

Abstract
Background and purpose:
Amitriptyline is a tricyclic antidepressant that is also widely used to treat neuropathic pain in humans, but the mechanism of this anti-hyperalgesic effect is unknown. Microglia in the mouse spinal cord become activated in neuropathic pain, and expression of P2X4 receptors by these microglia is increased. Antisense RNA targeting P2X4 receptors suppresses the development of tactile allodynia in rats. This suggests that blockade of P2X4 receptors might be the mechanism by which amitriptyline relieves neuropathic pain.
Experimental approach:
We expressed human, rat and mouse P2X receptors (P2X2, P2X4, P2X7) in human embryonic kidney cells and evoked inward currents by applying ATP. We compared the action of ATP on control cells and cells treated with amitriptyline.
Key results:
Amitriptyline (10 µM), either applied acutely or by pre-incubation for 2–6 h, had no effect on inward currents evoked by ATP (0.3–100 µM) at human P2X4 receptors. At rat and mouse receptors, amitriptyline (10 µM) caused a modest reduction in the maximum responses to ATP, without changes in EC50 values, but it had no effect at 1 µM. Amitriptyline also had no effects on currents evoked by ATP at rat P2X2 receptors, or at rat or human P2X7 receptors.
Conclusion and implications:
The results do not support the view that amitriptyline owes its pain-relieving actions in man to the direct blockade of P2X4 receptors.
Keywords: P2X receptors, human embryonic kidney cells, amitriptyline, tricyclic antidepressant, whole-cell recording
Introduction
The tricyclic antidepressant amitriptyline is widely used in the treatment of pain in man (Sindrup et al., 2005). In rodents, it produces behavioural consequences reported as anti-nociception in several models of neuropathic pain (Esser and Sawynok, 1999;Gerner et al., 2002; Lynch et al., 2005; Nagata et al., 2009; Su et al., 2009). Neuropathic pain is a complex chronic pain state as a consequence of tissue injury, and interest has focused recently on the possible relationship between endogenous ATP and P2X receptors (nomenclature follows Alexander et al., 2009) expressed by microglia in the parts of the dorsal horn where primary afferents terminate (McGaraughty and Jarvis, 2006).
Activation of P2X4 receptors (Tsuda et al., 2003; Zhang et al., 2006; Ulmann et al., 2008) and/or P2X7 receptors (Chessell et al., 2005) expressed by spinal microglia has been reported to mediate neuropathic pain. The expression of P2X4 receptors is enhanced in spinal microglia after peripheral nerve injury model (Tsuda et al., 2003). Neuropathic pain can be alleviated by certain P2 receptor antagonists such as 2′, 3′-O-(2,4,6-trinitrophenyl)-ATP (TNP-ATP) (Tsuda et al., 2003), even though TNP-ATP is only a weakly effective blocker of P2X4 receptors, compared with receptors containing P2X1 or P2X3 subunits (Virginio et al., 1998). It can also be inhibited by selectively reducing the expression of P2X4 receptors in microglia through intrathecal treatment with antisense oligonucleotides (Tsuda et al., 2003) or by disrupting the P2X4 receptor gene (Ulmann et al., 2008).
These results suggest the hypothesis that amitriptyline might relieve pain as a consequence of its blockade of P2X4 receptors. A recent attempt to address this question (Nagata et al., 2009) used calcium imaging in 1321N1 cells expressing P2X4 receptors. The authors found that paroxetine reduced ATP-evoked increases in intracellular calcium, but the study used rat receptors and effective concentrations were considerably higher than those associated with its use in humans (Nagata et al., 2009). There are pronounced differences in the sensitivity to blockers between human and rat P2X4 receptors (Jones et al., 2000), and the aim of the present study was to determine whether the more widely used tricyclic antidepressant amitriptyline blocks human P2X4 receptors at concentrations similar to those reached during its therapeutic use.
Methods
Cell culture and transfection
Human embryonic kidney (HEK)293 cells were maintained at 37°C and 5% CO2 in growth medium (Dulbecco's modified Eagle's medium, 10% fetal calf serum and 2 mM l-glutamine: Invitrogen, Paisley, UK). HEK293 cells were plated onto 35 mm dishes and allowed to attain a cell density of 105 cells·cm−2 before transfection. Cells were transfected for 4–6 h using lipofectamine 2000 (Invitrogen), with 0.5 µg·mL−1 receptor DNA and 0.05 µg·mL−1 of a plasmid encoding enhanced green fluorescent protein (pEGFP-N1). Cells were then trypsinized, washed, reconstituted in growth medium and plated out on glass coverslips at 1500 cells·cm−2. The efficiency of transfection was >60% as judged by epifluorescence microscopy. The coverslips were maintained at 37°C and 5% CO2 for 24–48 h prior to recording.
Whole-cell recording
The methods were as previously described (Sim et al., 2008). Coverslips with attached cells were placed in a recording chamber mounted on the stage of an Axiovert microscope (Carl Zeiss, Jena, Germany). The superfusing solution contained (mM): NaCl 147; KCl 3; MgCl2 1; CaCl2 2; HEPES 10 and d-glucose 13 (pH adjusted to 7.4 with NaOH, 300–315 mOsm·L−1) and was superfused at 5.5 mL·min−1. Whole-cell current recordings were made at a holding potential of −60 mV (EPC9 amplifier, HEKA, Lathbrecht, Germany) and at room temperature (20–23°C). Recording electrodes were pulled from borosilicate glass capillaries (Harvard Apparatus, Edenbridge, UK) on a vertical puller (HEKA) and had resistances of 6–9 MΩ, when filled with an intracellular solution containing (mM): NaCl 147; HEPES 10; EGTA 10 (pH adjusted to 7.3 with NaOH).
For concentration–response experiments using P2X4 receptors, ATP (0.3–300 µM) was applied for 2 s at 4–5 min intervals using an RSC-200 system (Bio-Logic Science Instruments, Claix, France), whereas ATP (0.01–10 mM, 2 s) was applied at 2 min intervals in the study with P2X7 receptors. Responses to ATP mediated by P2X4 receptors decline in amplitude with repeated applications, especially with concentrations >10 µM ATP: therefore, all concentration–responses curves in the present study were constructed using the mean values at each concentration taken from ascending and descending concentration–response experiments. For measurement of ‘rundown’ of responses, ATP (100 µM) was repeatedly applied for 2 s at intervals of 5 min.
For pretreatment experiments, transfected cells were pre-incubated with amitriptyline (1 µM or 10 µM) for a minimum period of 2 h and up to 6 h at 37°C, and recordings were made in the continuing presence of amitriptyline. Responses recorded after 6 h pretreatment were comparable to those after 2 h in all three P2X4 receptors tested.
In those studies, stock solutions of ATP were diluted into the extracellular solutions containing the appropriate concentration of amitriptyline. Furthermore, to ensure comparability between amitriptyline-treated and untreated cells the recordings from each class were alternated on a given day. In all experiments, we compared human, rat and mouse receptors from parallel transfections, and each group also received similar pretreatment periods. Results are expressed as current density (pA·pF−1).
Data analysis
Numerical data are means ± SEM, for the number of cells tested. Current traces were obtained using Axograph (Molecular Devices, Sunnyvale, CA, USA), Kaleidagraph (Synergy Software, Reading, PA, USA) and Canvas (ACD Systems, British Columbia, Canada) software. Concentration–response curves were fitted using the non-linear regression programme from Prism 4 (GraphPad, San Diego, CA, USA) to the mean values of the currents evoked at each concentration. In the case of human P2X7 receptors, the curves were fitted using an assumed maximum, as higher concentrations of ATP were not tested in this study. The agonist concentration required for the half-maximal responses (EC50) is expressed as its negative logarithm (pEC50) ± SEM. Tests for significant differences between numerical values were determined using unpaired t-tests and anova (InStat, GraphPad Software Inc., San Diego, CA, USA).
Materials
ATP and amitriptyline were purchased from Sigma Aldrich (Poole, UK).
Results
P2X4 receptors
ATP (100 µM, 2 s) evoked currents in cells expressing P2X4 receptors, as previously described (human: Fountain and North, 2006; rat: Buell et al., 1996; mouse: Townsend-Nicholson et al., 1999). Acute application of amitriptyline (10 µM) did not antagonize the ATP responses in either rat or human P2X4 receptors. Concentration–response curves were constructed from current amplitudes pooled for various concentrations of ATP from applications of increasing or decreasing (0.3, 1, 3, 10, 30 and 100 µM) concentrations. These concentration–response curves showed that pre-incubation with amitriptyline (1 and 10 µM: 2–6 h at 37°C) had no effect on the peak amplitude of the currents evoked by ATP in cells expressing human P2X4 receptors (Figure 1A). Responses evoked with 100 µM ATP in control (109 ± 24 pA·pF−1, n= 10) were also not significantly (P < 0.05) different from those evoked in the presence of 1 µM amitriptyline (108 ± 11 pA·pF−1, n= 11) and of 10 µM amitriptyline (109 ± 23 pA·pF−1, n= 9). pEC50 values were not significantly different from each other: control (5.3 ± 0.13, n= 6–13), 1 µM amitriptyline-treated (5.1 ± 0.14, n= 6–15) and 10 µM amitriptyline-treated (5.3 ± 0.14, n= 6–9).
Figure 1.
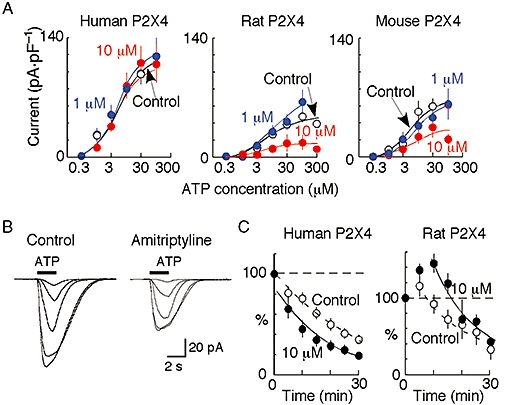
Amitriptyline action on ATP-evoked currents at human, rat and mouse P2X4 receptors. (A) Concentration–response curves show mean currents (±SEM) from pooled data in absence (control) and presence of 1 µM and 10 µM amitriptyline (n= 3–18 cells at each point; bars are SEM where this exceeds the symbol size). (B) Left, currents in response to increasing concentration of ATP (1, 3, 10, 30 and 100 µM) in cell expressing rat P2X4 receptors. Right, ATP-evoked currents (1, 3, 10, 30 and 100 µM) in another such cell that had been treated for 4 h with amitriptyline (10 µM). (C) Amplitude of currents evoked by ATP (100 µM, 2 s duration) applied repeatedly at 5 min intervals to cells expressing human or rat P2X4 receptors. Ordinate is expressed as percentage of amplitude of current evoked by first application of ATP. Open circles, control cells. Filled circles, after 2–6 h pre-incubation with amitriptyline.
Pre-incubation with 10 µM amitriptyline reduced the response to ATP in both rat and mouse P2X4 receptors (Figure 1). Representative currents for rat receptors are shown in Figure 1B. Concentration–response curves suggested that the reduction in the amplitude of the ATP currents was non-competitive (Figure 1A). The amplitude of the current evoked by 100 µM ATP was reduced by 35% [17 ± 9 pA·pF−1 (n= 9) compared with control 48 ± 7 pA·pF−1 (n= 18)] in the presence of 10 µM amitriptyline. However, in the presence of 1 µM amitriptyline, there was no significant (P= 0.29) change in the response to 100 µM ATP (Figure 1). The pEC50 values were not different among control (5.1 ± 0.20, n= 6–18), amitriptyline (1 µM; 4.8 ± 0.15, n= 6–8) and amitriptyline (10 µM; 5.2 ± 0.52, n= 6–12). At mouse receptors, 1 µM amitriptyline had no significant (P= 0.95) effect, whereas 10 µM amitriptyline reduced the current evoked by 100 µM ATP by 34% [21 ± 6 pA·pF−1 (n= 8) compared with control value of 62 ± 11 pA·pF−1 (n= 8)]. pEC50 values were not significantly different among control cells (5.2 ± 0.28, n= 6–12), cells treated with 1 µM amitriptyline (5.1 ± 0.16, n= 3–6) and cells treated with 10 µM amitriptyline (5.3 ± 0.67, n= 6–8).
ATP-evoked currents at P2X4 receptors show a prominent rundown in amplitude when ATP (100 µM, 2 s) is applied repeatedly at 1 min intervals (Fountain and North, 2006). In the present study, we tested the effect of repeated application of ATP (100 µM, 2 s) at 5 min intervals. The responses to ATP showed a similar degree of rundown in human, rat and mouse receptors. Responses at 30 min were reduced by 65 ± 4.7% (n= 3), 62 ± 17% (n= 3) and 66 ± 13% (n= 4) of the first response in human, rat and mouse P2X4 receptors respectively (Figure 1C). The rundown of the ATP-induced currents at human P2X4 receptors was also not significantly changed in the presence of amitriptyline (10 µM) (Figure 1C): the seventh response (at 30 min) was reduced in amplitude by 77 ± 6.0% (n= 3) (Figure 1C). At rat P2X4 receptors, the responses declined to a mean value of 70 ± 8.4% (n= 4) at 30 min (although there was an initial potentiation) (Figure 1C) and the corresponding value for mouse receptors was 68 ± 21% (n= 5) (data not shown).
P2X7 receptors
The effect of pretreatment with amitriptyline was evaluated on the ATP responses evoked in HEK cells expressing human or rat P2X7 receptors (Figure 2). Representative families of ATP currents evoked in human P2X7 receptors, in the absence and following pre-incubation of 10 µM amitriptyline, are shown in Figure 2A. There were no differences between the concentration–response curves in either human and rat P2X7 receptors. For the rat P2X7 receptor, pEC50 values were 2.5 ± 0.31 (n= 8–10) in amitriptyline and 3.0 ± 0.35 (n= 4–8) in control conditions. EC50 values for ATP could not be estimated reliably for human P2X7 receptors (Figure 2B).
Figure 2.
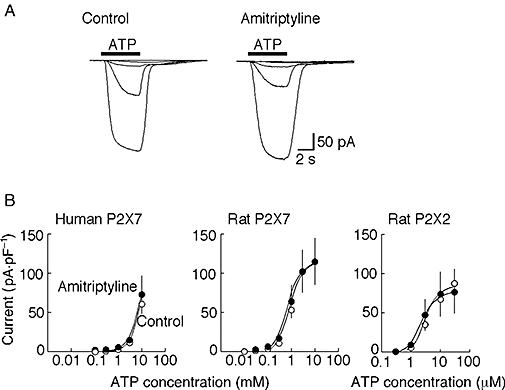
Lack of effect of amitriptyline at human and rat P2X7 receptors and at rat P2X2 receptors. (A) Left, currents in response to increasing concentration of ATP (0.1, 0.3, 1, 3 and 10 mM) in one human embryonic kidney cell expressing human P2X7 receptors. Right, ATP-evoked currents (0.1, 0.3, 1, 3 and 10 mM) in another such cell that had been treated for 4 h with amitriptyline (10 µM). (B) Concentration–response curves show mean currents (±SEM) from pooled data for cells expressing human P2X7, rat P2X7 (concentration in mM) and rat P2X2 receptors (concentration in µM) in absence (control) and presence of amitriptyline (10 µM) (n= 5–7 cells at each point; bars are SEM where this exceeds the symbol size).
P2X2 receptors
As an additional control for any non-specific effects of long incubations with amitriptyline, we tested the effect also on ATP-evoked currents at P2X2 receptors. There was no difference between the concentration–response curves for ATP acting at P2X2 receptors between control cells and cells pre-incubated with amitriptyline for 2–6 h. The respective pEC50 values were 5.3 ± 0.11 (n= 6) and 5.7 ± 0.23 (n= 6) (Figure 2).
Discussion
Our experiments show that acute application of amitriptyline (10 µM) does not change the currents elicited by ATP when applied to cells expressing P2X4 (human, rat or mouse), P2X7 (human or rat) or P2X2 (rat) receptors. When the amitriptyline was applied for a prolonged period (2–6 h) prior to recording, the currents elicited by ATP were not different for human P2X4 receptors, human or rat P2X7 receptors, or rat P2X2 receptors. For P2X4 receptors, the rundown observed with repeated ATP applications was not affected by amitriptyline. Rat and mouse P2X4 receptors showed a modest (35%) but significant (P < 0.05) reduction in amplitude with 10 µM but not with 1 µM amitriptyline. These effects of amitriptyline on rat and mouse P2X4 receptors seemed non-competitive, in that their EC50 for ATP was unchanged (Figure 1C). The results for the rat receptor are similar to those reported for another tricyclic antidepressant paroxetine (10 µM) by Nagata et al. (2009), but that study pre-applied paroxetine for 10 min and tested only a single ATP concentration (30 µM). The differences that we have found between human and rodent receptors are unsurprising in view of the known species differences in the actions of P2X receptor antagonists (North and Surprenant, 2000;Gever et al., 2006), most notably those for suramin and pyridoxal-phosphate-6-azophenyl-2′,4′-disulphonic acid at P2X4 receptors (Buell et al., 1996; Jones et al., 2000).
The concentrations of amitriptyline used in the present study are at least 10 times higher than those attained during its use in man for anti-hyperalgesia. Therapeutic efficacy as an antidepressant is associated with plasma concentrations of about 100 ng·mL−1 (300 nM) (Perry et al., 1994; Baldessarini, 1995), and Finnerup et al. (2005) note that similar doses are effective when it is used as an anti-hyperalgesic. Other ‘off-target’ actions of amitriptyline have been reported to occur at such concentrations. These include: (i) a block of the inactive state of the voltage-gated sodium channel NaV1.7, which is widely expressed on nociceptive primary afferent neurons (Dick et al., 2007); (ii) direct actions at opioid receptors (Benbouzid et al., 2008); (iii) an increase in adenosine release (Sawynok et al., 1999); and/or (iv) an up-regulation of excitatory amino acids transporters (Tai et al., 2006). Such actions of amitriptyline shown in rodents seem much more likely to account for the anti-hyperalgesic actions of amitriptyline in humans, than any block of P2X4 receptors.
P2X7 receptors are also expressed by microglia, and neuropathic pain is much reduced in mice lacking the P2X7 gene (Chessell et al., 2005). It therefore seemed reasonable to ask whether blockade of P2X7 receptor might contribute to the anti-hyperalgesic actions of amitriptyline. The present results show that there is no discernible block by 10 µM amitriptyline of ionic currents at either rat or human receptor (Figure 2). It remains conceivable, but seems unlikely that amitriptyline might block the cytokine release associated with P2X7 receptor activation while not blocking the ionic current.
In conclusion, our results show: (i) that prolonged application of amitriptyline can reduce ATP responses in a non-competitively manner at rat and mouse P2X4 receptors, but only at concentrations about 30 times higher than those likely to occur during therapeutic use; (ii) that they have no effect on ATP-evoked currents at human P2X4 receptors; and (iii) that they do not reduce currents at human or rat P2X7 receptors. They provide no support for the view that inhibition at P2X4 receptors underlies the anti-hyperalgesic actions of amitriptyline in humans.
Acknowledgments
This work was supported by the Wellcome Trust (GR074704) and by Brain Research Center of the 21st Century Frontier Research Program (Grant M103KV010015-06K2201-01510) funded by the Ministry of Science and Technology, Republic of Korea. We thank Laricia Bragg for tissue culture.
Glossary
Abbreviations:
- HEK
human embryonic kidney
- TNP-ATP
2′, 3′-O-(2,4,6-trinitrophenyl)-ATP
Conflicts of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldessarini RJ. Drugs and the treatment of psychiatric disorders: depression and mania. In: Hardman JG, Gilman A, Limbird LE, editors. Goodman and Gilman's the Pharmacological Basis of Therapeutics. New York: McGraw Hill; 1995. pp. 431–459. [Google Scholar]
- Benbouzid M, Choucair-Jaafar N, Yalcin I, Waltisperger E, Muller A, Freud-Mercier MJ, et al. Chronic, but not acute, tricyclic antidepressant treatment alleviates neuropathic allodynia after sciatic nerve cuffing in mice. Eur J Pain. 2008;12:1008–1017. doi: 10.1016/j.ejpain.2008.01.010. [DOI] [PubMed] [Google Scholar]
- Buell G, Lewis C, Collo G, North RA, Surprenant A. An antagonist-insensitive P2X receptor expressed in epithelia and brain. EMBO J. 1996;15:55–62. [PMC free article] [PubMed] [Google Scholar]
- Chessell IP, Hatcher JP, Bountra C, Michel AD, Hughes JP, Green P, et al. Disruption of the P2X7 purinoceptor gene abolishes chronic inflammatory and neuropathic pain. Pain. 2005;114:386–396. doi: 10.1016/j.pain.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Dick IE, Brochu RM, Purohit Y, Kaczorowski GJ, Martin WJ, Priest BT. Sodium channel blockade may contribute to the analgesic efficacy of antidepressants. J Pain. 2007;8:315–324. doi: 10.1016/j.jpain.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Esser MJ, Sawynok J. Acute amitriptyline in a rat model of neuropathic pain: differential symptom and route effects. Pain. 1999;80:643–653. doi: 10.1016/S0304-3959(98)00261-9. [DOI] [PubMed] [Google Scholar]
- Finnerup NB, Otto M, McQuay HJ, Jensen TS, Sindrup SH. Algorithm for neuropathic pain treatment: an evidence based proposal. Pain. 2005;118:289–305. doi: 10.1016/j.pain.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Fountain SJ, North RA. A C-terminal lysine that controls human P2X4 receptor desensitization. J Biol Chem. 2006;281:15044–15049. doi: 10.1074/jbc.M600442200. [DOI] [PubMed] [Google Scholar]
- Gerner P, Mujtaba M, Khan M, Sudoh Y, Vlassakov K, Anthony D, et al. N-phenylethyl amitriptyline in rat sciatic nerve blockade. Anesthesiology. 2002;96:1435–1442. doi: 10.1097/00000542-200206000-00024. [DOI] [PubMed] [Google Scholar]
- Gever JR, Cockayne DA, Dillon MP, Burnstock G, Ford AP. Pharmacology of P2X channels. Pflugers Arch. 2006;452:513–537. doi: 10.1007/s00424-006-0070-9. [DOI] [PubMed] [Google Scholar]
- Jones CA, Chessell IP, Simon J, Barnard EA, Miller KJ, Michel AD, et al. Functional characterization of the P2X4 receptor orthologues. Br J Pharmacol. 2000;129:388–394. doi: 10.1038/sj.bjp.0703059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch ME, Clark AJ, Sawynok J, Sullivan MJ. Topical amitriptyline and ketamine in neuropathic pain syndromes: an open-label study. J Pain. 2005;6:644–649. doi: 10.1016/j.jpain.2005.04.008. [DOI] [PubMed] [Google Scholar]
- McGaraughty S, Jarvis MF. Purinergic control of neuropathic pain. Drug Dev Res. 2006;67:376–388. [Google Scholar]
- Nagata K, Imai T, Yamashita T, Tsuda M, Tozaki-Saitoh H, Inoue K. Antidepressants inhibit P2X4 receptor function: a possible involvement in neuropathic pain relief. Mol Pain. 2009;5:20–32. doi: 10.1186/1744-8069-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RA, Surprenant A. Pharmacology of cloned P2X receptors. Annu Rev Pharmacol Toxicol. 2000;40:563–580. doi: 10.1146/annurev.pharmtox.40.1.563. [DOI] [PubMed] [Google Scholar]
- Perry PJ, Zeilmann C, Arndt S. Tricyclic antidepressant concentrations in plasma: an estimate of their sensitivity and specificity as a predictor of response. J Clin Psychopharmacol. 1994;14:230–240. [PubMed] [Google Scholar]
- Sawynok J, Reid AR, Esser MJ. Peripheral antinociceptive action of amitriptyline in the rat formalin test: involvement of adenosine. Pain. 1999;80:45–55. doi: 10.1016/s0304-3959(98)00195-x. [DOI] [PubMed] [Google Scholar]
- Sim JA, Broomhead HE, North RA. Ectodomain lysines and suramin block of P2X1 receptors. J Biol Chem. 2008;283:29841–29846. doi: 10.1074/jbc.M802523200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindrup SH, Otto M, Finnerrup NB, Jensen TS. Antidepressants in the treatment of neuropathic pain. Basic Clin Pharmacol Toxicol. 2005;96:399–409. doi: 10.1111/j.1742-7843.2005.pto_96696601.x. [DOI] [PubMed] [Google Scholar]
- Su X, Liang AH, Urban MO. The effect of amitriptyline on ectopic discharge of primary afferent fibers in the L5 dorsal root in a rat model of neuropathic pain. Anesth Analg. 2009;108:1671–1679. doi: 10.1213/ane.0b013e31819b0271. [DOI] [PubMed] [Google Scholar]
- Tai Y-H, Wang Y-H, Wang J-J, Toa P-L, Tung C-S, Wong C-S. Amitriptyline suppresses neuroinflammation and upregulates glutamate transporters in morphine-tolerant rats. Pain. 2006;124:77–86. doi: 10.1016/j.pain.2006.03.018. [DOI] [PubMed] [Google Scholar]
- Townsend-Nicholson A, King BF, Wildman SS, Burnstock G. Molecular cloning, functional characterization and possible cooperativity between the murine P2X4 and P2X4a receptors. Brain Res Mol Brain Res. 1999;64:246–254. doi: 10.1016/s0169-328x(98)00328-3. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter WM, et al. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- Ulmann L, Hatcher JP, Hughes JP, Chaumont S, Green PJ, Conquet F, et al. Up-regulation of P2X4 receptors in spinal microglia after peripheral nerve injury mediates BDNF release and neuropathic pain. J Neurosci. 2008;28:11263–11268. doi: 10.1523/JNEUROSCI.2308-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virginio C, Robertson G, Surprenant A, North RA. Trinitrophenyl-substituted nucleotides are potent antagonists selective for P2X1, P2X3, and heteromeric P2X2/3 receptors. Mol Pharmacol. 1998;53:969–973. [PubMed] [Google Scholar]
- Zhang Z, Artelt M, Burnet M, Trautmann K, Schluesener HJ. Lesional accumulation of P2X4 receptor+ monocytes following experimental traumatic brain injury. Exp Neurol. 2006;197:252–257. doi: 10.1016/j.expneurol.2005.09.015. [DOI] [PubMed] [Google Scholar]