

Abstract
Although nitrate therapy, used in the treatment of cardiovascular disorders, is frequently associated with side-effects, mainly headaches, the summaries of product characteristics of nitrate-containing medicines do not report detailed description of headaches and even do not highlight the possibility of nitrate-induced migraine. Two different types of nitrate-induced headaches have been described: (i) immediate headaches that develop within the first hour of the application, are mild or medium severity without characteristic symptoms for migraine, and ease spontaneously; and (ii) delayed, moderate or severe migraine-type headaches (occurring mainly in subjects with personal or family history of migraine), that develop 3–6 h after the intake of nitrates, with debilitating, long-lasting symptoms including nausea, vomiting, photo- and/or phono-phobia. These two types of headaches are remarkably different, not only in their timing and symptoms, but also in the persons who are at risk. Recent studies provide evidence that the two headache types are caused by different mechanisms: immediate headaches are connected to vasodilation caused by nitric oxide (NO) release, while migraines are triggered by other actions such as the release of calcitonin gene-related peptide or glutamate, or changes in ion channel function mediated by cyclic guanosine monophosphate or S-nitrosylation. Migraines usually need anti-attack medication, such as triptans, but these drugs are contraindicated in most medical conditions that are treated using nitrates. In conclusion, these data recommend the correction of summaries of nitrate product characteristics, and also suggest a need to develop new types of anti-migraine drugs, effective in migraine attacks, that could be used in patients with risk for angina pectoris.
Keywords: headache, migraine, vasodilators, NO donors, CGRP
Nitrate compounds have been used in clinical practice for decades; their application for angina pectoris started in the 19th century. The first medicines included amyl nitrite (Brunton, 1867), glycerol trinitrate (nitroglycerin) (Ebright, 1914), erythritol-tetranitrate and penta-erythrol-tetranitrate (Weitzman, 1953). Other nitrates have also been introduced into medicine such as sodium nitroprusside for hypertension emergency (Page et al., 1955), isosorbide dinitrate (Albert, 1961), isosorbide mononitrate (Abshagen and Sporl-Radun, 1981; Uberbacher et al., 1983) and molsidomine (Jansen and Klepzig, 1976). Currently their most common indication is for angina pectoris but nitrates can also be used to treat hypertensive crisis and occasionally congestive heart failure (Bussmann and Kaltenbach, 1976; Dupuis, 1994; Elkayam et al., 2008). The main basic mechanism of action of nitrates which is beneficial in the above mentioned pathological conditions is to reduce preload and afterload by vasodilation – vascular smooth muscle relaxation; thus nitrates decrease the strain on the heart and the cardiac oxygen demand. Nitrates are believed to exert their vasodilatory effect by releasing nitric oxide (NO), also known as endothelium-derived relaxing factor (Moncada et al., 1988; Esplugues, 2002).
Unfortunately, adverse effects such as headaches, postural hypotension and reflex tachycardia are frequently associated with nitrate therapy, regardless of the means of administration – be it sublingual, intravenous, oral or even transdermal (Thadani and Ripley, 2007). As cardiovascular diseases are very common in the human population and many patients receive nitrates, side-effects may be encountered by a wide variety of physicians but predominantly by general practitioners, cardiologists and emergency physicians. Yet detailed descriptions of the side-effects still do not differentiate between a common headache (due to vasodilation) and migraine which is a headache syndrome with specific symptoms and time-course that makes it distinctive from a common headache. Even the most thorough reviews do not provide a detailed discussion of nitrate-induced migraine (Thadani and Rodgers, 2006; Thadani and Ripley, 2007). Surprisingly, the possibility of migraine is not noted in the summaries of product characteristics (SmPC) of nitrate-containing medicinal products either (Table 1.).
Table 1.
Different nitrate preparations and the frequency of headache
| Medicine and its application | The frequency of headache in SmPC (EMC, 2010; Pharmindex, 2008) | The frequency of headache in Micromedex database (Micromedex, Updated periodically) |
|---|---|---|
| Nitroglycerin aerosol | Not properly specified, >10% | Any formulation: >60% |
| Nitroglycerin sublingual pill | Not properly specified, >10% | Migraine and cluster are mentioned |
| Nitroglycerin retard tablet | Not properly specified, >10% | |
| Nitroglycerin transdermal patch | very common >10% | |
| Isosorbid-mononitrate retard tablet | very common >10% | Between 25–40% |
| Isosorbid-dinitrate tablet | In the UK SmPC not specified Not marketed in Hungary. | Most frequent (>10%) |
| Sodium Nitroprusside | Not marketed in the UK and Hungary | Not specified |
| Pentaerythrol-tetranitrate tablet | Frequency is not specified. Not marketed in the UK | Frequency is not specified |
| Molsidomine tablet | Frequency is not specified. Not marketed in the UK | Between 10% and 25% |
A headache often develops after the administration of any nitrate. Its incidence, according to clinical studies, varies between 20% and 82% (Thadani and Rodgers, 2006; Elkayam et al., 2008). Approximately 10% of the exposed patients cannot tolerate the nitrate therapy because of unbearable headaches (Thadani and Rodgers, 2006), although it is unclear whether or not this intolerability is due to migraine. Usually the headache disappears 1–1.5 h after the administration of the nitrate and the symptoms do not resemble those of migraine without aura. However, other patients experience real migraine attacks several hours after taking the medicine (Bellantonio et al., 1997; Juhasz et al., 2004).
Our aim is to review the different types of nitrate-induced headaches and to discuss the possible mechanisms underlying these symptoms. We intend to point out that cardiovascular patients with headache syndrome, especially with migraine without aura, have a much greater risk of experiencing severe headache-type side-effects compared with non-migraineurs during nitrate therapy. We will discuss how to predict possible severe headache-type side-effects and consider treatments recommended by international guidelines.
The drug/molecular target nomenclature applied in the manuscript conforms to British Journal of Pharmacology's Guide to Receptors and Channels (Alexander et al., 2008).
Headaches are not uniform
In order to differentiate correctly between the specific types of headaches, it is important to mention the second version of the International Classification of Headache Disorders (Headache Classification Committee of the International Headache Society, 2004) that provides guidance for diagnosing headaches. Table 2 summarizes the lifetime prevalence, the characteristics, the duration and the related subtypes of the different primary headache groups. Syndromes of primary headaches are those headaches that are not attributed to other medical disorders or conditions based on medical history, physical, neurological and other examinations. To assign a particular headache diagnosis the patient must, in most cases, experience a minimum number of attacks.
Table 2.
Summary of primary headaches based on the second version of the International Classification of Headache Disorders (Headache Classification Committee of the International Headache Society, 2004)
| Tension | Migraine | Cluster | |
|---|---|---|---|
| Lifetime population prevalence | Male: 42% Female: 49% (Stovner et al., 2007) | Male: 10% Female: 22% (Stovner et al., 2007) | 0.12%, more prevalent in males (Fischera et al., 2008) |
| Headache characteristics | At least two of: 1.Bilateral 2.Pressing or tightening 3.Mild or moderate 4.NOT aggravated by routine physical activity | At least two of: 1.Unilateral 2.Pulsating 3.Moderate or severe 4.Aggravation by routine physical activity | 1.Strictly unilateral (periorbital, temporal) 2.Severe or very severe |
| Associated symptoms | Photophobia or phonophobia can be present (only one of them) | At least one of: 1.Photophobia, phonophobia 2.Nausea, vomiting | At least one of ipsilateral: 1.Conjunctival injection, lacrimation 2.Nasal conjection or rhinorrhoea 3.Eyelid oedema 4.Forehead, facial sweating 5.Miosis, ptosis 6.Restlessness, agitation |
| Duration | 30 min – 7 days, or may be continuous | 4–72 h | 15–180 min |
| Most frequent subtypes | Infrequent episodic tension-type headache Frequent episodic tension-type headache Chronic tension-type headache | Migraine without aura Migraine with aura | Cluster headache Other trigeminal autonomic cephalalgias |
Tension-type headache is the most prevalent in the population (Stovner et al., 2007) and if it is not frequent or chronic, medical consultation is rare (Goadsby, 2006b). It is often related to psychological stress or muscle tenderness and simple analgesics are the most effective class of drugs for acute treatment (Loder and Rizzoli, 2008).
Migraine is a common and disabling disorder. The main characteristics which separate migraine from the occasional tension-type headache are the associated features (Table 2.) that eventually lead to significant limitations in patients' lives (Goadsby, 2006b; Lipton and Bigal, 2007). Another important feature of migraine is its hereditary nature; several family clustering and twin studies suggest that genetic factors play an important role in the pathogenesis of migraine, with a heritability of around 50% (Wessman et al., 2007). Migraine attacks can be triggered by alteration in stress level, diet, sleep pattern or hormonal status (Goadsby, 2006b). Before a migraine attack, about half of the patients suffer from premonitory symptoms such as feeling tired or weary, difficulty concentrating or food cravings (Silberstein, 2004). A minority of migraineurs (approximately 30%) experience aura that consists of reversible focal neurological symptoms – most frequently visual or sensory – which precede, accompany, or (rarely) follow an attack (Silberstein, 2004; Goadsby, 2007b). Based on surveys at least 50% of patients with migraine have never been diagnosed, or have received an inappropriate diagnosis, and nearly three-quarters of potential migraineurs were only self-medicated (Brandes, 2008).
Cluster headache attacks typically occur in series (cluster periods) lasting for weeks or months and interrupted by remission periods usually lasting months or years (Headache Classification Committee of the International Headache Society, 2004). Although it is a rare type of headache, it is important to include it in this review because nitrate therapy may induce or exacerbate cluster attacks (Ekbom et al., 2004).
Characteristics of NO donor-induced headaches
As previously mentioned, the beneficial effects of nitrates relate to their NO-donating ability (Ignarro et al., 2002) and it has been suggested that NO release is also responsible for nitrate-induced headaches (Iversen, 1995). Subsequently, nitrate models of vascular headache have been developed, initiating a more systematic approach to the study of nitrate-induced headaches (Magis et al., 2007); a recent review (Olesen, 2008) summarizes the results of these studies in detail.
Healthy subjects
In healthy volunteers, glycerol trinitrate (GTN) infusion (max 0.5 mg·kg−1·min−1 over 15–20 min) caused a reproducible and immediate headache that eased rapidly after termination of the infusion. This headache was mild or moderate with pulsating quality and aggravated by physical activity but without any accompanying symptoms (e.g. photo-and phonophobia, nausea and vomiting) (Iversen, 1995). In the first 30–60 min following sublingual administration of GTN (0.5–0.9 mg), approximately 30% of healthy subjects experienced a non-specific headache that did not fulfil the International Headache Society (IHS) criteria for migraine without aura, lasted only a few minutes and disappeared spontaneously (Juhasz et al., 2003b; 2004; Sances et al., 2004). A delayed, migraine-type headache developed in healthy subjects only if they had a predisposition for migraine (e.g. family history of migraine) (Juhasz et al., 2003b; Afridi et al., 2004; Sances et al., 2004). However, extreme NO-donor exposure [5-isosorbide-mononitrate (5-ISMN) 30 mg three times daily] was able to provoke migraine even in healthy subjects without risk factors for migraine (Christiansen et al., 2000b).
Tension-type headache patients
Only one study reported the effect of GTN infusion (max 0.5 mg·kg−1·min−1 over 15–20 min) in episodic tension-type headache patients. Seven out of nine developed immediate headache with intensity and duration intermediate between those of migraineurs and controls (Olesen et al., 1993). In chronic tension-type headache patients, GTN infusion induced immediate headache similarly to episodic tension-type headache patients (Ashina et al., 2000). Furthermore, chronic tension-type headache patients experienced a delayed headache with similar characteristics to their usual headache. The intensity of this delayed pain reached its maximum about 8 h after the infusion and half of the patients had to use rescue medications (Ashina et al., 2000).
Migraine patients
Migraine sufferers are the most systematically investigated population regarding NO donor-induced headaches and the main findings of these studies have been reviewed recently (Magis et al., 2007). In this section we will focus on the characteristics of the induced headaches.
During GTN infusion (max 0.5 mg·kg−1·min−1 over 15–20 min), migraineurs experienced stronger immediate headaches (some with additional migraine-characteristic symptoms), compared with controls or to tension-type headache patients. However, in most cases this immediate headache did not fulfil the criteria for migraine because accompanying symptoms were not present (Thomsen and Olesen, 1997). In contrast to the healthy controls, approximately 80% of the migraineurs reported delayed headaches 3–6 h after the infusion, which they labelled as a typical migraine attack. These headaches fulfilled the IHS criteria for migraine without aura, were moderate or severe, with similar accompanying symptoms to those reported during spontaneous migraine attacks (Thomsen and Olesen, 1997; Christiansen et al., 2000a; Afridi et al., 2004).
Sublingual GTN (0.5 mg) administration provoked similar headache patterns to the GTN infusion in migraineurs. Twice as much migraineurs (67%) than controls experienced an immediate headache that developed within the first hour (mean latency: 10.5 ± 2.8 min; mean duration 30.0 ± 7.7 min); but these headaches did not fulfil the IHS criteria for migraine without aura and eased spontaneously. However, a typical migraine attack without aura subsequently developed in 71% of migraine patients with a peak headache intensity about 5–7 h after the GTN intake, but not in the controls without risk factors for migraine (Figure 1) (Juhasz et al., 2003b; 2004;). Using sublingual GTN in a higher dose (0.9 mg) even more migraineurs developed an immediate headache (83% with frequent migraine-characteristic symptoms), and 78% developed a delayed migraine-type headache with a mean latency of 140 min (Sances et al., 2004).
Figure 1.
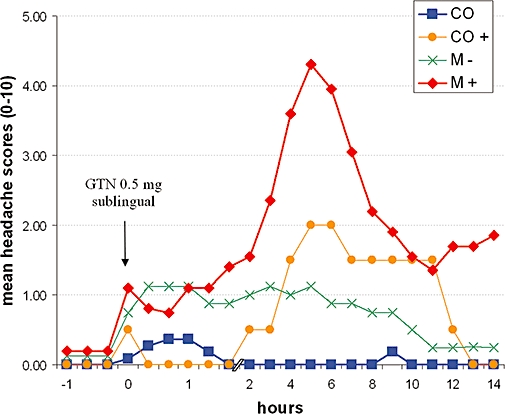
Headache intensity (0–10 verbal scale) after sublingual nitroglycerin (GTN, 0.5 mg) administration in controls (CO; n= 11), in controls with risk factor for migraine (CO+; n= 2), in migraineurs who did not develop delayed migraine type headache (M−; n= 8) and in migraineurs who developed typical delayed migraine without aura (M+; n= 20). (Combined data from Juhasz et al., 2003b; 2005;.)
It is interesting to note that GTN provocation only rarely (0–14%) caused typical aura symptoms (Christiansen et al., 1999; Bank, 2001; Sances et al., 2004; Afridi et al., 2005) and patients with familial hemiplegic migraine (FHM) did not develop a delayed migraine-type headache after GTN infusion (Hansen et al., 2008a,b;). FHM is a rare, severe, genetically heterogeneous autosomal dominant subtype of migraine with aura, characterized by at least some degree of weakness (hemiparesis) during the aura (Headache Classification Committee of the International Headache Society, 2004).
Cluster headache patients
Cluster attacks were induced by sublingual GTN only in the cluster headache patients whose disorder was in an active phase (i.e. in a cluster period, see paragraph about classification of headaches) (Fanciullacci et al., 1997; Ekbom et al., 2004). Cluster attacks started 30–45 min after GTN intake; pain became severe or very severe within 5–15 min and was indistinguishable from a spontaneous attack (Fanciullacci et al., 1997).
Nitrate-induced headaches in cardiovascular patients
Much less is known about nitrate-induced headaches as side-effects in clinical practice. Describing the safety profile of a medicine is not usually the main goal of a clinical study; the primary end-point is to examine the pharmacokinetics of the proposed medicine (phase I) or assess its effectiveness (phases II and III). Since 1949 several clinical trials have been designed and conducted to test the efficacy of nitrates in cardiovascular disorders (Dewar et al., 1959; Colditz et al., 1988; Yusuf et al., 1988; Scheidt, 1990; Held, 1992; Thadani and de Vane, 1992; Armstrong and Moe, 1993; Jugdutt, 1994; Abshagen, 1996; Parker, 1996; Thadani, 1997). Headache was reported as a common side-effect but without any deeper analysis (Thadani and Rodgers, 2006; Thadani and Ripley, 2007). Based on the estimation that the annual incidence rate of nitrate use (prescription of glyceryl trinitrate, isosorbide dinitrate and isosorbide mononitrate including sublingual, aerosol, transdermal and oral preparations for new patients) is about 1.5% in the European countries (Hemingway et al., 2006) and that 10% of nitrate-treated patients report unbearable headaches (Thadani and Rodgers, 2006) then in Europe alone more than 700 000 patients every year suffer from serious nitrate-induced headaches. However, there is no indication of the characteristics of the headache or of the time–intensity relationship.
Some studies have investigated the occurrence of nitrate-induced headaches in various pathological conditions. For example, the probability of suffering a GTN-induced headache was much lower in chest-pain patients with obstructive coronary artery disease, than in chest-pain patients with normal coronaries or with minimal coronary artery disease (Hsi et al., 2005); and using acute nitrate therapy the incidence of reported headache was low in acute myocardial infarction but high in unstable angina (Thadani and Ripley, 2007).
Migraine-like adverse effects after nitrate therapy have been reported only sparsely in the past; for example, Muller et al. reported hemicrania (Mueller and Meienberg, 1983) and Bank reported migraine with aura in a patient suffering from angina pectoris (Bank, 2001). Cluster headaches were also observed during nitrate therapy (Ekbom et al., 2004). In subjects with coexisting cluster headache and angina pectoris it has been shown that sustained nitrate therapy induced extra headache periods, but during active clusters angina pectoris became remitted (Ekbom et al., 2004).
Potential mechanisms of NO-provoked headache types
Vasodilation
The main effects of nitrates are attributed to their vasodilatory effect by releasing NO (Moncada et al., 1988). Although the relationship between NO and headaches has been the subject of considerable recent discussion and research, the exact mechanisms of the different types of NO-induced headaches are still not clear (van der Kuy and Lohman, 2003; Goadsby, 2007b; Olesen, 2008).
One hypothesis suggests that NO, as a powerful vasodilator, might be responsible for nitrate-induced migraine headaches through dilation of the large cranial arteries that have been reported during spontaneous migraine headaches (Iversen et al., 1990). However, some recent data contradict any direct relation between NO-induced migraine attacks and NO-evoked vasodilation (Akerman et al., 2002b; Goadsby, 2006b; Juhasz et al., 2007); a human 3T magnetic resonance angiography study found that migraine attacks were not associated with vasodilation of cerebral or meningeal blood vessels (Schoonman et al., 2008). Furthermore, sildenafil induces migraine without any change in middle cerebral artery diameter through activation of the cyclic guanosine monophosphate (cGMP) pathway, which is part of the NO-mediated signalling cascade (Kruuse et al., 2003), while the potent vasodilator vasoactive intestinal polypeptide causes a weak immediate headache in healthy volunteers (Hansen et al., 2006) but does not trigger a migraine attack in migraine patients (Rahmann et al., 2008).
Other data provide further evidence for the dissociation of vasodilator and migraine-inducing effect of nitrates. The 5-HT2 receptor agonist meta-chlorophenylpiperazine (m-CPP) caused migraine-type headaches with similar characteristics to those induced by nitrates in the same patient group at risk for NO donor-induced migraine. However, in contrast to nitrates, this compound causes vasoconstriction and sympathoadrenal activation rather than vasodilation (Bagdy et al., 1988; Bagdy, 1998)
Taken together these observations suggest that although NO plays a role in some aspects of the pathophysiology of migraine it is unlikely simply to be a vascular effect. A more likely explanation of the mechanism of action of NO in migraine could be, for example, through the release of calcitonin gene-related peptide (CGRP) (Juhasz et al., 2003b; 2005;). This will be discussed in the next section.
Previous studies have suggested that immediate headaches may be related to vasodilation caused by direct activation of the NO-cGMP pathway (Figure 3) (Akerman et al., 2002b; Juhasz et al., 2003b). Through protein kinase G (PKG) the NO-cGMP pathway is able to control the function of various ion channels, including potassium channels that modify the smooth muscle contractility and vascular tone (Sobey, 2001). It is well documented that during NO-donor administration, meningeal and cerebral vasodilation occur both in animals (Dong et al., 1998; Bergerot et al., 2006) and in humans (Schoonman et al., 2008) and human subjects also report immediate headaches paralleling the time-course of the vasodilation (Hansen et al., 2007). It was observed recently that NO-donor GTN-induced dural and pial vasodilation involved the opening of calcium-activated potassium (KCa1.1 also known as BK or Slo1) channels (Gozalov et al., 2007), which suggests that the KCa1.1 channels may play an important role in the NO-induced immediate headaches.
Figure 3.
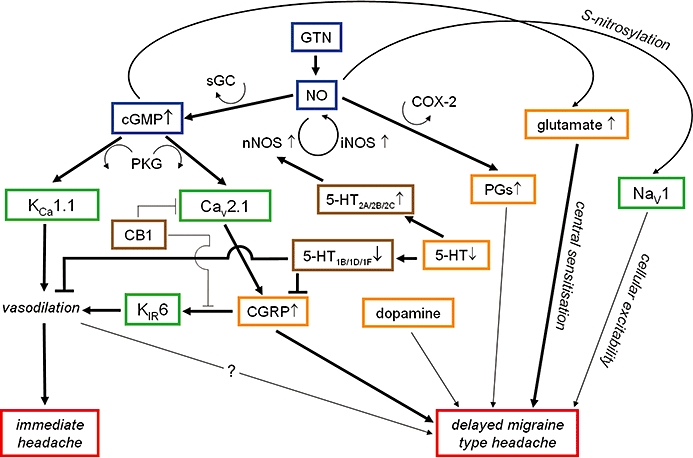
Nitrate derived NO actions relevant to nitroglycerin (GTN)-induced immediate and delayed (migraine-type) headaches. The NO donor GTN activates the NO-cGMP pathway via the soluble guanylate cyclase enzyme (sGC). Through protein kinase G the NO-cGMP pathway is able to control the function of various ion channels, including the calcium-activated potassium channels KCa1.1 (also known as BK or Slo1) that modify smooth muscle contractility and may play an important role in immediate headaches. The NO-cGMP pathway also increases calcitonin gene-related peptide (CGRP) release mainly by activation of Cav2.1 (P/Q-type) voltage-gated calcium channels. CGRP can cause vasodilation via KIR6 channels (also known as KATP) and can cause delayed migraine-type headaches via mechanisms which are not fully understood yet. The CB1 receptor is able to inhibit both CGRP- and NO-induced dural vessel dilation. It is possible that this is partially due to the inhibition of Cav2.1 (P/Q-type) voltage-gated calcium channels but the mechanism is still unclear. The trigeminovascular activation, both vasodilation and CGRP release, can be blocked via the activation of 5-HT1B/1D/1F receptors. In chronic migraine patients up-regulation of 5-HT2 receptors due to the low baseline 5-HT level may occur, and an increase in nNOS activity and elevated NO release due to an acute increase in 5-HT release could be expected. The NO-cGMP pathway also increases the release of glutamate that has major role in central sensitization and can lead to migraine attacks. Dopamine, prostaglandins and sodium channels also play a role in NO-induced migraine-type headaches but further studies are needed to elucidate the exact mechanism. Red box: different type of headaches, green boxes: ion channels, orange boxes: neurotransmitters, brown boxes: neurotransmitters and modulators, blue boxes: GTN-NO-cGMP pathway, arrows: activation lines: blockade of action, thick lines: evidence based pathways, thin lines: hypothesized pathways.
Calcitonin gene-related peptide (CGRP)
Elevated CGRP levels have been found in patients during spontaneous migraine (Goadsby et al., 1990; Gallai et al., 1995; Bellamy et al., 2006b) and cluster headache attacks (Goadsby and Edvinsson, 1994; Fanciullacci et al., 1995), which suggests the activation of the trigeminovascular system (Goadsby, 2007b). The proven efficacy of CGRP antagonists in migraine attacks supports the pivotal role of CGRP in migraine pain (Doods et al., 2007; Edvinsson, 2008). Besides CGRP antagonists, animal studies indicate a potential usefulness of CGRP antibodies in the treatment of migraine as these antibodies showed a long inhibitory effect (Zeller et al., 2008).
GTN-induced migraine is the most studied human migraine model and shows considerable similarities with spontaneous migraine attacks (Olesen, 2008). Indeed, previous studies have demonstrated that peripheral plasma CGRP concentration increased significantly also during the GTN-induced delayed migraine attack (Figure 2) (Juhasz et al., 2003b; 2005;), which provides additional support for the hypothesis that migraine attacks are a result of trigeminovascular activation (Akerman et al., 2002b).
Figure 2.
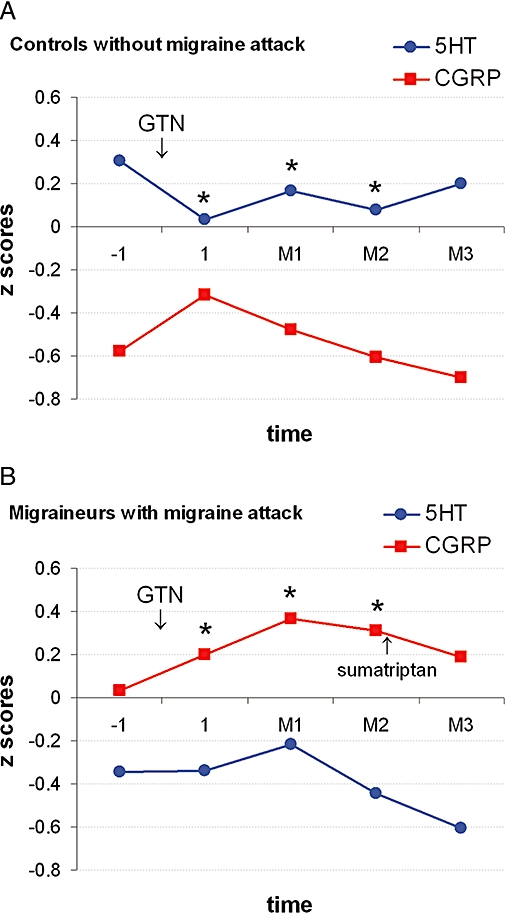
Effects of nitroglycerin (GTN, 0.5 mg sublingual) on plasma calcitonin gene-related peptide (CGRP) and platelet 5-HT concentrations in controls without delayed migraine attack (n= 10; A) and in migraineurs with delayed migraine attack (n= 19; B). We calculated z scores for plasma CGRP and platelet 5-HT concentrations [(subgroup mean − grand mean)/subgroup SD] to compare their changes during the GTN challenge. Baseline blood samples were collected at 7.00 a.m. (−1). A secondary blood sample was taken 1 h after sublingual application of GTN, at 9.00 a.m. (1). The next three blood samples were taken 60 min (M1), 120 min (M2) and 180 min (M3) after the beginning of the migraine attack. In controls, similar time schedules were used based on preliminary data (M1: 5 h, M2: 6 h and M3: 7 h after GTN respectively). Migraine patients took 20 mg sumatriptan nasal spray immediately after the M2 blood sampling. *Significant changes after sublingual GTN compared with baseline (P < 0.05). (Combined data from Juhasz et al., 2003b; 2005;.)
In contrast, the plasma CGRP concentration did not increase significantly during the NO-induced immediate headache (Ashina et al., 2001; Juhasz et al., 2003b). These data support the hypothesis that the initial headache may be related to a direct action of the NO-cGMP pathway via vasodilation, independently of the CGRP release (Ashina et al., 2001; Akerman et al., 2002b) as we discussed in the previous section. It seems that GTN triggers a cascade of events, initially leading to vasodilation, and subsequently to the release of CGRP which is involved in the causation of migraine in subjects who are at risk.
The mechanism of NO-induced delayed migraine is not clear. CGRP receptors can be found in the trigeminocervical complex (Storer et al., 2004) and CGRP and neuronal NO synthase (nNOS) are frequently co-localized in neurons of the trigeminal ganglion (Hou et al., 2001) and NO regulates the expression of the CGRP gene (Bellamy et al., 2006a). Animal studies have demonstrated that NO donors caused biphasic activation of the trigeminal nucleus caudalis measured by c-fos expression (Tassorelli et al., 2000; Akerman et al., 2002b). The second phase reached its maximum after 4 h (Pardutz et al., 2000; Tassorelli et al., 2000) similar to the latency of delayed headache attacks. At the same time (4 h after NO-donor administration) both neuronal (Pardutz et al., 2000) and inducible (Reuter et al., 2001) NO synthase (nNOS and iNOS) expressions were up-regulated. This cascade of events eventually leads to CGRP release from the trigeminal ganglion (Pardutz et al., 2000; Tassorelli et al., 2000; Akerman et al., 2002b; Greco et al., 2008b). The CGRP release seems to involve activation of mainly Cav2.1 (P/Q-type) and Cav3 (T-type) voltage-gated Ca2+ channels (Bellamy et al., 2006a; Xiao et al., 2008). On the other hand, blockers of Cav1, Cav2.1 and Cav3 channels could inhibit CGRP release and consequently the dural vasodilation indicating an important role of these channels in trigeminovascular nociception (Akerman et al., 2003). In addition, both endogenous and exogenous CGRP-induced vasodilations have been attenuated by the KIR6 (also known as KATP) channel inhibitor glibenclamide (Gozalov et al., 2008), which suggests that KIR6 channels, under certain circumstances, are involved in the CGRP-induced actions and might have a role in migraine pain (Figure 3).
Serotonin (5-HT)
Serotonin has been implicated in migraine pathophysiology for several years. In our previous studies, platelet serotonin concentration was significantly lower in migraineurs (Juhasz et al., 2003a), and after GTN administration, migraineurs developed delayed migraine-type headaches much more frequently than did controls (Juhasz et al., 2003b). Chronically low 5-HT levels in plasma and brain have been reported to increase the sensitivity of the trigeminovascular pathway (Hamel, 2007), and serotonin depletion to increase nNOS activity (Tagliaferro et al., 2001). In animal studies, hyposerotoninergic conditions enhanced the vascular effects of GTN, especially 30–60 min after administration (Srikiatkhachorn et al., 2000). In addition, up-regulation of 5-HT2 receptors led to increased NO production, through activation of nNOS (Figure 3), and this process increased the chemically induced pain sensitivity in animals and might be responsible for chronic headaches in humans (Srikiatkhachorn et al., 2002; Mehrotra et al., 2008). These data support the observation that migraineurs are hypersensitive to NO donors.
In our GTN challenge study we could not demonstrate significant changes in platelet serotonin concentration in those who developed delayed migraine, but in those who remained migraine-free we observed an early decrease in platelet serotonin concentration, suggesting release (Figure 2) (Juhasz et al., 2003b). Our results suggest that a significant release of serotonin in subjects who did not develop migraine attack may act on 5-HT1B/1D receptors, as do triptans, and thus prevent trigeminovascular activation (Juhasz et al., 2003b; 2005;). Indeed, triptans were effective in NO donor-induced migraine attacks (Juhasz et al., 2005), prevented NO-induced immediate headache in healthy controls (Iversen and Olesen, 1996) and also blocked NO-induced blood vessel dilatation and trigeminovascular activation in an animal model (Akerman et al., 2002a).
Other neurotransmitters
In general, NO is a modulator of several neuronal functions in the brain and these effects are mainly mediated by cGMP (Prast and Philippu, 2001; Esplugues, 2002; Garthwaite, 2008). The potential mechanisms of NO-provoked headache types are summarized in Figure 3.
It has been demonstrated that NO donors increase the release of glutamate (via the NO-cGMP pathway), while NOS inhibitors decrease glutamate output (Prast and Philippu, 2001; Garthwaite, 2008). The elevated glutamate level might contribute to the central neuronal sensitization to incoming sensory signals, which is an important factor in migraine (Calabresi et al., 2007). A previous study showed that cerebrospinal and plasma glutamate levels were increased in chronic migraine patients (Peres et al., 2004). Antiepileptic drugs which are effective in migraine prevention target the glutamate-mediated excitatory and/or GABA-mediated inhibitory systems in conjunction with a modulation of voltage-gated sodium (Nav1) and calcium (Cav) channels; indeed chronic administration of valproic acid prevented GTN-induced delayed migraine attack (Tvedskov et al., 2004). However, further studies are needed to investigate the role of central sensitization and glutamate in NO-induced headaches and migraine.
Dopamine is also believed to play an important role in the pathophysiology of migraine although there are several conflicting results. Migraineurs are hypersensitive to dopamine agonists, while dopamine antagonists have beneficial effects in migraine treatment (Akerman and Goadsby, 2007). Dopamine is also implicated in premonitory symptoms, such as yawning and nausea, and may also contribute to hypotensive changes (Akerman and Goadsby, 2007). Currently there is little available information concerning dopamine and GTN-induced headaches although, in animals, intact central dopaminergic neurotransmission is essential for the GTN-induced activation of brain areas involved in migraine attacks, as well as for the hyperalgesic response to painful stimuli elicited by the GTN (Greco et al., 2008a). It is important to note that dopamine neurotransmission interacts robustly with the 5-HT system. Serotonin receptors, such as 5-HT1A, 5-HT1B, 5-HT2A and 5-HT2C, that have been implicated in migraine pathophysiology (Hamel, 2007), also modulate dopamine release (Alex and Pehek, 2007).
Finally, we recently showed that variations in the cannabinoid receptor 1 gene were significantly associated with migraine headaches (Juhasz et al., 2009). Akerman et al. demonstrated that anandamide, an endogenous ligand to the cannabinoid CB1 receptor, decreases CGRP and NO induced dural vasodilations by 30% and 40%, respectively, in animal models (Akerman et al., 2004). The exact mechanism of this action is not fully understood. However, as the CB1 receptor is able to inhibit voltage-gated calcium channels and activate inwardly rectifying potassium channels it is possible that ion channels have a major role in its effect.
Cyclooxygenase-2 (COX-2) – prostaglandin pathway
A previous study reported that, in the jugular venous blood, the level of algogen prostaglandins (PGs), namely the PGE2 and the stable product of prostacyclin (PGF1α), significantly increased during the late phase (2–4 h after the onset) of spontaneous migraine attacks (Sarchielli et al., 2000). In animal models of migraine, GTN activated the COX-2 – PGE2 pathway in the brainstem 4 h after GTN administration (Tassorelli et al., 2007). However, it has been demonstrated that GTN-induced vasodilation in vivo is independent of the PG system (Ahlner et al., 1991) thus it is unlikely that the COX-2 – PGE2 pathway has a major role in immediate GTN-induced headaches but its role in delayed migraine-type headaches warrants further studies.
NO-induced ion channel modifications
NO modifies the function of ion channels responsible for excitability, predominantly in two ways (Ahern et al., 2002) – indirectly and directly. According to the classical view, all the actions of NO are mediated by cGMP; the three principal targets of which are PKG, cyclic nucleotide gated channels and cyclic nucleotide phosphodiesterase. Of these, PKG provides the broadest means for controlling ion-channel functions, as these proteins contain PKG phosphorylation sites that are strongly conserved. This classical view was overturned following findings that NO could modify proteins through direct chemical reactions. One such modification, termed S-nitrosylation, occurs at the thiol side-chains of cysteine residues through a complex chemical mechanism without the assistance of enzymes. However, S-nitrosylation requires higher concentrations of NO than the activation of the cGMP pathway. S-nitrosylation of various ion channels can be mediated not only directly by NO, but also by NO metabolites, peroxynitrite and other reactive oxidant species (e.g. NO2, OH, H2O2) (Kang et al., 2007; Liu et al., 2007; Ashki et al., 2008; Dyachenko et al., 2009). Nevertheless it was supposed that the nitrosative and oxidative actions of peroxynitrite may play a role both in regulation of normal cellular functions and in its well-known cytotoxic effects (Ferdinandy, 2006). Table 3 summarizes NO actions on various ion channels, focusing mainly on those that are thought to play a role in migraine pain.
Table 3.
Ion channels involved in NO effects (modified from Ahern et al., 2002)
| Ion channel | Site | Effect | References |
|---|---|---|---|
| cGMP-modulated channels | |||
| Nav channel | Olfactory receptors | ↑ (PKG) | Kawai and Miyachi (2001) |
| Cardiomyocytes | ↓ (PKG) | Ahmmed et al. (2001) | |
| KCa1.1 channel | Pituitary nerve | ↑ (PKG) | Klyachko et al. (2001) |
| Pituitary cell line | ↑ (PKG) | White (1999) | |
| Smooth muscle | ↑ (PKG) | Pfeifer et al. (1998) | |
| Dermal fibroblast | ↑ | Roh et al. (2007) | |
| Endothelial cell | ↑ | Dong et al. (2008) | |
| Kv1.5 channel | Cardiomyocytes | ↓ | Nunez et al. (2006) |
| Kv4.3 channel | Cardiomyocytes | ↓ | Gomez et al. (2008) |
| KIR6 channel | Cardiomyocytes | ↑ (PKG) | Han et al. (2001) |
| Cav1 channel | Cardiomyocytes | Multiple actions | Fischmeister and Mery (1996) |
| Cardiomyocytes | ↓ | Bai et al. (2004) | |
| Hippocampal neuron | ↓ (PKG) | Doerner and Alger (1988) | |
| Cav2.2 channel | Retinal ganglion cell | ↓ (PKG) | Hirooka et al. (2000) |
| Dorsal root ganglion | ↓ (PKG) | Yoshimura et al. (2001) | |
| Cav3 channel | Olfactory receptor | ↑ (PKG) | Kawai and Miyachi (2001) |
| S-nitrosylated channels | |||
| Nav channel | Nodose ganglia | ↓ | Li et al. (1998) |
| Posterior pituitary | Persistence | Ahern et al. (2000) | |
| Cardiomyocytes | Persistence | Ahern et al. (2000) | |
| Hippocampus | Persistence | Hammarstrom and Gage (1999) | |
| Spinal cord neuron | ↓ | Ashki et al. (2008) | |
| KCa1.1 channel | Brain | ↑ | Shin et al. (1997) |
| Posterior pituitary | ↑ | Ahern et al. (1999) | |
| Hippocampal neuron | ↑ | Tjong et al. (2007; 2008;) | |
| Smooth muscle | ↑ | Bolotina et al. (1994); Lang and Watson (1998) | |
| Kv1.5 channel | Cardiomyocytes | ↓ | Nunez et al. (2006) |
| Kv2.1 channel | Cardiomyocytes | ↑ | Gomez et al. (2009) |
| Kv4.3 channel | Hippocampus | ↓ | Liu et al. (2007) |
| Cav1 channel | Cardiomyocytes | ↓ | Hu et al. (1997) |
| Cav1.2 channel | Smooth muscle | ↓ | Kang et al. (2007) |
Sodium channels
The primary route by which NO modulates Nav1 channels appears to be directly via S-nitrosylation although Nav1 channels of sensory neurons differentially respond to NO (Hammarstrom and Gage, 1999; Ahern et al., 2002). In one class of sensory neurons (nodose ganglion) S-nitrosylation inhibits both tetrodotoxin (TTX)-sensitive and -insensitive Nav1 channels (Li et al., 1998), while in dorsal root ganglia neither TTX-sensitive nor -resistant action potentials were affected by NO (Yoshimura et al., 2001).
A different form of Nav1 channel modulation has been described for the ‘persistent’ Na+ current: many excitable tissues have a component of Na+ current that is resistant to inactivation. This ‘persistent’ Na+ current is believed to play an important role in the integration of synaptic inputs, the generation of rhythmic oscillations, and the pathological changes in electrical excitability associated with many disease states including cardiac arrhythmias, ischaemic stroke, epilepsy and probably migraine (Segal and Douglas, 1997). NO donors have been shown to increase persistent Na+ current in posterior pituitary nerve terminals (Ahern et al., 2000) and hippocampal neurons (Hammarstrom and Gage, 1999). This action of NO on Nav1 channels is thought to be a direct chemical reaction with a protein thiol (S-nitrosylation) as described above (Hammarstrom and Gage, 1999; Ahern et al., 2000; Evans and Bielefeldt, 2000). In neurohypophysial nerve terminals and in ventricular myocytes NO reduced Na+ current inactivation thus inducing persistent Na+ currents. This effect was independent of cGMP, and was blocked by the alkylating agent N-ethylmaleimide thus, apparently NO acts directly on the channel or on a closely associated protein. Persistent Na+ current could also be induced by endogenous NO, generated enzymatically by nNOS (activated via Ca2+ loading). Both rises in cellular NO and modulation of Na+ channels were blocked by NOS inhibitors. These findings suggest that NO is a potential physiological regulator of Nav1 channel persistence and this is likely to have important consequences for the regulation of cellular excitability (Ahern et al., 2000).
The potential role of Nav1 channels in the pathophysiology of migraine is supported by the fact that among the well-known prophylactic drugs amytriptyline, valproate and topiramate also affect Nav1 channel function (Meldrum and Rogawski, 2007), and also by the association between Nav1 channel mutations and FHM (see section ‘Migraine as a channelopathy’).
Calcium channels
cGMP differentially modulates a variety of Cav channels depending on tissue, species and age. cGMP depresses Ca2+ current in hippocampal neurons (Doerner and Alger, 1988), inhibits the activation of Cav2.2 (N-type Ca2+) channels in dorsal root ganglion neurons (Yoshimura et al., 2001), but has no effect on Cav2.2 and Cav1 (l-type) channels in pituitary nerve terminals (Klyachko et al., 2001). NO can also inhibit cardiac Cav1 channels directly, via S-nitrosylation (Hu et al., 1997). High voltage-activated (HVA) Ca2+ channels (Cav1, Cav2.1, Cav2.2 and Cav3) play a prominent role in trigeminovascular nociception as they are localized to trigeminal presynaptic nerve terminal in the dura and the trigeminal nucleus caudalis. It is likely that mainly Cav2.1 channels are involved with initiation of migraine attacks and/or aura symptoms as well as with nociceptive transmission because they participate in controlling the release of CGRP (Xiao et al., 2008). Indeed, mutations in Cav2.1 channels are among the established genetic risk factors for FHM (see section ‘Migraine as a channelopathy’). Several Ca2+ channel blockers (dihydropyridines, flunarizine and anti-epileptics with Ca2+ channel inhibitory activity) are used as prophylactic drugs against migraine (Evers, 2008; Meldrum and Rogawski, 2007). Recently, it has become apparent that the molecular targets for gabapentin and pregabalin are α2-δ proteins of HVA Ca2+ channels (Davies et al., 2007); this interaction may explain the anti-epileptic and antinociceptive actions of these drugs (Davies et al., 2007).
Ca2+-activated K+ (KCa1.1) channels
Large-conductance, calcium-activated potassium (KCa1.1) channels are expressed in both excitable and non-excitable cells and are involved in many cellular functions, such as action potential repolarization, neuronal excitability, transmitter release and hormone secretion. KCa1.1 channels are special among K+ channels, being sensitive to both intracellular Ca2+ concentrations and voltage. These features make KCa1.1 channels ideal negative feedback regulators in many cell types: by hyperpolarizing the membrane they decrease voltage-dependent Ca2+ entry. Located in dendrites, axons and synaptic terminals, KCa1.1 channels thus play an important role in controlling the excitability of neurons (Ghatta et al., 2006; Nardi and Olesen, 2008). Both PKG and S-nitrosylation enhance the activity of KCa1.1 channels. In smooth muscle the cGMP-dependent pathway predominates (Pfeifer et al., 1998) but S-nitrosylation also seems to be involved (Bolotina et al., 1994; Lang and Watson, 1998), and both pathways are important in nerve terminals (Ahern et al., 2002).
As we mentioned previously, animal studies suggest that KCa1.1 channels are involved in the NO-donor GTN-induced dural and pial vasodilation or acetylcholine induced rabbit middle cerebral artery dilation and thus may play an important role in the NO-induced immediate headaches (Dong et al., 1998; Bergerot et al., 2006; Gozalov et al., 2007).
Other potassium channels
The NO-cGMP cascade regulates several other potassium channels. S-nitrosylation enhances the activity of Kv channel in arterial smooth muscle (Yuan et al., 1996). PKG modulates the neuronal voltage-gated potassium channels Kv3.1 and Kv3.2 (Moreno et al., 2001). In vestibular hair cells cGMP inhibits the delayed rectifier K+ current and shifts its activation curve to more positive direction (Behrend et al., 1997). In hippocampal CA1 neurons peroxynitrite donor caused an inhibition of transient outward potassium current (Ito, Kv 4.3 channel) and delayed rectifier potassium current (IK) (Liu et al., 2007). PKG enhances the activity of cardiac KIR6 channels (Han et al., 2001). In cardiac cells NO modifies K+ currents in a complex fashion. NO activates the slow component of the delayed rectifier current (IKs) (Bai et al., 2004) and the inwardly rectifying K+ current (IK1, KIR2.1 channel) (Gomez et al., 2009), but inhibits the fast component (IKr) (Taglialatela et al., 1999), the hKv1.5 channel which generates the ultrarapid delayed rectifier current (IKur) (Nunez et al., 2006) and the transient outward current (Ito) (Kv4.3 channel) (Gomez et al., 2008).
There is growing evidence that Kv7 (KCNQ) potassium channels, especially the neuronal Kv7.2 and Kv7.3, play a role in neurological diseases (Miceli et al., 2008). Neuronal Kv7 gene defects have been implicated in two rare forms of genetically determined human channelopathies, namely benign familial neonatal seizures and non-syndromic autosomal-dominant hearing loss. Compounds acting as direct activators of neuronal Kv7 channels have been approved recently for clinical use as analgesics and anticonvulsants (Gribkoff, 2008; Miceli et al., 2008). As neuronal hyperexcitability is a marker of other neurologic diseases, the role of this type of potassium channel has been suggested in migraine also, although direct evidence is lacking.
Nitrate tolerance and headaches
The main limitation of nitrate therapy, especially when continuous application is required, is nitrate tolerance. Nitrate tolerance is characterized by the decrease of vasodilator effect of organic nitrates that can reach complete loss of vasodilatation within 24–48 h (Mayer and Beretta, 2008). This is a complex, still not fully understood process that has been related to molecular changes in intrinsic vascular processing, such as desensitization of soluble guanylyl cyclase, oxidative stress, uncoupling of endothelial NO synthase reduced GTN activation because of vascular thiol depletion and inactivation of mitochondrial aldehyde dehydrogenase (ALDH-2) (see detailed reviews from Mayer and Beretta, 2008 and Daiber et al., 2008). Neurohormonal responses, such as sympathoadrenal axis activation and/or renin–angiotensin system activation, also contribute to the decreased nitrate efficacy (Daiber et al., 2008; Mayer and Beretta, 2008).
Regarding GTN-induced headaches attenuation can be seen after 5–7 days of the initiation of therapy (Ahlner et al., 1991). However, this timeframe does not coincide with the vascular nitrate tolerance mentioned above. Christiansen et al. showed that in healthy volunteers daily 3 × 30 mg 5-ISMN provoked the most frequent and intense headaches in the first 3 days, followed by gradual decrease in headache symptoms and tolerance had developed by the sixth day (Christiansen et al., 2000b; 2008;). They proposed that extracerebral arteries, which showed only partial nitrate tolerance after 24 h, might contribute to GTN-induced headaches besides other mechanisms. On the other hand, there is no available data about those patients whose headache was initially unbearable. Indeed headache side-effects are the major cause of discontinuation of nitrate therapy (Ahlner et al., 1991) and detailed data on these patients' headaches are not reported so far. As nitrates exert diverse effects in different tissues – for example myocardial anti-ischaemic effect has been preserved even after vascular nitrate tolerance (Csont and Ferdinandy, 2005) – it can be hypothesized that tolerance to GTN-induced severe headaches are limited or absent in these patients.
Genetic and inherited vulnerability to NO-induced headaches
To the best of our knowledge the genetic risk factors for NO-induced delayed migraine or unbearable headache have not been investigated yet. However, healthy subjects with a family history of migraine are more sensitive to NO-provoked headaches (Juhasz et al., 2003b; 2004; Afridi et al., 2004; Sances et al., 2004), which supports the hypothesis that inherited vulnerability factors play a significant role. Therefore in this section we will summarize the relevant results from genetic studies about migraine, but for detailed reviews see the work of van den Maagdenberg et al. (van den Maagdenberg et al., 2007) and Wessman et al. (Wessman et al., 2007).
Migraine as a channelopathy
Migraine is a complex genetic neurovascular disorder (Goadsby, 2007b). Many chromosomal regions are reported to be potentially involved, but mutations in the three genes for FHM – CACNA1A, ATP1A2 and SCNA1A – form the only established molecular knowledge of migraine (van den Maagdenberg et al., 2007). From a clinical point of view, FHM and migraine may be part of the same spectrum and may share some pathogenetic mechanisms. Therefore, FHM seems a valid model to study genetic factors of migraine in general.
FHM1 (CACNA1A gene)
This gene encodes the pore-forming α1A subunit of Cav2.1 calcium channels (Ophoff et al., 1998) which modulate release of neurotransmitters at peripheral and particularly central excitatory synapses. Many CACNA1A mutations have been analysed with electrophysiological techniques in neuronal and non-neuronal cell models (Pietrobon, 2005; Jeng et al., 2006; Pietrobon, 2007). Because of the different experimental circumstances, varying and conflicting results have been obtained (Pietrobon, 2007). While the consistent change found with FHM1 mutations was an enhanced single channel Ca2+ influx with an increased channel open probability producing a gain-of-function of Cav2.1 channels (Hans et al., 1999; Tottene et al., 2002; 2005;), other data obtained from transfected cells indicated the opposite effect – a loss-of-function (Cao et al., 2004; Jeng et al., 2006). Theoretically, the observed gain-of-function of single channels should lead to an easier opening of channels in neurons, resulting in increased Cav2.1-dependent neurotransmitter release from cortical neurons.
FHM2 (ATP1A2 gene)
This gene encodes the α2 subunit of sodium–potassium pump ATPase (De fusco et al., 2003). Glial and neuronal Na+/K+ ATPase modulate the re-uptake of K+ and glutamate from the synaptic cleft into neurons and astrocytes. Molecular studies of a few FHM2 mutations show different functional changes from a complete loss of function to a reduced function in the Na+/K+ ATPase activity to variable degrees. However, the common consequences of these mutations are reduced reuptake of K+ and glutamate from the synaptic cleft (see review by Pietrobon, 2007). This decreased clearance of K+ and glutamate by astrocytes during cortical neuronal activity could depolarize neurons resulting in an impaired recovery from neuronal excitation.
FHM3 (SCNA1 gene)
This gene encodes the α1 subunit of neuronal voltage-gated sodium (Nav1.1) channels that play an important role in the generation and propagation of action potentials. Only a few mutations (Q1489K, T1174S, L1649Q) have so far been identified, confirming the relationship between SCNA1 and FHM3 (Dichgans et al., 2005; Gargus and Tournay, 2007; Vanmolkot et al., 2008). However, mutation scanning of a large number of other FHM families suggests that the SCNA1 gene is a rare cause of FHM. Mutation Q1489K, located in the cytoplasmic linker between domains IIIS6 and IVS1 which is critical for fast inactivation, has been studied for its functional effects (Dichgans et al., 2005). This mutation causes a two to four times faster recovery from fast inactivation. These findings suggest a gain-of-function mechanism in FHM3 with predicted enhanced neuronal excitability and release of neurotransmitters.
Common consequence of FHM mutations
The common consequence of FHM1, FHM2 and FHM3 mutations seems to lead to increased levels of glutamate and K+ in the synaptic cleft causing an increased propensity for cortical spreading depression (CSD) (Ferrari and Goadsby, 2006). There is accumulating evidence that NO production is markedly augmented during CSD (Olesen, 2008). In this way, although mutations in FHM genes (CACNA1A in FHM-1 and ATP1A2 in FHM-2) are not associated with hypersensitivity to NO in GTN-induced headache models (Hansen et al., 2008a,b;), both CSD and GTN provocation result in higher NO levels in the central nervous system and may act on a common pathological pathway. In conclusion, even though the FHM genes are probably not directly involved in common migraines, the study of the cellular mechanisms of enhanced susceptibility to CSD and enhanced cortical excitability in FHM knock-in animal models may provide unique insights into possible mechanisms of common migraines (Pietrobon, 2007) and so also help to understand NO-induced headaches.
Nitric oxide synthase and other genes
Hypersensitivity of migraineurs to NO in the GTN-induced headache model directed research interest towards NOS genes. However, there is no confirmation that they play a major role in the vulnerability to these headaches (van den Maagdenberg et al., 2007; Wessman et al., 2007; Montagna, 2008; Olesen, 2008). Furthermore, there are no convincing replicated results that genetic variations in the serotonergic, dopaminergic, or other plausible pathways/systems are associated with common forms of migraine – with or without aura (van den Maagdenberg et al., 2007; Wessman et al., 2007; Montagna, 2008). One possible explanation for this failure is that headache diagnoses based on the IHS criteria (1988; 2004) might not represent biological pathways influenced by specific genetic variations (Anttila et al., 2006; Russell, 2007; van den Maagdenberg et al., 2007; Wessman et al., 2007). However, the episodic nature of migraine suggests an ionopathic disturbance (Goadsby, 2007a) that can be primary (as in FHM) or secondary – namely dysfunction in the ion channels' controlling networks.
Treatment of NO donor-induced migraine
Based on experimental headache provocation studies, GTN-induced headaches respond to the same drugs that are used to treat primary headache disorders (Fanciullacci et al., 1997; Ashina et al., 2000; Tvedskov et al., 2004; Juhasz et al., 2005; Magis et al., 2007). Thus, guidelines for treating primary headache patients can be adapted, after taking into account individual circumstances such as general medical condition and age (Evers et al., 2006; Goadsby, 2006a; Martelletti et al., 2008). In general, triptans should be avoided in patients with a history of coronary vascular pathology or multiple risk factors for cardiovascular disease, although the evidence suggests that they are generally safe and well-tolerated (Dodick et al., 2004).
An accurate headache diagnosis and the recognition of the possible role of nitrates depend on taking a careful medical history. Unfortunately, headaches at emergency circumstances are frequently under-diagnosed and/or under-treated, both in Europe and in the United States (Gupta et al., 2007; Martelletti et al., 2008). After exclusion of other secondary headaches, the first choice of treatment is oral or venous non-steroidal anti-inflammatory drugs. Analgesics with evidence of efficacy on the acute migraine treatment and their recommended doses can be found in the European Federation of Neurological Societies guideline, e.g. acetylsalicylic acid 1000 mg, ibuprofen 200–800 mg, naproxen 500–1000 mg, diclofenac 50–100 mg, paracetamol 1000 mg (Evers et al., 2006). Depending on the symptoms anti-emetic drugs might be necessary, e.g. metoclopramide: 10–20 mg oral, 10 mg i.v., i.m. or s.c., or 20 mg suppository; domperidon 20–30 mg oral (Ashina et al., 2000; Evers et al., 2006; Gupta et al., 2007; Martelletti et al., 2008). It is hoped that in future, new specific drugs without cardiovascular side-effects, such as CGRP antagonists, might be available for relevant at-risk patients (Doods et al., 2007).
Previous studies demonstrated that in most cases tolerance developed to nitrate-induced headaches; this tolerance was independent of the type of headache (whether it fulfilled the IHS criteria for migraine without aura or not) (Christiansen et al., 2000b; Thadani and Rodgers, 2006). However, a revised treatment plan was necessary for about 10% of patients whose nitrate-induced headache was unbearable (Thadani and Rodgers, 2006). Migraine and cluster headache patients are more susceptible to NO-induced severe headaches, so it is possible that patients with unacceptable headaches may suffer from migraine, or in some cases from cluster headaches, although there is no published evidence for this hypothesis to the best of our knowledge. Thus further studies are needed to investigate which subgroup of cardiovascular patients are not likely to tolerate nitrate therapy due to severe headache side-effects. Future research could also study the possible diagnostic and prognostic value of nitrate-induced headaches.
Conclusions
Nitrate compounds are frequently used as therapeutic drugs, despite evidence that they often trigger serious migraine attack in migraine patients (although not in healthy persons devoid of primary headache in their medical history). However, migraine is neither listed as an adverse effect of these compounds, nor noted in the summaries of product characteristics. Headache is a known side-effect of nitrates, but it is not generally known that NO donors may cause two markedly different types of headaches; (i) immediate headaches, which can occur in anyone (not just patients at risk of migraine); although these are uncomfortable, they are not serious and disappear spontaneously, and (ii) migraine attacks, which occur in migraineurs (or those at risk), usually start several hours after initial drug administration, are serious, debilitating, and of long duration needing special treatment – usually anti-attack drugs, such as triptans. However, nitrates are commonly used to treat angina pectoris, and triptans are contraindicated for this condition. Recent studies regarding the mechanism of action and side-effects of nitrates provide evidence that the two headaches are caused by different mechanisms: immediate headaches are associated with vasodilation caused by NO release, while migraines are triggered by other actions such as CGRP release, cGMP-modulated or S-nitrosylation-mediated changes in ion channel functions.
In conclusion, these data suggest that correction of summaries of nitrate product characteristics is desirable. In addition, there is an urgent need to develop new types of anti-migraine drugs, effective in migraine attacks that could be used in patients at risk for angina pectoris.
Acknowledgments
The study was supported by the Sixth Framework Program of the EU, NewMood, LSHM-CT-2004-503474; by the Hungarian Research Fund Grant T022256/1997, T032398/2000; and Postdoctoral PhD Fellowship Program (G.J., 1999–2002) of the Semmelweis University, Ministry of Culture and Education, Hungary. The authors are indebted to Dr Laszlo Csanady for critically reading the manuscript.
Glossary
Abbreviations:
- 5-HT
serotonin
- 5-ISMN
5-isosorbide-mononitrate
- ATP1A2
gene encoding the α2 subunit of sodium–potassium pump ATPases
- BK channels
calcium-activated potassium channels
- CACNA1A
gene encoding the α1A subunit of Cav2.1 (P/Q-type) voltage-gated neuronal calcium channels
- CB1
cannabinoid receptor 1
- cGMP
cyclic guanosine monophosphate
- CGRP
calcitonin gene-related peptide
- CNGCs
cyclic nucleotide gated channels
- CSD
cortical spreading depression
- EFNS
European Federation of Neurological Societies
- FHM
familial hemiplegic migraine
- GABA
gamma-amino-butyric acid
- hKv1.5
ultrarapid delayed rectifier potassium channel
- HVA
high voltage activated
- IHS
International Headache Society
- IKr
rapid component of delayed outward potassium current
- IKs
slow component of delayed outward potassium current
- IKur
ultrarapid delayed rectifier potassium current
- Ito
transient outward current
- KATP channel
ATP-dependent potassium channel
- Kv3.1 and Kv3.2
neuronal voltage-gated potassium channels
- Kv4.3
transient outward potassium channel
- Kv7(KCNQ)
Kv7 subfamily of voltage-gated potassium channels (also called KCNQ or M-channels)
- Na+/K+ ATPase
sodium/potassium ATPase
- NO
nitric oxide
- NOS
nitric oxide synthase
- PKG
protein kinase G
- SCNA1A
gene encoding the α1 subunit of neuronal voltage-gated sodium (Nav1.1) channels
- SmPC
summary of product characteristics
- TTX
tetrodotoxin
Conflict of Interest
The authors declare that, except for income received from their primary employer, no financial support or compensation has been received from any individual or corporate entity over the past three years for research or professional service and there are no personal financial holdings that could be perceived as constituting a potential conflict of interest. Dr Pál Riba occasionally has acted as an expert for the Hungarian drug regulatory agency (National Institute of Pharmacy, Hungary).
References
- Abshagen U. [Controlled clinical studies of tolerance development and dosing problems in nitrate therapy] Herz. 1996;21(Suppl 1):23–30. [PubMed] [Google Scholar]
- Abshagen U, Sporl-Radun S. First data on effects and pharmacokinetics of isosorbide-5-mononitrate in normal man. Eur J Clin Pharmacol. 1981;19:423–429. doi: 10.1007/BF00548586. [DOI] [PubMed] [Google Scholar]
- Afridi SK, Kaube H, Goadsby PJ. Glyceryl trinitrate triggers premonitory symptoms in migraineurs. Pain. 2004;110:675–680. doi: 10.1016/j.pain.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Afridi S, Kaube H, Goadsby PJ. Occipital activation in glyceryl trinitrate induced migraine with visual aura. J Neurol Neurosurg Psychiatry. 2005;76:1158–1160. doi: 10.1136/jnnp.2004.050633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahern GP, Hsu SF, Jackson MB. Direct actions of nitric oxide on rat neurohypophysial K+ channels. J Physiol. 1999;520(Pt 1):165–176. doi: 10.1111/j.1469-7793.1999.00165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahern GP, Hsu SF, Klyachko VA, Jackson MB. Induction of persistent sodium current by exogenous and endogenous nitric oxide. J Biol Chem. 2000;275:28810–28815. doi: 10.1074/jbc.M003090200. [DOI] [PubMed] [Google Scholar]
- Ahern GP, Klyachko VA, Jackson MB. cGMP and S-nitrosylation: two routes for modulation of neuronal excitability by NO. Trends Neurosci. 2002;25:510–517. doi: 10.1016/s0166-2236(02)02254-3. [DOI] [PubMed] [Google Scholar]
- Ahlner J, Andersson RG, Torfgard K, Axelsson KL. Organic nitrate esters: clinical use and mechanisms of actions. Pharmacol Rev. 1991;43:351–423. [PubMed] [Google Scholar]
- Ahmmed GU, Xu Y, Hong Dong P, Zhang Z, Eiserich J, Chiamvimonvat N. Nitric oxide modulates cardiac Na(+) channel via protein kinase A and protein kinase G. Circ Res. 2001;89:1005–1013. doi: 10.1161/hh2301.100801. [DOI] [PubMed] [Google Scholar]
- Akerman S, Goadsby PJ. Dopamine and migraine: biology and clinical implications. Cephalalgia. 2007;27:1308–1314. doi: 10.1111/j.1468-2982.2007.01478.x. [DOI] [PubMed] [Google Scholar]
- Akerman S, Williamson DJ, Kaube H, Goadsby PJ. The effect of anti-migraine compounds on nitric oxide-induced dilation of dural meningeal vessels. Eur J Pharmacol. 2002a;452:223–228. doi: 10.1016/s0014-2999(02)02307-5. [DOI] [PubMed] [Google Scholar]
- Akerman S, Williamson DJ, Kaube H, Goadsby PJ. Nitric oxide synthase inhibitors can antagonize neurogenic and calcitonin gene-related peptide induced dilation of dural meningeal vessels. Br J Pharmacol. 2002b;137:62–68. doi: 10.1038/sj.bjp.0704842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerman S, Williamson DJ, Goadsby PJ. Voltage-dependent calcium channels are involved in neurogenic dural vasodilatation via a presynaptic transmitter release mechanism. Br J Pharmacol. 2003;140:558–566. doi: 10.1038/sj.bjp.0705456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerman S, Kaube H, Goadsby PJ. Anandamide is able to inhibit trigeminal neurons using an in vivo model of trigeminovascular-mediated nociception. J Pharmacol Exp Ther. 2004;309:56–63. doi: 10.1124/jpet.103.059808. [DOI] [PubMed] [Google Scholar]
- Albert A. Isosorbide dinitrate in treatment of angina pectoris. Preliminary clinical impression. J Lancet. 1961;81:112–114. [PubMed] [Google Scholar]
- Alex KD, Pehek EA. Pharmacologic mechanisms of serotonergic regulation of dopamine neurotransmission. Pharmacol Ther. 2007;113:296–320. doi: 10.1016/j.pharmthera.2006.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl. 2):S1–209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anttila V, Kallela M, Oswell G, Kaunisto MA, Nyholt DR, Hamalainen E, et al. Trait components provide tools to dissect the genetic susceptibility of migraine. Am J Human Genetics. 2006;79:85–99. doi: 10.1086/504814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong PW, Moe GW. Medical advances in the treatment of congestive heart failure. Circulation. 1993;88:2941–2952. doi: 10.1161/01.cir.88.6.2941. [DOI] [PubMed] [Google Scholar]
- Ashina M, Bendtsen L, Jensen R, Olesen J. Nitric oxide-induced headache in patients with chronic tension-type headache. Brain. 2000;123(Pt 9):1830–1837. doi: 10.1093/brain/123.9.1830. [DOI] [PubMed] [Google Scholar]
- Ashina M, Bendtsen L, Jensen R, Schifter S, Olesen J. Calcitonin gene-related peptide levels during nitric oxide-induced headache in patients with chronic tension-type headache. Eur J Neurol. 2001;8:173–178. doi: 10.1046/j.1468-1331.2001.00191.x. [DOI] [PubMed] [Google Scholar]
- Ashki N, Hayes KC, Bao F. The peroxynitrite donor 3-morpholinosydnonimine induces reversible changes in electrophysiological properties of neurons of the guinea-pig spinal cord. Neuroscience. 2008;156:107–117. doi: 10.1016/j.neuroscience.2008.06.050. [DOI] [PubMed] [Google Scholar]
- Bagdy G. Serotonin, anxiety, and stress hormones. Focus on 5-HT receptor subtypes, species and gender differences. Ann N Y Acad Sci. 1998;851:357–363. doi: 10.1111/j.1749-6632.1998.tb09009.x. [DOI] [PubMed] [Google Scholar]
- Bagdy G, Szemeredi K, Hill JL, Murphy DL. The serotonin agonist, M-chlorophenylpiperazine, markedly increases levels of plasma catecholamines in the conscious rat. Neuropharmacology. 1988;27:975–980. doi: 10.1016/0028-3908(88)90055-x. [DOI] [PubMed] [Google Scholar]
- Bai CX, Takahashi K, Masumiya H, Sawanobori T, Furukawa T. Nitric oxide-dependent modulation of the delayed rectifier K+ current and the l-type Ca2+ current by ginsenoside Re, an ingredient of Panax ginseng, in guinea-pig cardiomyocytes. Br J Pharmacol. 2004;142:567–575. doi: 10.1038/sj.bjp.0705814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bank J. Migraine with aura after administration of sublingual nitroglycerin tablets. Headache. 2001;41:84–87. doi: 10.1046/j.1526-4610.2001.111006084.x. [DOI] [PubMed] [Google Scholar]
- Behrend O, Schwark C, Kunihiro T, Strupp M. Cyclic GMP inhibits and shifts the activation curve of the delayed-rectifier (I[K1]) of type I mammalian vestibular hair cells. Neuroreport. 1997;8:2687–2690. doi: 10.1097/00001756-199708180-00010. [DOI] [PubMed] [Google Scholar]
- Bellamy J, Bowen EJ, Russo AF, Durham PL. Nitric oxide regulation of calcitonin gene-related peptide gene expression in rat trigeminal ganglia neurons. Eur J Neurosci. 2006a;23:2057–2066. doi: 10.1111/j.1460-9568.2006.04742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellamy JL, Cady RK, Durham PL. Salivary levels of CGRP and VIP in rhinosinusitis and migraine patients. Headache. 2006b;46:24–33. doi: 10.1111/j.1526-4610.2006.00294.x. [DOI] [PubMed] [Google Scholar]
- Bellantonio P, Micieli G, Buzzi MG, Marcheselli S, Castellano AE, Rossi F, et al. Haemodynamic correlates of early and delayed responses to sublingual administration of isosorbide dinitrate in migraine patients: a transcranial Doppler study. Cephalalgia. 1997;17:183–187. doi: 10.1046/j.1468-2982.1997.1703183.x. [DOI] [PubMed] [Google Scholar]
- Bergerot A, Holland PR, Akerman S, Bartsch T, Ahn AH, MaassenVanDenBrink A, et al. Animal models of migraine: looking at the component parts of a complex disorder. Eur J Neurosci. 2006;24:1517–1534. doi: 10.1111/j.1460-9568.2006.05036.x. [DOI] [PubMed] [Google Scholar]
- Bolotina VM, Najibi S, Palacino JJ, Pagano PJ, Cohen RA. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;368:850–853. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- Brandes JL. The migraine cycle: patient burden of migraine during and between migraine attacks. Headache. 2008;48:430–441. doi: 10.1111/j.1526-4610.2007.01004.x. [DOI] [PubMed] [Google Scholar]
- Brunton TL. Use of nitrate of amyl in angina pectoris. Lancet. 1867;2:628–629. [Google Scholar]
- Bussmann WD, Kaltenbach M. Sublingual nitroglycerin in the treatment of left ventricular failure and pulmonary edema. Eur J Cardiol. 1976;4:327–333. [PubMed] [Google Scholar]
- Calabresi P, Galletti F, Rossi C, Sarchielli P, Cupini LM. Antiepileptic drugs in migraine: from clinical aspects to cellular mechanisms. Trends Pharmacol Sci. 2007;28:188–195. doi: 10.1016/j.tips.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Cao YQ, Piedras-Renteria ES, Smith GB, Chen G, Harata NC, Tsien RW. Presynaptic Ca2+ channels compete for channel type-preferring slots in altered neurotransmission arising from Ca2+ channelopathy. Neuron. 2004;43:387–400. doi: 10.1016/j.neuron.2004.07.014. [DOI] [PubMed] [Google Scholar]
- Christiansen I, Thomsen LL, Daugaard D, Ulrich V, Olesen J. Glyceryl trinitrate induces attacks of migraine without aura in sufferers of migraine with aura. Cephalalgia. 1999;19:660–667. doi: 10.1046/j.1468-2982.1999.019007660.x. discussion 626. [DOI] [PubMed] [Google Scholar]
- Christiansen I, Daugaard D, Lykke Thomsen L, Olesen J. Glyceryl trinitrate induced headache in migraineurs – relation to attack frequency. Eur J Neurol. 2000a;7:405–411. doi: 10.1046/j.1468-1331.2000.00094.x. [DOI] [PubMed] [Google Scholar]
- Christiansen I, Iversen HK, Olesen J. Headache characteristics during the development of tolerance to nitrates: pathophysiological implications. Cephalalgia. 2000b;20:437–444. doi: 10.1046/j.1468-2982.2000.00064.x. [DOI] [PubMed] [Google Scholar]
- Christiansen I, Iversen HK, Olesen J, Tfelt-Hansen P. Nitric oxide-induced headache may arise from extracerebral arteries as judged from tolerance to isosorbide-5-mononitrate. J Headache Pain. 2008;9:215–220. doi: 10.1007/s10194-008-0043-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colditz GA, Halvorsen KT, Goldhaber SZ. Randomized clinical trials of transdermal nitroglycerin systems for the treatment of angina: a meta-analysis. Am Heart J. 1988;116(1):174–180. doi: 10.1016/0002-8703(88)90263-3. Pt 1. [DOI] [PubMed] [Google Scholar]
- Csont T, Ferdinandy P. Cardioprotective effects of glyceryl trinitrate: beyond vascular nitrate tolerance. Pharmacol Ther. 2005;105:57–68. doi: 10.1016/j.pharmthera.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Daiber A, Wenzel P, Oelze M, Munzel T. New insights into bioactivation of organic nitrates, nitrate tolerance and cross-tolerance. Clin Res Cardiol. 2008;97:12–20. doi: 10.1007/s00392-007-0588-7. [DOI] [PubMed] [Google Scholar]
- Davies A, Hendrich J, Van Minh AT, Wratten J, Douglas L, Dolphin AC. Functional biology of the alpha(2)delta subunits of voltage-gated calcium channels. Trends Pharmacol Sci. 2007;28:220–228. doi: 10.1016/j.tips.2007.03.005. [DOI] [PubMed] [Google Scholar]
- De Fusco M, Marconi R, Silvestri L, Atorino L, Rampoldi L, Morgante L, et al. Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump alpha2 subunit associated with familial hemiplegic migraine type 2. Nature Genetics. 2003;33:192–196. doi: 10.1038/ng1081. [DOI] [PubMed] [Google Scholar]
- Dewar HA, Horler AR, Newell DJ. A clinical trial of penta-erythritol tetranitrate, a khellin derivative (recordil), and iproniazid in angina of effort. Br Heart J. 1959;21:315–322. doi: 10.1136/hrt.21.3.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dichgans M, Freilinger T, Eckstein G, Babini E, Lorenz-Depiereux B, Biskup S, et al. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet. 2005;366:371–377. doi: 10.1016/S0140-6736(05)66786-4. [DOI] [PubMed] [Google Scholar]
- Dodick D, Lipton RB, Martin V, Papademetriou V, Rosamond W, MaassenVanDenBrink A, et al. Consensus statement: cardiovascular safety profile of triptans (5-HT agonists) in the acute treatment of migraine. Headache. 2004;44:414–425. doi: 10.1111/j.1526-4610.2004.04078.x. [DOI] [PubMed] [Google Scholar]
- Doerner D, Alger BE. Cyclic GMP depresses hippocampal Ca2+ current through a mechanism independent of cGMP-dependent protein kinase. Neuron. 1988;1:693–699. doi: 10.1016/0896-6273(88)90168-7. [DOI] [PubMed] [Google Scholar]
- Dong DL, Yue P, Yang BF, Wang WH. Hydrogen peroxide stimulates the Ca(2+)-activated big-conductance K channels (BK) through cGMP signaling pathway in cultured human endothelial cells. Cell Physiol Biochem. 2008;22:119–126. doi: 10.1159/000149789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Waldron GJ, Cole WC, Triggle CR. Roles of calcium-activated and voltage-gated delayed rectifier potassium channels in endothelium-dependent vasorelaxation of the rabbit middle cerebral artery. Br J Pharmacol. 1998;123:821–832. doi: 10.1038/sj.bjp.0701680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doods H, Arndt K, Rudolf K, Just S. CGRP antagonists: unravelling the role of CGRP in migraine. Trends Pharmacol Sci. 2007;28:580–587. doi: 10.1016/j.tips.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Dupuis J. Nitrates in congestive heart failure. Cardiovasc Drugs Ther. 1994;8:501–507. doi: 10.1007/BF00877928. [DOI] [PubMed] [Google Scholar]
- Dyachenko V, Rueckschloss U, Isenberg G. Modulation of cardiac mechanosensitive ion channels involves superoxide, nitric oxide and peroxynitrite. Cell Calcium. 2009;45:55–64. doi: 10.1016/j.ceca.2008.06.002. [DOI] [PubMed] [Google Scholar]
- Ebright GE. The effects of nitroglycerin on those engaged in its manufacture. JAMA. 1914;62:201. [Google Scholar]
- Edvinsson L. CGRP blockers in migraine therapy: where do they act? Br J Pharmacol. 2008;155:967–969. doi: 10.1038/bjp.2008.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekbom K, Sjostrand C, Svensson DA, Waldenlind E. Periods of cluster headache induced by nitrate therapy and spontaneous remission of angina pectoris during active clusters. Cephalalgia. 2004;24:92–98. doi: 10.1111/j.1468-2982.2004.00634.x. [DOI] [PubMed] [Google Scholar]
- Elkayam U, Janmohamed M, Habib M, Hatamizadeh P. Vasodilators in the management of acute heart failure. Crit Care Med. 2008;36(1) Suppl:S95–105. doi: 10.1097/01.CCM.0000297161.41559.93. [DOI] [PubMed] [Google Scholar]
- EMC Electronic Medicines Compendium UK. 2010. http://emc.medicines.org.uk (accessed October 2010.
- Esplugues JV. NO as a signalling molecule in the nervous system. Br J Pharmacol. 2002;135:1079–1095. doi: 10.1038/sj.bjp.0704569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JR, Bielefeldt K. Regulation of sodium currents through oxidation and reduction of thiol residues. Neuroscience. 2000;101:229–236. doi: 10.1016/s0306-4522(00)00367-5. [DOI] [PubMed] [Google Scholar]
- Evers S. Treatment of migraine with prophylactic drugs. Expert Opin Pharmacother. 2008;9:2565–2573. doi: 10.1517/14656566.9.15.2565. [DOI] [PubMed] [Google Scholar]
- Evers S, Afra J, Frese A, Goadsby PJ, Linde M, May A, et al. EFNS guideline on the drug treatment of migraine – report of an EFNS task force. Eur J Neurol. 2006;13:560–572. doi: 10.1111/j.1468-1331.2006.01411.x. [DOI] [PubMed] [Google Scholar]
- Fanciullacci M, Alessandri M, Figini M, Geppetti P, Michelacci S. Increase in plasma calcitonin gene-related peptide from the extracerebral circulation during nitroglycerin-induced cluster headache attack. Pain. 1995;60:119–123. doi: 10.1016/0304-3959(94)00097-X. [DOI] [PubMed] [Google Scholar]
- Fanciullacci M, Alessandri M, Sicuteri R, Marabini S. Responsiveness of the trigeminovascular system to nitroglycerine in cluster headache patients. Brain. 1997;120(Pt 2):283–288. doi: 10.1093/brain/120.2.283. [DOI] [PubMed] [Google Scholar]
- Ferdinandy P. Peroxynitrite: just an oxidative/nitrosative stressor or a physiological regulator as well? Br J Pharmacol. 2006;148:1–3. doi: 10.1038/sj.bjp.0706693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari MD, Goadsby PJ. Migraine as a cerebral ionopathy with abnormal central sensory processing. In: Gilman S, Pedley T, editors. Neurobiology of Disease. New York: Elsevier; 2006. pp. 333–348. [Google Scholar]
- Fischera M, Marziniak M, Gralow I, Evers S. The incidence and prevalence of cluster headache: a meta-analysis of population-based studies. Cephalalgia. 2008;28:614–618. doi: 10.1111/j.1468-2982.2008.01592.x. [DOI] [PubMed] [Google Scholar]
- Fischmeister R, Mery PF. Regulation of cardiac calcium channels by cGMP and NO. In: Morad M, Ebashi S, Trautwein W, Kurachi Y, editors. Molecular Physiology and Pharmacology of Cardiac Ion Channels and Transporters. Dordrecht, The Netherlands: Kluwer Acad. Publ; 1996. pp. 93–105. [Google Scholar]
- Gallai V, Sarchielli P, Floridi A, Franceschini M, Codini M, Glioti G, et al. Vasoactive peptide levels in the plasma of young migraine patients with and without aura assessed both interictally and ictally. Cephalalgia. 1995;15:384–390. doi: 10.1046/j.1468-2982.1995.1505384.x. [DOI] [PubMed] [Google Scholar]
- Gargus JJ, Tournay A. Novel mutation confirms seizure locus SCN1A is also familial hemiplegic migraine locus FHM3. Pediatr Neurol. 2007;37:407–410. doi: 10.1016/j.pediatrneurol.2007.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite J. Concepts of neural nitric oxide-mediated transmission. Eur J Neurosci. 2008;27:2783–2802. doi: 10.1111/j.1460-9568.2008.06285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghatta S, Nimmagadda D, Xu X, O'Rourke ST. Large-conductance, calcium-activated potassium channels: structural and functional implications. Pharmacol Ther. 2006;110:103–116. doi: 10.1016/j.pharmthera.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Goadsby PJ. Migraine: emerging treatment options for preventive and acute attack therapy. Expert Opinion Emerg Drugs. 2006a;11:419–427. doi: 10.1517/14728214.11.3.419. [DOI] [PubMed] [Google Scholar]
- Goadsby PJ. Recent advances in the diagnosis and management of migraine. BMJ. 2006b;332:25–29. doi: 10.1136/bmj.332.7532.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goadsby PJ. Emerging therapies for migraine. Nat Clin Pract Neurol. 2007a;3:610–619. doi: 10.1038/ncpneuro0639. [DOI] [PubMed] [Google Scholar]
- Goadsby PJ. Recent advances in understanding migraine mechanisms, molecules and therapeutics. Trends Mol Med. 2007b;13:39–44. doi: 10.1016/j.molmed.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Goadsby PJ, Edvinsson L. Human in vivo evidence for trigeminovascular activation in cluster headache. Neuropeptide changes and effects of acute attacks therapies. Brain. 1994;117(Pt 3):427–434. doi: 10.1093/brain/117.3.427. [DOI] [PubMed] [Google Scholar]
- Goadsby PJ, Edvinsson L, Ekman R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurology. 1990;28:183–187. doi: 10.1002/ana.410280213. [DOI] [PubMed] [Google Scholar]
- Gomez R, Nunez L, Vaquero M, Amoros I, Barana A, de Prada T, et al. Nitric oxide inhibits Kv4.3 and human cardiac transient outward potassium current (Ito1) Cardiovasc Res. 2008;80:375–384. doi: 10.1093/cvr/cvn205. [DOI] [PubMed] [Google Scholar]
- Gomez R, Caballero R, Barana A, Amoros I, Calvo E, Lopez JA, et al. Nitric oxide increases cardiac IK1 by nitrosylation of cysteine 76 of Kir2.1 channels. Circ Res. 2009;105:383–392. doi: 10.1161/CIRCRESAHA.109.197558. [DOI] [PubMed] [Google Scholar]
- Gozalov A, Jansen-Olesen I, Klaerke D, Olesen J. Role of BK(Ca) channels in cephalic vasodilation induced by CGRP, NO and transcranial electrical stimulation in the rat. Cephalalgia. 2007;27:1120–1127. doi: 10.1111/j.1468-2982.2007.01409.x. [DOI] [PubMed] [Google Scholar]
- Gozalov A, Jansen-Olesen I, Klaerke D, Olesen J. Role of K ATP channels in cephalic vasodilatation induced by calcitonin gene-related peptide, nitric oxide, and transcranial electrical stimulation in the rat. Headache. 2008;48:1202–1213. doi: 10.1111/j.1526-4610.2008.01205.x. [DOI] [PubMed] [Google Scholar]
- Greco R, Tassorelli C, Armentero MT, Sandrini G, Nappi G, Blandini F. Role of central dopaminergic circuitry in pain processing and nitroglycerin-induced hyperalgesia. Brain Res. 2008a;1238:215–223. doi: 10.1016/j.brainres.2008.08.022. [DOI] [PubMed] [Google Scholar]
- Greco R, Tassorelli C, Sandrini G, Di Bella P, Buscone S, Nappi G. Role of calcitonin gene-related peptide and substance P in different models of pain. Cephalalgia. 2008b;28:114–126. doi: 10.1111/j.1468-2982.2007.01468.x. [DOI] [PubMed] [Google Scholar]
- Gribkoff VK. The therapeutic potential of neuronal K V 7 (KCNQ) channel modulators: an update. Expert Opin Ther Targets. 2008;12:565–581. doi: 10.1517/14728222.12.5.565. [DOI] [PubMed] [Google Scholar]
- Gupta MX, Silberstein SD, Young WB, Hopkins M, Lopez BL, Samsa GP. Less is not more: underutilization of headache medications in a university hospital emergency department. Headache. 2007;47:1125–1133. doi: 10.1111/j.1526-4610.2007.00846.x. [DOI] [PubMed] [Google Scholar]
- Hamel E. Serotonin and migraine: biology and clinical implications. Cephalalgia. 2007;27:1293–1300. doi: 10.1111/j.1468-2982.2007.01476.x. [DOI] [PubMed] [Google Scholar]
- Hammarstrom AK, Gage PW. Nitric oxide increases persistent sodium current in rat hippocampal neurons. J Physiolo. 1999;520(Pt 2):451–461. doi: 10.1111/j.1469-7793.1999.t01-1-00451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Kim N, Kim E, Ho WK, Earm YE. Modulation of ATP-sensitive potassium channels by cGMP-dependent protein kinase in rabbit ventricular myocytes. J Biol Chem. 2001;276:22140–22147. doi: 10.1074/jbc.M010103200. [DOI] [PubMed] [Google Scholar]
- Hans M, Luvisetto S, Williams ME, Spagnolo M, Urrutia A, Tottene A, et al. Functional consequences of mutations in the human alpha1A calcium channel subunit linked to familial hemiplegic migraine. J Neurosci. 1999;19:1610–1619. doi: 10.1523/JNEUROSCI.19-05-01610.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen JM, Sitarz J, Birk S, Rahmann AM, Oturai PS, Fahrenkrug J, et al. Vasoactive intestinal polypeptide evokes only a minimal headache in healthy volunteers. Cephalalgia. 2006;26:992–1003. doi: 10.1111/j.1468-2982.2006.01149.x. [DOI] [PubMed] [Google Scholar]
- Hansen JM, Pedersen D, Larsen VA, Sanchez-del-Rio M, Alvarez Linera JR, Olesen J, et al. Magnetic resonance angiography shows dilatation of the middle cerebral artery after infusion of glyceryl trinitrate in healthy volunteers. Cephalalgia. 2007;27:118–127. doi: 10.1111/j.1468-2982.2006.01257.x. [DOI] [PubMed] [Google Scholar]
- Hansen JM, Thomsen LL, Marconi R, Casari G, Olesen J, Ashina M. Familial hemiplegic migraine type 2 does not share hypersensitivity to nitric oxide with common types of migraine. Cephalalgia. 2008a;28:367–375. doi: 10.1111/j.1468-2982.2008.01542.x. [DOI] [PubMed] [Google Scholar]
- Hansen JM, Thomsen LL, Olesen J, Ashina M. Familial hemiplegic migraine type 1 shows no hypersensitivity to nitric oxide. Cephalalgia. 2008b;28:496–505. doi: 10.1111/j.1468-2982.2008.01559.x. [DOI] [PubMed] [Google Scholar]
- Headache Classification Subcommittee of the International Headache Society. The International Classification of Headache Disorders: 2nd edition. Cephalalgia. 2004;24(Suppl 1):9–160. doi: 10.1111/j.1468-2982.2003.00824.x. [DOI] [PubMed] [Google Scholar]
- Held P. Effects of nitrates on mortality in acute myocardial infarction and in heart failure. Br J Clin Pharmacol. 1992;34(Suppl. 1):25S–28S. doi: 10.1111/j.1365-2125.1992.tb04145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemingway H, McCallum A, Shipley M, Manderbacka K, Martikainen P, Keskimaki I. Incidence and prognostic implications of stable angina pectoris among women and men. JAMA. 2006;295:1404–1411. doi: 10.1001/jama.295.12.1404. [DOI] [PubMed] [Google Scholar]
- Hirooka K, Kourennyi DE, Barnes S. Calcium channel activation facilitated by nitric oxide in retinal ganglion cells. J Neurophysiol. 2000;83:198–206. doi: 10.1152/jn.2000.83.1.198. [DOI] [PubMed] [Google Scholar]
- Hou M, Kanje M, Longmore J, Tajti J, Uddman R, Edvinsson L. 5-HT(1B) and 5-HT(1D) receptors in the human trigeminal ganglion: co-localization with calcitonin gene-related peptide, substance P and nitric oxide synthase. Brain Res. 2001;909:112–120. doi: 10.1016/s0006-8993(01)02645-2. [DOI] [PubMed] [Google Scholar]
- Hsi DH, Roshandel A, Singh N, Szombathy T, Meszaros ZS. Headache response to glyceryl trinitrate in patients with and without obstructive coronary artery disease. Heart (Br Cardiac Soc) 2005;91:1164–1166. doi: 10.1136/hrt.2004.035295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Chiamvimonvat N, Yamagishi T, Marban E. Direct inhibition of expressed cardiac l-type Ca2+ channels by S-nitrosothiol nitric oxide donors. Circ Res. 1997;81:742–752. doi: 10.1161/01.res.81.5.742. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ, Napoli C, Loscalzo J. Nitric oxide donors and cardiovascular agents modulating the bioactivity of nitric oxide: an overview. Circ Res. 2002;90:21–28. doi: 10.1161/hh0102.102330. [DOI] [PubMed] [Google Scholar]
- Iversen HK. Experimental headache in humans. Cephalalgia. 1995;15:281–287. doi: 10.1046/j.1468-2982.1995.1504281.x. [DOI] [PubMed] [Google Scholar]
- Iversen HK, Olesen J. Headache induced by a nitric oxide donor (nitroglycerin) responds to sumatriptan. A human model for development of migraine drugs. Cephalalgia. 1996;16:412–418. doi: 10.1046/j.1468-2982.1996.1606412.x. [DOI] [PubMed] [Google Scholar]
- Iversen HK, Nielsen TH, Olesen J, Tfelt-Hansen P. Arterial responses during migraine headache. Lancet. 1990;336:837–839. doi: 10.1016/0140-6736(90)92339-j. [DOI] [PubMed] [Google Scholar]
- Jansen E, Klepzig H. [Studies on the influence of Molsidomin on coronary heart disease (author's transl)] Med Klin. 1976;71:2072–2076. [PubMed] [Google Scholar]
- Jeng CJ, Chen YT, Chen YW, Tang CY. Dominant-negative effects of human P/Q-type Ca2+ channel mutations associated with episodic ataxia type 2. Am J Physiol. 2006;290:C1209–C1220. doi: 10.1152/ajpcell.00247.2005. [DOI] [PubMed] [Google Scholar]
- Jugdutt BI. Nitrates in myocardial infarction. Cardiovasc Drugs Ther. 1994;8:635–646. doi: 10.1007/BF00877417. [DOI] [PubMed] [Google Scholar]
- Juhasz G, Zsombok T, Laszik A, Jakus R, Faludi G, Sotonyi P, et al. Despite the general correlation of the serotonin transporter gene regulatory region polymorphism (5-HTTLPR) and platelet serotonin concentration, lower platelet serotonin concentration in migraine patients is independent of the 5-HTTLPR variants. Neurosci Lett. 2003a;350:56–60. doi: 10.1016/s0304-3940(03)00834-6. [DOI] [PubMed] [Google Scholar]
- Juhasz G, Zsombok T, Modos EA, Olajos S, Jakab B, Nemeth J, et al. NO-induced migraine attack: strong increase in plasma calcitonin gene-related peptide (CGRP) concentration and negative correlation with platelet serotonin release. Pain. 2003b;106:461–470. doi: 10.1016/j.pain.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Juhasz G, Zsombok T, Gonda X, Bagdy G. [Nitroglycerin-induced headaches] Orv Hetil. 2004;145:2323–2328. [PubMed] [Google Scholar]
- Juhasz G, Zsombok T, Jakab B, Nemeth J, Szolcsanyi J, Bagdy G. Sumatriptan causes parallel decrease in plasma calcitonin gene-related peptide (CGRP) concentration and migraine headache during nitroglycerin induced migraine attack. Cephalalgia. 2005;25:179–183. doi: 10.1111/j.1468-2982.2005.00836.x. [DOI] [PubMed] [Google Scholar]
- Juhasz G, Zsombok T, Gonda X, Nagyne N, Modosne E, Bagdy G. Effects of autogenic training on nitroglycerin-induced headaches. Headache. 2007;47:371–383. doi: 10.1111/j.1526-4610.2006.00718.x. [DOI] [PubMed] [Google Scholar]
- Juhasz G, Lazary J, Chase D, Pegg E, Downey D, Toth ZG, et al. Variations in the cannabinoid receptor 1 gene predispose to migraine. Neurosci Lett. 2009;461:116–120. doi: 10.1016/j.neulet.2009.06.021. [DOI] [PubMed] [Google Scholar]
- Kang M, Ross GR, Akbarali HI. COOH-terminal association of human smooth muscle calcium channel Ca(v)1.2b with Src kinase protein binding domains: effect of nitrotyrosylation. Am J Physiol. 2007;293:1983–1990. doi: 10.1152/ajpcell.00308.2007. [DOI] [PubMed] [Google Scholar]
- Kawai F, Miyachi E. Modulation by cGMP of the voltage-gated currents in newt olfactory receptor cells. Neurosc Res. 2001;39:327–337. doi: 10.1016/s0168-0102(00)00236-4. [DOI] [PubMed] [Google Scholar]
- Klyachko VA, Ahern GP, Jackson MB. cGMP-mediated facilitation in nerve terminals by enhancement of the spike afterhyperpolarization. Neuron. 2001;31:1015–1025. doi: 10.1016/s0896-6273(01)00449-4. [DOI] [PubMed] [Google Scholar]
- Kruuse C, Thomsen LL, Birk S, Olesen J. Migraine can be induced by sildenafil without changes in middle cerebral artery diameter. Brain. 2003;126:241–247. doi: 10.1093/brain/awg009. Pt 1. [DOI] [PubMed] [Google Scholar]
- van der Kuy PH, Lohman JJ. The role of nitric oxide in vascular headache. Pharm World Sci. 2003;25:146–151. doi: 10.1023/a:1024800512790. [DOI] [PubMed] [Google Scholar]
- Lang RJ, Watson MJ. Effects of nitric oxide donors, S-nitroso-L-cysteine and sodium nitroprusside, on the whole-cell and single channel currents in single myocytes of the guinea-pig proximal colon. Br J Pharmacol. 1998;123:505–517. doi: 10.1038/sj.bjp.0701605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Chapleau MW, Bates JN, Bielefeldt K, Lee HC, Abboud FM. Nitric oxide as an autocrine regulator of sodium currents in baroreceptor neurons. Neuron. 1998;20:1039–1049. doi: 10.1016/s0896-6273(00)80484-5. [DOI] [PubMed] [Google Scholar]
- Lipton RB, Bigal ME. Ten lessons on the epidemiology of migraine. Headache. 2007;47(Suppl 1):S2–S9. doi: 10.1111/j.1526-4610.2007.00671.x. [DOI] [PubMed] [Google Scholar]
- Liu ZW, Lei T, Zhang T, Yang Z. Peroxynitrite donor impairs excitability of hippocampal CA1 neurons by inhibiting voltage-gated potassium currents. Toxicol Lett. 2007;175:8–15. doi: 10.1016/j.toxlet.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Loder E, Rizzoli P. Tension-type headache. BMJ. 2008;336:88–92. doi: 10.1136/bmj.39412.705868.AD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Maagdenberg AM, Haan J, Terwindt GM, Ferrari MD. Migraine: gene mutations and functional consequences. Curr Opin Neurol. 2007;20:299–305. doi: 10.1097/WCO.0b013e3281338d1f. [DOI] [PubMed] [Google Scholar]
- Magis D, Bendtsen L, Goadsby PJ, May A, Sanchez del Rio M, Sandor PS, et al. Evaluation and proposal for optimization of neurophysiological tests in migraine: part 2 – neuroimaging and the nitroglycerin test. Cephalalgia. 2007;27:1339–1359. doi: 10.1111/j.1468-2982.2007.01435.x. [DOI] [PubMed] [Google Scholar]
- Martelletti P, Farinelli I, Steiner TJ. Acute migraine in the Emergency Department: extending European principles of management. Intern Emerg Med. 2008;3(Suppl. 1):S17–S24. doi: 10.1007/s11739-008-0188-1. [DOI] [PubMed] [Google Scholar]
- Mayer B, Beretta M. The enigma of nitroglycerin bioactivation and nitrate tolerance: news, views and troubles. Br J Pharmacol. 2008;155:170–184. doi: 10.1038/bjp.2008.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrotra S, Gupta S, Chan KY, Villalon CM, Centurion D, Saxena PR, et al. Current and prospective pharmacological targets in relation to antimigraine action. Naunyn Schmiedebergs Arch Pharmacol. 2008;378:371–394. doi: 10.1007/s00210-008-0322-7. [DOI] [PubMed] [Google Scholar]
- Meldrum BS, Rogawski MA. Molecular targets for antiepileptic drug development. Neurotherapeutics. 2007;4:18–61. doi: 10.1016/j.nurt.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miceli F, Soldovieri MV, Martire M, Taglialatela M. Molecular pharmacology and therapeutic potential of neuronal Kv7-modulating drugs. Curr Opin Pharmacol. 2008;8:65–74. doi: 10.1016/j.coph.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Micromedex® Healthcare Series [Internet Database] Greenwood Village, CO, USA: Thomson Reuters (Healthcare); [Google Scholar]
- Moncada S, Palmer RM, Higgs EA. The discovery of nitric oxide as the endogenous nitrovasodilator. Hypertension. 1988;12:365–372. doi: 10.1161/01.hyp.12.4.365. [DOI] [PubMed] [Google Scholar]
- Montagna P. The primary headaches: genetics, epigenetics and a behavioural genetic model. J Headache Pain. 2008;9:57–69. doi: 10.1007/s10194-008-0026-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno H, Vega-Saenz de Miera E, Nadal MS, Amarillo Y, Rudy B. Modulation of Kv3 potassium channels expressed in CHO cells by a nitric oxide-activated phosphatase. J Physiol. 2001;530(Pt 3):345–358. doi: 10.1111/j.1469-7793.2001.0345k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller RA, Meienberg O. Hemicrania with oculosympathetic paresis from isosorbide dinitrate. N Engl J Med. 1983;308:458–459. [PubMed] [Google Scholar]
- Nardi A, Olesen SP. BK channel modulators: a comprehensive overview. Current Medicinal Chem. 2008;15:1126–1146. doi: 10.2174/092986708784221412. [DOI] [PubMed] [Google Scholar]
- Nunez L, Vaquero M, Gomez R, Caballero R, Mateos-Caceres P, Macaya C, et al. Nitric oxide blocks hKv1.5 channels by S-nitrosylation and by a cyclic GMP-dependent mechanism. Cardiovasc Res. 2006;72:80–89. doi: 10.1016/j.cardiores.2006.06.021. [DOI] [PubMed] [Google Scholar]
- Olesen J. The role of nitric oxide (NO) in migraine, tension-type headache and cluster headache. Pharmacol Ther. 2008;120:157–171. doi: 10.1016/j.pharmthera.2008.08.003. [DOI] [PubMed] [Google Scholar]
- Olesen J, Iversen HK, Thomsen LL. Nitric oxide supersensitivity: a possible molecular mechanism of migraine pain. Neuroreport. 1993;4:1027–1030. doi: 10.1097/00001756-199308000-00008. [DOI] [PubMed] [Google Scholar]
- Ophoff RA, Terwindt GM, Frants RR, Ferrari MD. P/Q-type Ca2+ channel defects in migraine, ataxia and epilepsy. Trends Pharmacol Sci. 1998;19:121–127. doi: 10.1016/s0165-6147(98)01182-1. [DOI] [PubMed] [Google Scholar]
- Page IH, Corcoran AC, Dustan HP, Koppanyi T. Cardiovascular actions of sodium nitroprusside in animals and hypertensive patients. Circulation. 1955;11:188–198. doi: 10.1161/01.cir.11.2.188. [DOI] [PubMed] [Google Scholar]
- Pardutz A, Krizbai I, Multon S, Vecsei L, Schoenen J. Systemic nitroglycerin increases nNOS levels in rat trigeminal nucleus caudalis. Neuroreport. 2000;11:3071–3075. doi: 10.1097/00001756-200009280-00008. [DOI] [PubMed] [Google Scholar]
- Parker JA. Organic nitrates: new formulations and their clinical advantages. Am J Cardiol. 1996;77:38C–40C. doi: 10.1016/s0002-9149(96)00187-7. [DOI] [PubMed] [Google Scholar]
- Peres MF, Zukerman E, Senne Soares CA, Alonso EO, Santos BF, Faulhaber MH. Cerebrospinal fluid glutamate levels in chronic migraine. Cephalalgia. 2004;24:735–739. doi: 10.1111/j.1468-2982.2004.00750.x. [DOI] [PubMed] [Google Scholar]
- Pfeifer A, Klatt P, Massberg S, Ny L, Sausbier M, Hirneiss C, et al. Defective smooth muscle regulation in cGMP kinase I-deficient mice. EMBO J. 1998;17:3045–3051. doi: 10.1093/emboj/17.11.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pharmindex. Budapest, Hungary: CMPMedica; 2008. [Google Scholar]
- Pietrobon D. Migraine: new molecular mechanisms. Neuroscientist. 2005;11:373–386. doi: 10.1177/1073858405275554. [DOI] [PubMed] [Google Scholar]
- Pietrobon D. Familial hemiplegic migraine. Neurotherapeutics. 2007;4:274–284. doi: 10.1016/j.nurt.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Prast H, Philippu A. Nitric oxide as modulator of neuronal function. Prog Neurobiol. 2001;64:51–68. doi: 10.1016/s0301-0082(00)00044-7. [DOI] [PubMed] [Google Scholar]
- Rahmann A, Wienecke T, Hansen JM, Fahrenkrug J, Olesen J, Ashina M. Vasoactive intestinal peptide causes marked cephalic vasodilation, but does not induce migraine. Cephalalgia. 2008;28:226–236. doi: 10.1111/j.1468-2982.2007.01497.x. [DOI] [PubMed] [Google Scholar]
- Reuter U, Bolay H, Jansen-Olesen I, Chiarugi A, Sanchez del Rio M, Letourneau R, et al. Delayed inflammation in rat meninges: implications for migraine pathophysiology. Brain. 2001;124(Pt 12):2490–2502. doi: 10.1093/brain/124.12.2490. [DOI] [PubMed] [Google Scholar]
- Roh S, Choi S, Lim I. Involvement of protein kinase A in nitric oxide stimulating effect on a BK(Ca) channel of human dermal fibroblasts. J Invest Dermatol. 2007;127:2533–2538. doi: 10.1038/sj.jid.5700907. [DOI] [PubMed] [Google Scholar]
- Russell MB. Genetics in primary headaches. J Headache Pain. 2007;8:190–195. doi: 10.1007/s10194-007-0389-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sances G, Tassorelli C, Pucci E, Ghiotto N, Sandrini G, Nappi G. Reliability of the nitroglycerin provocative test in the diagnosis of neurovascular headaches. Cephalalgia. 2004;24:110–119. doi: 10.1111/j.1468-2982.2004.00639.x. [DOI] [PubMed] [Google Scholar]
- Sarchielli P, Alberti A, Codini M, Floridi A, Gallai V. Nitric oxide metabolites, prostaglandins and trigeminal vasoactive peptides in internal jugular vein blood during spontaneous migraine attacks. Cephalalgia. 2000;20:907–918. doi: 10.1046/j.1468-2982.2000.00146.x. [DOI] [PubMed] [Google Scholar]
- Scheidt S. Angina: evolution of the role of nitrates. Am Heart J. 1990;120:757–761. doi: 10.1016/0002-8703(90)90049-4. discussion 769–772. [DOI] [PubMed] [Google Scholar]
- Schoonman GG, van der Grond J, Kortmann C, van der Geest RJ, Terwindt GM, Ferrari MD. Migraine headache is not associated with cerebral or meningeal vasodilatation – a 3T magnetic resonance angiography study. Brain. 2008;131(Pt 8):2192–2200. doi: 10.1093/brain/awn094. [DOI] [PubMed] [Google Scholar]
- Segal MM, Douglas AF. Late sodium channel openings underlying epileptiform activity are preferentially diminished by the anticonvulsant phenytoin. J Neurophysiol. 1997;77:3021–3034. doi: 10.1152/jn.1997.77.6.3021. [DOI] [PubMed] [Google Scholar]
- Shin JH, Chung S, Park EJ, Uhm DY, Suh CK. Nitric oxide directly activates calcium-activated potassium channels from rat brain reconstituted into planar lipid bilayer. FEBS Lett. 1997;415:299–302. doi: 10.1016/s0014-5793(97)01144-7. [DOI] [PubMed] [Google Scholar]
- Silberstein SD. Migraine. Lancet. 2004;363:381–391. doi: 10.1016/S0140-6736(04)15440-8. [DOI] [PubMed] [Google Scholar]
- Sobey CG. Potassium channel function in vascular disease. Arterioscler Thromb Vasc Biol. 2001;21:28–38. doi: 10.1161/01.atv.21.1.28. [DOI] [PubMed] [Google Scholar]
- Srikiatkhachorn A, Anuntasethakul T, Maneesri S, Phansuwan-Pujito P, Patumraj S, Kasantikul V. Hyposerotonin-induced nitric oxide supersensitivity in the cerebral microcirculation. Headache. 2000;40:267–275. doi: 10.1046/j.1526-4610.2000.00040.x. [DOI] [PubMed] [Google Scholar]
- Srikiatkhachorn A, Suwattanasophon C, Ruangpattanatawee U, Phansuwan-Pujito P. 2002 Wolff Award. 5 -HT2A receptor activation and nitric oxide synthesis: a possible mechanism determining migraine attacks. Headache. 2002;42:566–574. doi: 10.1046/j.1526-4610.2002.02142.x. [DOI] [PubMed] [Google Scholar]
- Storer RJ, Akerman S, Goadsby PJ. Calcitonin gene-related peptide (CGRP) modulates nociceptive trigeminovascular transmission in the cat. Br J Pharmacol. 2004;142:1171–1181. doi: 10.1038/sj.bjp.0705807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stovner L, Hagen K, Jensen R, Katsarava Z, Lipton R, Scher A, et al. The global burden of headache: a documentation of headache prevalence and disability worldwide. Cephalalgia. 2007;27:193–210. doi: 10.1111/j.1468-2982.2007.01288.x. [DOI] [PubMed] [Google Scholar]
- Tagliaferro P, Ramos AJ, Lopez-Costa JJ, Lopez EM, Saavedra JP, Brusco A. Increased nitric oxide synthase activity in a model of serotonin depletion. Brain Res Bull. 2001;54:199–205. doi: 10.1016/s0361-9230(00)00450-0. [DOI] [PubMed] [Google Scholar]
- Taglialatela M, Pannaccione A, Iossa S, Castaldo P, Annunziato L. Modulation of the K(+) channels encoded by the human ether-a-gogo-related gene-1 (hERG1) by nitric oxide. Mol Pharmacol. 1999;56:1298–1308. doi: 10.1124/mol.56.6.1298. [DOI] [PubMed] [Google Scholar]
- Tassorelli C, Costa A, Blandini F, Joseph SA, Nappi G. Effect of nitric oxide donors on the central nervous system – nitroglycerin studies in the rat. Func Neurol. 2000;15(Suppl 3):19–27. [PubMed] [Google Scholar]
- Tassorelli C, Greco R, Armentero MT, Blandini F, Sandrini G, Nappi G. A role for brain cyclooxygenase-2 and prostaglandin-E2 in migraine: effects of nitroglycerin. Int Rev Neurobiol. 2007;82:373–382. doi: 10.1016/S0074-7742(07)82020-4. [DOI] [PubMed] [Google Scholar]
- Thadani U. Oral nitrates: more than symptomatic therapy in coronary artery disease? Cardiovasc Drugs Ther. 1997;11(Suppl. 1):213–218. doi: 10.1023/a:1007750706831. [DOI] [PubMed] [Google Scholar]
- Thadani U, Ripley TL. Side effects of using nitrates to treat heart failure and the acute coronary syndromes, unstable angina and acute myocardial infarction. Expert Opin Drug Saf. 2007;6:385–396. doi: 10.1517/14740338.6.4.385. [DOI] [PubMed] [Google Scholar]
- Thadani U, Rodgers T. Side effects of using nitrates to treat angina. Expert Opin Drug Saf. 2006;5:667–674. doi: 10.1517/14740338.5.5.667. [DOI] [PubMed] [Google Scholar]
- Thadani U, de Vane PJ. Efficacy of isosorbide mononitrate in angina pectoris. Am J Cardiol. 1992;70:67G–71G. doi: 10.1016/0002-9149(92)90029-x. [DOI] [PubMed] [Google Scholar]
- Thomsen LL, Olesen J. A pivotal role of nitric oxide in migraine pain. Ann N Y Acad Sci. 1997;835:363–372. doi: 10.1111/j.1749-6632.1997.tb48642.x. [DOI] [PubMed] [Google Scholar]
- Tjong YW, Jian K, Li M, Chen M, Gao TM, Fung ML. Elevated endogenous nitric oxide increases Ca2+ flux via l-type Ca2+ channels by S-nitrosylation in rat hippocampal neurons during severe hypoxia and in vitro ischemia. Free Radic Biol Med. 2007;42:52–63. doi: 10.1016/j.freeradbiomed.2006.09.020. [DOI] [PubMed] [Google Scholar]
- Tjong YW, Li M, Hung MW, Wang K, Fung ML. Nitric oxide deficit in chronic intermittent hypoxia impairs large conductance calcium-activated potassium channel activity in rat hippocampal neurons. Free Radic Biol Med. 2008;44:547–557. doi: 10.1016/j.freeradbiomed.2007.10.033. [DOI] [PubMed] [Google Scholar]
- Tottene A, Fellin T, Pagnutti S, Luvisetto S, Striessnig J, Fletcher C, et al. Familial hemiplegic migraine mutations increase Ca(2+) influx through single human CaV2.1 channels and decrease maximal CaV2.1 current density in neurons. Proc Natl Acad Sci USA. 2002;99:13284–13289. doi: 10.1073/pnas.192242399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tottene A, Pivotto F, Fellin T, Cesetti T, Maagdenberg AM, Pietrobon D. Specific kinetic alterations of human CaV2.1 calcium channels produced by mutation S218L causing familial hemiplegic migraine and delayed cerebral edema and coma after minor head trauma. J Biol Chem. 2005;280:17678–17686. doi: 10.1074/jbc.M501110200. [DOI] [PubMed] [Google Scholar]
- Tvedskov JF, Thomsen LL, Iversen HK, Gibson A, Wiliams P, Olesen J. The prophylactic effect of valproate on glyceryltrinitrate induced migraine. Cephalalgia. 2004;24:576–585. doi: 10.1111/j.1468-2982.2003.00720.x. [DOI] [PubMed] [Google Scholar]
- Uberbacher HJ, Steinorth G, Glocke M, Abshagen U. An open, long-term, multi-centre study of the anti-anginal efficacy and safety of isosorbide 5-mononitrate at low doses in patients with coronary heart disease. Pharmatherapeutica. 1983;3:331–341. [PubMed] [Google Scholar]
- Vanmolkot KR, Terwindt GM, Frants RR, Haan J, Maagdenberg AM, Ferrari MD. A gene for a new monogenic neurovascular migraine syndrome: a next step in unravelling molecular pathways for migraine? Cephalalgia. 2008;28:471–473. doi: 10.1111/j.1468-2982.2007.01511.x. [DOI] [PubMed] [Google Scholar]
- Weitzman D. Penta-erythritol tetranitrate in treatment of angina. Br Med J. 1953;2:1409–1412. doi: 10.1136/bmj.2.4851.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessman M, Terwindt GM, Kaunisto MA, Palotie A, Ophoff RA. Migraine: a complex genetic disorder. Lancet Neurol. 2007;6:521–532. doi: 10.1016/S1474-4422(07)70126-6. [DOI] [PubMed] [Google Scholar]
- White RE. Cyclic GMP and ion channel regulation. Adv Second Messenger Phosphoprotein Res. 1999;33:251–277. doi: 10.1016/s1040-7952(99)80013-5. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Richter JA, Hurley JH. Release of glutamate and CGRP from trigeminal ganglion neurons: Role of calcium channels and 5-HT1 receptor signaling. Mol Pain. 2008;4:12. doi: 10.1186/1744-8069-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura N, Seki S, de Groat WC. Nitric oxide modulates Ca(2+) channels in dorsal root ganglion neurons innervating rat urinary bladder. J Neurophysiol. 2001;86:304–311. doi: 10.1152/jn.2001.86.1.304. [DOI] [PubMed] [Google Scholar]
- Yuan XJ, Tod ML, Rubin LJ, Blaustein MP. NO hyperpolarizes pulmonary artery smooth muscle cells and decreases the intracellular Ca2+ concentration by activating voltage-gated K+ channels. Proc Natl Acad Sci USA. 1996;93:10489–10494. doi: 10.1073/pnas.93.19.10489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusuf S, Collins R, MacMahon S, Peto R. Effect of intravenous nitrates on mortality in acute myocardial infarction: an overview of the randomised trials. Lancet. 1988;1:1088–1092. doi: 10.1016/s0140-6736(88)91906-x. [DOI] [PubMed] [Google Scholar]
- Zeller J, Poulsen KT, Sutton JE, Abdiche YN, Collier S, Chopra R, et al. CGRP function-blocking antibodies inhibit neurogenic vasodilatation without affecting heart rate or arterial blood pressure in the rat. Br J Pharmacol. 2008;155:1093–1103. doi: 10.1038/bjp.2008.334. [DOI] [PMC free article] [PubMed] [Google Scholar]