

Abstract
Glycogen synthase kinase-3 beta (GSK3β) is a multifunctional serine/threonine kinase which was originally identified as a regulator of glycogen metabolism. It plays a key role in the regulation of numerous signalling pathways including cellular process such as cell cycle, inflammation and cell proliferation. Over the last few years there is a considerable rise in the number of journals and patents publication by different workers worldwide. Many pharmaceutical companies are focusing on GSK3β as a therapeutic target for the treatment of disease conditions. The present review is focused on signalling pathways of different disease conditions where GSK3β is implicated. In this review, we present a comprehensive map of GSK3β signalling pathways in disease physiologies. Structural analysis of GSK3β along with molecular modelling reports from numerous workers are reviewed in context of design and development of GSK3β inhibitors. Patent landscape of the small molecule modulators is profiled. The chemo space for small molecule modulators extracted from public and proprietary Kinase Chembiobase for GSK3β are discussed. Compounds in different clinical phases of discovery are analysed. The review ends with the overall status of this important therapeutic target and challenges in development of its modulators.
Keywords: development of modulators, GSK3β, therapeutic landscape
Introduction
Glycogen synthase kinase-3 beta (GSK3β) – also known as human tau protein kinase (TPK I) is a serine/threonine protein kinase discovered in 1980. It plays a key role in transduction of regulatory and proliferative signals arising out of the cells, cytoskeletal proteins and transcription factors. Early interest on GSK3β as a diabetes target arose from its potential to modulate blood glucose levels (Embi et al., 1980). GSK3β gained prominence as a potential drug target when its importance in type 2 diabetes and obesity was determined by in vitro and in vivo experiments (Gum et al., 2003; Ring et al., 2003). The target gained further interest when it was discovered that it plays various roles in other disease physiologies. It has implications in Alzheimer's disease (AD) and mood disorders (Hsiung et al., 2003). Recently, it has been associated with Osteoporosis (Smith and Frenkel, 2005), Atherosclerosis (Robertson et al., 2006) and Cancer (Inoki et al., 2006). It was also reported to play a role in Cardiac Hypertrophy (Morisco et al., 2001). It is unique in that it is constitutively active in cells and its inhibition is responsible for cell signalling (Inoki et al., 2006). Phosphorylation of the residue Tyrosine in 216th position results in the constitutive activity of GSK3β and believed to be important target for signal transduction (Bhat et al., 2000; Sayas et al., 2006).
Structure elucidation of GSK3β and mode of interactions with ligands has gained considerable interest. The protein data bank (PDB) (Berman et al., 2000) has documented several X-ray GSK3β-ligand complexes. In addition to these structures in the public domain, an apo X-ray crystal structure is reported in patent literature (Bussiere et al., 2002).
The availability of the structural information has led to the molecular modelling studies on this target. Several molecular modelling studies of GSK3β-ligand complexes have been reported. Virtual high-throughput screening, Structure Activity Relationship and 3D-Quantitative Structure Activity Relationship studies were reported by many workers (Martinez et al., 2005; Polgar et al., 2005; Lather et al., 2008; Prasanna et al., 2009). Modelling studies from our group, reported the binding mode of the protein-ligand complexes of GSK3β (Gadakar et al., 2007). This study focused on cross-docking experiments of ligands with the structures in PDB. Also reported in this study are the results of enrichment studies for the retrieval of known actives from a decoy data set obtained from Jubilant's KinaseChembiobase™.
Even though GSK3β is an interesting target for drug discovery, its multiple roles in different signalling pathways raises the issue of druggability and selectivity. Cohen (2001) pointed out a danger, that the prolonged use of GSK3β inhibitor could be potentially oncogenic. This becomes a critical bottleneck in the design of GSK3β inhibitors for lifelong diseases like diabetes and neurodegenerative disorders. However, Frame and co-workers in 2001 had reported a novel approach to the design of GSK3β inhibition without effecting the wingless integration (WNT) signalling which is primarily responsible for the cause of cancer. They are of the view that mutation of Arg-96 abolishes the phosphorylation of ‘primed’ glycogen synthase as well as inhibition by PKB-mediated phosphorylation of Ser-9. Hence, the phosphorylated N terminus acts as a pseudosubstrate, occupying the same phosphate binding site used by primed substrates. Significantly, this mutation does not affect phosphorylation of ‘nonprimed’ substrates in the WNT signalling pathway (Frame et al., 2001). This hypothesis avoids the oncogenic potential of GSK3β inhibitors and has led to new approaches to the design and development of more selective GSK3β inhibitors for the treatment of diabetes without causing cancer. A similar approach was also reported by Martinez and Perez for the design and development of GSK3β inhibitor for AD avoiding the oncogenic potential of GSK3β inhibitors (Martinez and Perez, 2008).
Glycogen synthase kinase-3 beta as a key drug target is getting established, with some compounds in clinical phase of drug discovery and development. In this review we present a comprehensive role of GSK3β in metabolic and other therapeutic indications to elucidate a full spectrum of its function (Figure 1). The data were mined and analysed from different public and commercial databases like KEGG, Biocarta, Pathart™. Analysis of these databases demonstrates that GSK3β is important in various diseases causing mechanisms like different types of cancer, diabetes, cardiac hyperthrophy, Alzheimer and other central nervous system (CNS) disorders. We have also analysed and discussed the chemo-space for the development of modulators for GSK3β and have highlighted the present status of compounds in clinical phase of drug development.
Figure 1.
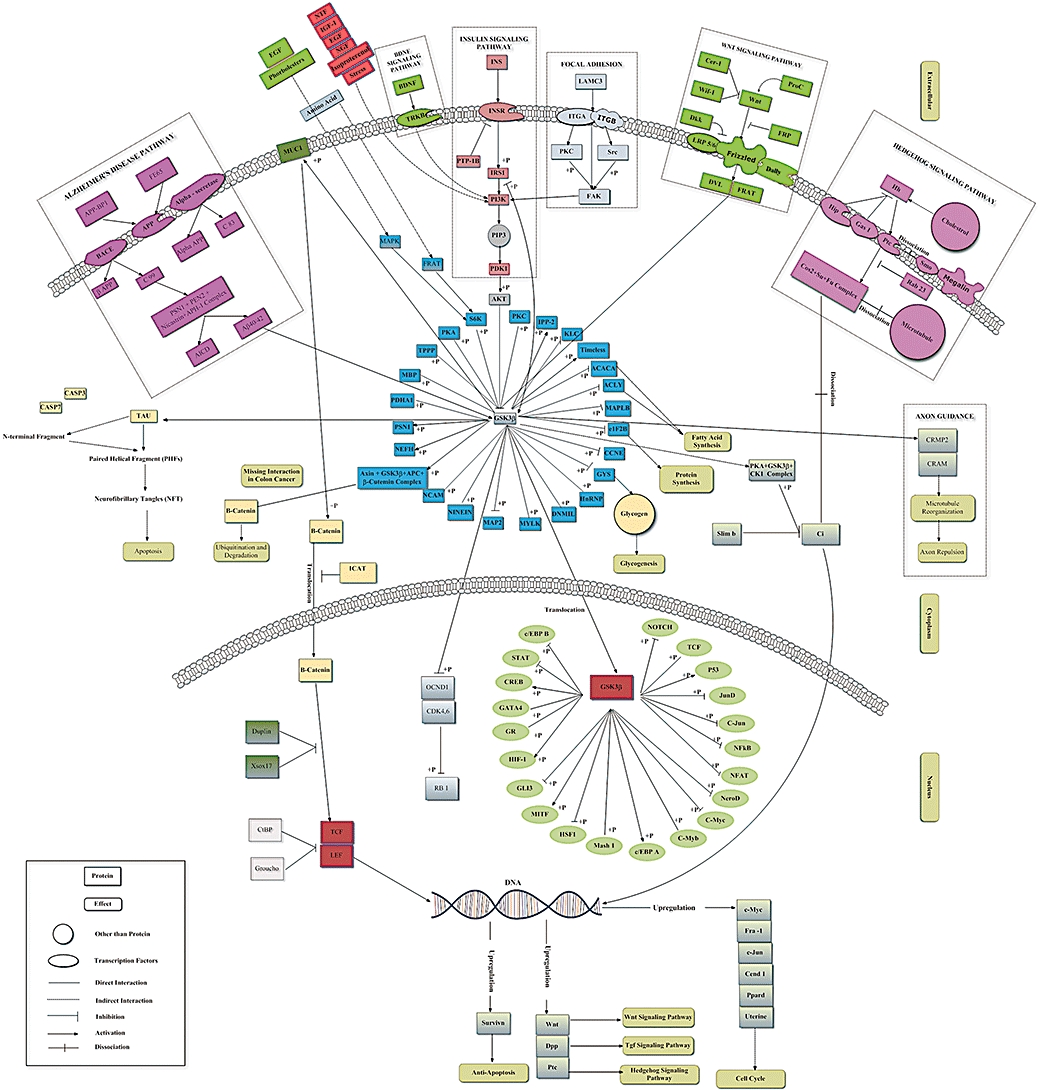
Glycogen synthase kinase-3 beta (GSK3β) signalling pathway map. The interaction of GSK3β with different substrates along with its involvement in different disease causing pathway is shown. Also various cytosolic and nuclear substrates are also shown.
Structural features of GSK3β
Due to the growing significance of GSK3β as a therapeutic target in various disease areas, we have undertaken a study of the different features of GSK3β with respect to its sequences and X-ray crystal structure data. The X-ray crystal structures of GSK3β are of moderate to high resolution (from 3.2 Å to 1.8 Å). The ligands binding to the protein belongs to different chemical scaffolds (Gadakar et al., 2007). Multiple sequence analysis – MSA (Thompson et al., 1994) of the reported structures in PDB revealed that the sequences are across the species and show exceptional conservation at binding site with few exceptions in the N and C terminal regions. A molecular overlay of these structures based on the Cα coordinates for understanding the different ligand binding pockets was carried out (Figure 2). It was observed that GSK3β has three binding sites (Figure 2A): (i) ATP site [Leu132, Tyr134, Val135, Pro136, Arg141 (Figure 2B)]; (ii) Axin Binding site [Lys85, Asp133, Val135, Lys183, Asp200 (Figure 2C)]; and (iii) Priming site Arg96 [Arg180, Ser203, Lys205, Val214 (Figure 2D)]. The Axin site is the smallest of the three pockets and can be also termed as a general extension of the ATP binding active site.
Figure 2.
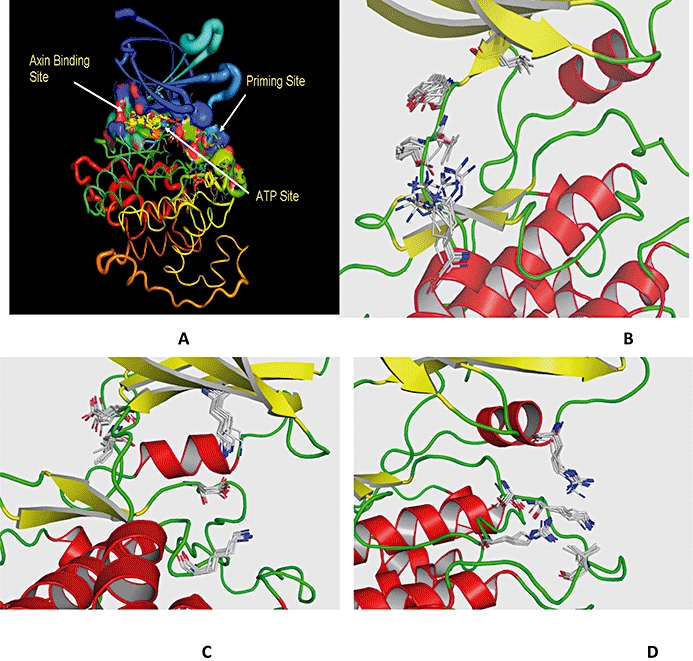
Different binding pockets of GSK3β. (A) Overlay of X-ray structures at Cα depicting different binding sites for GSK3β; (B) ATP site; (C) axin binding site; (D) priming site. The residues of each site are overlaid to understand the mobility of each residue. The helices are denoted in red, β-sheets in yellow and loops in green colour. The residues are shown in elemental colours. GSK3β, glycogen synthase kinase-3 beta.
We have carried out an analysis of the main chain and side chain dihedral angles of GSK3β in these structures. The analysis has thrown light on the rigidity/flexibility of the protein's backbone at the Cα and preferred side chain Cβ, Cγ positions. These studies were carried to understand the mobility and major geometries of the ligand binding pockets to assess the available 3D space for design specific modulators of GSK3β. It was observed that in majority of the cases, the backbone is rigid while the side chains show considerable mobility. We observed considerable protein mobility in the ligand binding pockets and this depends on the ligands and its binding modes (unpublished data). These geometries are of considerable interest for the design and development of GSK3β specific modulators. Any mutations at the residues adjacent to the binding site residues also influence the mobility of the backbone as evident from these studies.
GSK3β and human disease
There have been periodic reviews on the role of GSK3β in human disease physiologies over the years. Frame and Cohen (2001), elaborately reviewed the role of GSK3β in disease signalling pathways. Recent reports based on the disease signalling mechanism of GSK3β have thrown light on the significance of this target in various human diseases and the importance of understanding its full signalling spectrum (Doble and Woodgett, 2003). It was observed in these reports that since its discovery in the involvement of type 2 diabetes, GSK3β was discovered to play a role in many other disease indications making it a lucrative target as well as a complex one. Some of the important signalling pathways in which GSK3β plays a critical role resulting in the disease conditions in humans are discussed as under.
Insulin signalling pathway
Malfunctioning of insulin signalling pathway leads to diabetes. Dent et al. (1990) has described the molecular mechanism by which insulin stimulates glycogen synthesis in mammalian skeletal muscle. Type 2 diabetes was the first disease condition implicated to GSK3β, due to its negative regulation of several aspects of insulin signalling pathway (Embi et al., 1980). In this pathway 3-phosphoinositide-dependent protein kinase activates AKT which in turn, inactivates GSK3β. This inactivation of GSK3β leads to the dephosphorylation and activation of glycogen synthase which helps glycogen synthesis. (Cohen et al., 1997).
Sung and co-workers in 1998 had reported that non selective GSK3β inhibitors may be useful as therapeutics for the treatment of the insulin resistance in type 2 diabetes (Sung et al., 1998). Later on, it was reported that the inhibition of GSK3β improves insulin action and glucose metabolism in human skeletal muscle (Nikoulina et al., 2002). Inhibitors of GSK3β are expected to affect lowering of plasma glucose similar to insulin, making GSK3β an attractive target for the treatment of type 2 diabetes (Dokken et al., 2005). Treatment with GSK3β inhibitors caused improvement in glucose disposal, which could be attributed to an approximate two-fold increase in liver glycogen synthesis. This result was observed in oral glucose tolerance tests and euglycemic-insulinemic clamp studies in Zucker diabetic fatty (fa/fa) rats (Cline et al., 2002). Non-Selective GSK3β inhibition enhanced insulin action in insulin-resistant skeletal muscle of the prediabetic obese Zucker rat, in part by relieving the deleterious effects of GSK3β action on post-insulin receptor insulin signalling (Qu et al., 2006). Other signalling pathways also implicates GSK3β as a negative regulator of insulin signalling pathway. For, e.g. Protein tyrosine phosphatase 1B (PTP1B) acts as a negative regulator of insulin signal transduction by activating GSK3β. This phenomenon was highlighted when Hong and Lee in 1997 had reported that the reduction of PTP1B increases insulin-dependent metabolic signalling and improve insulin sensitivity in diabetic animal model (Hong and Lee, 1997). However, this approach had taken a back seat as the design and development of modulators for the negative regulators are quite challenging.
AD pathway
Glycogen synthase kinase-3 beta is involved in the abnormal phosphorylation of pathological tau in AD (Hanger et al., 1992; Mazanetz and Fischer, 2007). GSK3β is activated in Amyloidβ-peptide pathway (Figure 1). It is well studied in human, mouse and rat. Cedazo-minguez et al. (2003) proved that early activation of GSK3β induces apolipoprotein(APP)-E4 and β-amyloid, could lead to apoptosis and tau hyperphosphorylation. Among other aspect of AD, Hernández and Avila (2008) had also reported the relevance of activation of GSK3β at molecular level. It also plays an important role for axonal elongation. APP intracellular domain transgenic mice showed activation of GSK3β and phosphorylation of Collapsin response mediating protein-2 protein – a GSK3β substrate that plays a crucial role in Semaphorin3a-mediated axonal guidance (Ryan and Pimplikar, 2005). Their report on the potential of GSK3β inhibitors for the therapy of AD was supported by the fact that the transgene shutdown in symptomatic mice leads to normal GSK3β activity, normal phospho-tau levels, diminished neuronal death and suppression of the cognitive deficit. These data highlights the important role for GSK3β in axonal elongation. It also plays a pivotal role in the development of AD by its involvement in the formation of (PHF)-tau, which is an integral component of neurofibrillary tangle deposits that disrupt neuronal function, and is a marker of neurodegeneration in AD (Takashima, 2006).
Martinez and Perez (2008) has provided a good rational for the development of GSK3β inhibitor for treatment of AD. The data from their study clearly identified GSK3β inhibitors as one of the most promising approaches for the future treatment of AD. A reduction of the aberrant over activity of GSK3β might decrease several aspects of the neuronal pathology in AD.
Although these reports implicated GSK3β in neurological disorders; its roles in normal CNS functions have not yet been determined. To examine the potential role for GSK3β in synaptic plasticity, Peineau et al. (2007) investigated the effects of inhibition of the enzyme on induction of long-term depression (LTD) and long-term potentiation (LTP). This was studied in rat hippocampal slices. Inhibition of LTD by GSK3β inhibitor lithium was observed regardless of whether the inhibitor was applied before or after induction of LTD. This phenomenon suggests that GSK3β might function in LTD maintenance (Peineau et al., 2007). The role of GSK3β in synaptic plasticity was again determined by Peineau in 2008 where they had reported that the blockade of GSK3β influence on the processes that underlie learning and memory mechanisms. From their various results on the study of LTP and LTD mechanism, it is being believed that GSK3β is required for LTD and provides a mechanism by which LTP can inhibit LTD (Peineau et al., 2008).
BDNF signalling pathway
Glycogen synthase kinase-3 beta is an intracellular enzyme implicated as a critical component in many neuronal signalling pathways. Several intracellular signalling cascades and several neurotransmitter systems including serotonergic, dopaminergic, cholinergic and glutamatergic converge on GSK3β to modulate its activity. Due to the changes in these neurotransmitter systems,GSK3β has been linked to the mood and bipolar disorders, depression, and schizophrenia. GSK3β regulation may represent a novel target to treat mood disorders (Jope and Roh, 2006). Dysregulated GSK3β in bipolar disorder, depression and schizophrenia could have multiple effects that could impair neural plasticity, such as modulation of neuronal architecture, neurogenesis, gene expression and the ability of neurons to respond to stressful, potentially lethal conditions (Chen et al., 1999). There are also existing reports from the genetic perspective supporting the role of GSK3β in the disease physiology of bipolar mood disorder (Gould, 2006)
The role of GSK3β in mood disorder was highlighted by the study of lithium and valproate (Klein and Melton, 1996; Ryves et al., 2002); both of which are GSK3β inhibitors and are used to treat mood disorders. GSK3β has host of substrates of which more than dozen are transcription factors (Kannoji et al., 2008). Most of them get inhibited upon phosphorylation. Of these transcription factors, CREB is an important one which plays a critical role in mood disorder (Blendy, 2006).
Mai et al. in 2002 has reported that brain-derived neurotrophic factor (BDNF) increases the serine9-phosphorylation of GSK3β which inhibits its activity. This is in addition to the increased phosphorylation of Fork-head transcription factors (FKHRL1). Over expression of GSK3β did not affect BDNF-induced phosphorylation of AKT, extracellular signal-regulated kinases 1/2 (ERK1/2) or FKHRL1, but abolished CREB phosphorylation (Mai et al., 2002). The inhibition of BDNF-induced CREB phosphorylation in GSK3β-overexpressing SH-SY5Y cells were blocked by treatment with lithium.
It was reported by Emamian et al. (2004), that alterations in AKT1-GSK3β signalling contribute to schizophrenia pathogenesis. They observed a decrease in AKT1 protein levels and its phosphorylation of GSK3β at Serine-9 in the peripheral lymphocytes and brains of individuals with schizophrenia. It was also reported that schizophrenia with deficiency of AKT1, is more sensitive to the sensorimotor gating-disruptive effect of amphetamine (Smith and Frenkel, 2005). Although preclinical data from biochemical, behavioural and human genetic studies supports the therapeutic role of GSK3β in mood disorder, but proof of concept in the clinical trials is yet to be established as emphasized by Gould (2006).
Oncology pathway
Glycogen synthase kinase-3 beta's role in cancer is a well-accepted phenomenon where it acts as a negative regulator for cell growth. Of all the different pathways leading to cancer, WNT signalling is the predominant ones. WNT signalling pathway is involved in disease physiologies like Osteoporosis and Cancer. It stimulates translation and cell growth by activating the tuberous sclerosis complex (TSC) – a mammalian target of rapamycin (mTOR) pathway via GSK3β (Shao et al., 2005). Inhibition of GSK3β by AKT positively regulated the G1/S cell cycle progression. Growth hormone activates AKT, which in turn inhibits the GSK3β activity. This leads to increase in cyclin D1 and inhibition of Forkhead family transcription factors and the tumour suppressor tuberin (TSC2) (Liang and Slingerland, 2003). GSK3β is inactivated after phosphorylation when stimulated with IL-6. Inhibition of GSK3β activity is sufficient to suppress cell growth and induce apoptosis thus overriding the effects of IL-6 in myeloma cells (Inoki et al., 2006). GSK3β also control oncogenicity via B-cell CLL/lymphoma 3 (BCL-3). GSK3β phosphorylate BCL-3 and modulates its degradation and its oncogenicity (Viatour et al., 2004). ERK1/2 plays important in cell proliferation. GSK3β phosphorylation is dependent on the activation of ERK1/2 induced by tungstate (Gomez-Ramos et al., 2006).
Although till now any mutation in GSK3β is not found which can be related to disease conditions, but mutations in downstream substrates have been reported (Sagae et al., 1999). For e.g. β-catenin gene mutation at potential GSK3β phosphorylation sites results in accumulation of β-catenin protein within the cells and its translocation to nuclei. Accumulated β-catenin protein may be involved in the development of endometrioid-type ovarian carcinomas (Ohira et al., 2003). Due to the pronounced importance of the role of GSK3β in cancer, in the following text we discuss its role in various types of cancer.
Lung cancer
Wingless integration inhibits GSK3β, leading to increase free β-catenin and up-regulation of E-cadherin in lung cancer cells. This phenomenon was reported by Ohira et al. in 2003. Their finding supports that E-cadherin induction by WNT/β-catenin signalling is an evolutionarily conserved pathway in lung cancer cells. Loss of WNT7a expression may be important in lung cancer development or progression by its effects on E-cadherin (Ohira et al., 2003). The possible role of GSK3β in causing lung cancer was also studied by Tian et al. (2006) where they reported that in vitro cigarette smoke components notably inhibited GSK3β similar to lithium or SB216763. The inhibition of GSK3β either with cigarette smoke or GSK3β inhibitors like lithium and SB216763 significantly enhanced involucrin expression in cultured porcine tracheobronchial epithelial cells probably via negative regulation of AP-1 activity leading to squamous differentiation. These studies throw light on the probable role of GSK3β in causing lung cancer.
Breast cancer
Erythroblastic leukemia viral oncogene homolog 2 (ErbB2) can cooperate in mammary epithelial cell transformation. Activation of MAPK and PI3K/AKT, indirectly inhibits GSK3β, leading to the production of excess ErbB2 signals. These ErbB2 signals can modulate cyclin D1 and p27 and dysregulates the G1-to-S transition of cell cycle leading to mammary epithelial cell transformation (Lenferink et al., 2001). Hyperactive WNT signalling is associated with the development and progression of human breast cancer. Jong et al. (2006) demonstrated that WNT signalling engages tumour cell dedifferentiation and tissue-invasive activity through an Axin2-dependent pathway. Axin2 regulates epithelial mesenchymal transition (EMT) by acting as a nucleocytoplasmic chaperone for GSK3β. GSK3β is responsible for controlling Snail protein turnover and activity. The identification of a β-catenin–T cell factor (TCF)-regulated Axin2–GSK3β–Snail1 axis provides new mechanistic insights into cancer-associated EMT programmes (Jong et al., 2006). The clinical evidence for the first time in support of the WNT/beta-catenin signalling in breast cancer was recently reported by Prasad et al. (2009). They reported the WNT/beta-catenin signalling up-regulation in invasive ductal carcinomas and key components of this pathway – E-cadherin, Slug and GSK3β with beta-catenin in the development of EMT. Their studies demonstrated significant correlation between GSK3β nuclear localization and tumour grade (P= 0.02), suggesting its association with tumour progression.
Colon cancer
In a study on colon cancer cells by Shakoori et al. (2005), it was observed that the inhibition of GSK3β activity by chemical inhibitors and its expression by RNA interference induced apoptosis and attenuated proliferation. This phenomenon suggests that there is a role of GSK3β in promoting tumour cell survival and proliferation. It was also observed by others that through the inhibition of GSK3β, Prostaglandin E2 transactivated the β-catenin/TCF-dependent transcription induction of TCF-4 in colon cancer cells (Liao et al., 2004). Inhibition of GSK-3, a downstream target of active AKT, completely blocked the induction of TNF-related apoptosis-inducing ligand (TRAIL) by wortmannin. This was reported by Wang et al. (2002). Their study demonstrated the induction of the TRAIL by inhibition of PI 3-kinase in colon cancer cell lines. The studies further opened up the opportunity for understanding the role of PI 3-kinase/AKT/GSK-3 pathway in intestinal cell homeostasis. In 2008 Kang and co-workers had demonstrated that the inhibition of GSK3β in human tumour cell regulates the ubiquitin-mediated proteolysis of Cdc25A during early phases of the cell cycle. The overproduction of Cdc25A during the G1 phase of cell cycle strongly correlated with GSK3β inactivation. The PI-3K/AKT pathway negatively regulates both GSK3β and CHK1 and this in turn promotes cell cycle advancement by elevating Cdc25A levels (Kang et al., 2008).
Ovarian cancer
GSK3β plays an important role as a positive regulator of human ovarian cancer cells (Cao et al., 2006). The mutational studies implicating the role of GSK3β in ovarian cancer were reported way back in 1999 (Kim et al., 1999). The over expression of the GSK3β in ovarian cancer cells led Cao and coworker to study the role of GSK3β in ovarian cancer. When treated with inhibitors of GSK3β in ovarian cancer cell lines they reported that there were marked reductions in the tumour growth. Similar reduction in tumour volume and size was also observed in nude mice having cancer xenograft model when treated with GSK3β inhibitor LiCl. It was also observed that the GSK3β inhibition via integrin-linked kinase (ILK)-mediated signalling pathway leads to the stabilization of Snail and β-catenin and regulation of transcriptional programs that control ovarian cancer (Rosano et al., 2005).
Prostate cancer
Glycogen synthase kinase-3 beta activity is required for androgen-stimulated gene expression in prostate cancer cells (Thiel et al., 2006). β-catenin mediates the cross-talk between PI3K/AKT and androgen pathways. Liang and Slingerland (2003) had reported the multiple roles of PI3K/PK in cell cycle progression. The PI3K/AKT signal induces phosphorylation and inactivation of GSK3β resulting in increased nuclear levels of β-catenin. Consequently, increased β-catenin elevates androgen receptor activity to stimulate prostate cell growth and survival (Sharma et al., 2002). GSK3β suppression sensitizes prostate cancer cells to TRAIL-induced apoptosis that is dependent on caspase-8 activities but independent of NF-κB activation. This suggests that a mechanism involving GSK3β activation may be responsible for TRAIL resistance in prostate cancer cells (Koul et al., 2005).
Other types of cancer
In addition to the types of cancers discussed above GSK3β is also reported to be involved in the cause of other cancers like stomach cancer, melanoma and glioblastoma. It was also reported that inhibition of GSK3β by AKT in the pathway leads to stomach cancer (Krymsky et al., 2001). Thiel et al. (2006) had also observed that Phorbol 12-myristate 13-acetate (PMA) induced cyclooxygenase-2 (COX-2) protein expression was mediated through PKC/PI3K/AKT pathway where GSK3β is the downstream target. According to them this type of GSK3β inhibition leads to stomach cancer.
Inhibition of GSK3β by the activated AKT in the IGF-1 signalling pathway leads to the stabilization of β-catenin leading to melanoma. Stabilized β-catenin then translates into nucleus and promotes the transduction of the cell proliferation leading to melanoma. This phenomenon was reported by Diehl et al. (1998). In 2008, Bellei et al. had reported the effect of inhibiting GSK3β activity on the regulation of melanocyte differentiation. They studied the effect of GSK3β specific inhibitors (SB216763, SB415286, BIO and LiCl) in murine melanoma cell line B16 and normal human melanocytes. It was observed that there is a dose dependent accumulation of β-catenin leading to melanoma. This study almost confirms the role of GSK3β in the development of melanoma.
Results of the work carried out by Welcker et al. (2003) exhibited that blocking the ILK/AKT pathway is a potential strategy for molecular targeted therapy for gliomas. Downstream regulation of GSK3β phosphorylation through AKT, mTOR in glioblastoma cells leads to the decreased cell proliferation, invasion and angiogenesis. The role of GSK3β in glioblastoma was further validated by the recent study on the role of lithium in the treatment of glioblastoma (Nowicki et al., 2008). They reported that the examination of known targets of lithium showed that inositol monophosphatase inhibition had no effect on glioma migration, whereas notable effect was observed on glioma migration during the inhibition of GSK3β. According to them specific pharmacological GSK3β inhibitors and siRNA knockdown of GSK3β isoforms both reduced cell motility. These results not only throws light into the previously unidentified biochemical pathways in glioblastomas, but also guides in the development of inhibitors for the treatment of glioblastoma.
Osteoporosis
It was reported that dexamethasone (DEX) activates GSK3β and inhibits a differentiation-related cell cycle that occurs at a commitment stage immediately after confluence. Inhibition of a PI3K/AKT/GSK3β/β-catenin/Lymphoid enhancer binding factor (LEF) axis, and stimulation of histone deacetylase 1 (HDAC1) cooperates to mediate the inhibitory effect of DEX on WNT signalling and the osteoblast differentiation-related cell cycle (GA et al., 2002). Osteoporosis also develops due to the disruption of the balance between the adipocytes and osteoblast. These imbalance occurs when adipocytes develops at the expense of the osteoblasts (Cohen and Frame, 2001). In this context Laure-Emmanuelle et al. (2008) has reported the effect of GSK3β inhibitors in control of adipocytes. Their studies demonstrated the potential of GSK3β as target to control adipocytes and hence control osteoporosis.
Cardiac hypertrophy
Recent years has witnessed a host of reports by various workers on the possible role of GSK3β in cardiac hyperthrophy. In 2002, Hardt and Sadoshima, and in 2007, Kerkela et al. reported the pivotal role of GSK3β in cardiac hypertrophy. It was reported by Robertson et al. (2006) that the inhibition of GSK3β by β-adrenergic stimulation abrogates GSK3β-induced nuclear export of GATA binding protein 4 (GATA4). This phenomenon may represent an important GSK3β signalling mechanism mediating cardiac hypertrophy. Although the involvement of GSK3β in cardiac hypertrophy was initially reported in 2001 by Morisco et al., till recently there is very less clarity of the mechanism by which GSK3β plays a role in cardiac process. Sugden et al. (2008) has elaborately reviewed the probable role of GSK3β in cardiac process. However, due to various conflicting reports, it is opined that still there is a dilemma whether the activation or inhibitory effect of GSK3β would lead to therapeutically acceptable molecule to treat cardiac hypertrophy (Sugden et al., 2008). However, a latter report from the Sadoshima's group (2008) reported that maintaining the unphosphorylated S9 of GSK3β in Knockin mice prevents cardiac decompensation on cardiac hypertrophy and function during pressure overload (Matsuda et al., 2008). These reports further validates the role of GSK3β in cardiac hypertrophy. However, from these studies it is observed that to have an efficient modulator it is important to have isoform selectivity.
Hypertension
The role of GSK3β in causing hypertension in animal models has been reported. In hypertensive rats, enhanced GSK3β activity modestly augments the effects of PI3K but does not appear to contribute greatly to the altered arterial reactivity in deoxycorticosterone-salt hypertension (Loberg et al., 2003). GSK3β-mediated regulation of mitochondrial permeability transition pore formation in the ischemic reperfused heart was studied by Mozaffari and Schaffer (2008). They found that the treatment with GSK3β inhibitors, LiCl and SB-216763 reduced infarct size at both pressures, with the effect more marked at the higher perfusion pressure. This study emphasized the role of GSK3β in controlling hypertension. However, more studies are required to validate this as a target in drug discovery space.
Cross-talk in signalling pathways implicating GSK3β
Since the discovery of GSK3β and its implications in various disease physiologies, it has been studied in isolation w.r.t its effect in various signalling pathways. However, last few years have seen a change in the approach to the study of the role of GSK3β in relation to disease physiologies. In addition to the individual signalling pathways – as discussed earlier in this review – it was observed from various reports that GSK3β signalling pathways are implicated in crosstalk with other signalling pathways in disease physiologies. These reports from different workers highlight the importance of the cross-talk between these cascades. One of the common signalling pathway involved in the crosstalk during a variety of cellular processes is the WNT signalling pathway. It was reported by Ross et al. in 2000 that WNT signalling, mediated by WNT-10b, is a molecular switch that governs adipogenesis. From their experimental results they have found that disruption of WNT signalling also causes trans-differentiation of myoblasts into adipocytes in vitro. This finding highlights the importance of this pathway not only in adipocyte differentiation but also in mesodermal cell fate determination (Ross et al., 2000). It was also reported by Framer in 2005 that phosphorylation of C/EBPbeta at a consensus ERK/GSK3β site is required for the PPARgamma-associated expression of adiponectin during the terminal stages of adipogenesis.GSK3β also influences PPARγ activity by regulating the turnover and subcellular localization of beta-catenin, which is a potent transcriptional activator of WNT signalling. They had also reported a crosstalk between PPARγ and beta-catenin signalling. Activation of PPARgamma induces the degradation of beta-catenin during preadipocyte differentiation by mechanisms that require GSK3β and the proteasome (Farmer, 2005). Kotoh and Katoh (2006) has reported that cross-talk between WNT and FGF signalling pathways at GSK3β leading to the regulation of β-catenin and SNAIL signalling cascades which has implication in malenoma. The involvement of GSK3β in the regulation of Snail is of considerable interest, as GSK3β is involved in the regulation of WNT and hedgehog pathways. These pathways are already known to control cell fate and morphogenesis during development and tumourigenesis (Zhou and Hung, 2005). WNT signalling has also been linked to type 2 diabetes. It is been reported that carriers of variants of the transcription factor 7-like 2 gene, an important component of the WNT pathway, are at enhanced risk for developing type 2 diabetes (Bordonaro, 2009). Earlier, Yi et al. (2008) had demonstrated the cross-talk between insulin signalling and WNT from intestinal endocrine L cells. In their study they found that in the GLUTag cells, insulin-induced activation of glu expression occurred through the same TCF site that mediates cat/TCF activation. This simulation was attenuated by Phosphatidylinositol 3-kinase inhibition. In addition to it, nuclear beta-catenin content in the intestinal L cells was increased by insulin. According to them, these findings indicate that enhancement of beta-catenin nuclear translocation and cat/TCF binding are among the mechanisms underlying the cross-talk between the insulin and WNT signalling pathways in intestinal endocrine L cells (Yi et al., 2008). In AD pathogenesis GSK3β is reported to have a dynamic cross-talk with CDK5. Although these two proteins were analysed from their individual roles in AD, Engmann and Giese in 2005 had reported that there is a dynamic cross-talk between the two proteins. Evidence from the p25 transgenic mice suggests that during aging or prolonged over activation of CDK5, GSK3β activity may alter in favour of AD pathogenesis (Engmann and Giese, 2009). GSK3β pathway is reported to be one of the common pathway disrupted by insulin resistance and amyloid beta. This was reported by Zhao and Townsend (2009) where they studied the process of type 2 diabetes and AD. Studying these processes at the cellular level they suggested that the insulin resistance and Amyloid beta aggregation may not be the consequence of excitotoxicity, aberrant Ca2+ signals and pro-inflammatory cytokines built in can also promote these pathological effectors. These reports on the cross-talks of GSK3β with different proteins and signalling pathways throws open a new vista of understanding the disease physiologies/mechanism. Further studies on this area would lead to new concepts and approaches to the design and development of modulators for GSK3β.
Knockout studies
There are reports regarding the knockout studies carried out in the context GSK3β as a target. Kim et al. (2006) found no gross morphological deficits in nervous system development in GSK3β null mice. In contrast, it is also reported that disruption of the murine GSK3β gene results in embryonic lethality caused by severe liver degeneration during mid-gestation (Hoeflich et al., 2000). GSK3α/β double-knockout mouse embryonic stem cell displayed hyperactivated WNT/β-catenin signalling and were severely compromised in their ability to differentiate, but could be rescued to normality by re-expression of functional GSK3β (Doble et al., 2007). The rheostatic regulation of GSK3β highlights the importance of considering the contributions of both homologs when studying GSK3β functions in mammalian systems (Michelon et al., 2006).
Single nucleotide polymorphism (SNP) in GSK3β
Single nucleotide polymorphism, which is the major cause of abnormalities in the metabolic behaviour in the human, was also identified in case of GSK3β. An SNP (-50 T/C) falling into the effective promoter region (nt –171 to +29) of the gene coding for GSK3β has been linked with different age at onset of bipolar illness and with different antidepressant effects of total sleep deprivation (Benedetti et al., 2005). Two functional SNPs in the GSK3β gene viz 50T-C (rs334558) – a promoter polymorphism were identified,in which the T allele has greater transcriptional activity in vitro, and 157T-C in intron 5 (rs6438552), which is an intronic polymorphism were identified by Kwok et al. (2005). In this study the T allele is observed to be associated with increased levels of GSK3β lacking exons 9 and 11. Increased levels of GSK3β lacking exons 9 and 11 was correlated with enhanced phosphorylation of MAPT in vitro concluding that GSK3β polymorphisms interact with MAPT haplotypes to modify disease risk in Parkinson Disease (Kwok et al., 2005).
siRNA
siRNA technology is used to study the signal transduction process for GSK3β. Use of this technology to determine the role of GSK3β for various disease conditions are reported recently by various workers. It was used to exhibit that burn injury stimulates GSK3β activity in skeletal muscle and that GSK3β may regulate glucocorticoid-mediated muscle wasting (Fang et al., 2007). GSK3β and h-prune cooperatively regulate the disassembly of focal adhesions to promote cell migration and that h-prune is useful as a marker for tumour aggressiveness (Kobayashi et al., 2006). GSK3β is required for 17β-estradiol (E2)-induced estrogen receptor-alpha (ERalpha) phosphorylation at Ser-118 and full transcriptional activity of the receptor upon E2 stimulation (Grisouard et al., 2007). In 2008 Nowicki et al. through siRNA knockdown of GSK3β had demonstrated the role of GSK3β in glioblastoma.
Patent landscape of GSK3β
Although GSK3β was discovered 28 years back, the interest in it as potential drug targets became prominent only in the beginning of this century. To evaluate the importance of this target in the therapeutic patents, GSK3β patent analysis revealed that the number of patents published had increased nearly three times from merely 4 in 1999 to 20 in 2001 reaching a peak of 136 publication in 2008 (Figure 3). GSK3β has found prominence in the different therapeutic area with Alzheimer's being the most prominent ones followed by Cancer and Diabetes (Figure 4). Of the total number of patents published around 153 are in the area of Alzheimer's with Cancer and Diabetes around 170 publications. This clearly indicates the growing importance of GSK3β in Pharmaceutical industry. But interestingly in spite of this increasing number of patents published in recent years for GSK3β inhibitors, the number of compounds in the different phases of clinics or discovery phase of research is very minimal. A company wise distribution of global patent publication exhibits that Vertex pharmaceuticals, Astra Zeneca, Glaxo-SmithKlime along with Sanofi are the major players in the arena (Figure 5).
Figure 3.
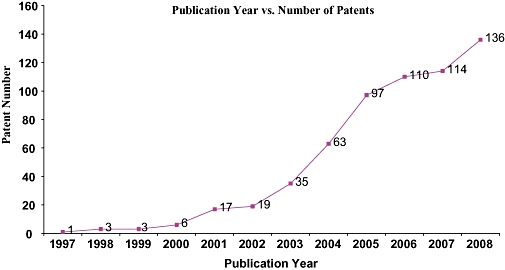
Glycogen synthase kinase-3 beta (GSK3β) patents filed in last 10 years. The gradual increase in the number of patents shows the importance of GSK3β as an emerging drug target.
Figure 4.
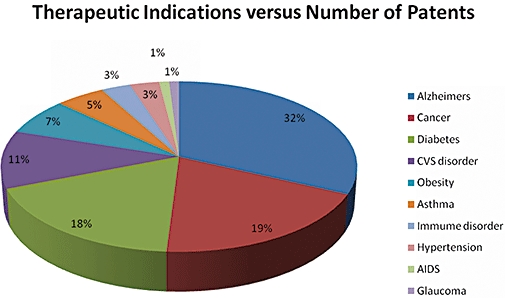
Different area of therapeutic indications affected by GSK3β. Alzheimer being the most preferred once with 32% of the total published patents falling in this therapeutic area followed by Obeisity. GSK3β, glycogen synthase kinase-3 beta.
Figure 5.
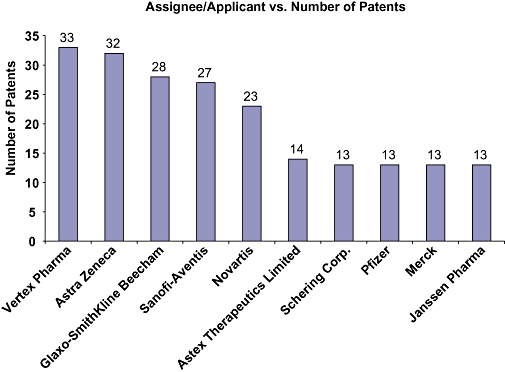
A comparison of worldwide patent activity of major companies which are actively filing patent applications related to GSK3β. Vertex Pharma, AstraZeneca and Glaxo-SmithKline stand at the top in this domain followed by Sanofi-Aventis, Novartis, Astex Therapeutics, Pfizer. GSK3β, glycogen synthase kinase-3 beta.
Development of GSK3β inhibitors
It is promising that GSK3β inhibition throws a great hope in the modulation of many disease pathways. With newer information on the different aspect of GSK3β in the disease physiologies, priority on the search of its inhibitors has been high and many reports (journal literature and patents) have been published. Many different chemo spaces are being explored to design specific as well as non-specific GSK3β inhibitors.
Lithium and GSK3β inhibition
Lithium is a well known inhibitor of GSK3β, which was discovered in 1996 3β. Lithium alone and with some other drugs is in clinical phase of discovery time space, to study its inhibitory effect on GSK3β (discussed later). Therapeutic plasma lithium concentration ranges from 0.6–1.2 mM to inhibit the GSK3β activity (Phiel and Klein, 2001).
Lithium has shown to increase phosphorylation of GSK3β at serine-9 site in the treated cells (De sarno et al., 2002) and inhibits GSK3β by competition for magnesium (Mg2+), but not ATP or substrate (Ryves et al., 2002).
Lithium protects tau from hyperphosphorylation and may rescue memory retention in the rats by inhibiting GSK3β activity (Qu et al., 2006). Specific inhibition of cellular GSK3β by lithium or GSK3β antisense, elicits a reduction in β-amyloid (Aβ). Ryder et al. in 2003 had reported that the oral administration of lithium significantly reduces Aβ production whereas direct intracerebroventricular administration of roscovitine augmented Aβ production in the brains of PDAPP [APP (V717F)] mice. The data supports a function for GSK3β in APP processing, further implicating the GSK3β in the pathogenesis of AD (Ryder et al., 2003). Recently, Mendes et al. (2009) evaluated the transcriptional regulation of GSK3β in lithium treated primary cultures of rat and hypocampal neurons and found that there was a dose dependent reduction in the number with the expression of GSK3β which was further confirmed by in vivo experiments in different brain regions. This study reinforces the potential of lithium for inhibition of pathological pathways in AD. The regulation of behavioural effect of lithium was reviewed by Beaulieu and Caron (2008). They have explained the molecular mechanism involved in the behavioural effect brought about by the effect of lithium.
Other GSK3β inhibitors
The inhibitors of GSK3β for its chemo-space has been analysed using KinaseChemBioBase™. A search in the KinaseChemBioBase™ exhibited that inhibitors of various efficacy are reported. Although there are 45 520 compounds reported in the published literature and patents as GSK3β inhibitors only 8193 of them have been reported with activity values. Most of the compounds (2540) were reported to be at the range of 10 nM–1 µM activity. Similarly, 1525 number of compounds was reported to have the activity value between 1 µM and 100 µM. Around 12 compounds (Raymond et al., 1999; Patricia et al., 2003; Thierry et al., 2003; Joong et al., 2004; Pascal et al., 2004; Shudong et al., 2004; Toshiyuki et al., 2004; Shudong et al., 2005) are reported to have a very high potency of <1 nM against GSK3β where a substantial compounds amounting to 485 in numbers are reported to have shown potency between 1 and 100 nM. Although majority of the compounds from the published patents is reported to have a micro-molar potency (Table 1), there are several high nanomolar inhibitors of GSK3β.
Table 1.
Chemotypes present along with the number of molecules GSK3β inhibitors
| Chemotype structure and name |
No. of molecules with various IC50 ranges |
Assay type | Assay method | ||||||
|---|---|---|---|---|---|---|---|---|---|
| <1 nM | 1 nM–10 nM | 10 nM–<100 nM | 100 nM–1 µM | 1 µM–10 µM | 10 µM–100 µM | >100 µM | |||
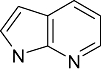 Azaindole Azaindole |
0 | 10 | 36 | 29 | 4 | 0 | 0 | In vitro | Scintillation counter |
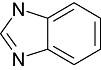 Benzimidazole Benzimidazole |
2 | 16 | 42 | 61 | 2 | 17 | 0 | In vitro | Standard coupled enzyme assay, liquid scintillation counter |
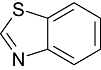 Benzthiazole Benzthiazole |
0 | 0 | 0 | 1 | 0 | 9 | 0 | In vitro | Standard coupled enzyme assay, liquid scintillation counter |
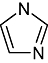 Imidazole Imidazole |
1 | 11 | 17 | 299 | 101 | 47 | 1 | In vitro | Scintillation counter, HTRF assay |
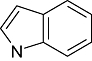 Indole Indole |
1 | 24 | 80 | 614 | 366 | 21 | 3 | In vitro | Liquid scintillation counter |
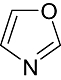 Oxazole Oxazole |
0 | 0 | 1 | 1 | 2 | 5 | 0 | In vitro | Liquid scintillation counter, microtiterplate scintillation counter |
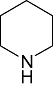 Piperidine Piperidine |
1 | 6 | 12 | 81 | 97 | 74 | 0 | In vitro | Scintillation counter |
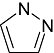 Pyrazole Pyrazole |
1 | 10 | 56 | 403 | 33 | 86 | 0 | In vitro | Scintillation counter |
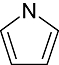
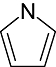 Pyrrole Pyrrole |
0 | 5 | 15 | 11 | 25 | 9 | 0 | In vitro | Liquid scintillation counter |
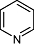 Pyridine Pyridine |
2 | 32 | 87 | 340 | 713 | 251 | 7 | In vitro | Liquid scintillation spectrometry, western blotting |
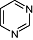 Pyrimidine Pyrimidine |
4 | 30 | 129 | 153 | 557 | 600 | 9 | In vitro | Spectrophotometric coupled enzyme assay, scintillation counter |
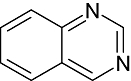
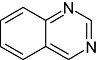 Quinazoline Quinazoline |
0 | 0 | 0 | 1 | 1 | 1 | 0 | In vitro | Standard couple enzyme assay, trilux wallac scintillation counter, ELISA |
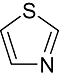
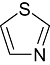 Thiazole Thiazole |
0 | 0 | 4 | 14 | 29 | 20 | 6 | In vitro | Scintillation counter |
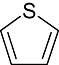
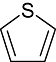 Thiophene Thiophene |
0 | 4 | 16 | 30 | 11 | 7 | 0 | In vitro | Scintillation counter, wallac counter, standard coupled, microbeta microplate counter |
The in various IC50 range along with the assay methods and types are also indicated.
GSK3β, glycogen synthase kinase-3 beta.
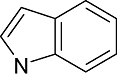
Various classes of compounds are reported to occupy the chemo space as inhibitors of GSK3β (Table 2). It was observed that among various classes of compounds, Pyrimidine class has the highest number of compounds followed by compounds of Pyridine derivatives. Pyrazoles and Thiazole along with the Indole and Imidazole have a significant amount of compounds among all the classes of compounds reported as GSK3β inhibitors. However, compounds like Thiophene, Benzthiazole, Benzoxazole and others chemotypes are not much explored for the inhibition of GSK3β Compounds from the chemotypes of Benzimidazole, Pyrimidine, Pyridine, Pyrazole and Imidazole had been reported to have high potency GSK3β inhibitor with <1 nM activity (Table 1). It was observed that the molecules containing the groups like Pyrrolidine-2-one, Pyrrolidine-2,5-Dione, Piperidin-2-one or Piperidin-2,6-Dione were showing high to moderate (0.2 µM–50 µM) inhibition of GSK3β. It was observed that Benzimidazole is also an important moiety for the high activity against GSK3β. Janssen Pharmaceutical has reported a unique moiety of the type pyrido(3,2-b)azepin-6-one which exhibits a potency of <100 nm activity for GSK3β.
Table 2.
Various chemotypes present in GSK3β inhibitor molecules
| Chemotype structure | Chemotype name | No. of molecules | Off-target kinases |
|---|---|---|---|
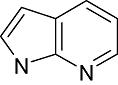 |
Azaindole | 733 | PKA, Erk2, EGFR, CK1, CDK1, VEGFR, PKCalpha, PKCbeta, PKCgamma, CDK1/Cyclin B, c-Met, TAK1A, c-Kit, Flt3, KDR, Jak3, Syk |
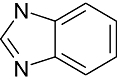 |
Benzimidazole | 880 | CDK2/Cyclin A, Src, Aur2, Lck, EGFR, VEGFR2, erbB2 |
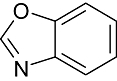 |
Benzoxazole | 18 | CDK5/p25, CDK2/Cyclin A, CDK9/Cyclin T1 |
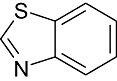 |
Benzthiazole | 460 | Src, AKT, CDK2/Cyclin A, CDK5/p25, CDK9/Cyclin T1, ILK1, AUR2 |
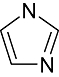 |
Imidazole | 3 751 | Aur2, CDK5/p25, CK1, CK2, VEGFR, PKA, EGFR, Erk2, HER2 |
 |
Indole | 2 075 | PKCalpha, PKCgamma, PKCbeta, CDK1, CDK5, ILK1, MSK1, MEK1, CK2, Chk1, Chk2, p38, AKT1, PDK1, AurA, AurB, Fak, Tie2, EGFR, VEGFR2, ABL1, Src, JNK |
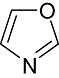 |
Oxazole | 2 861 | N/A |
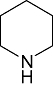 |
Piperidine | 2 814 | Erk2, Aur2, CDK5/p25, EGFR, VEGFR2, CK1, CK2, PKA, CDK1/Cyclin B, CDK1, Src, Syk, MK-2, PRAK, ROCK1, JNK3 |
 |
Purine | 11 | CDK2/Cyclin A, Aur 2 |
 |
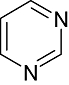
Pyrazine | 1 378 | CDK5/p25, CDK1/Cyclin B, PIM1, CK1, CK2, PKG, PKA, JNK, MAPKK, cRaf, Erk1, Erk2, Src, Srk, Aur2, Syk, VEGFR2 |
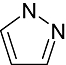 |
Pyrazole | 4 571 | AUR2, Erk2, Src, AKT, CDK2/Cyclin A, Syk, MK-2, PRAK |
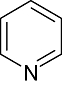 |
Pyridine | 12 944 | Aur2, JNK3, CDK2/Cyclin A, Lck, CDK2, EGFR, CK1, CK2, PKA, Erk2, HER2 |
 |
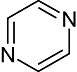
Pyrimidine | 14 880 | Aur2, JNK3, Lck, Src, Erk2 |
 |
Pyrrole | 935 | CDK5/p25, Erk2, CDK2/Cyclin A, Lck, CDK1, CDK5, JNK3, c-met, CDK4/Cyclin D1, MEK1, CK1 |
 |
Quinazoline | 1 378 | Aur2, CDK2/Cyclin A, Src, AKT, Src, Erk, PRAK, MK-2, Syk, Lck, ROCK2, Chk1, PDK1, PKA, JNK, MAPK, SAPK |
 |
Thiazole | 4 364 | CDK5/p25, JNK3, Erk2, VEGFR, CDK1, PKA, EGFR, SYK, Aur2 |
 |
Thiophene | 3 338 | EGFR, VEGFR, PDGFR, HER2, Erk2, CK1, CK2, CDK1/Cyclin B, PKA |
GSK3β, glycogen synthase kinase-3 beta.
Further a detailed analysis of all the 485 molecules which were showing less than 100 nanomolar activities from different companies revealed that these molecules belong to 17 different classes of scaffolds and many compounds are having resemblance and might be an analog of the highly nonselective kinase inhibitor Staurosporine. In spite of achieving potent GSK3β inhibitors, the selectivity may be hindering development. Less diversity of functional groups is observed in the high nano-molar inhibitors (Figures 6 and 7).
Figure 6.
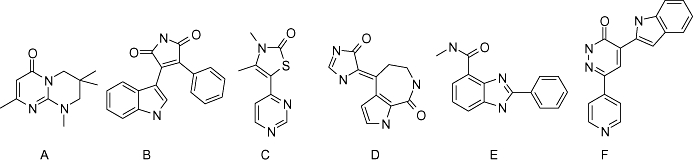
Moieties and groups for active inhibitors. (A) Pyrimido[1,2-a]pyrimidin-4-one, (B) pyrrole-2,5-dione, (C) thiazo-1,2-one, (D) pyrrolo[2,3-c]azepin-8-one, (E) 2-phenyl-benzoimidazole, (F) pyridazin-3-one.
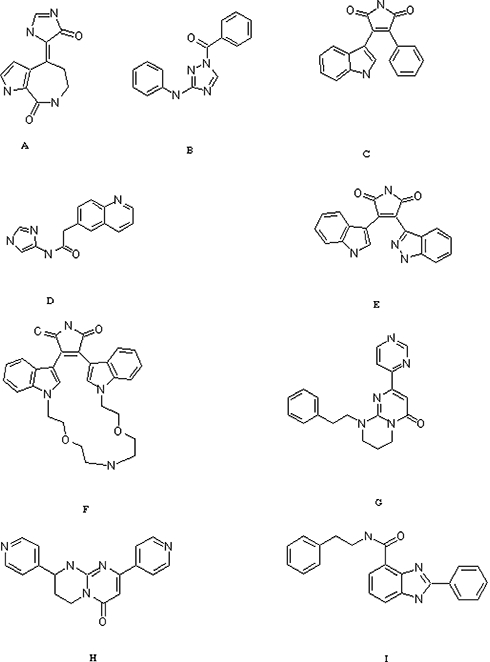
Glycogen synthase kinase-3 beta (GSK3β) inhibitors from different companies. The patents and the affiliations are (A) WO 04/103958, A2, Michigan State University, (B) WO 02/057240, A1, Ortho Mc Neil Pharmaceutical, (C) WO 03/076398, A2, Eli Lilly, (D) WO 03/027116, A2, Safoni-syntheLab, (E) WO 05/00836, A1, Janssen Pharmaceutical, (F) WO 02/46197, A1, Ortho Mc Neil Pharmaceutical, (G) WO 05/042525, A2, Cyclacel Ltd, (H) WO 04/072062, A2, Novartis AG, (I) WO 03/072579, A1, Crystal Genomics.
Clinical status of GSK3β
The published patents on GSK3β inhibitors are increasing exponentially. Majority of the compounds are still in the discovery phase and few are in the clinics. Some of the candidates are studied for the secondary outcome measure studies instead of direct effect of GSK3β. Of the clinical candidates as GSK3β inhibitors majority of them are in the area of Cancer. In a Phase II trial, Genentech and MD Anderson Cancer centre is investigating the role of Erlotinib (Tarceva) in the transitional cell carcinoma. One of the objectives of this study is to evaluate the downstream signalling pathway of GSK3β to gain more information on the biological response on a given tumour. GSK3β assessment as a biomarker in relevance to the drug Enzastaurin is under Phase I study by Eli Lilly and National Cancer Institute. This is in addition to the pharmacological safety studies carried out for the combination therapy of Enzastaurin and Bevacizumab with patients having Metastatic Cancer. Enzastaurin in combination with the Carboplatin is being studied as a Phase I trial for the indication of CNS Tumours. In this study the activation of GSK3β in the peripheral mononuclear blood cells are correlated with the clinical outcome. Moreover, recently in a combination study of Enzastaurin with other targeted agents,Vogl et al. (2009) has reported that Sorafenib and Sunitinib in combination with Enzastaurin showed synergistic reduction of viable renal cell carcinoma. This was due to the inhibition of cell growth through the inhibition of phosphor-S6-kinase and GSK3β
Inhibitory role of GSK3β in AD is conducted by the National Institute of Neurological Disorders and Stroke (NINDS) with lithium and with the combination of the drug Divalporex. Lithium or in combination with Divalporex is known to inhibit GSK3β. This might reduce the phosphorylation of the Tau to reduce the pathogenic effect of Alzheimer. For progressive Supranuclear Palsy (PSP), another neurodegenerative disease, the inhibitory effect of GSK3β by lithium and valporic acid is under study. Nates University Hospital is studying the effect of Valporic acid in the inhibition of GSK3β and the reduction of tau phosphorylation for a neuroprotective effect. Westat and NINDS analysed the effect of lithium on GSK3β which reduced tau phosphorylation as a secondary outcome measure for PSP.
Neuropharma have reported moving GSK3β inhibitors into the Phase I of clinical studies but details of the studies are not available. Also many pharmaceutical companies like Pfizer, Chiron, Vertex and others are focusing on GSK3β inhibitors for various therapeutic conditions (Table 3).
Table 3.
Status of different molecules at clinical phase
| Structure | Code/name | Company | Therapy areas | Status |
|---|---|---|---|---|
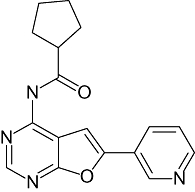 |
SmithKline Beecham Pharmaceuticals | Neurodegenerative disease; non-insulin dependent diabetes | Discovery | |
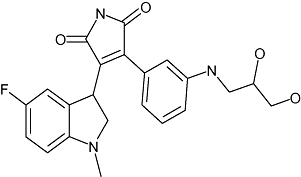 |
Roche Holding AG | Osteoporosis | Discovery | |
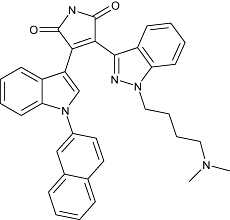 |
Johnson & Johnson | Cardiovascular disease | Discovery | |
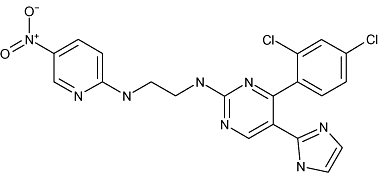 |
CHIR-98023, CHIR-73911, CHIR-98014 | Chiron Corp | Cerebrovascular ischemia; non-insulin dependent diabetes | Discovery |
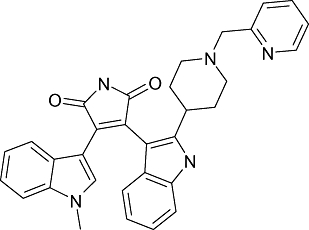 |
LY-317615 (Enzastaurin) | Eli Lilly & Co | Glioma; non-small-cell lung cancer; diffuse large B-cell lymphoma; carcinoma; cancer; solid tumour; ovary tumour | Phase I clinical trial |
 |
NP-12 (NP-031112) | Neuropharma SA (Zeltia SA) | Alzheimer's disease; central nervous system disease | Phase I clinical trial |
| Neu-120 (Structure not available) | Neu-120 | Neurim Pharmaceuticals | NMDA receptor modulator; MAO B inhibitor; antiparkinsonian; glycogen synthase; kinase-3 beta inhibitor | Phase 1 clinical trial |
| CP-70949 (Structure not available) | CP-70949 | Pfizer Inc | Diabetes mellitus | Discovery |
| SAR-502250 (Structure not available) | SAR-502250 | Sanofi-aventis | Alzheimer's disease; non-insulin dependent diabetes | Discovery |
| VX-608 (Structure not available) | VX-608 | Vertex Pharmaceuticals Inc | Cerebrovascular ischemia; non-insulin dependent diabetes | No development reported |
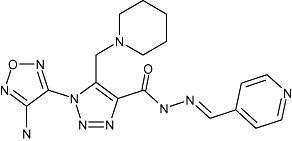 |
Novo Nordisk A/S | Non-insulin dependent diabetes | No development reported | |
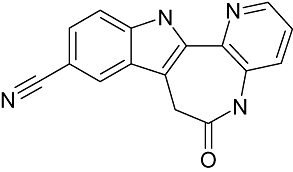 |
KUSTU-144 (cazpaullone) | DeveloGen AG | Diabetes mellitus | Discovery |
Different companies are pursuing this target for different disease indications.
N/A, not available; SAR, Structure Activity Relationship.
Conclusion
The journey of three decades for GSK3β in its therapeutic landscape has seen many interesting phenomenon – from its initial discovery it is a target for the treatment of diabetes including a recent report of proof of concept (Frame and Zheleva, 2006). Being a kinase there are issues relating to the selectivity with respect to other kinases as well as its own isoforms. However, there are reports of compounds like AR-A014418 to be a selective GSK3β inhibitor with its moderate ability to cross blood–brain barrier (Bhat et al., 2003). In addition to it, Meijer et al. (2003) had also reported a GSK3β selective inhibitor from the chemical class Tyrian purple indirubins. Structure-based approaches are also gaining prime importance in the design of specific inhibitors for this target (Patel et al., 2007). Inspite of all apprehensions, it is evident from exponential increase in reports (both patents and journal literature) that it is an important and viable target. Preclinical data indicates that its inhibition can play a vital role in the treatment of neurological disorders (Gould, 2006). Although the role of GSK3β in Cardiac hypotrophy is still not very clear, it can be a viable target with short-term administration. There are apprehensions in certain quarters that the prolonged use of GSK3β inhibitors might leads to cancer. In this regard, Frame and Zheleva (2006) opinioned that it is more overemphasized keeping in view of the data from various experiments. Patel and Woodgett (2008) have also opined that the fear of GSK3β inhibitors repressing beta catenin has now given a new dimension. According to them there has been a reversal of such effect. The trend in the clinical studies for GSK3β inhibition will throw out more light into this interesting but complex target. With the advent in structural activity data, the probability of many GSK3β inhibitors entering to clinic looks promising.
References
- Figure 7.Beaulieu JM, Caron MG. Looking at lithium: molecular moods and complex behavior. Mol Interv. 2008;8:230–241. doi: 10.1124/mi.8.5.8. [DOI] [PubMed] [Google Scholar]
- Bellei B, Flori E, Izzo E, Maresca V, Picardo M. GSK3β inhibition promotes melanogenesis in mouse B16 melanoma cells and normal human melanocytes. Cell Signalling. 2008;20:1750–1761. doi: 10.1016/j.cellsig.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Benedetti F, Serretti A, Pontiggia A, Bernasconi A, Lorenzi C, Colombo C, et al. Long-term response to lithium salts in bipolar illness is influenced by the glycogen synthase kinase 3-beta -50 T/C SNP. Neurosci Lett. 2005;376:51–55. doi: 10.1016/j.neulet.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat RV, Shanley J, Correll MP. l Regulationand localization of tyrosine216 phosphorylation of glycogen synthase kinase-3beta in cellular and animal models of neuronal degeneration. Proc Natl Acad Sci U S A. 2000;97:11074–11079. doi: 10.1073/pnas.190297597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat R, Xue Y, Berg S, Hellberg S, Ormo M, Nilsson Y, et al. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J Biol Chem. 2003;278:45937–45945. doi: 10.1074/jbc.M306268200. [DOI] [PubMed] [Google Scholar]
- Blendy JA. The role of CREB in depression and antidepressant treatment. Biol Psychiatry. 2006;59:1144–1150. doi: 10.1016/j.biopsych.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Bordonaro M. Role of Wnt signaling in the development of type 2 diabetes. Vitam Horm. 2009;80:563–581. doi: 10.1016/S0083-6729(08)00619-5. [DOI] [PubMed] [Google Scholar]
- Bussiere DE, He M, Le VP, Jansen JM, Chin S, Martin E. Charaterisation of GSK3β protein and methods of use thereof. 2002. WO 02/24893 A2.
- Cao Q, Lu X, Feng YJ. Glycogen synthase kinase-3beta positively regulates the proliferation of human ovarian cancer cells. Cell Res. 2006;16:671–677. doi: 10.1038/sj.cr.7310078. [DOI] [PubMed] [Google Scholar]
- Cedazo-Minguez A, Popescu BO, Blanco-Millan JM, Akterin S, Pei JJ, Winblad B, et al. Apolipoprotein E and beta-amyloid (1-42) regulation of glycogen synthase kinase-3beta. J Neurochem. 2003;87:1152–1164. doi: 10.1046/j.1471-4159.2003.02088.x. [DOI] [PubMed] [Google Scholar]
- Chen G, Huang LD, Jiang YM, Manji HK. The mood-stabilizing agent valproate inhibits the activity of glycogen synthase kinase-3. J Neurochem. 1999;72:1327–1330. doi: 10.1046/j.1471-4159.2000.0721327.x. [DOI] [PubMed] [Google Scholar]
- Cline GW, Johnson K, Regittnig W, Perret P, Tozzo E, Xiao L, et al. Effects of a novel glycogen synthase kinase-3 inhibitor on insulin-stimulated glucose metabolism in Zucker diabetic fatty (fa/fa) rats. Diabetes. 2002;51:2903–2910. doi: 10.2337/diabetes.51.10.2903. [DOI] [PubMed] [Google Scholar]
- Cohen P. The role of protein phosphorylation in human health and disease. The Sir Hans Krebs Medal Lecture. Eur J Biochem. 2001;268:5001–5010. doi: 10.1046/j.0014-2956.2001.02473.x. [DOI] [PubMed] [Google Scholar]
- Cohen P, Alessi DR, Cross DA. PDK1, one of the missing links in insulin signal transduction? FEBS Lett. 1997;410:3–10. doi: 10.1016/s0014-5793(97)00490-0. [DOI] [PubMed] [Google Scholar]
- Cohen P, Frame S. The renaissance of GSK3β. Nat Rev Mol Cell Biol. 2001;2:769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- De Sarno P, Li X, Jope RS. Regulation of Akt and glycogen synthase kinase-3 beta phosphorylation by sodium valproate and lithium. Neuropharmacology. 2002;43:1158–1164. doi: 10.1016/s0028-3908(02)00215-0. [DOI] [PubMed] [Google Scholar]
- Dent P, Lavoinne A, Nakielny S, Caudwell FB, Watt P, Cohen P. The molecular mechanism by which insulin stimulates glycogen synthesis in mammalian skeletal muscle. Nature. 1990;348:302–308. doi: 10.1038/348302a0. [DOI] [PubMed] [Google Scholar]
- Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble BW, Patel S, Wood GA, Kockeritz LK, Woodgett JR. Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev Cell. 2007;12:957–971. doi: 10.1016/j.devcel.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokken BB, Sloniger JA, Henriksen EJ. Acute selective glycogen synthase kinase-3 inhibition enhances insulin signaling in prediabetic insulin-resistant rat skeletal muscle. Am J Physiol Endocrinol Metab. 2005;288:E1188–E1194. doi: 10.1152/ajpendo.00547.2004. [DOI] [PubMed] [Google Scholar]
- Emamian ES, Hall D, Birnbaum MJ, Karayiorgou M, Gogos JA. Convergent evidence for impaired AKT1-GSK3β signaling in schizophrenia. Nat Genet. 2004;36:131–137. doi: 10.1038/ng1296. [DOI] [PubMed] [Google Scholar]
- Embi N, Rylatt DB, Cohen P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochem. 1980;107:519–527. [PubMed] [Google Scholar]
- Engmann O, Giese KP. Crosstalk between Cdk5 and GSK3beta: Implications for Alzheimer's. Disease Front Mol Neurosci. 2009;2:2. doi: 10.3389/neuro.02.002.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang CH, Li B, James JH, Yahya A, Kadeer N, Guo X, et al. GSK-3{beta} activity is increased in skeletal muscle after burn injury in rats. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1545–R1551. doi: 10.1152/ajpregu.00244.2007. [DOI] [PubMed] [Google Scholar]
- Farmer SR. Regulation of PPARgamma activity during adipogenesis. Int J Obes (Lond) 2005;29(Suppl. 1):S13–S16. doi: 10.1038/sj.ijo.0802907. [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P. GSK3β takes centre stage more than 20 years after its discovery. Biochem J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. Pt 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame S, Zheleva D. Targeting Glycogen synthase kinase-3 in insulin signaling. Expert Opin Ther Targets. 2006;10:429–444. doi: 10.1517/14728222.10.3.429. [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P, Biondi RM. A common phosphate binding site explains the unique substrate specificity of GSK3β and its inactivation by phosphorylation. Mol Cell. 2001;7:1321–1327. doi: 10.1016/s1097-2765(01)00253-2. [DOI] [PubMed] [Google Scholar]
- GA M, Uddin S, Mahmud D, Damacela I, Lavelle D, Ahmed M, et al. Regulation of myeloma cell growth through Akt/GSK3β/forkhead signaling pathway. Biochem Biophys Res Commun. 2002;297:760–764. doi: 10.1016/s0006-291x(02)02278-7. [DOI] [PubMed] [Google Scholar]
- Gadakar PK, Phukan S, Dattatreya P, Balaji VN. Pose prediction accuracy in docking studies and enrichment of actives in the active site of GSK-3beta. J Chem Inf Model. 2007;47:1446–1459. doi: 10.1021/ci6005036. [DOI] [PubMed] [Google Scholar]
- Gomez-Ramos A, Dominguez J, Zafra D, Corominola H, Gomis R, Guinovart JJ, et al. Sodium tungstate decreases the phosphorylation of tau through GSK3β inactivation. J Neurosci Res. 2006;83:264–273. doi: 10.1002/jnr.20726. [DOI] [PubMed] [Google Scholar]
- Gould TD. Targeting glycogen synthase Kinase-3 as an approach to develop novel mood-stabilizing medications. Expert Opin Ther Targets. 2006;10:377–392. doi: 10.1517/14728222.10.3.377. [DOI] [PubMed] [Google Scholar]
- Grisouard J, Medunjanin S, Hermani A, Shukla A, Mayer D. Glycogen synthase kinase-3 protects estrogen receptor alpha from proteasomal degradation and is required for full transcriptional activity of the receptor. Mol Endocrinol. 2007;21:2427–2439. doi: 10.1210/me.2007-0129. [DOI] [PubMed] [Google Scholar]
- Gum RJ, Gaede LL, Koterski SL, Heindel M, Clampit JE, Zinker BA, et al. Reduction of protein tyrosine phosphatase 1B increases insulin-dependent signaling in ob/ob mice. Diabetes. 2003;52:21–28. doi: 10.2337/diabetes.52.1.21. [DOI] [PubMed] [Google Scholar]
- Hanger DP, Hughes K, Woodgett JR, Brion JP, Anderton BH. Glycogen synthase kinase-3 induces Alzheimer's disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett. 1992;147:58–62. doi: 10.1016/0304-3940(92)90774-2. [DOI] [PubMed] [Google Scholar]
- Hardt SE, Sadoshima J. Glycogen synthase kinase 3b: a novel regulator of cardiac hypertrophy and development. Circ Res. 2002;90:1055–1063. doi: 10.1161/01.res.0000018952.70505.f1. [DOI] [PubMed] [Google Scholar]
- Hernández F, Avila J. The role of glycogen synthase kinase 3 in the early stages of Alzheimers' disease. FEBS Lett. 2008;582:3848–3854. doi: 10.1016/j.febslet.2008.10.026. [DOI] [PubMed] [Google Scholar]
- Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- Hong M, Lee VM. Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons. J Biol Chem. 1997;272:19547–19553. doi: 10.1074/jbc.272.31.19547. [DOI] [PubMed] [Google Scholar]
- Hsiung SC, Adlersberg M, Arango V, Mann JJ, Tamir H, Liu KP. Attenuated 5-HT1A receptor signaling in brains of suicide victims: involvement of adenylyl cyclase, phosphatidylinositol 3-kinase, Akt and mitogen-activated protein kinase. J Neurochem. 2003;87:182–194. doi: 10.1046/j.1471-4159.2003.01987.x. [DOI] [PubMed] [Google Scholar]
- Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3β to regulate cell growth. Cell. 2006;126:955–968. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- Jong IY, Li X-Y, Ota I, Hu C, Kim HS, Kim NH, et al. A Wnt–Axin2–GSK3β cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol. 2006;8:1398–1406. doi: 10.1038/ncb1508. [DOI] [PubMed] [Google Scholar]
- Joong MC, Ro S, Lee TG, Lee KJ, Shin D, Hyun Y-L, et al. Glycogen synthase kinase 3beta inhibitor, composition and process for the preparation thereof. 2004. WO 04/065370 A1.
- Jope RS, Roh MS. Glycogen synthase kinase-3 (GSK3β) in psychiatric diseases and therapeutic interventions. Curr Drug Targets. 2006;7:1421–1434. doi: 10.2174/1389450110607011421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang T, Wei Y, Honaker Y, Yamaguchi H, Appella E, Hung MC, et al. GSK-3b targets Cdc25A for ubiquitin-mediated proteolysis and GSK-3b inactivation correlates with Cdc25A overproduction in human cancers. Cancer Cell. 2008;13:36–47. doi: 10.1016/j.ccr.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannoji A, Phukan S, Babu VS, Balaji VN. GSK3β: a master switch and a promising target. Expert Opin Ther Target. 2008;12:1443–1455. doi: 10.1517/14728222.12.11.1443. [DOI] [PubMed] [Google Scholar]
- Kerkela R, Woulfe K, Force T. Glycogen synthase kinase-3b – actively inhibiting hypertrophy. Trends Cardiovasc Med. 2007;17:91–96. doi: 10.1016/j.tcm.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Kim W, Wilson P, Morland S, Campbell I, Walsh M, Hurst T, et al. β-Catenin mutation and expression analysis in ovarian cancer: exon 3 mutations and nuclear translocation in 16% of endometrioid tumours. Int J Cancer. 1999;82:625–629. doi: 10.1002/(sici)1097-0215(19990827)82:5<625::aid-ijc1>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Kim WY, Zhou FQ, Zhou J, Yokota Y, Wang YM, Yoshimura T, et al. Essential roles for GSK-3s and GSK-3-primed substrates in neurotrophin-induced and hippocampal axon growth. Neuron. 2006;52:981–996. doi: 10.1016/j.neuron.2006.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Hino S, Oue N, Asahara T, Zollo M, Yasui W, et al. Glycogen synthase kinase 3 and h-prune regulate cell migration by modulating focal adhesions. Mol Cell Biol. 2006;26:898–911. doi: 10.1128/MCB.26.3.898-911.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotoh M, Katoh M. Cross-talk of WNT and FGF signaling pathways at GSK3β to regulate β-catenin and SNAIL signaling cacades. Cancer Biology & Therapy. 2006;5:1059–1064. doi: 10.4161/cbt.5.9.3151. [DOI] [PubMed] [Google Scholar]
- Koul D, Shen R, Bergh S, Lu Y, de Groot JF, Liu TJ, et al. Targeting integrin-linked kinase inhibits Akt signaling pathways and decreases tumor progression of human glioblastoma. Mol Cancer Ther. 2005;4:1681–1688. doi: 10.1158/1535-7163.MCT-05-0258. [DOI] [PubMed] [Google Scholar]
- Krymsky MA, Kudryashov DS, Shirinsky VP, Lukas TJ, Watterson DM, Vorotnikov AV. Phosphorylation of kinase-related protein (telokin) in tonic and phasic smooth muscles. J Muscle Res Cell Motil. 2001;22:425–437. doi: 10.1023/a:1014503604270. [DOI] [PubMed] [Google Scholar]
- Kwok JB, Hallupp M, Loy CT, Chan DK, Woo J, Mellick GD, et al. GSK3β polymorphisms alter transcription and splicing in Parkinson's disease. Ann Neurol. 2005;58:829–839. doi: 10.1002/ana.20691. [DOI] [PubMed] [Google Scholar]
- Lather V, Jagmohan SS, Kristam R, Karthikeyan NA, Balaji VN. QSAR models for prediction of glycogen synthase kinase-3beta inhibitory activity of indirubin derivatives. QSAR Combi Sci. 2008;27:718–728. [Google Scholar]
- Laure-Emmanuelle Z, Wdziekonski B, Fontaine C, Villageois P, Peraldi P, Dani C. Effects of GSK3β inhibitors on in vitro expansion and differentiation of human adipose-derived stem cells into adipocytes. BMC Cell Biol. 2008;9:11. doi: 10.1186/1471-2121-9-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenferink AE, Busse D, Flanagan WM, Yakes FM, Arteaga CL. ErbB2/neu kinase modulates cellular p27(Kip1) and cyclin D1 through multiple signaling pathways. Cancer Res. 2001;61:6583–6591. [PubMed] [Google Scholar]
- Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle. 2003;2:339–345. [PubMed] [Google Scholar]
- Liao X, Thrasher JB, Holzbeierlein J, Stanley S, Li B. Glycogen synthase kinase-3beta activity is required for androgen-stimulated gene expression in prostate cancer. Endocrinology. 2004;145:2941–2949. doi: 10.1210/en.2003-1519. [DOI] [PubMed] [Google Scholar]
- Loberg RD, Northcott CA, Watts SW, Brosius FC., 3rd PI3-kinase-induced hyperreactivity in DOCA-salt hypertension is independent of GSK-3 activity. Hypertension. 2003;41:898–902. doi: 10.1161/01.HYP.0000061762.58873.2F. [DOI] [PubMed] [Google Scholar]
- Mai L, Jope RS, Li X. BDNF-mediated signal transduction is modulated by GSK3β and mood stabilizing agents. J Neurochem. 2002;82:75–83. doi: 10.1046/j.1471-4159.2002.00939.x. [DOI] [PubMed] [Google Scholar]
- Martinez A, Alonso M, Castro A, Dorronsoro I, Gelpi JL, Luque FJ, et al. SAR and 3D-QSAR studies on thiadiazolidinone derivatives: exploration of structural requirements for glycogen synthase kinase 3 inhibitors. J Med Chem. 2005;48:7103–7112. doi: 10.1021/jm040895g. [DOI] [PubMed] [Google Scholar]
- Martinez A, Perez DI. GSK-3 inhibitors: a ray of hope for the treatment of Alzheimer's disease? J Alzheimers Dis. 2008;15:181–191. doi: 10.3233/jad-2008-15204. [DOI] [PubMed] [Google Scholar]
- Matsuda T, Zhai P, Maejima Y, Hong C, Gao S, Tian B, et al. Distinct roles of GSK-3alpha and GSK-3beta phosphorylation in the heart under pressure overload. Proc Natl Acad Sci. 2008;105:20900–20905. doi: 10.1073/pnas.0808315106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazanetz MP, Fischer PM. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat Rev Drug Discov. 2007;6:464–479. doi: 10.1038/nrd2111. [DOI] [PubMed] [Google Scholar]
- Meijer L, Skaltsounis AL, Magiatis P, Polychronopoulos P, Knockaert M, Leost M, et al. GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem Biol. 2003;10:1255–1266. doi: 10.1016/j.chembiol.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Mendes CT, Mury FB, de Sá Moreira E, Alberto FL, Forlenza OV, Dias-Neto E, et al. Lithium reduces GSK3β mRNA levels: implications for Alzheirmer Disease. Eur Arch Psychiatry Clin Neurosci. 2009;259:16–22. doi: 10.1007/s00406-008-0828-5. [DOI] [PubMed] [Google Scholar]
- Michelon L, Meira-Lima I, Cordeiro Q, Miguita K, Breen G, Collier D, et al. Association study of the INPP1, 5HTT, BDNF, AP-2beta and GSK-3beta GENE variants and restrospectively scored response to lithium prophylaxis in bipolar disorder. Neurosci Lett. 2006;403:288–293. doi: 10.1016/j.neulet.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Morisco C, Seta K, Hardt SE, Lee Y, Vatner SF, Sadoshima J. Glycogen synthase kinase 3beta regulates GATA4 in cardiac myocytes. J Biol Chem. 2001;276:28586–28597. doi: 10.1074/jbc.M103166200. [DOI] [PubMed] [Google Scholar]
- Mozaffari MS, Schaffer SW. Effect of Pressure Overload on Cardioprotection of Mitochondrial KATP Channels and GSK-3β: Interaction With the MPT Pore. Am J Hypertension. 2008;21:570–575. doi: 10.1038/ajh.2008.25. [DOI] [PubMed] [Google Scholar]
- Nikoulina SE, Ciaraldi TP, Mudaliar S, Carter L, Johnson K, Henry RR. Inhibition of glycogen synthase kinase 3 improves insulin action and glucose metabolism in human skeletal muscle. Diabetes. 2002;51:2190–2198. doi: 10.2337/diabetes.51.7.2190. [DOI] [PubMed] [Google Scholar]
- Nowicki M, Dmitrieva N, Stein AM, Cutter JL, Godlewski J, Saeki Y, et al. Lithium inhibits invasion of glioma cells; possible involvement of glycogen synthase kinase-3. Neuro Oncol. 2008;10:690–699. doi: 10.1215/15228517-2008-041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohira T, Gemmill RM, Ferguson K, Kusy S, Roche J, Brambilla E, et al. WNT7a induces E-cadherin in lung cancer cells. Proc Natl Acad Sci U S A. 2003;100:10429–10434. doi: 10.1073/pnas.1734137100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascal G, Alistair L, Mourad S, Franck S, Philippe Y. Substituted 8′-pyridinyl-dihydrospiro-(cycloalkyl)-pyrimido (1,2-a)pyrimidin-6-one and 8′-pyrimidinyl-dihydrospiro-(cycloalkyl)-pyrimido (1,2-a) pyrimidin-6-one derivatives and their use against neurogenerative diseases. 2004. WO 04/078760 A1.
- Patricia A, Day L, Leyi G. Use of a GSK3β inhibitor in the manufacture of a medicament for increasing bone formation methods for increasing bone formation. 2003. WO 03/057202 A1.
- Patel DS, Dessalew N, Iqbal P, Bharatam PV. Structure-based approaches in the design of GSK-3 selective inhibitors. Curr Protein Pept Sci. 2007;8:352–364. doi: 10.2174/138920307781369409. [DOI] [PubMed] [Google Scholar]
- Patel S, Woodgett J. Glycogen synthase kinase-3 and cancer: good cop, bad cop? Cancer Cell. 2008;14:351–353. doi: 10.1016/j.ccr.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peineau S, Bradley C, Taghibiglou C, Doherty A, Bortolotto AZ, Wang YT, et al. The role of GSK-3 in synaptic plasticity. Br J Pharmacol. 2008;153(S1):S428–S437. doi: 10.1038/bjp.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, et al. LTP inhibits LTD in the hippocampus via regulation of GSK3β. Neuron. 2007;53:703–717. doi: 10.1016/j.neuron.2007.01.029. [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Klein PS. Molecular targets of lithium action. Annu Rev Pharmacol Toxicol. 2001;41:789–813. doi: 10.1146/annurev.pharmtox.41.1.789. [DOI] [PubMed] [Google Scholar]
- Polgar T, Baki A, Szendrei GI, Keseru GM. Comparative virtual and experimental high-throughput screening for glycogen synthase kinase-3beta inhibitors. J Med Chem. 2005;48:7946–7959. doi: 10.1021/jm050504d. [DOI] [PubMed] [Google Scholar]
- Prasad CP, Rath G, Mathur S, Bhatnagar D, Parshad R, Ralhan R. Expression analysis of E-cadherin, Slug and GSK3β in Invasive ductal carcinoma of breast. BMC Cancer. 2009;9:325. doi: 10.1186/1471-2407-9-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanna S, Daga PR, Xie A, Doerksen RJ. Glycogen synthase kinase-3 inhibition by 3-anilino-4-phenylmaleimides: insights from 3D-QSAR and docking. J Comput Aided Mol Des. 2009;23:113–127. doi: 10.1007/s10822-008-9244-1. [DOI] [PubMed] [Google Scholar]
- Qu ZS, Tian Q, Zhou XW, Wang Q, Zhang Q, Wang JZ. Mechanism of tau hyperphosphorylation in brain cortex of diabetic rats and effect of LiCl. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2006;28:244–248. [PubMed] [Google Scholar]
- Raymond JA, Michel R, Jasbinder S, Danny L. Granulatimide derivatives for use in cancer treatment. 1999. WO 99/47522 A1.
- Ring DB, Johnson KW, Henriksen EJ, Nuss JM, Goff D, Kinnick TR, et al. Selective glycogen synthase kinase 3 inhibitors potentiate insulin activation of glucose transport and utilization in vitro and in vivo. Diabetes. 2003;52:588–595. doi: 10.2337/diabetes.52.3.588. [DOI] [PubMed] [Google Scholar]
- Robertson LA, Kim AJ, Werstuck GH. Mechanisms linking diabetes mellitus to the development of atherosclerosis: a role for endoplasmic reticulum stress and glycogen synthase kinase-3. Can J Physiol Pharmacol. 2006;84:39–48. doi: 10.1139/Y05-142. [DOI] [PubMed] [Google Scholar]
- Rosano L, Spinella F, Di Castro V, Nicotra MR, Dedhar S, de Herreros AG, et al. Endothelin-1 promotes epithelial-to-mesenchymal transition in human ovarian cancer cells. Cancer Res. 2005;65:11649–11657. doi: 10.1158/0008-5472.CAN-05-2123. [DOI] [PubMed] [Google Scholar]
- Ross SE, Hemati N, Longo KA, Bennett CN, Lucas PC, Erickson RL, et al. Inhibition of adipogenesis by Wnt signaling. Science. 2000;289:950–953. doi: 10.1126/science.289.5481.950. [DOI] [PubMed] [Google Scholar]
- Ryan KA, Pimplikar SW. Activation of GSK-3 and phosphorylation of CRMP2 in transgenic mice expressing APP intracellular domain. J Cell Biol. 2005;171:327–335. doi: 10.1083/jcb.200505078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryder J, Su Y, Liu F, Li B, Zhou Y, Ni B. Divergent roles of GSK3βand CDK5 in APP processing. Biochem Biophys Res Commun. 2003;312:922–929. doi: 10.1016/j.bbrc.2003.11.014. [DOI] [PubMed] [Google Scholar]
- Ryves WJ, Dajani R, Pearl L, Harwood AJ. Glycogen synthase kinase-3 inhibition by lithium and beryllium suggests the presence of two magnesium binding sites. Biochem Biophys Res Commun. 2002;290:967–972. doi: 10.1006/bbrc.2001.6305. [DOI] [PubMed] [Google Scholar]
- Sagae S, Kobayashi K, Nishioka Y, Sugimura M, Ishioka S, Nagata M, et al. Mutational analysis of beta-catenin gene in Japanese ovarian carcinomas: frequent mutations in endometrioid carcinomas. Jpn J Cancer Res. 1999;90:510–515. doi: 10.1111/j.1349-7006.1999.tb00777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayas CL, Ariaens A, Ponsioen B, Moolenaar WH. GSK-3 is activated by the tyrosine kinase Pyk2 during LPA1-mediated neurite retraction. Mol Biol Cell. 2006;17:1834–1844. doi: 10.1091/mbc.E05-07-0688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakoori A, Ougolkov A, Yu ZW, Zhang B, Modarressi MH, Billadeau DD, et al. Deregulated GSK3β activity in colorectal cancer: its association with tumor cell survival and proliferation. Biochem Biophys Res Commun. 2005;334:1365–1373. doi: 10.1016/j.bbrc.2005.07.041. [DOI] [PubMed] [Google Scholar]
- Shao J, Jung C, Liu C, Sheng H. Prostaglandin E2 Stimulates the beta-catenin/T cell factor-dependent transcription in colon cancer. J Biol Chem. 2005;280:26565–26572. doi: 10.1074/jbc.M413056200. [DOI] [PubMed] [Google Scholar]
- Sharma M, Chuang WW, Sun Z. Phosphatidylinositol 3-kinase/Akt stimulates androgen pathway through GSK3_ inhibition and nuclear _-catenin accumulation. J Biol Chem. 2002;277:30935–30941. doi: 10.1074/jbc.M201919200. [DOI] [PubMed] [Google Scholar]
- Shudong W, Christopher M, Gavin W, Kenneth D, Daniella Z, Peter F. Therapeutic applications of 2-substituted 4-heteroarylpyrimidines. 2004. WO 04/056368 A1.
- Shudong W, Janice M, Darren G, Ashley C, Nicholas T, Peter F. 2-Aminophenyl-4-phenylpyrimidines as kinase inhibitors. 2005. WO 05/012262 A1.
- Smith E, Frenkel B. Glucocorticoids inhibit the transcriptional activity of LEF/TCF in differentiating osteoblasts in a glycogen synthase kinase-3beta-dependent and -independent manner. J Biol Chem. 2005;280:2388–2394. doi: 10.1074/jbc.M406294200. [DOI] [PubMed] [Google Scholar]
- Sugden PH, Fuller SJ, Weiss SC, Clerk A. Glycogen synthase kinase 3 (GSK3β) in the heart: a point of integration in hypertrophic signaling and a therapeutic target? A critical analysis. Br J Pharmacol. 2008;153:S137–S153. doi: 10.1038/sj.bjp.0707659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung CK, Choi WS, Scalia P. Insulin-stimulated glycogen synthesis in cultured hepatoma cells: differential effects of inhibitors of insulin signaling molecules. J Recept Signal Transduct Res. 1998;18:243–263. doi: 10.3109/10799899809047746. [DOI] [PubMed] [Google Scholar]
- Takashima A. GSK-3 is essential in the pathogenesis of Alzheimer's disease. J Alzheimers Dis. 2006;9:309–317. doi: 10.3233/jad-2006-9s335. [DOI] [PubMed] [Google Scholar]
- Thiel A, Heinonen M, Rintahaka J, Hallikainen T, Hemmes A, Dixon DA, et al. Expression of cyclooxygenase-2 is regulated by glycogen synthase kinase-3beta in gastric cancer cells. J Biol Chem. 2006;281:4564–4569. doi: 10.1074/jbc.M512722200. [DOI] [PubMed] [Google Scholar]
- Thierry G, Patrick L, Alistair L, Severine M, Alain N, Mourad S, et al. Substituted-2-pyridinyl-6,7,8,9-tetrahydropyrimido(1,2-a)pyrimidin-4-one and 7-pyridinyl-2,3-dihydroimidazo(1,2-a)pyrimidin-5(1H)one derivatives. 2003. WO 03/027116 A2.
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian D, Zhu M, Chen WS, Li JS, Wu RL, Wang X. Role of glycogen synthase kinase 3 in squamous differentiation induced by cigarette smoke in porcine tracheobronchial epithelial cells. Food Chem Toxicol. 2006;44:1590–1596. doi: 10.1016/j.fct.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Toshiyuki K, Kenji F, Tokushi H. Dihydropyrazolopyridine compounds. 2004. WO 04/014910 A1.
- Viatour P, Dejardin E, Warnier M, Lair F, Claudio E, Bureau F, et al. GSK3β-mediated BCL-3 phosphorylation modulates its degradation and its oncogenicity. Mol Cell. 2004;16:35–45. doi: 10.1016/j.molcel.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Vogl UM, Berger W, Micksche M, Pirker C, Lamm W, Pichelmeyer O, et al. Synergistic effect of Sorafenib and Sunitinib with Enzastaurin, a selective protein kinase C inhibitor in renal cell carcinoma cell lines. Cancer Lett. 2009;277:218–226. doi: 10.1016/j.canlet.2008.12.013. [DOI] [PubMed] [Google Scholar]
- Wang Q, Wang X, Hernandez A, Hellmich MR, Gatalica Z, Evers BM. Regulation of TRAIL expression by the phosphatidylinositol 3-kinase/Akt/GSK-3 pathway in human colon cancer cells. J Biol Chem. 2002;277:36602–36610. doi: 10.1074/jbc.M206306200. [DOI] [PubMed] [Google Scholar]
- Welcker M, Singer J, Loeb KR, Grim J, Bloecher A, Gurien-West M, et al. Multisite phosphorylation by Cdk2 and GSK3β controls cyclin E degradation. Mol Cell. 2003;12:381–392. doi: 10.1016/s1097-2765(03)00287-9. [DOI] [PubMed] [Google Scholar]
- Yi F, Sun J, Lim GE, Fantus IG, Brubaker PL, Jin T. Cross talk between the insulin and Wnt signaling pathways: evidence from intestinal endocrine L cells. Endocrinology. 2008;149:2341–2351. doi: 10.1210/en.2007-1142. [DOI] [PubMed] [Google Scholar]
- Zhao WQ, Townsend M. Insulin resistance and amyloidogenesis as common molecular foundation for type 2 diabetes and Alzheimer's disease. Biochim Biophys Acta. 2009;1792:482–496. doi: 10.1016/j.bbadis.2008.10.014. [DOI] [PubMed] [Google Scholar]
- Zhou BP, Hung MC. Wnt, hedgehog and snail: sister pathways that control by GSK-3beta and beta-Trcp in the regulation of metastasis. Cell Cycle. 2005;4:772–776. doi: 10.4161/cc.4.6.1744. [DOI] [PubMed] [Google Scholar]