the reference), 26% are insertions of unlinked DNA, 9% are tandem duplications, and 6% are inversions. To investigate the origins of SV, we characterized 3316 breakpoint sequences at single-nucleotide resolution. We find that ∼16% of non-transposon SVs have complex breakpoint patterns consistent with template switching during DNA replication or repair, and that this process appears to preferentially generate certain classes of complex variants. Moreover, we find that SVs are significantly enriched in regions of segmental duplication, but that this effect is largely independent of DNA sequence homology and thus cannot be explained by non-allelic homologous recombination (NAHR) alone. This result suggests that the genetic instability of such regions is often the cause rather than the consequence of duplicated genomic architecture.
In the six years since the first genome-wide analyses revealed extensive DNA copy number variation (CNV) among human individuals (Iafrate et al. 2004; Sebat et al. 2004), numerous studies have extended this observation in scope and scale with increasingly powerful genomic tools. It is now widely recognized that structural variation (SV), which includes duplications, deletions, inversions, transpositions, and other genomic rearrangements, is an abundant and functionally important class of genetic variation in mammals (Zhang et al. 2009a). Besides the emerging role of inherited variants in complex disease, new structural mutations contribute to sporadic human disorders, are a hallmark of tumor genomes, and drive the evolution of genes and species. For all of these reasons, it is important to generate accurate SV maps in many different organisms and cellular contexts, so that the biological consequences of SV may be assessed, and so that the molecular mechanisms that generate new variation may be fully understood.
Several technical challenges have precluded a more complete understanding of the patterns and origins of SV. First, most studies have used array comparative genome hybridization (aCGH), which has limited resolution, cannot detect balanced rearrangements or reconstruct locus architecture, and has limited ability to detect SVs composed of multi-copy elements such as segmental duplications (SDs) or transposable elements (TEs). Second, sequence-based methods such as paired-end mapping (PEM) have emerged as a potent alternative to aCGH (Raphael et al. 2003; Tuzun et al. 2005; Korbel et al. 2007; Lee et al. 2008), but their practical utility has been limited by the high cost of “long-read” sequencing, and the computational difficulties associated with interpreting “short-read” sequence data from complex genomes. Thus, while a number of PEM-based algorithms have been developed to identify SV from short-read sequence data (Chen et al. 2009; Hormozdiari et al. 2009; Korbel et al. 2009; Medvedev et al. 2009) and newer methods have been devised to map SVs at higher resolution (Lee et al. 2009; Sindi et al. 2009), all short-read PEM studies except one (Hormozdiari et al. 2009) have restricted their analyses to paired-end reads that map uniquely to the reference genome. This approach is not ideal given that SVs often involve repeated sequences such as segmental duplications and transposons. Finally, it has been difficult to evaluate structural mutation mechanisms in an unbiased way because genome-wide studies have thus far characterized relatively few breakpoints at single-nucleotide resolution (Korbel et al. 2007; Kidd et al. 2008; Kim et al. 2008; Perry et al. 2008), and the relative contribution of different molecular mechanisms remains a matter of debate.
Despite rapid advances in DNA sequencing technologies, affordable and accurate assembly of entire mammalian genomes remains years away. Indeed, even traditional methods have difficulty resolving complex genomic regions. In the interim, we argue that the optimal solution for breakpoint detection is a hybrid approach that combines PEM and local de novo assembly. Here we describe a general approach for unbiased detection, assembly, and mechanistic interpretation of SV breakpoints using both short and long reads, and apply it to whole-genome sequence data from two widely used inbred mouse strains. We show that our algorithms accurately identify diverse classes of SV, capture an unprecedented number of variants, and reveal novel breakpoint features. Of mechanistic significance, we report an abundance of complex SVs that appear to be derived from template switching during DNA replication or repair, and a propensity for duplicated genomic regions to generate new variants through mechanism(s) other than non-allelic homologous recombination (NAHR). A unique strength of this study is our choice of the mouse genome; because the reference genome is derived from an established inbred line (C57BL/6J), we were able to sequence an animal whose genome should be essentially identical to the reference. This important methodological control, which has not been present in any other PEM study, allowed us to distinguish true genetic variation from technical “noise” and poorly assembled genomic regions.
Results
Sequence data
The sequence data for this study come from two independent sources (Table 1). First, we used Illumina paired-end sequencing (Bentley et al. 2008) to generate roughly 130 million and 75 million paired-end reads (which we refer to as “matepairs”) each from the DBA/2J and C57BL/6J strains (hereafter referred to as DBA and B6, respectively). Matepairs had a median fragment size of 432 and 457 bp, resulting in 13.4 and 8.3 mean physical coverage for DBA and B6, respectively (Supplemental Figs. S1, S2; Table 1). We aligned reads with the BWA algorithm (Li and Durbin 2009), which identified ∼88% of DBA and 95% of B6 matepairs as “concordant,” meaning that they mapped to the reference genome with the expected orientation and size (i.e., median fragment size ± 10 median absolute deviations). We then remapped the remaining “discordant” matepairs with the more sensitive NOVOALIGN algorithm (C Hercus, unpubl.) to identify additional concordant matepairs and all discordant mapping positions. This two-tiered mapping approach provides high sensitivity with reasonable speed (Supplemental Fig. S3). We recorded all discordant alignments for matepairs without a single concordant mapping and retained those with 1000 or fewer mapping combinations. These mappings serve as the starting point for breakpoint prediction with HYDRA.
Table 1.
Summary of sequencing data

The number of Illumina fragments represents the sequences that passed Illumina's quality filters. WGS long-read counts represent the number of sequences for which accurate quality and clipping coordinates were available. The median Illumina size is the median mapping distance for pairs that aligned concordantly to the reference genome, whereas the WGS long-read figure reflects the median length of the sequences after quality and vector clipping. Median Illumina read length is the median length of the sequence on each end of each matepair. Observed coverage was computed empirically as the mean number of concordant pairs that spanned each base in the genome. The fraction aligned represents the proportion of reads that aligned somewhere in the genome, requiring that each end of Illumina reads aligned.
NA, Not applicable.
We obtained 8.0 million whole-genome shotgun (WGS) sequence reads (long-reads) from DBA (Mural et al. 2002) and 34.6 million from B6 (Mouse Genome Sequencing Consortium 2002) from the NCBI Trace Archive. These reads were generated by traditional Sanger sequencing and have a median size of 674 bp. We mapped long-reads to the reference genome using BLAT and recorded all possible mapping positions. We then classified reads as concordant or discordant, with concordant defined as reads with one or more mapping positions wherein at least 90% of the read aligned with no less than 90% identity. These data represent 1.9-fold sequence coverage of the DBA genome and 8.6-fold of B6, and allow for SV identification when distinct segments of the same discordant read map to disparate genomic positions. We refer to this approach as “split-read” mapping. Since the WGS data are derived from the same inbred lines used for Illumina sequencing, they provide an independent means to validate and assemble SV breakpoints predicted by HYDRA.
SV identification with HYDRA
The principles of PEM have been described in detail elsewhere (Medvedev et al. 2009). The fundamental notion is that variant breakpoints are apparent by the relative distance and orientation of discordant mapping positions (e.g., Fig. 1A). A more subtle yet crucial point is that in order to detect SVs arising from multi-copy sequences, one must examine discordant matepairs that have many possible mapping locations. This is a necessary consideration for mapping SV within segmental duplications, which are often unstable and hypervariable, and for mapping transposon insertions. Accordingly, our algorithm, HYDRA, is designed to localize SV breakpoints from discordant matepair mapping positions, where multiple mappings can be considered for each matepair.
Figure 1.

Overview of structural variation discovery pipeline. (A) Paired-end mapping signatures are shown for five different classes of structural variation as detected by paired-end mapping. Notably, each end of a given discordant matepair will map to each copy of a segmental duplication (blue, bottom left panel) when a mutation arises in one copy. In the case of a new transposon insertion (gray rectangle with red arrowhead, bottom right panel), ends of discordant matepairs that originated from the newly inserted sequence will map to all other similar elements in the genome. (Exp.) Experimental genome; (Disc.) discordant pairs from experimental genome; (Ref.) reference genome; (SD) segmental duplication. (B) Matepairs from the DBA strain are aligned to the mouse reference genome. (C) Clusters of discordant matepairs (often with multiple possible mapping combinations) are identified by HYDRA as putative variants (a deletion is shown). (D) Discordant WGS long-reads that corroborate the HYDRA call are assembled into breakpoint contigs (“breaktigs”) with phrap. The red asterisk indicates the nucleotide at which the SV breakpoint occurred. (E) Breaktigs are then aligned to the reference genome with MEGABLAST using very sensitive settings. The observed sequence homology (evident as alignment “overlap” at the breakpoint) in the resulting alignments is a hallmark of the causal SV mechanism, where negative overlap indicates the presence of an insertion or small-scale rearrangements directly at the breakpoint. (NAHR) Non-allelic homologous recombination.
HYDRA uses a heuristic approach to identify SV from matepairs with one or more mappings (see Methods). HYDRA compares discordant mappings to each other and identifies collections of matepairs, each derived from an independent DNA fragment, whose mappings corroborate a common variant. Two mappings “support” each other when they span the same genomic interval, have consistent orientations, and span a relative genomic distance consistent with the size of the input sequencing libraries. For each putative variant, HYDRA examines the supporting mappings and chooses the single mapping (the “seed”) that is supported by the most other mappings. Subsequent mappings are integrated into the variant call in decreasing order of their overlap with the seed. This process maximizes the number of mutually supporting mappings that define a variant. Each matepair is allowed to support only a single variant, and when multiple possible variants exist, HYDRA selects the one with the most supporting mappings.
HYDRA is designed to detect novel DNA junctions (breakpoints) in a “test” genome relative to a reference genome and can, in theory, detect any genetic event that generates a breakpoint, provided that both reads of a discordant matepair are aligned and span the breakpoint. Such events include deletions, duplications, inversions, insertions of arbitrary length in either the reference or the test genome, and large rearrangements such as translocations. HYDRA can detect breakpoints composed of either unique or repetitive sequence. In contrast, most existing algorithms are limited to uniquely mapped reads (Chen et al. 2009; Korbel et al. 2009; Sindi et al. 2009) or traditional clone-based Sanger data sets (Raphael et al. 2003; Tuzun et al. 2005; Lee et al. 2008). HYDRA does not classify variants nor group multiple breakpoints into a single variant call; however, it does optionally allow for matepairs that span the two breakpoints of an inversion to be integrated into a single call. This simple approach reduces assumptions about variant structure and increases sensitivity, but necessitates a subsequent classification step, which we performed using BEDTools (Quinlan and Hall 2010) and genome annotations.
HYDRA made 15,690 variant calls for the DBA strain and 1189 for the B6 strain, both relative to the reference genome (Supplemental Table S1). Based on the size distribution of the sequencing libraries, this data set has ∼400-bp resolution for insertions and deletions, and ∼100-bp resolution for duplications and inversions.
Although developed independently, HYDRA uses a similar clustering strategy to VariationHunter-SC (VH) (Hormozdiari et al. 2009), which is the only other published algorithm of which we are aware that, by design, detects multi-copy variants from next-generation sequence data. To compare the two algorithms, we ran VH on our DBA data set. The results were strikingly similar for the variant classes detected by both algorithms; VH detected 6366 deletions and 525 inversions, HYDRA called 6331 deletions and 495 inversions, and ∼95% of each algorithm's calls were reported by the other (Supplemental Fig. S4). However, HYDRA also made an additional 9359 calls that were not detected by VH since the current version of VH does not attempt to identify tandem duplications or insertions other than “basic” (or “spanned”) insertions (Hormozdiari et al. 2009; Medvedev et al. 2009). One major difference between the two algorithms is that, whereas HYDRA uses simple heuristics, VH uses a more sophisticated approach based on maximum parsimony. However, HYDRA is about 13 times faster than VH and, unlike VH, reports the alternate loci for variants in multicopy sequence, which is useful for genotyping SVs (since different loci may be chosen in different experiments).
Validation
We sought to evaluate the accuracy of HYDRA with WGS split-read mappings. Long-reads that span an SV breakpoint will map to the reference genome in split fashion (Mills et al. 2006; Ye et al. 2009), and when correctly oriented split-read mappings define a breakpoint in the same small interval predicted by HYDRA, this provides independent evidence of an SV (Figs. 1C, 2A) (see Methods). The B6 long-reads in this study were used to assemble the reference genome itself and thus serve as a control to ensure that split-reads are the product of genetic variation, not read-mapping artifacts or other sources of noise. This is a rigorous control given that the B6 data represent 8.6-fold genomic coverage. Initial validation experiments revealed 59% of variants in DBA to be false positives; however, we noticed that most resulted from matepairs that were judged to be discordant merely because neither BWA nor NOVOALIGN found the concordant mapping location(s). This effect persisted despite using very sensitive alignment settings. However, after mapping discordant matepairs with MEGABLAST and removing “low-confidence” HYDRA calls that contained concordant matepairs, the validation rate of “high-confidence” calls improved to ∼90% (Fig. 2B). The sensitivity of short-read alignment thus presents an obstacle for accurate SV detection in complex genomes. We note, however, that more sensitive alignment will likely improve the performance of all PEM algorithms, not just our own, and that this problem should be greatly ameliorated with longer, more accurate reads. Importantly, roughly half of all validated SVs include matepairs that map to multiple genomic positions, and validation rates are similar between HYDRA variants identified by either unique or repetitive matepairs, as well as among different SV classes (Fig. 2C,D).
Figure 2.

Validation of HYDRA calls. (A) HYDRA breakpoint calls in DBA were compared to split-read (S/R) alignments of WGS long-reads from both DBA and B6. Calls in DBA are corroborated by split-read mapping(s) from DBA that map within the predicted breakpoint interval in an orientation that is consistent with the HYDRA call. However, if one or more split-reads from B6 supports the call, it is refuted under the assumption that it originated from a mapping artifact or an error in the reference genome assembly. Cases in which split-reads were observed in neither strain were deemed inconclusive. Due to the relatively low coverage of DBA WGS long-reads, many HYDRA calls were inconclusive. (B) The number and validation rate for high-confidence and low-confidence HYDRA calls are shown for DBA. The 7784 final HYDRA calls represent the high-confidence calls that were not refuted by split-reads plus the low-confidence calls that were confirmed. (LSV) Local SVs, such as duplications, deletions, and inversions; (TEV) transposable element variants; (DI) “distant” insertions of non-transposon DNA from >1 Mb away (including retrogenes). For a detailed table describing the different SV classes and their validation rates, see Supplemental Table S1. (C) The validation rate (blue) and number (gray) of HYDRA SV calls is shown as a function of the mean number of mapping combinations observed among the supporting matepairs. (D) A comparison of the validation rate of HYDRA SV calls by the type of variant.
Reference genome “noise”
Our analysis of a B6 individual that is at most 30 generations separated from the reference genome (see Methods) represents a unique test of the specificity of short-read PEM. HYDRA reported 405 high-confidence breakpoint calls between our B6 sample and the reference, and this level of divergence is incompatible with such a short period of pedigreed inbreeding (Egan et al. 2007). One obvious source of “noise” is persistent read mapping artifacts. This appears to be a relatively minor source of false positives since 70% of the HYDRA calls in B6 were validated by long-reads (see above). False SV calls can also stem from loci that are poorly assembled in the reference genome or from discordant matepairs that originate from genomic regions that are absent from the reference (e.g., centromeres, telomeres, short arms, and gaps), yet systematically map back to incorrect genomic positions. Three lines of evidence point to these effects as a major source of noise. First, 73% of the B6 calls are also present in the DBA strain, which identifies the reference genome as the outlier. Second, 27% of the 405 SVs map to assembly gaps or unplaced contigs, and 59% map to segmental duplications, which are often difficult to assemble (Eichler 2001). Finally, only seven of the 405 (1.7%) calls in B6 are due to TE insertions in the reference genome. This is in stark contrast to the 37% of DBA SV calls that correspond to TE insertions in the reference (see below) and supports the argument that only about 20 B6 SV calls actually represent recent mutations.
To further assess the reference assembly, we used depth of coverage analysis (DOC) (Alkan et al. 2009; Chiang et al. 2009; Yoon et al. 2009) to identify 124 copy number “differences” between our B6 sample and the reference genome (Supplemental Fig. S5) (see Methods). These loci encompass 1.3% of the mouse genome (36 Mb) and colocalize with 41% of the B6 HYDRA calls (see above). The majority (77%) have more copies than the reference, which is consistent with the propensity of genome assemblers to collapse recent duplicates. The presence of misassembled loci is not entirely surprising given their correlation with known gaps and segmental duplications, yet some are dramatic. For example, the Sfi1 gene, which functions in spindle assembly and chromosome segregation (Salisbury 2004), is present in the reference genome as a single copy; instead, based on read depth, we estimate that the mouse genome has 20–30 copies of this gene. Increased coverage depth of Sanger reads has also been observed at this locus in the B6 strain (She et al. 2008). Thus, even a high-quality reference genome from an inbred organism is far from complete.
Identification of 7196 SVs between two “classical” inbred mouse strains
Despite a number of aCGH studies, the landscape of SV in the mouse genome remains poorly defined. Remarkably, we observed 7196 SVs between DBA and B6 (Table 2). These represent a nonredundant set of the 7784 “final” HYDRA breakpoint calls (Fig. 2B), which include high-confidence calls with a validation rate of ∼90% as well as low-confidence calls that were directly validated by split-read mapping. We did not consider SVs in DBA that were also identified in B6, and we excluded 348 calls that appeared to result from simple sequence repeat (SSR) length expansions or contractions, since these are generally not considered SV. The 7196 SVs affect the copy number or structure of 1709 genes, including 395 with known phenotypic effects (Bult et al. 2008), and may contribute in no small degree to the numerous phenotypic differences between the B6 and DBA strains.
Table 2.
Summary of final SVs

Intrachromosomal (LSV) calls represent variants where the ends of the HYDRA call were ≤1 Mb apart from one another on the same chromosome. TEV calls represent the union of variable transposon insertions in the B6 and DBA genomes. Ambiguous TEVs are cases in which the exact transposon class is uncertain because a recent mobile element was annotated on both ends of the HYDRA call. Distant insertions are either interchromosomal insertions or insertions of “distant” DNA (i.e., >1 Mb away) from the same chromosome. Gene features were classified as potentially affected if the HYDRA call overlapped with the annotation by at least 1 bp. Overlap with SDs was called if either end of the HYDRA call or 50% of its genomic span overlapped with an SD annotation. Note that unlike Figure 2B, the numbers in this table represent the final number of nonredundant variants, not the number of HYDRA breakpoint calls.
Most variation is caused by retrotransposons
Nearly 78% of SVs are insertions of DNA from distant loci, which we define as interchromosomal or >1 Mb away, and in most cases (∼90%) the inserted DNA corresponds to an annotated TE. The preponderance of transposable element variation (TEV) is not entirely surprising given that TE insertions account for 10% of spontaneous mouse mutants (Kazazian 2004), and that a previous study identified roughly 6700 variable long interspersed nuclear element (LINE1 [L1]) insertions in the reference using WGS data from multiple strains (Akagi et al. 2008). However, our identification of 5029 TEVs in a single strain comparison greatly exceeds previous reports. Of the 4412 TEVs that correspond to a single TE annotation, 43% are L1s, 52% are long terminal repeats (LTRs), and 5% are short interspersed nuclear elements (SINEs). In addition to disrupting functional elements, TEVs have regulatory potential due to their ability to attract epigenetic silencing factors and to serve as alternate promoters. In this context the TEVs that we report will be a valuable resource for probing the genetic basis of gene expression variation. As expected, roughly half of TEVs are insertions in the DBA genome. Unlike TE insertions in B6, which appear as “deletions” in DBA and are easily identified, virtually all TE insertions in the DBA genome are identified by matepairs with multiple mappings between distant loci and would be missed by algorithms that focus on uniquely mapped reads (Chen et al. 2009; Korbel et al. 2009; Sindi et al. 2009) or simple variants (Hormozdiari et al. 2009).
L1-encoded factors can also act on host mRNA transcripts, leading to duplicated genes that lack introns and promoters (retrogenes). We discovered 55 retrogene insertions in DBA, apparent as “deletions” that span introns. We identified an additional 438 insertions of DNA originating from distant loci that were not annotated as TEs in the reference genome. Some distant insertions appear to be unannotated TEs, and others retrotransposed copies of noncoding transcripts or genes lacking introns. Still others map to RNA, satellite or telomeric repeats, or high-copy segmental duplications. At present it is difficult to interpret these variants, but their validation rate is similar to other SV classes (Fig. 2). The prevalence of TEV, retrogenes, and distant insertions demonstrates that the most powerful force generating SV in the mouse genome is retrotransposition. While TEs are known to be more active in mouse than man (Kazazian 2004), this result nevertheless suggests that TE-mediated SV may also be more prevalent in the human genome than presently recognized (Xing et al. 2009).
Extent and distribution of local duplications, deletions, and inversions (LSVs)
The remaining 1610 SVs are non-transposon variants involving intrachromosomal genomic segments separated by <1 Mb (LSVs). Of these, 80% are deletions, 12% are tandem duplications, and 8% are inversions. The majority of LSVs are small (Supplemental Fig. S6), as has been observed in previous studies with similar resolution (Bentley et al. 2008; Ahn et al. 2009; Hormozdiari et al. 2009). While PEM alone cannot distinguish between a deletion in DBA and an insertion in the reference, manual inspection of read depth indicates that ∼90% of this class appear to indeed be deletions. The prevalence of deletions is consistent with aCGH studies, although, whereas this has previously been explained by aCGH detection bias (McCarroll et al. 2008; Cahan et al. 2009), our data demonstrate that deletions are, in fact, much more common than duplications and inversions. This may be caused by a propensity for nonhomologous end-joining (NHEJ) to generate simple deletions, which require just one chromosomal breakage, rather than duplications and inversions, which require two.
Next, we examined the genomic distribution of the 1610 LSVs. LSVs are found throughout the genome (Supplemental Fig. S7) but are not distributed evenly. Consistent with previous reports, we observe a twofold enrichment of LSVs at segmental duplications (Table 2), which comprise ∼5% of the genome (She et al. 2008). As expected, this enrichment is not observed for TEVs. Our data also show that this effect is stronger for duplications (∼34%) than for inversions (∼12%) and deletions (∼10%). This may reflect mechanistic differences in how distinct LSV classes are generated and suggests that duplicated regions may be especially prone to spawning new duplications.
To estimate the extent of LSV between the two strains, we compared our data set to the highest-resolution genome-wide aCGH study to date (Cahan et al. 2009), to another that targeted segmental duplications (She et al. 2008), and to the 197 CNVs we identified by DOC analysis. To account for the substantially lower resolution of the CNV data sets relative to HYDRA, we used a lenient measure of colocalization (10% reciprocal overlap). Our high-confidence LSV data set contains 30% of the CNVs reported by Cahan et al. (2009), 8% of those from She et al. (2008), and 21% of those identified by DOC. When considering a more inclusive event list consisting of alternate genomic locations for multicopy LSVs as well as low-confidence HYDRA calls (see Methods), this improves to 44%, 35%, and 59% for the three CNV data sets, respectively. In contrast, only 13% of the HYDRA SVs were reported as a CNV, and the 87% that are novel have a similar validation rate to those that are not. Therefore, our data significantly extend knowledge of LSV in the mouse genome. The moderate levels of overlap indicate that HYDRA captures a relatively distinct class of variation from aCGH and DOC. This appears to be predominantly due to CNVs that are present as tandem arrays or flanked by large repeats. Such variants are difficult (and sometimes impossible) to detect by PEM-based methods. This effect has been widely predicted (e.g., see Zhang et al. 2009a) and is supported by the prevalence of tandem duplications (She et al. 2008) and recurrent CNVs with indistinguishable breakpoints (Egan et al. 2007) in the mouse genome.
Merging of the SVs reported in this study and the two aCGH studies indicates two classical inbred strains differ by roughly 1900 LSVs larger than 1 kb in size, and that these comprise 1.9% of the mouse genome.
LSVs are often present in clusters
In addition to their colocalization with segmental duplications, LSVs have a markedly nonrandom genomic distribution and are often present in clusters of multiple adjacent or overlapping variants (Supplemental Fig. S8). Remarkably, 10.6% of HYDRA calls are found within 1 kb of another call, while only 1.6% are expected by chance. This effect is apparent at single-copy loci (66%) and at segmental duplications (34%). Such SV clusters could result when multiple independent mutations arise in close proximity, perhaps at an unstable genomic region, or at complex variants formed by multiple template switches at a stalled (FoSTeS) (Lee et al. 2007) or broken (MMBIR) (Smith et al. 2007; Hastings et al. 2009a) replication fork. Complex variants are often not directly supported by coverage depth, which raises a very important point: Multistep rearrangements can give rise to discordant read mapping patterns that suggest a duplication or deletion, yet do not involve substantial loss or gain of sequence. This exemplifies the inherent difficulty of reconstructing locus structure by PEM alone.
Characterization of 3316 breakpoint sequences at single-nucleotide resolution
Assessment of mechanism requires that SV breakpoints are mapped to single-nucleotide resolution, and historically this step has been a major bottleneck. Ideally, breakpoints would be assembled from the same reads used to predict SV, but our PEM data set has insufficient coverage for this purpose; instead, we used DBA long-reads obtained by WGS, a strategy that is analogous to mixed Illumina/454 Life Sciences (Roche) sequencing and applicable to forthcoming long-read platforms. For each validated SV we extracted the long-reads with split-read mappings within predicted breakpoint intervals (Fig. 2) and assembled them with phrap (P Green, unpubl.). We aligned the resulting breakpoint-containing contigs (henceforth referred to as “breaktigs”) to the reference genome using MEGABLAST and inspected/interpreted alignments using the PARASIGHT software (J Bailey and E Eichler, unpubl.). We retained breaktigs that unequivocally confirmed the SV predicted by HYDRA. The final data set is comprised of 3316 breaktigs, including 2145 TEVs and 1171 LSVs.
We first assessed the degree of alignment “overlap” present between the DNA segments adjacent to each breakpoint (Figs. 1E, 3). Overlap measures homology and thus suggests mechanism; extensive overlap indicates that an SV likely arose by NAHR, while little or no overlap implies that the variant arose through a mechanism that requires little or no homology, like NHEJ or template switching. In contrast, negative overlap indicates unaligned sequence at the breakpoint, as can occur due to inserted DNA or small-scale rearrangements.
Figure 3.

Characterization of 3316 breakpoint sequences. (A) Histogram of the alignment overlap at all 3316 assembled breakpoint sequences (breaktigs). Positive overlap indicates homology at the breakpoint, while negative overlap indicates the presence of an unaligned segment at the breakpoint, suggesting an insertion or small-scale rearrangement. (B) Histogram of the subset (2145 of 3316) of breakpoints that were determined to be transposon insertions (TEVs) based on TE annotations. Note that the majority of the breakpoints in A showing 3–10-bp and 10–20-bp overlap are explained by target-site duplications from LTR and LINE insertions, respectively. (C) Histogram of the 1171 duplication, deletion, and inversion (LSV) breakpoints. (D) For each of four different ranges (dashed lines) of observed homology at LSV breakpoints, the fraction of breakpoints that overlapped with six different repeat annotations is shown. In all four observed homology ranges, the observed overlap with segmental duplications is higher than the ∼5% null expectation. Whereas breakpoints having little or no homology (two left pie charts) typically only overlapped with SDs, breakpoints having >10 bp of homology overlapped more frequently with SDs and dispersed repeats. (Seg. dup.) Segmental duplications; (LINE) long interspersed nuclear elements; (LTR) long terminal repeats ; (SINE) short interspersed nuclear elements; (DNA trans.) DNA transposons; (SSR) simple sequence repeats. (NHEJ) nonhomologous end-joining; (NAHR) non-allelic homologous recombination. (E) Detailed histograms of C reflecting simple and complex LSV breakpoints, respectively, as defined in the text. (F) The distribution of observed combinations of breakpoints (at least one breakpoint of each type at a given complex locus) at complex loci. (del) Deletion; (dup) duplication; (ins) insertion; (inv) inversion.
When the entire data set is considered there are three predominant peaks in the distribution (Fig. 3A). The two peaks centered at ∼5 bp and ∼15 bp are explained by LTR and L1 insertions, respectively, since these sizes correspond to the target site duplications generated by their machinery (Galun 2003), and since annotated TEs account for the vast majority of these classes (Fig. 3B). The third peak is centered on 2–3 bp of overlap (Fig. 3C); most of these breakpoints represent LSVs and presumably result from a combination of end-joining and template-switching. Of the LSV breakpoints, 25% show microhomology (2–10 bp), 16% show no homology (0 or 1 bp overlap), and 33% show inserted DNA at the breakpoint (<0 bp overlap). Most breakpoint insertions are too small to discern whether they originate from distinct genomic loci or template-independent addition of nucleotides, as has been reported for NHEJ (Gu et al. 2007). We also note that apparent insertions can result from alignment artifacts caused by low quality or polymorphic reads. However, of the 28 breakpoint insertions larger than 20 bp, all but three appear to be derived from elsewhere in the genome (see below). These are best explained by a replication-based mechanism, not NHEJ. A small number of LSV breakpoints (19) show the 10–20 bp of homology indicative of L1-mediated retrotransposition, which is expected given the existence of B6 retrogene insertions in our LSV data set, but some breakpoints from this class could also be due to microhomology-mediated end-joining (MMEJ) (Yan et al. 2007) or microhomology-mediated break-induced replication (MMBIR) (Hastings et al. 2009a).
Interestingly, NAHR-derived breakpoints are very rare in our data set. Even using a lenient threshold for homology (>20 bp), only ∼4.3% of LSVs were generated by NAHR, with SINEs and SSRs the most common repeats found at the breakpoints (Fig. 3D). We caution that this is an underestimate of NAHR since our data set is strongly biased against variants formed by exchange between large (>500 bp) repeats. Nevertheless, these data show that a substantial amount of variation stems from mechanisms that require little or no DNA homology and that NAHR between small repeats is a minor source of SV.
Complex variants
Given our observation of clustered HYDRA variants (Supplemental Fig. S8) and the proposition that complex replication-mediated rearrangements might be common in the human genome (Hastings et al. 2009b; Zhang et al. 2009b), we examined our breakpoint data for evidence of complexity. The simplest definition of complexity is the presence of multiple breakpoints in close proximity. We identified 129 breaktigs that contained multiple breakpoints, each involving a distinct split-read mapping spanning >100 bp in the reference, and 22 loci at which breakpoints captured by distinct breaktigs mapped to within 1 kb of each other. We further identified an additional 28 breakpoints with insertions >20 bp in length. Of these, three were insertions of simple sequence, which may reflect template-independent synthesis during NHEJ. However, the remainder appear to entail complex rearrangements; six are insertions of DNA from <10 kb away, 14 are insertions from distant loci, and five are small-scale rearrangements that appear as insertions because they perturb alignment of the breakpoint region. Similar small-scale insertions and rearrangements have recently been reported at breakpoints in a tumor genome (Hampton et al. 2009), and at several large-scale rearrangement breakpoints identified among human individuals (Gajecka et al. 2008) and in the gibbon lineage (Girirajan et al. 2009).
While NHEJ could in theory produce the complex breakpoint patterns that we describe, this would require the multi-step formation of adjacent DNA lesions. In contrast, DNA replication-based mechanisms can entail multiple template switches per single broken/stalled replication fork (Lee et al. 2007; Smith et al. 2007), and are thus a more plausible explanation (Hastings et al. 2009b). Taken together, these data indicate that 16% of LSVs in the mouse genome have complex breakpoint patterns that are best explained by replication-based mechanisms such as MMBIR. We note that this may be an underestimate since this estimate only includes LSVs that were validated by long-reads, and since our measure of complexity does not include breaktigs separated by more than 1 kb of intervening DNA. Moreover, many LSVs caused by MMBIR may not result in detectably complex breakpoints and would be indistinguishable from NHEJ. However, it is also likely that some complex variants arose through independent mutation. For example, only 27% of breakpoints in complex LSVs clearly show microhomology (2–10 b), and some show characteristics of NAHR or retrotransposition (Fig. 3E).
We examined the composition of complex variants and noticed that some classes are more common than others. For example, despite the fact that deletions comprise 79% of breakpoints and duplications just 12%, adjacent deletion/duplication breakpoint combinations are far more common than deletion/deletion (Fig. 3F). This pattern is caused by several different types of rearrangements. The most common appears to be cases in which duplicated genomic segments are separated by intervening nonduplicated sequence, sometimes accompanying an actual deletion and sometimes not. Other common patterns include adjacent inversion breakpoints, deletions where one or more internal DNA segments are not deleted but rather transposed to the “edge” of the deleted segment (Fig. 4), and deletions where sequence has been inserted directly into the breakpoint. These patterns are difficult to explain with current models (Hastings et al. 2009b), but suggest that certain template switches are more common during replication than others.
Figure 4.

Visualizing a complex SV in a promoter region. (A) A snapshot of aligned sequence data at a validated SV locus from our local mirror of the UCSC Genome Browser (chr9: 98,880,333–98,889,602). At this locus, HYDRA detected one deletion and two inversion breakpoints in the DBA strain from the aligned discordant matepairs (red, those suggesting a deletion; blue, those suggesting an inversion), where F denotes a read mapping to the forward, or plus strand, and R the reverse strand. The dearth of uniquely aligned concordant matepairs (dark green) corroborates the deletion call. Note that a single concordant matepair is aligned within the span of the putative deletion where the two inversion breakpoints overlap, indicating that this segment is not deleted. Three WGS split-reads (gray) from the DBA strain also confirm the HYDRA calls and the observed complexity. (B) The three WGS split-reads were assembled into a 712-bp breakpoint sequence (breaktig) that was then aligned to the reference genome. The image displayed (using PARASIGHT) is representative of the 3316 such images we used to inspect assembled breakpoints. Aligned sections in black are in the same orientation in the breaktig and the reference genome, and the alignments in orange are in the opposite orientation. The complex variant involves two adjacent deletions of 2.5 kb and 0.9 kb, which are separated by an intervening ∼300-bp segment that was not deleted but, rather, inverted. An additional 15-bp deletion is present between the two rightmost alignments to the reference, but is difficult to see at this scale.
On the role of segmental duplications
Nearly all previous SV mapping studies have reported a correlation between structural variation and segmental duplications in the reference genome (Marques-Bonet et al. 2009), and this has generally been explained by the propensity of local repeats to undergo NAHR. We also observe this correlation, but in our data set it cannot be explained by NAHR alone. We define colocalization as an SD touching a breakpoint or encompassing 50% of the variant's span in the reference genome. We observe a twofold enrichment of SDs at LSV breakpoints. Notably, this effect is more pronounced for simple variants (2.1-fold, P < 0.001) than for complex variants with multiple breakpoints (1.5-fold, P < 0.001) (Supplemental Table S2). While the 90 simple breakpoints represent a twofold enrichment over the 43 that are expected by chance, only two contain the homology required for NAHR. A caveat is that some of this enrichment is caused by recent L1-mediated insertions in the reference genome that are annotated as SDs. To account for this effect, we removed 19 insertions that contained target-site duplications characteristic of L1 (10–20 bp). The resulting enrichment is milder but persists in a highly significant fashion (1.9-fold, P < 0.001). Thus, in our data set the enrichment of segmental duplications at LSV breakpoints cannot be explained by NAHR. This result suggests that localized genetic instability may often be the cause rather than the consequence of duplicated genome architecture.
Discussion
We have developed a genome-wide approach for identifying, assembling, and interpreting SV breakpoints, and applied it to two inbred mouse genomes. Our strategy combines PEM with short paired-end reads (HYDRA) and breakpoint definition with long-reads. Until accurate whole-genome assembly becomes feasible, it is likely that this approach (or some variant of it) will serve as the most powerful and economical means to study structural variation landscapes. We refined our method using data from the reference genome itself, and we demonstrated their accuracy at both unique and repetitive loci. We documented 7196 high-confidence SVs between the two strains and inspected 3316 breakpoint sequences at single-nucleotide resolution. These efforts resulted in five main findings: (1) most variation is caused by retrotransposition of LTRs, LINEs, and host-transcripts; (2) deletions are far more common than duplications and inversions; (3) most LSV breakpoints in our data set are explained by mechanisms requiring little or no homology, such as NHEJ or MMBIR; (4) 16% of LSVs are found in clusters or have breakpoints that have undergone small-scale rearrangement, and are therefore complex in nature; and (5) LSV breakpoints are significantly enriched at segmental duplications, but this effect cannot be explained solely by NAHR.
The lessons learned from this study can inform future sequence-based studies of SV. The fact that we re-sequenced a B6 individual that is so closely related to the reference genome allowed us to clearly distinguish bona fide genetic variation from spurious differences. In addition to known sequencing artifacts (Quail et al. 2008), as well as others that we have identified (see Methods), this unique control identified read-mapping and reference genome quality as major sources of false positives. These results underscore the need for continued efforts to improve reference genome assemblies for key species and illustrate that sacrificing short-read alignment sensitivity in the interest of speed and data storage comes at the cost of a substantial increase in false positives. While we note that longer and more accurate reads should mitigate the current alignment bottleneck, the challenge presented by interpreting multiple mappings will remain until read lengths span large genomic duplications. This will be especially true for efforts to resolve SV in complex genomic regions and highly rearranged cancer genomes. However, our results show that even with short-reads it is possible to accurately map SV in duplicated and repetitive genomic regions, provided that reads are mapped in a sensitive fashion and many possible mapping combinations are examined. Indeed, nearly half of the variants we report would have been missed were matepairs with multiple mappings excluded from our analysis. We acknowledge, however, that the discovery and interpretation of SV is easier in an inbred genome such as mouse, and that modifications may be necessary to achieve similar validation rates in studies of the human genome.
Our data provide a genome-wide demonstration that complex rearrangements are a major source of structural variation in mammalian genomes. We show that ∼16% of LSVs have complex breakpoint patterns and that certain complex variants are more common than others. These findings are consistent with the observation of multi-step template switching during break-induced replication (Smith et al. 2007), and with the replication-based models (FoSTeS and MMBIR) (Lee et al. 2007; Hastings et al. 2009a) that have been proposed to explain complex disease-causing mutations at several loci in the human genome (Lee et al. 2007; Carvalho et al. 2009; Zhang et al. 2009b). This observation is important because complex rearrangements can shuffle regulatory elements and lead to very different functional outcomes than simple duplication and deletion (Fig. 4; Zhang et al. 2009b). Moreover, complex variants may often arise during mitosis rather than meiosis and may be affected by cellular stress (Hastings et al. 2009a), which raises fundamental questions about their contribution to genome variation and disease.
Finally, our observation that the enrichment of segmental duplications at LSV breakpoints is largely independent of NAHR provides genome-wide evidence that localized genomic instability may often be caused by predisposing features other than repeats per se. It is noteworthy that a similar effect has been observed at non-NAHR breakpoints in a tumor genome (Hampton et al. 2009), and suggested by nonrecurrent disease-causing rearrangements that arise at or near SDs (Stankiewicz et al. 2003; Lee et al. 2006; Bauters et al. 2008; Carvalho et al. 2009) and by the prevalence of recent SDs that appear to be caused by NHEJ (Kim et al. 2008). A noncausative relationship has also been proposed to explain the correlation between segmental duplications and breakpoint reuse during mammalian genome evolution (Bailey et al. 2004). The underlying cause of this effect is not clear, but reports of instability caused by genome architecture (Stankiewicz et al. 2003), fragile sites (Durkin and Glover 2007), physical structure (Zhou and Mishra 2005), non-B DNA at palindromes (Inagaki et al. 2009), and specific elements (Myers et al. 2008) raise a host of possibilities. We expect that future application of genome-wide breakpoint characterization methods similar to those we describe will shed considerable light on this subject.
Methods
See Supplemental material for a detailed version of the Methods.
Illumina paired-end sequence data
We obtained one female mouse each from the C57BL/6J (B6) and DBA/2J (DBA) inbred strains from the Jackson Laboratory (Bar Harbor, ME). The B6 individual was obtained in January 2006. This mouse was a retired foundation stock breeder (F226) and is thus derived from the colony nucleus and should be minimally diverged from the reference genome (Mouse Genome Sequencing Consortium 2002). The DBA mouse was obtained in December 2004. We constructed paired-end sequencing libraries according to the manufacturer's protocols, as described (Bentley et al. 2008).
Paired-end sequence alignment and classification
We aligned all matepairs that passed our quality and complexity filters with BWA (Li and Durbin 2009). Since we found that BWA provides reasonable alignment sensitivity while using minimal computer memory and disk space, we use it as an initial screen to identify matepairs that are concordant with the mm9 reference sequence. Concordant matepairs were used for CNV detection via depth of coverage (DOC) analysis. All remaining matepairs that were either discordant with or did not align to the reference genome were realigned with NOVOALIGN (C Hercus, unpubl., http://www.novocraft.com/products.html#novoalign). We used NOVOALIGN as a secondary screen to identify additional concordant matepairs that were missed by BWA. We separately aligned each end of each remaining pair and recorded all possible mapping locations. We paired all possible NOVOALIGN alignments for each end of each pair using custom scripts and screened for combinations that proved to be concordant with the reference genome. For those matepairs that were still found to be discordant, we computed all possible mapping combinations.
We realigned each discordant matepair from each putative SV call with MEGABLAST (Zhang et al. 2000) and asked if any of the matepairs in an SV call were found to be concordant. If so, we classified the SV call as a low-confidence variant owing to the possibility that it was observed merely because of a lack of alignment sensitivity. SV calls where no matepairs were found to be concordant were classified as high-confidence variants.
WGS long-read alignment
All vector and quality trimmed WGS long-reads were aligned to mm9 with BLAT (Kent 2002). Long-reads were classified as either mapping concordantly or discordantly with the reference genome. In order to be classified as concordant, we required that a given read have at least one mapping where 90% of the read aligned in a single block and that 90% of the bases in the aligned portion matched the bases in the reference genome. All WGS reads that failed this check were classified as discordant.
Structural variation discovery with HYDRA
HYDRA is designed to compare discordant matepair mappings to one another, and to identify putative SVs as those that have a minimal number of matepairs with corroborating genomic positions, sizes, and read orientations. For the present study, we imposed three limits on read-mapping information: (1) we excluded known sequencing artifacts (see Supplemental Methods); (2) we excluded discordant matepairs with more than 1000 mapping combinations; (3) we excluded matepairs where both reads mapped to SSRs.
Preliminary screening for putative SV
HYDRA's speed comes largely by performing an efficient initial screen of all discordant mappings in search of evidence for potential SV. The four primary steps in this screening process are as follows:
We first determine which discordant mappings from each matepair should be retained for further SV discovery. Hydra allows one to retain (1) the mappings with the least combined (read1 + read2) edit distance (termed “best” mappings), (2) all mappings within a user-defined edit distance of the “best” mappings, or (3) all mappings regardless of edit distance. In this study, we retained only the “best” mappings.
We then group all remaining discordant mappings where the ends of the matepairs are aligned to the same chromosome(s) and in the same orientation(s). This preliminary screen segregates similar discordant mappings that together would corroborate a potential SV, thereby greatly reducing the number of mappings that must be directly compared to one another in order to detect an SV “cluster.”
- We then sort each group of mappings from Step 2 by the mapping distance between each end of the mapping (i.e., the mapping “length”). Once the mappings are sorted by length, we collect mappings whose lengths differ by no more than a user-specified “length deviation” (termed “maxLengthDev”) parameter, which is based on the insert size variation of the sequencing library. Specifically, for any two mappings i and j, we require:
All mappings whose lengths meet this restriction are grouped into putative SV clusters. At the end of this step, HYDRA has constructed clusters of mappings whose chromosome(s), orientation(s), and mapping lengths suggest potential SV.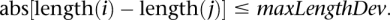
- The mappings within each putative cluster created in Step 3 are then sorted by their genomic coordinates. This step further refines putative clusters by requiring that discordant mappings localize to the same genomic region(s) and thus support the same putative SV breakpoint. Once mappings in each cluster are sorted by their genomic coordinates, HYDRA refines putative clusters by screening for mappings that span a common genomic interval and do not exceed a user-specified “nonoverlap” (termed “maxNonOverlap”) parameter, which is based on the insert size variation of the sequencing library. Specifically, for any two mappings i and j in a putative cluster, we require:
where leftStart is the leftmost coordinate of each mapping and rightEnd is the rightmost coordinate of each mapping. As illustrated in Supplemental Figure S9, this restriction is designed to prevent the clustering of discordant mappings that have similar lengths yet do not support the same SV breakpoint. At the end of this step, HYDRA has identified putative SV clusters from mappings that have similar lengths and orientations and support the same potential SV.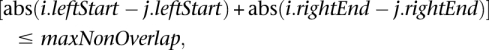
Refining SV breakpoints
After the preliminary screening for putative SV, clusters having a sufficient number of supporting matepairs (in this study, two) are further processed in an effort to choose the best set of mappings with which to describe the SV breakpoint. First, HYDRA compares each mapping (i) in each cluster to all the other mappings (j) in that cluster and tabulates how many other mappings meet both the maxLengthDev and maxNonOverlap restrictions with respect to the ith mapping. Mappings that meet both restrictions with respect to i are classified as “supporting” the ith mapping.
HYDRA chooses the mapping that has the most “support” from the other mappings in the cluster as the “seed” mapping for the variant. Proper seed selection maximizes the resolution of the putative breakpoint by incorporating the most supporting mappings. The variant is refined by iteratively adding the mapping with the next most support until we encounter a mapping that does not support all of the previously added mappings. We are ultimately left with a set of discordant mappings that mutually corroborate the same SV and whose mappings collectively define the breakpoint of the variant as precisely as possible with the available data.
Resolving ambiguities arising from multiple mappings
Since HYDRA may interrogate multiple mappings per discordant matepair, there are cases in which the mappings for a given discordant pair support multiple structural variants. In such cases, we select the SV call that is supported by the most discordant mappings. In cases in which multiple competing SV calls have the same level of mapping support, we select the variant with the least number of mismatches and gaps among all the supporting mappings. In cases of a tie, a variant mapping location is selected randomly; thus, the final set of putative variants are those with the strongest support from the discordant mappings. Importantly, we also report those variants whose supporting mappings were re-distributed to other more well-supported variants so that intersample variant comparisons can be made.
A cautionary note
HYDRA may report more false-positive SV calls than observed in this study if artifactual discordant matepair mappings are not cleansed from the input file. We advise that potential users of this algorithm carefully read the detailed Supplemental Methods.
Validation
True HYDRA SV calls between DBA and B6 should be corroborated by DBA WGS long-reads but not B6 long-reads. Specifically, bona fide SV in DBA should be supported by one or more DBA long-reads with a so-called “split-read” alignment. We developed a pipeline to screen each putative DBA SV for supporting split-read alignments from both DBA and B6. Since HYDRA SV calls typically do not map the exact breakpoint(s) of a given SV whereas the split-reads do, we assessed split-read mappings in the predicted breakpoint-containing interval. This interval is defined by the insert size of the Illumina sequencing libraries. We then required any observed split-read in B6 and DBA to have 90% overlap with the predicted breakpoint interval in order to be included in our SV validation scheme.
Breakpoint assembly and visualization
Breakpoint contig (“breaktig”) assembly with long-reads
The HYDRA variant calls that were confirmed by DBA split-reads were further characterized in an effort to identify the exact nucleotide at which the SV breakpoint(s) occurred. In such cases, we assembled the corroborating long-reads with phrap (P Green, unpubl., http://www.phrap.org) using default parameters. When a single read confirmed the HYDRA call, we attempted to identify the breakpoint from that single read. The resulting breaktigs were then aligned to the variant locus with MEGABLAST.
Calling and annotating SV breakpoints
The best alignments between the assembled breaktig and the reference locus were used to identify the type and precise breakpoint for each SV. TE insertions were identified as apparent deletions in the DBA genome (both flanking alignments were in the correct orientation) (see Fig. 3B) where the supposedly deleted region was at least 50% comprised of recent TE annotations in the reference genome (see below). The remaining breakpoints were classified as deletions, duplications, or inversions.
Visualization and homology estimates
We visualized alignments between each breaktig and the reference genome with a custom pipeline built on the PARASIGHT alignment visualization software (J Bailey and E Eichler, unpubl., http://eichlerlab.gs.washington.edu/jeff/parasight). We developed a script to compute the amount of homology (overlap) observed at each breakpoint. When no overlap was observed, we classified the SV as a “flush” breakpoint. When negative overlap exceeding 20 bp was observed (i.e., there was a gap in the breaktig between two adjacent alignments to the reference genome) (see Figs. 1E, 3C), we investigated the possibility that DNA was inserted in the DBA genome at the breakpoint. Positive overlap indicates local sequence homology at the breakpoint. Substantial (e.g., ≥20 bp) sequence homology is indicative of NAHR, while <20 bp of homology indicates either NHEJ, target-site duplication (TSD) caused by TE insertions, or MMBIR.
Comparing HYDRA SV to genome annotations
We developed a new software suite (BEDTools, http://code.google.com/p/bedtools) (Quinlan and Hall 2010), to facilitate the annotation and functional characterization of the SVs discovered in this study with respect to genome annotations in the UCSC Genome Browser's BED format (Kent et al. 2002). We used BEDTools to compute the amount of overlap between our variants and existing and custom genome annotations. Precise definitions for overlap with various genomic features are available in the Supplemental Methods.
Acknowledgments
We thank A. Prorock for assistance with DNA sequencing; W.R. Pearson and S. Bekiranov for useful discussions; and G. Marth, C. Stewart, D. Schones, and S. Nuñez for comments on the manuscript. The work was funded by an NRSA postdoctoral fellowship 1F32HG005197-01 (A.Q.), a Burroughs Wellcome Fund Career Award (I.H.), an NIH Director's New Innovator Award DP2OD006493-01 (I.H.), and by the UVA School of Medicine (M.L.L.).
Footnotes
[Supplemental material is available online at http://www.genome.org. The sequence data generated for this study have been submitted to the Short Read Archive (http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi) under accession no. SRA010027. Structural variant calls have been submitted to dbVAR (http://www.ncbi.nlm.nih.gov/projects/dbvar/) under accession no. nsdt19. Source code for the HYDRA algorithm is available at http://code.google.com/p/hydra-sv/.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.102970.109.
References
- Ahn SM, Kim TH, Lee S, Kim D, Ghang H, Kim BC, Kim SY, Kim WY, Kim C, Park D, et al. 2009. The first Korean genome sequence and analysis: Full genome sequencing for a socio-ethnic group. Genome Res 19: 1622–1629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akagi K, Li J, Stephens RM, Volfovsky N, Symer DE 2008. Extensive variation between inbred mouse strains due to endogenous L1 retrotransposition. Genome Res 18: 869–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan C, Kidd JM, Marques-Bonet T, Aksay G, Antonacci F, Hormozdiari F, Kitzman JO, Baker C, Malig M, Mutlu O, et al. 2009. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet 41: 1061–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Baertsch R, Kent WJ, Haussler D, Eichler EE 2004. Hotspots of mammalian chromosomal evolution. Genome Biol 5: R23 http://genomebiology.com/2004/5/4/R23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauters M, Van Esch H, Friez MJ, Boespflug-Tanguy O, Zenker M, Vianna-Morgante AM, Rosenberg C, Ignatius J, Raynaud M, Hollanders K, et al. 2008. Nonrecurrent MECP2 duplications mediated by genomic architecture-driven DNA breaks and break-induced replication repair. Genome Res 18: 847–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, et al. 2008. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456: 53–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bult CJ, Eppig JT, Kadin JA, Richardson JE, Blake JA 2008. The Mouse Genome Database (MGD): Mouse biology and model systems. Nucleic Acids Res 36: D724–D728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahan P, Li Y, Izumi M, Graubert TA 2009. The impact of copy number variation on local gene expression in mouse hematopoietic stem and progenitor cells. Nat Genet 41: 430–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Zhang F, Liu P, Patel A, Sahoo T, Bacino CA, Shaw C, Peacock S, Pursley A, Tavyev YJ, et al. 2009. Complex rearrangements in patients with duplications of MECP2 can occur by fork stalling and template switching. Hum Mol Genet 18: 2188–2203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP, et al. 2009. BreakDancer: An algorithm for high-resolution mapping of genomic structural variation. Nat Methods 6: 677–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang DY, Getz G, Jaffe DB, O'Kelly MJ, Zhao X, Carter SL, Russ C, Nusbaum C, Meyerson M, Lander ES 2009. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat Methods 6: 99–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durkin SG, Glover TW 2007. Chromosome fragile sites. Annu Rev Genet 41: 169–192 [DOI] [PubMed] [Google Scholar]
- Egan CM, Sridhar S, Wigler M, Hall IM 2007. Recurrent DNA copy number variation in the laboratory mouse. Nat Genet 39: 1384–1389 [DOI] [PubMed] [Google Scholar]
- Eichler EE 2001. Segmental duplications: What's missing, misassigned, and misassembled and should we care? Genome Res 11: 653–656 [DOI] [PubMed] [Google Scholar]
- Gajecka M, Gentles AJ, Tsai A, Chitayat D, Mackay KL, Glotzbach CD, Lieber MR, Shaffer LG 2008. Unexpected complexity at breakpoint junctions in phenotypically normal individuals and mechanisms involved in generating balanced translocations t(1;22)(p36;q13). Genome Res 18: 1733–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galun E 2003. Retroelements Kluwer Academic Publishers, Dordrecht, The Netherlands [Google Scholar]
- Girirajan S, Chen L, Graves T, Marques-Bonet T, Ventura M, Fronick C, Fulton L, Rocchi M, Fulton RS, Wilson RK, et al. 2009. Sequencing human-gibbon breakpoints of synteny reveals mosaic new insertions at rearrangement sites. Genome Res 19: 178–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Lu H, Tippin B, Shimazaki N, Goodman MF, Lieber MR 2007. XRCC4:DNA ligase IV can ligate incompatible DNA ends and can ligate across gaps. EMBO J 26: 1010–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton OA, Den Hollander P, Miller CA, Delgado DA, Li J, Coarfa C, Harris RA, Richards S, Scherer SE, Muzny DM, et al. 2009. A sequence-level map of chromosomal breakpoints in the MCF-7 breast cancer cell line yields insights into the evolution of a cancer genome. Genome Res 19: 167–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, Lupski JR 2009a. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet 5: e1000327. doi: 10.1371/journal.pgen.1000327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Lupski JR, Rosenberg SM, Ira G 2009b. Mechanisms of change in gene copy number. Nat Rev Genet 10: 551–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari F, Alkan C, Eichler EE, Sahinalp SC 2009. Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes. Genome Res 19: 1270–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C 2004. Detection of large-scale variation in the human genome. Nat Genet 36: 949–951 [DOI] [PubMed] [Google Scholar]
- Inagaki H, Ohye T, Kogo H, Kato T, Bolor H, Taniguchi M, Shaikh TH, Emanuel BS, Kurahashi H 2009. Chromosomal instability mediated by non-B DNA: Cruciform conformation and not DNA sequence is responsible for recurrent translocation in humans. Genome Res 19: 191–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian HH Jr 2004. Mobile elements: Drivers of genome evolution. Science 303: 1626–1632 [DOI] [PubMed] [Google Scholar]
- Kent WJ 2002. BLAT—the BLAST-like alignment tool. Genome Res 12: 656–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D 2002. The Human Genome Browser at UCSC. Genome Res 12: 996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, Hansen N, Teague B, Alkan C, Antonacci F, et al. 2008. Mapping and sequencing of structural variation from eight human genomes. Nature 453: 56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim PM, Lam HY, Urban AE, Korbel JO, Affourtit J, Grubert F, Chen X, Weissman S, Snyder M, Gerstein MB 2008. Analysis of copy number variants and segmental duplications in the human genome: Evidence for a change in the process of formation in recent evolutionary history. Genome Res 18: 1865–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, Kim PM, Palejev D, Carriero NJ, Du L, et al. 2007. Paired-end mapping reveals extensive structural variation in the human genome. Science 318: 420–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel JO, Abyzov A, Mu XJ, Carriero N, Cayting P, Zhang Z, Snyder M, Gerstein MB 2009. PEMer: A computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data. Genome Biol 10: R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Inoue K, Cheung SW, Shaw CA, Stankiewicz P, Lupski JR 2006. Role of genomic architecture in PLP1 duplication causing Pelizaeus-Merzbacher disease. Hum Mol Genet 15: 2250–2265 [DOI] [PubMed] [Google Scholar]
- Lee JA, Carvalho CM, Lupski JR 2007. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131: 1235–1247 [DOI] [PubMed] [Google Scholar]
- Lee S, Cheran E, Brudno M 2008. A robust framework for detecting structural variations in a genome. Bioinformatics 24: i59–i67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Hormozdiari F, Alkan C, Brudno M 2009. MoDIL: Detecting small indels from clone-end sequencing with mixtures of distributions. Nat Methods 6: 473–474 [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques-Bonet T, Girirajan S, Eichler EE 2009. The origins and impact of primate segmental duplications. Trends Genet 25: 443–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wysoker A, Shapero MH, de Bakker PI, Maller JB, Kirby A, et al. 2008. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet 40: 1166–1174 [DOI] [PubMed] [Google Scholar]
- Medvedev P, Stanciu M, Brudno M 2009. Computational methods for discovering structural variation with next-generation sequencing. Nat Methods 6: S13–S20 [DOI] [PubMed] [Google Scholar]
- Mills RE, Luttig CT, Larkins CE, Beauchamp A, Tsui C, Pittard WS, Devine SE 2006. An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res 16: 1182–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouse Genome Sequencing Consortium 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562 [DOI] [PubMed] [Google Scholar]
- Mural RJ, Adams MD, Myers EW, Smith HO, Miklos GL, Wides R, Halpern A, Li PW, Sutton GG, Nadeau J, et al. 2002. A comparison of whole-genome shotgun-derived mouse chromosome 16 and the human genome. Science 296: 1661–1671 [DOI] [PubMed] [Google Scholar]
- Myers S, Freeman C, Auton A, Donnelly P, McVean G 2008. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat Genet 40: 1124–1129 [DOI] [PubMed] [Google Scholar]
- Perry GH, Ben-Dor A, Tsalenko A, Sampas N, Rodriguez-Revenga L, Tran CW, Scheffer A, Steinfeld I, Tsang P, Yamada NA, et al. 2008. The fine-scale and complex architecture of human copy-number variation. Am J Hum Genet 82: 685–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail MA, Kozarewa I, Smith F, Scally A, Stephens PJ, Durbin R, Swerdlow H, Turner DJ 2008. A large genome center's improvements to the Illumina sequencing system. Nat Methods 5: 1005–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM 2010. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raphael BJ, Volik S, Collins C, Pevzner PA 2003. Reconstructing tumor genome architectures. Bioinformatics 19: ii162–ii171 [DOI] [PubMed] [Google Scholar]
- Salisbury JL 2004. Centrosomes: Sfi1p and centrin unravel a structural riddle. Curr Biol 14: R27–R29 [DOI] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Maner S, Massa H, Walker M, Chi M, et al. 2004. Large-scale copy number polymorphism in the human genome. Science 305: 525–528 [DOI] [PubMed] [Google Scholar]
- She X, Cheng Z, Zollner S, Church DM, Eichler EE 2008. Mouse segmental duplication and copy number variation. Nat Genet 40: 909–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindi S, Helman E, Bashir A, Raphael BJ 2009. A geometric approach for classification and comparison of structural variants. Bioinformatics 25: i222–i230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CE, Llorente B, Symington LS 2007. Template switching during break-induced replication. Nature 447: 102–105 [DOI] [PubMed] [Google Scholar]
- Stankiewicz P, Shaw CJ, Dapper JD, Wakui K, Shaffer LG, Withers M, Elizondo L, Park SS, Lupski JR 2003. Genome architecture catalyzes nonrecurrent chromosomal rearrangements. Am J Hum Genet 72: 1101–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, Pertz LM, Haugen E, Hayden H, Albertson D, Pinkel D, et al. 2005. Fine-scale structural variation of the human genome. Nat Genet 37: 727–732 [DOI] [PubMed] [Google Scholar]
- Xing J, Zhang Y, Han K, Salem AH, Sen SK, Huff CD, Zhou Q, Kirkness EF, Levy S, Batzer MA, et al. 2009. Mobile elements create structural variation: Analysis of a complete human genome. Genome Res 19: 1516–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, Gumaste S, Geyer M, Zarrin AA, Manis JP, et al. 2007. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature 449: 478–482 [DOI] [PubMed] [Google Scholar]
- Ye K, Schulz MH, Long Q, Apweiler R, Ning Z 2009. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25: 2865–2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S, Xuan Z, Makarov V, Ye K, Sebat J 2009. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res 19: 1586–1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Schwartz S, Wagner L, Miller W 2000. A greedy algorithm for aligning DNA sequences. J Comput Biol 7: 203–214 [DOI] [PubMed] [Google Scholar]
- Zhang F, Gu W, Hurles ME, Lupski JR 2009a. Copy number variation in human health, disease, and evolution. Annu Rev Genomics Hum Genet 10: 451–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Khajavi M, Connolly AM, Towne CF, Batish SD, Lupski JR 2009b. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat Genet 41: 849–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Mishra B 2005. Quantifying the mechanisms for segmental duplications in mammalian genomes by statistical analysis and modeling. Proc Natl Acad Sci 102: 4051–4056 [DOI] [PMC free article] [PubMed] [Google Scholar]