

Abstract
Background and purpose:
Adenosine is an endogenous modulator, interacting with four G-protein coupled receptors (A1, A2A, A2B and A3) and acts as a potent inhibitor of inflammatory processes in several tissues. So far, the functional effects modulated by adenosine receptors on human synoviocytes have not been investigated in detail. We evaluated mRNA, the protein levels, the functional role of adenosine receptors and their pharmacological modulation in human synoviocytes.
Experimental approach:
mRNA, Western blotting, saturation and competition binding experiments, cyclic AMP, p38 mitogen-activated protein kinases (MAPKs) and nuclear factor (NF)-κB activation, tumour necrosis factor α (TNF-α) and interleukin-8 (IL-8) release were assessed in human synoviocytes isolated from patients with osteoarthritis.
Key results:
mRNA and protein for A1, A2A, A2B and A3 adenosine receptors are expressed in human synoviocytes. Standard adenosine agonists and antagonists showed affinity values in the nanomolar range and were coupled to stimulation or inhibition of adenylyl cyclase. Activation of A2A and A3 adenosine receptors inhibited p38 MAPK and NF-κB pathways, an effect abolished by selective adenosine antagonists. A2A and A3 receptor agonists decreased TNF-α and IL-8 production. The phosphoinositide 3-kinase or Gs pathways were involved in the functional responses of A3 or A2A adenosine receptors. Synoviocyte A1 and A2B adenosine receptors were not implicated in the inflammatory process whereas stimulation of A2A and A3 adenosine receptors was closely associated with a down-regulation of the inflammatory status.
Conclusions and implications:
These results indicate that A2A and A3 adenosine receptors may represent a potential target in therapeutic modulation of joint inflammation.
Keywords: adenosine receptors, human synoviocytes, mRNA, Western blotting, receptor binding, cAMP, MAPK p38, NF-κB, TNF-α, IL-8
Introduction
Human synoviocytes play a central role in the pathogenesis of joint destruction, primarily through their secretion of a wide range of pro-inflammatory mediators including cytokines, growth factors and lipid mediators of inflammation. Pro-inflammatory agents produced by synoviocytes are detrimental to articular cartilage in different joint diseases such as osteoarthritis (OA) and rheumatoid arthritis (Abeles and Pillinger, 2006; De Mattei et al., 2009). OA is the most common form of arthritis, and is the single most important cause of disability in older adults (Benito et al., 2005; Goldring and Goldring, 2007). At the present the current recommended treatment of OA involves weight loss, physical therapy and the use of pain relievers (Altman and Barkin, 2009). However, these drugs do not reverse the degenerative process in OA and show some adverse effect on cartilage metabolism (Zhang et al., 2008).
Adenosine is a modulator which interact with four cell surface receptor subtypes, A1, A2A, A2B and A3 adenosine receptors (nomenclature follows Alexander et al., 2009) which are coupled to different G-proteins (Burnstock, 2008). A1 and A3 adenosine receptors, through Gi proteins, mediate inhibition of the adenylate cyclase activity, while A2A and A2B adenosine receptors, via Gs proteins, stimulate cAMP production (Haskòet al., 2008). Modulation of adenosine receptors has an important role in the regulation of the inflammatory processes (Palmer and Trevethick, 2008; Gessi et al., 2008; Ham and Rees, 2008). Understanding how cytokine release is regulated endogenously can give important insight in various disease pathologies. It is well-known that mitogen-activated protein kinases (MAPKs) like p38 are involved in controlling cellular responses as the release of pro-inflammatory cytokines (Fotheringham et al., 2004). The cell signalling pathways initiated by pro-inflammatory events converge on activation of the nuclear factor kappaB (NF-κB) which drives cytokine transcription and production (Wen et al., 2006). Notably, p38 MAPK is one of the kinases implicated in the phosphorylation of NF-κB inhibitors (IkBs) (Westra and Limburg, 2006). Once phosphorylated, IkBs undergo polyubiquitination and ultimately proteosomic degradation, allowing NF-κB to enter the nucleus and promote the transcription of inflammatory genes, such as tumour necrosis factor α (TNF-α) and interleukin-8 (IL-8) (Barnes and Karin, 1997). A role of adenosine in modulating the activity of bovine chondrocytes and synoviocytes has been documented by previous studies of our group. In bovine synoviocytes, adenosine receptors have been characterized from saturation, competition binding experiments and Western blotting analysis (Varani et al., 2008). Functional studies suggested an anti-inflammatory effect consequent on activation of A1 and A2A adenosine receptors in LPS-induced prostaglandin E2 production, mediated by a down-regulation of tumour necrosis factor (TNF)-α and cyclooxygenase-2 mRNA expression (De Mattei et al., 2009). It has been recently reported that in synoviocytes from rheumatoid arthritis patients, A3 adenosine receptors are over-expressed and stimulation of these receptors mediated a reduction in inflammation as a decrease in NF-κB and TNF-α release (Ochaion et al., 2008). Furthermore it has been demonstrated that in different cells or tissues, adenosine is a regulator of NF-κB and MAPK signalling through its interaction with its various receptors (Majumdar and Aggarwal, 2003; Schulte and Fredholm, 2003; Jijon et al., 2005).
With this background, the aim of this study was to investigate the presence of adenosine receptors in primary cultures of human synovial cells from OA patients using mRNA and Western blotting assays. Saturation binding experiments were performed to evaluate affinity (KD) and density (Bmax) of A1, A2A, A2B and A3 adenosine receptors. Affinity values (Ki) of adenosine receptor agonists and antagonists were determined by using competition binding experiments. In order to complete the pharmacological characterization, adenosine receptors were evaluated from a functional point of view. Thus the effect of adenosine receptor agonists and antagonists was investigated on cAMP production. We also assessed the involvement of adenosine receptors on signal transduction pathways including p38 MAPK and NF-κB. Consequently, the effect of adenosine receptor agonists on TNF-α and IL-8 secretion were analysed. The involvement of adenosine receptors was further confirmed using adenosine receptor antagonists to block the effect of the agonists. Finally Gi, Gs and phosphoinositide 3-kinase (PI3K) pathways were investigated to analyse adenosine receptor signalling.
Methods
Subjects
Human samples were collected with approved informed consent in accordance with the principles of the Declaration of Helsinki. The study protocol was approved by the local Ethics Committee of the University of Ferrara and the subjects provided written consent after receiving detailed verbal and written explanations of the study. Synovial tissues were obtained from patients with end-stage OA undergoing total joint replacement surgery. The diagnosis was based on clinical and radiological criteria. All patients (n= 35, F/M: 23/12; age: 63.7 ± 3.4 years) enrolled in this study were recruited from the Department of Biomedical Sciences and Advanced Therapies, Orthopaedic Clinic of the University Hospital of Ferrara, Italy. After providing a full medical history, each patient was given a physical examination, electrocardiogram and routine blood tests. Mean (±SEM) disease duration was 6 ± 1 years. 80% of the enrolled patients were receiving non steroidal anti-inflammatory drugs (NSAIDs) and 20% had no medications.
Cell culture
Primary lines of surface adherent synoviocytes were isolated by enzymatic digestion of synovial tissues for 2–3 h at 37°C in Dulbecco's modified Eagle's medium (DMEM) containing 1.5 mg·mL−1 of collagenase type I-A and 1 mg·mL−1 of hyaluronidase (Sigma-Aldrich, Milan, Italy). After digestion the cells were recovered by centrifugation and plated in T25 culture flasks (Miyashita et al., 2004). Human synoviocytes were maintained in culture in DMEM, 10% fetal calf serum, penicillin (100 U·mL−1), streptomycin (100 µg·mL−1), l-glutamine (2 mmol·L−1), passaged when reaching confluence and used at the 3rd to 4th passages for binding and functional experiments.
CHO or HEK 293 cells transfected with human A1, A2A, A2B and A3 adenosine receptors were prepared as previously described. Cell membranes were prepared for the competition binding experiments, as previously described (Varani et al., 2000; 2005;).
Human synoviocyte characterization
Immunofluorescence with the primary monoclonal antibody specific for human vimentin (Sigma Aldrich, St Louis, MO) was used to evaluate the expression of this marker of fibroblasts, in primary cultures of human synovial cells, as previously described. Nuclei were stained with the selective DNA dye, DAPI (4′,6-diamidino-2-phenylindole) (0.1 mg·mL−1 in PBS-EGTA) for 10 min. Fluorescence was visualized using the Nikon Eclipse TE 2000-E microscope equipped with a digital camera DXM 1200F (Nikon Instruments, Firenze, Italy).
To exclude the presence of contaminating macrophages or endothelial cells, synoviocyte cultures were analysed for CD14 and von-Willebrand factor (vWF) expression by reverse transcription polymerase chain reaction (RT-PCR). Briefly, 2 µl cDNA have been amplified by specific oligonucleotide primers for CD14 (for-5′-CTG GAA GCC GGC G-3′; rev5′-AGC TGA GCA GGA ACC TGT GC-3′) and for vWF (for-5′-TGG CCA GAC CTT GCT GAA GA-3′; rev-5′-CCA TTA TGG AGA ATC ACC TCC A-3′). Cycling parameters have been as follows: 1 min at 94°C; 1 min at the specific annealing temperature (62°C for CD14 and 55°C for vWF); and 1 min at 72°C. PCR product sizes are 405 bp for CD14 and 252 bp for vWF. mRNA from human macrophages and endothelial cells have been used as a positive control for CD14 and vWF expression respectively (Miyashita et al., 2004).
Real-time RT-PCR experiments
Total cytoplasmic RNA was extracted by the acid guanidinium thiocyanate phenol method. Quantitative real-time RT-PCR assay (Gessi et al., 2004) of A1, A2A, A2B and A3 receptor mRNAs was carried out using gene-specific fluorescently labelled TaqMan MGB probe (minor groove binder) in a ABI Prism 7700 Sequence Detection System (Applied Biosystems, Warrington Cheshire, UK). For the real-time RT-PCR of A1, A2A, A2B and A3 adenosine receptors the assays-on-demand™ Gene expression Products NM 000674, NM 000675, NM 000676 and NM 000677 were used respectively. For the real-time RT-PCR of the reference gene the endogenous control human GAPDH kits was used, and the probe was fluorescent-labelled with VIC™ (Applied Biosystems, Warrington Cheshire, UK). Genomic contamination was ruled out by including an RT-negative sample in each PCR set as a control.
Western blotting analysis
Human synoviocytes were washed with ice-cold phosphate buffer saline containing 1 mmol·L−1 sodium orthovanadate, 104 mmol·L−1 4-(2-aminoethyl)-benzenesulfonyl fluoride, 0.08 mmol·L−1 aprotinin, 2 mmol·L−1 leupeptin, 4 mmol·L−1 bestatin, 1.5 mmol·L−1 pepstatin A, 1.4 mmol·L−1 E-64 (Sigma Aldrich, St Louis, MO, USA). Cells were then lysed in Triton lysis buffer and the protein concentration was determined using BCA protein assay kit (Pierce, Rockford, IL, USA). Aliquots of total protein sample (50 µg) were analysed using antibodies specific for human A1, A2A, A2B and A3 adenosine receptors (1 µg·mL−1 dilution, Alpha Diagnostic, San Antonio, TX, USA) and P-p38 (1 µg·mL−1 dilution, Cell Signaling Technology, Danvers, MA, USA) (Merighi et al., 2002). Filters were washed and incubated for 1 h at room temperature with peroxidase-conjugated secondary antibodies (1:2000 dilution). Specific reactions were revealed with enhanced chemiluminescence Western blotting detection reagent (GE Healthcare, UK). Western blotting assays were also normalized against the housekeeping protein β-actin.
Saturation and competition binding experiments to A1, A2A, A2B and A3 adenosine receptors
To obtain membrane preparations, the culture medium was removed and the cells were washed with PBS and scraped off T75 flasks in ice-cold hypotonic buffer (5 mmol·L−1 Tris HCl, 2 mmol·L−1 EDTA, pH 7.4). The cell suspension was homogenized by using a Polytron, centrifuged for 30 min at 100 000 g and used in the saturation and competition binding experiments.
Saturation binding experiments on human synoviocyte membranes were performed by using [3H]-1,3-dipropyl-8-cyclopentyl-xanthine ([3H]-DPCPX), [3H]-4-(2-[7-amino-2-(2-furyl) [1,2,4]-triazolo [2,3-a][1,3,5] triazin-5-ylamino] ethyl)phenol ([3H]-ZM 241385), [3H]-N-benzo[1,3]dioxol-5-yl-2-[5-(2,6-dioxo-1,3-dipropyl-2,3,6,7-tetrahydro-1H-purin-8-yl)-1-methyl-1H-pyrazol-3-yl-oxy]-acetamide ([3H]-MRE 2029F20) and [3H]-5N-(4-methoxyphenylcarbamoyl) amino-8-propyl-2-(2-furyl) pyrazolo [4,3-e]-1,2,4-triazolo [1,5-c]pyrimidine ([3H]-MRE 3008F20) as radioligands to study the presence of A1, A2A, A2B and A3 adenosine receptors respectively (Borea et al., 1994; Varani et al., 1998; Varani et al., 2000; Varani et al., 2005). Briefly, these radioligands at different concentrations (0.01–20 nmol·L−1 or 0.01–30 nmol·L−1) were incubated with 100 µg of protein per assay of membrane suspension for 90 min at 25°C (A1 adenosine receptors) or 60 min at 4°C (A2A and A2B adenosine receptors) or 150 min at 4°C (A3 adenosine receptors).
Competition binding experiments with 1 nmol·L−1[3H]-DPCPX, [3H]-ZM 241385, [3H]-MRE 2029F20 or [3H]-MRE 3008F20 were carried out to determine the affinity values of the selective adenosine agonists and antagonists for the A1, A2A, A2B and A3 adenosine receptors in synoviocytes. In these assays, human synoviocyte membranes (100 µg protein per assay) were incubated with different concentrations of the examined agonists [N6-cyclohexyladenosine (CHA), 2-[p-(2-carboxyethyl)-phenethyl-amino]-5′-N-ethyl-carboxamidoadenosine (CGS 21680), 5′-N-ethylcarboxamidoadenosine (NECA), and N6-(3-iodo-benzyl)-2-chloro-adenosine-5′-N-methyluronamide (Cl-IB-MECA)] and antagonists [1,3-dipropyl-8-cyclopentyl-xanthine (DPCPX), 5-amino-7-(phenylethyl)-2-(2-furyl)-pyrazolo[4,3-e]-1,2,4,triazolo[1,5-c] pyrimidine (SCH 58261), N-benzo[1,3] dioxol-5-yl-2-[5-(2,6-dioxo-1,3-dipropyl-2,3,6,7-tetrahydro-1H-purin-8-yl)-1-methyl-1H-pyrazol-3-yloxy]-acetamide (MRE 2029F20) and 5-n-(4-methoxy phenyl-carbamoyl) amino-8-propyl-2-(2-furyl) pyrazolo [4,3-e]-1,2,4-triazolo [1,5-c] pyrimidine (MRE 3008F20)].
Non-specific binding was determined in the presence of DPCPX, ZM 241385, MRE 2029F20 or MRE 3008F20 at 1 µmol·L−1, respectively, and was always <25% of the total binding. Similar experiments were also performed by using A1, A2A, A2B and A3 adenosine receptor agonists such as CHA, CGS 21680, NECA and Cl-IB-MECA at 1 µmol·L−1, respectively, and was always <30% of the total binding. At the end of the incubation, bound and free radioactivity were separated by filtering, in a Brandel cell harvester, the assay mixture through Whatman GF/B glass-fibre filters. The filter bound radioactivity was counted in a liquid Scintillation Counter Tri Carb Packard 2500 TR (Perkin-Elmer Life and Analytical Sciences, Boston, MA, USA).
Similar competition binding experiments were performed in CHO cells transfected with human A1, A2A or A3 adenosine receptors and A2BHEK 293 cells to evaluate affinity and selectivity of adenosine receptor agonists and antagonists used in functional assays.
Measurement of cyclic AMP levels
Human synoviocytes (106 cells per sample) were suspended in 0.5 mL incubation mixture Krebs Ringer phosphate buffer, containing 1.0 IU·mL−1 adenosine deaminase (Sigma, St Louis, MO, USA) and preincubated for 10 min in a shaking bath at 37°C. Then the effect of selected adenosine agonists was studied by using CHA, CGS 21680, NECA and Cl-IB-MECA at 1 µmol·L−1. To better investigate the inhibitory effect of CHA and Cl-IB-MECA the cells were also incubated with forskolin (1 µmol·L−1) and/or 0.5 mmol·L−1 of 4-(3-butoxy-4-methoxybenzyl)-2-imidazolidinone (Ro 20-1724), a phosphodiesterase inhibitor. Selective A1, A2A, A2B and A3 adenosine receptor antagonists as DPCPX, SCH 58261 MRE 2029F20 and MRE 3008F20 at 1 µmol·L−1 were also used to verify the specific involvement of these subtypes in cAMP production. The final aqueous solution was used to evaluate cAMP levels, using a competition protein binding assay with [3H]-cAMP, Trizma base 0.1 mol·L−1, aminophylline 8.0 mmol·L−1, mercaptoethanol 6.0 mmol·L−1, pH 7.4 (Varani et al., 2008). At the end of the incubation time (150 min at 4°C) and after the addition of charcoal, the samples were centrifuged at 2000×g for 10 min and the clear supernatant was counted in a liquid Scintillation Counter Tri Carb Packard 2500 TR (Perkin-Elmer Life and Analytical Sciences, Boston, MA, USA).
NF-κB activation
Nuclear extracts from human synoviocytes were obtained by using a nuclear extract kit (Active Motif, Carlsbad, CA, USA) following the manufacturer's instructions. NF-κB activation was evaluated by detecting phosphorylated p50 and p65 proteins in nuclear extracts by using the TransAM NF-κB kit (Active Motif, Carlsbad, CA, USA). Phosphorylated NF-κB subunits specifically bind to the immobilized oligonucleotides containing the NF-κB consensus site (5′-GGGACTTTCC-3′). The primary antibody used to detect NF-κB recognized an epitope on each subunit that is accessible only when activated and bound to its DNA target. A horseradish peroxidase (HRP)-conjugated secondary antibody provided a sensitive colorimetric readout that was quantified by spectrophotometry at 450 nm (Gomez Cabrera et al., 2006).
TNF-α and IL-8 release
Tumour necrosis factor α and IL-8 levels were measured in human synoviocytes by using a highly sensitive enzyme linked immunosorbent assay (R and D Systems, Minneapolis, USA) in accordance with the manufacturer's instructions (Forrest et al., 2005).
Data and statistical analysis
The protein concentration was determined according to a Bio-Rad method with bovine albumin as reference standard (Bradford, 1976). A weighted non linear least-squares curve fitting program Ligand was used for computer analysis of saturation and competition binding experiments (Munson and Rodbard, 1980). Functional experiments were calculated by non-linear regression analysis using the equation for a sigmoid concentration-response curve (GraphPAD Prism, San Diego, CA, USA). Analysis of data was performed by repeated measures analysis of variance (anova) followed by Bonferroni's test that was used for multiple comparisons of data sets and was considered significant at a value of P < 0.02. All data are reported as mean ± SEM of independent experiments and are indicated in the figure legends. Each experiment was performed by using the synoviocytes derived from one single donors, and was performed in duplicate (for binding experiments) or in triplicate (for functional experiments). The experiments were repeated at least three or four times as indicated from n-values that represent the number of patients used.
Materials
[3H]-DPCPX, specific activity 120 Ci·mmol−1 was purchased from Perkin Elmer Life and Analytical Sciences (Boston, MA, USA). [3H]-ZM 241385, specific activity 27 Ci·mmol−1 was obtained from Biotrend (Cologne, Germany). [3H]-MRE 2029 F20, specific activity 123 Ci·mmol−1 and [3H]-MRE 3008F20, specific activity 67 Ci·mmol−1 were synthesized from Amersham International Chemical Laboratories (Buckinghamshire, UK). [3H]-cAMP, specific activity 21 Ci·mmol−1 was purchased from GE Healthcare, UK. CHA, CGS 21680, NECA, Cl-IB-MECA, DPCPX, forskolin, Ro 20-1724 and Pertussis toxin were obtained from Sigma, St Louis, MO. LY294002 was purchased from Calbiochem, San Diego, CA. SCH 58261, MRE 2029F20 and MRE3008F20 were a kind gift from Prof Pier Giovanni Baraldi (Dep. of Pharmaceutical Sciences, University of Ferrara, Italy). All other reagents were of analytical grade and obtained from commercial sources.
Results
Phenotypic characterization of human synoviocytes
Cells isolated from synovium of OA patients were a homogenous population as demonstrated by their fibroblast-like morphology (Figure 1A). Primary cultures of human synovial cells also showed the expression of vimentin, a specific cell marker for mesenchymal cells and synovial fibroblasts (Figure 1B). RT-PCR data showed that, under our experimental conditions, mRNA for CD14 and vWF were not amplified, suggesting the absence of macrophage and endothelial cell contamination in synovial cell cultures (Figure 1C).
Figure 1.
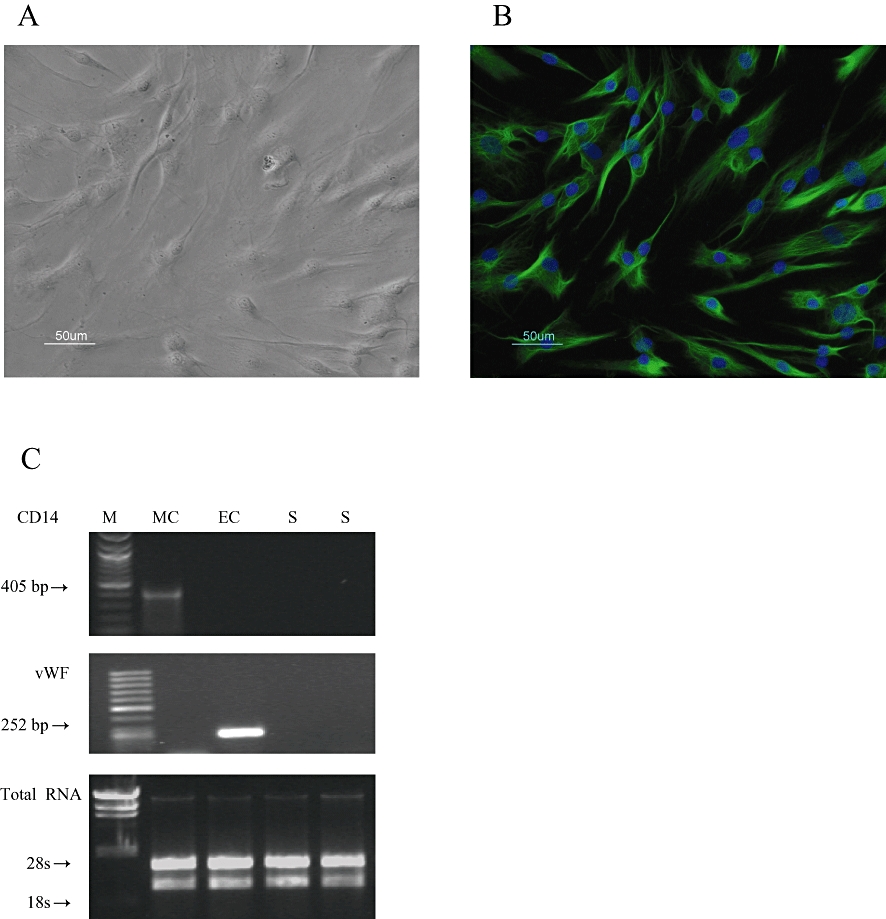
Culture of human synoviocytes. (A) phase contrast and (B) vimentin expression by immunofluorescence. Nuclei were counterstained in blue with DAPI. Original magnification, ×200. (C) CD14 and vWF mRNA expression in macrophages (MC), endothelial cells (EC) and in synoviocytes (S). One microgram of total RNA has been loaded and stained with ethidium bromide to confirm equal RNA quantity. M = DNA ladder marker (Biolabs, Ipswich, MA, USA).
Evaluation of the mRNA and protein levels of A1, A2A, A2B and A3 adenosine receptors
Figure 2A shows adenosine receptor mRNA in human synoviocytes by using real-time quantitative RT-PCR. The present analysis performed with primers specifically designed for the various cloned human adenosine receptors revealed the expression of mRNA for A1, A2A, A2B and A3 adenosine receptors. In particular, high levels of A2A and A3 mRNA were found in human synoviocytes. The presence of adenosine receptors was also confirmed by WWestern blot analysis (Figure 2B). In human synoviocytes, A2A and A3 adenosine receptors were present with a higher expression than A1 and A2B adenosine receptors, as demonstrated by the densitometric analysis shown in Figure 2C.
Figure 2.
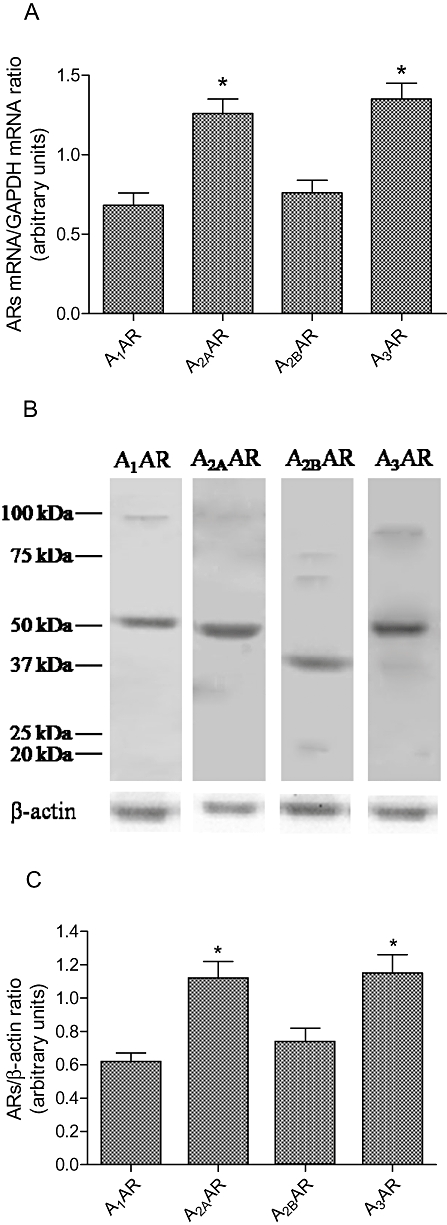
(A) mRNA expression (n= 4) of A1, A2A, A2B and A3 adenosine receptors (AR) and (B) representative Western blotting analysis in human synoviocytes. Densitometric analysis (n= 4) for adenosine receptors were also shown (C).
Saturation and competition binding experiments
Saturation binding experiments in primary cultures of human synovial membranes were performed to evaluate affinity (KD) and receptor density (Bmax) of adenosine receptors. Figure 3A illustrate saturation binding curves relative to A1, A2A, A2B and A3 adenosine receptors showing affinity in the nanomolar range and different receptor density. Scatchard plot analysis revealed the presence of an high affinity binding site as suggested by the linearity of the lines (Figure 3B). Computer analysis of the data failed to show a significantly better fit to a two site than to a one site binding model, indicating that, under our experimental conditions, there was, primarily, a single class of high affinity binding site. Competition binding experiments to A1, A2A, A2B and A3 adenosine receptors by using selective adenosine receptor agonists and antagonists in human synoviocyte membranes were performed (Figure 4). As expected, CHA and Cl-IB-MECA showed biphasic competition binding curves for A1 and A3 adenosine receptors, respectively, as suggested by a significantly better fit to a two site binding model and by an Hill coefficient less than unity (0.54 and 0.63 respectively). Their competition binding curves were best described by the existence of one high affinity (KH) and low affinity (KL) agonist-receptor binding state (Figure 4A,D). Moreover, CGS 21680 and NECA agonists revealed good affinity values for A2A and A2B adenosine receptors confirming a tight coupling between the receptors and G protein (Varani et al., 1998). For CGS 21680 and NECA, Hill coefficients were close to unity excluding the involvement of multiple coupling affinity states (Table 1). In addition, competition binding experiments were also carried out studying selected adenosine receptor antagonists as DPCPX, SCH 58261, MRE 2029F20 and MRE 3008F20 revealing Ki values in the nanomolar range (Table 1).
Figure 3.
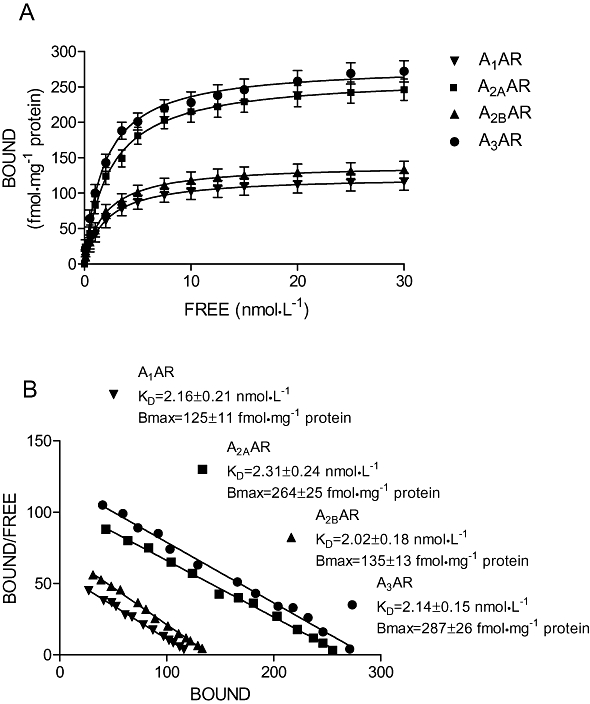
Saturation curves (A) and Scatchard plot (B) of [3H]DPCPX, [3H]ZM 241385, [3H]MRE 2029F20, [3H]MRE 3008F20 binding to A1, A2A, A2B and A3 adenosine receptors (AR) in human synoviocytes respectively. Each value represents the mean ± SEM of four separate experiments performed in duplicate.
Figure 4.
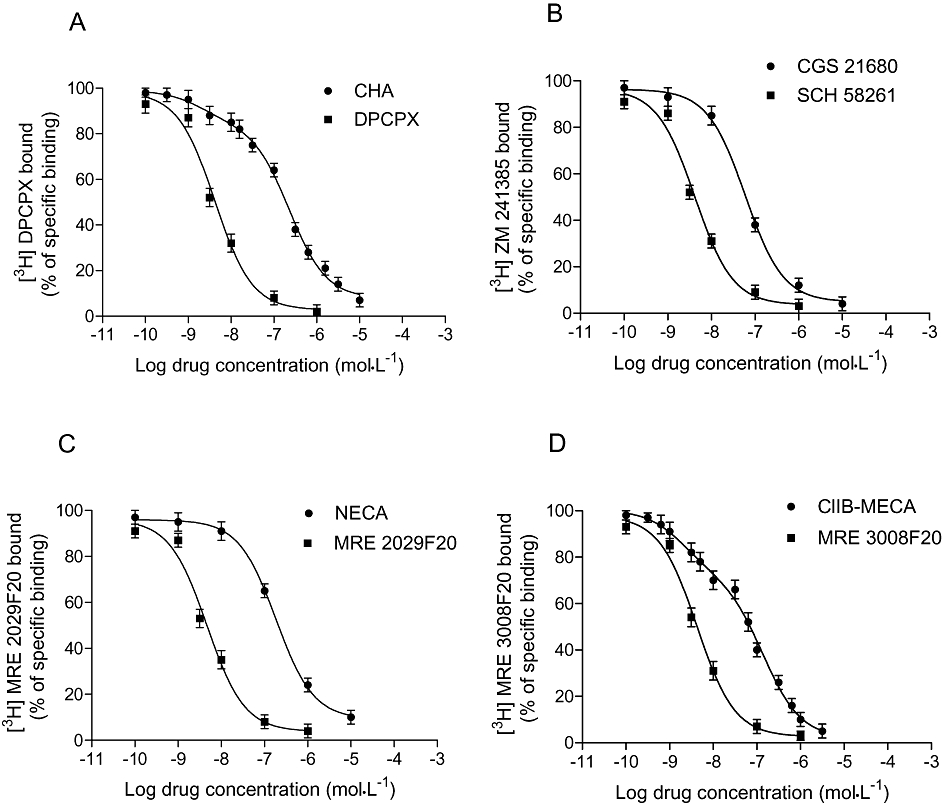
Affinity values of selected A1, A2A, A2B and A3 adenosine receptor agonists and antagonists obtained from competition binding experiments for A1 (A), A2A (B), A2B (C) and A3 (D) adenosine receptors. Each value represents the mean ± SEM of four separate experiments performed in duplicate.
Table 1.
Affinities of standard adenosine agonists and antagonists to human A1, A2A, A2B and A3 adenosine receptors, in human synoviocytes or in transfected CHO or HEK cells
| Compounds |
[3H] DPCPX binding to hA1CHO cells |
[3H] ZM 241385 binding to hA2ACHO cells |
[3H] MRE 2029F20 binding to hA2BHEK 293 cells |
[3H] MRE 3008F20 binding to hA3CHO cells |
Human synoviocytes |
|
|---|---|---|---|---|---|---|
| pKH, pKL or pKi | pKi | pKi | pKH, pKL or pKi | pKH, pKL or pKi | nH | |
| CHA | 8.96 ± 0.04* | 6.09 ± 0.03 | <5.3 | 7.19 ± 0.05* | 9.08 ± 0.03*a | 0.54 ± 0.05 |
| A1 > A3 > A2A | 6.76 ± 0.03# | 5.57 ± 0.04# | 6.94 ± 0.04# | |||
| CGS 21680 | <5.3 | 7.92 ± 0.03 | <5.3 | 5.91 ± 0.04 | 7.55 ± 0.05b | 1.04 ± 0.09 |
| A2A >> A3 | ||||||
| NECA | 7.70 ± 0.04 | 8.08 ± 0.04 | 6.74 ± 0.03 | 7.44 ± 0.05 | 7.02 ± 0.04c | 0.91 ± 0.08 |
| A2A > A1 > A3 > A2B | ||||||
| Cl-IBMECA | <5.3 | 6.19 ± 0.04 | <5.3 | 8.89 ± 0.05* | 9.04 ± 0.04*d | 0.63 ± 0.05 |
| A3 >> A2A | 6.80 ± 0.04# | 7.06 ± 0.03# | ||||
| DPCPX | 8.80 ± 0.03 | 6.58 ± 0.04 | 7.40 ± 0.04 | 5.94 ± 0.06 | 8.65 ± 0.06a | 1.14 ± 0.10 |
| A1 > A2B > A2A > A3 | ||||||
| SCH 58261 | 6.26 ± 0.04 | 8.60 ± 0.05 | <5.3 | <5.3 | 8.56 ± 0.03b | 1.06 ± 0.09 |
| A2A >> A1 | ||||||
| MRE 2029F20 | 6.55 ± 0.03 | <5.3 | 8.46 ± 0.05 | <5.3 | 8.57 ± 0.06c | 1.11 ± 0.11 |
| A2B >> A1 | ||||||
| MRE 3008F20 | 5.93 ± 0.04 | 6.72 ± 0.03 | 5.83 ± 0.03 | 9.05 ± 0.05 | 8.55 ± 0.04d | 0.99 ± 0.08 |
| A3 >> A2A > A1 > A2B | ||||||
The data are expressed as mean ± SEM.
Competition binding experiments of CHA and DPCPX were performed by using [3H]-DPCPX as radioligand;
Competition binding experiments of CGS 21680 and SCH 58261 were performed by using [3H]-ZM 241385 as radioligand;
Competition binding experiments of NECA and MRE2029F20 were performed by using [3H]-MRE2029F20 as radioligand;
Competition binding experiments of Cl-IB-MECA and MRE 3008F20 were performed by using [3H]-MRE 3008F20 as radioligand.
Affinity values are represented as pKH (*), pKL (#) or pKi values; nH= Hill coefficient.
The affinity of the adenosine receptor agonists and antagonists for human A1, A2A, A2B and A3 adenosine receptors expressed in CHO or HEK 293 cells was reported and compared with human synoviocytes in Table 1. CHA showed a high affinity for A1 adenosine receptors, similar to that observed in human synoviocytes. Our experimental data revealed a good affinity of CGS 21680 and NECA for A2A adenosine receptors. Cl-IB-MECA presented a very high affinity for human A3 adenosine receptors in synoviocytes. DPCPX, SCH 58261, MRE 2029F20 and MRE3008F20 showed an high affinity for A1, A2A, A2B and A3 adenosine receptors respectively (Table 1). The high affinity in human synoviocytes of these adenosine receptor antagonists confirmed the binding with A1, A2A, A2B and A3 adenosine receptors respectively (Table 1).
cAMP assays in human synoviocytes
The A2A and A2B adenosine receptors are coupled to stimulation of adenylate cyclase via Gs proteins which mediate an increase of cAMP production. CGS 21680 and NECA at 1 µmol·L−1 mediated a significant increase in cAMP formation reaching 65–90 pmoles per 106 cells respectively. The presence of a selective A2A receptor antagonist, SCH 58261 completely blocked cAMP production (Figure 5A). MRE 2029F290, an A2B adenosine antagonist, was only able partially to inhibit NECA-stimulated cAMP levels, because NECA activates both A2A and A2B adenosine receptors (Figure 5A). The effect of A1 and A3 receptor agonists such as CHA and Cl-IB-MECA (1 µmol·L−1) was evaluated in the presence of 1 µmol·L−1 forskolin and 0.5 mmol·L−1 Ro 20-1724. This experimental condition was chosen because the basal levels (15–20 pmol cAMP per assay) are too low to evaluate, reliably, a direct inhibitory effect. In our experimental conditions CHA and Cl-IB-MECA were able to decrease cAMP levels by 70%. DPCPX was not able to block completely the inhibitory effect of CHA, probably because of the activation of A3 adenosine receptors. On the other hand, MRE 3008F20 blocked the inhibitory effect induced by the selective A3 agonist.
Figure 5.
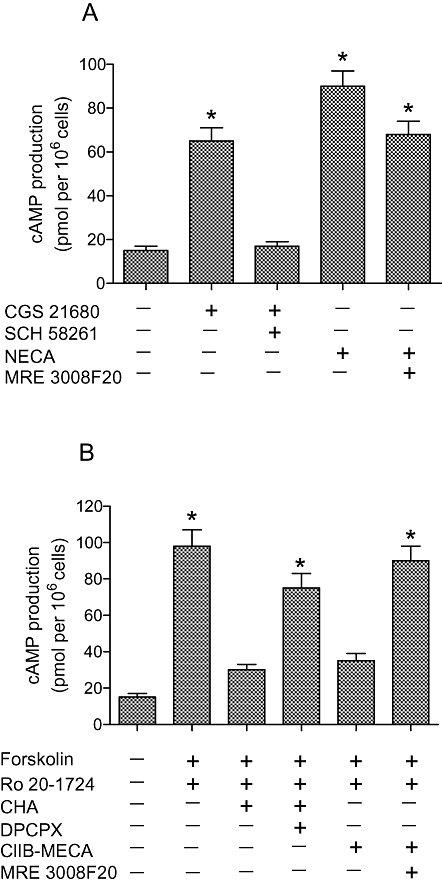
Stimulatory effect on cAMP levels of CGS 21680 (1 µmol·L−1) and NECA (1 µmol·L−1) in the absence and in the presence of SCH 58261 (1 µmol·L−1) and MRE 2029F20 (1 µmol·L−1) respectively (A). Inhibitory effect of CHA (1 µmol·L−1) and Cl-IB-MECA (1 µmol·L−1) in the absence and in the presence of DPCPX (1 µmol·L−1) and MRE 3008F20 (1 µmol·L−1) respectively (B). Each value represents the mean ± SEM of three separate experiments performed in triplicate. *P < 0.01 versus control conditions.
p38 MAPK activation
Western blotting analysis showed that LPS (10 µg·mL−1) was able to increase phospho-p38 levels of 72% in comparison with basal levels. All adenosine agonists investigated at 100 nmol·L−1 did not significantly modify phospho-p38 levels (data not shown). Interestingly, at 1 µmol·L−1 adenosine receptor agonists such as CGS 21680 and NECA were able to inhibit significantly the LPS-stimulated P-p38 levels (Figure 6A,B). Moreover, Cl-IB-MECA (1 µmol·L−1) was also able to inhibit the LPS-stimulated P-p38 levels, although to a lesser extent (Figure 6A,B). SCH 58261 and MRE 3008F20 blocked the inhibitory effect of CGS 21680 and Cl-IB-MECA respectively. On the contrary, MRE 2029F20 was not able to block the effects of NECA, probably due to the persistent modulation of A2A and A3 receptors. CHA (1 µmol·L−1) in the absence and in the presence of DPCPX (1 µmol·L−1) had no effect in the modulation of P-p38 levels suggesting that the A1 adenosine receptors were not involved in the activation of p38.
Figure 6.
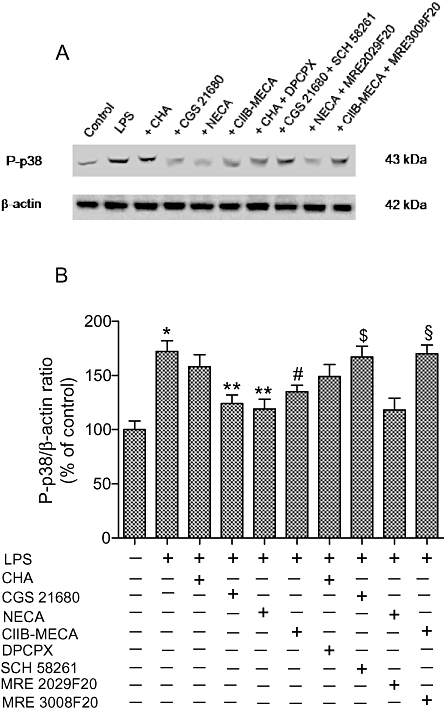
Western blotting analysis of phosphorylated p38 (P-p38) in the absence and in the presence of LPS (10 µg·mL−1). The effect of examined adenosine receptor agonists (1 µmol·L−1) and antagonists (1 µmol·L−1) was also evaluated (A). Densitometric analysis (n= 4) of the bands obtained were also shown (B). *P < 0.01 versus control conditions; **P < 0.01 versus LPS conditions; #P < 0.02 versus LPS conditions; $P < 0.01 versus CGS 21680; §P < 0.02 versus Cl-IB-MECA.
NF-κB activation in human synoviocytes
NF-κB levels were evaluated studying the activation of the p50 (Figure 7A) and p65 (Figure 7B) subunits. In primary cultures of human synoviocytes, adenosine agonists examined were not able to decrease the LPS-stimulated NF-κB levels, at 100 nmol·L−1 with the exception of Cl-IB-MECA, which mediated a reduction in p50 and p65 levels by 33 ± 3% and 24 ± 3% respectively (P < 0.05). All adenosine agonists investigated at the higher concentration 1 µmol·L−1 were able to inhibit the LPS-stimulated NF-κB levels. The inhibitory effect of CHA was reversed only by the A3 receptor antagonist MRE 3008F20. In addition, SCH 58261, but not MRE 3008F20, was able to block the reduction of NF-κB levels mediated by CGS 21680 because of the high or low affinity of this agonist for A2A or A3 adenosine receptors respectively (Table 1). The inhibitory effect of the pan-adenosine agonist NECA was partially blocked (P < 0.05) only by SCH 58261 and MRE 3008F20 suggesting the involvement of A2A and A3 adenosine receptors. Interestingly, the effect of NECA was completely prevented by simultaneous incubation with SCH 58261 and MRE 3008F20. The reduction in p50 and p65 activation mediated by Cl-IB-MECA was reversed only by MRE 3008F20 and not by SCH 58261. This effect was most likely due to the low affinity of the A3 receptor agonist, Cl-IB-MECA for A2A adenosine receptors (Table 1, Figure 7A,B).
Figure 7.
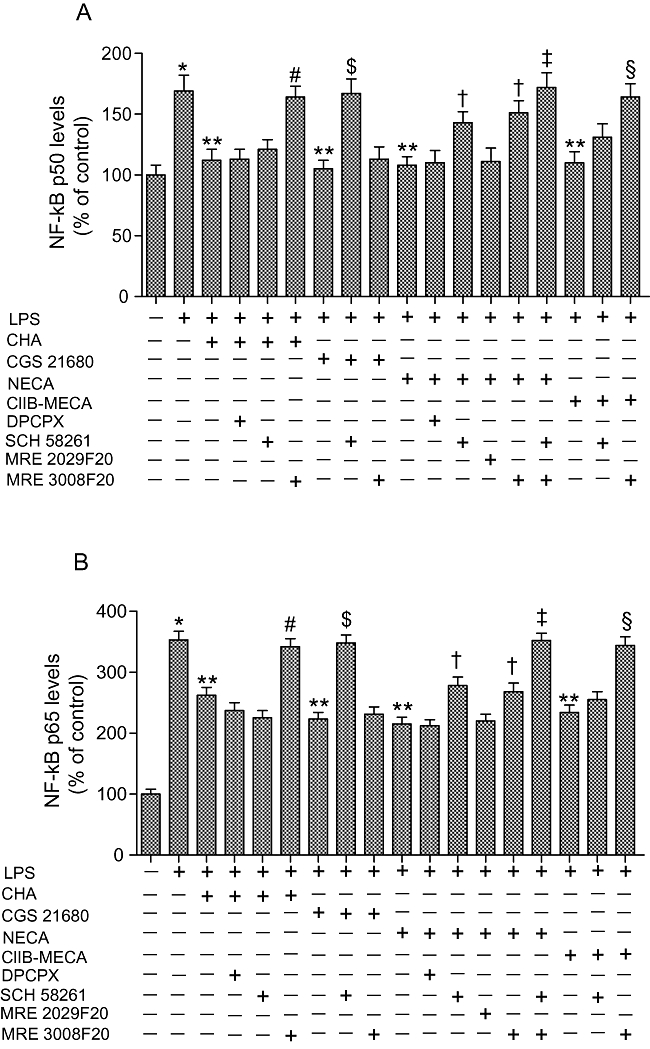
Effect of adenosine receptor agonists (1 µmol·L−1) and antagonists (1 µmol·L−1) in human synoviocytes on NF-κB activation which was evaluated by detecting phosphorylated p50 (A) and p65 (B) proteins in nuclear extracts (n= 4). *P < 0.01 versus control conditions. **P < 0.01 versus LPS conditions; #P < 0.01 versus CHA; $P < 0.01 versus CGS 21680; †P < 0.05 versus NECA; ‡P < 0.01 versus NECA; §P < 0.01 versus Cl-IB-MECA.
TNF-α and IL-8 production
In human synoviocytes, the effect of adenosine agonists and antagonists on TNF-α (Figure 8A) and IL-8 (Figure 8B) release was evaluated in the presence of LPS (10 µg·mL−1). Of the agonists examined at 100 nmol·L−1 only Cl-IB-MECA was able to decrease significantly TNF-α and IL-8 production by 35 ± 3% and 28 ± 2% respectively (P < 0.05). All adenosine agonists investigated at 1 µmol·L−1 concentration were able to inhibit LPS-stimulated release of TNF-α and IL-8. The inhibitory effect of CHA was counteracted by MRE 3008F20 and not by DPCPX or SCH 58261 suggesting the involvement of A3 adenosine receptors (Table 1). The effect of CGS 21680 was only blocked by SCH 58261 and not by MRE 3008F20 probably because of the low affinity of CGS 21680 for A3 adenosine receptors. NECA was able to significantly decrease the LPS-stimulated release of TNF-α and IL-8, an effect blocked by SCH 58261 or MRE 3008F20 (P < 0.05), but not by DPCPX or MRE 2029F20. The simultaneous use of SCH 58261 and MRE 3008F20 completely reversed the inhibitory effect of NECA (P < 0.01). Finally, only MRE 3008F20 was able to block the effect of Cl-IB-MECA on TNF-α and IL-8 release.
Figure 8.
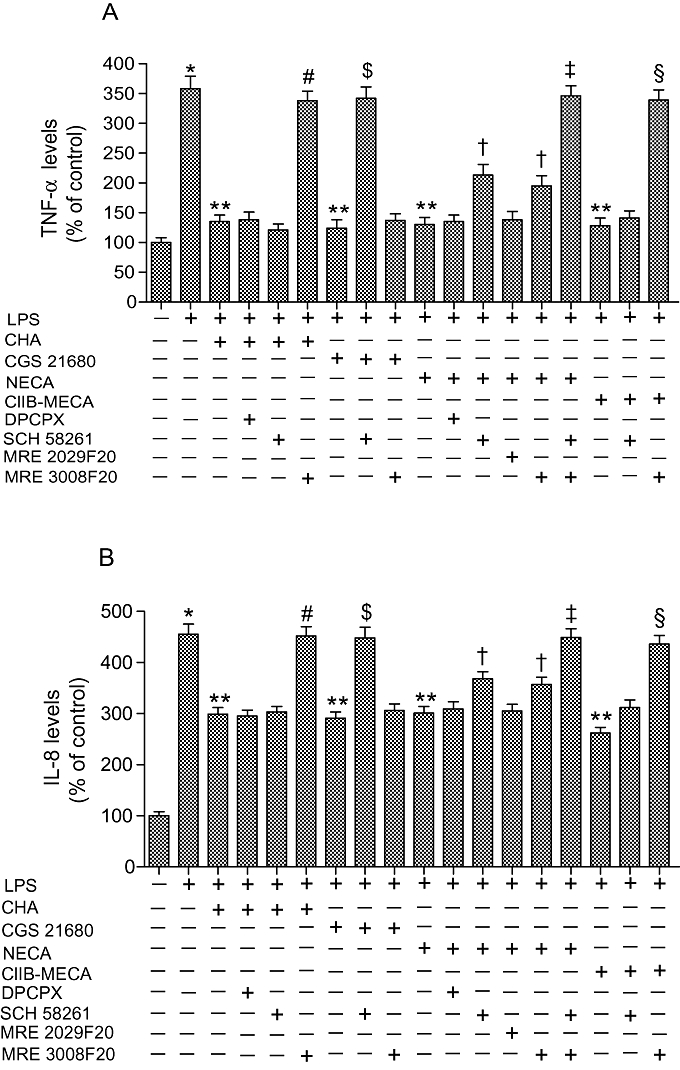
Tumour necrosis factor α (TNF-α) (A) and interleukin-8 (IL-8) (B) levels in human synoviocytes in control conditions and stimulated by LPS (10 µg·mL−1). TNF-α and IL-8 levels were also calculated (n= 4) in the presence of adenosine receptor agonists (1 µmol·L−1) and antagonists (1 µmol·L−1). *P < 0.01 versus control conditions. **P < 0.01 versus LPS conditions; #P < 0.01 versus CHA; $P < 0.01 versus CGS 21680; †P < 0.05 versus NECA; ‡P < 0.01 versus NECA; §P < 0.01 versus Cl-IB-MECA.
Modulation of Gi, Gs and PI3K pathways
To determine whether the Gi pathway or the PI3K pathway was involved in the response to activation of A3 adenosine receptors, human synoviocytes were preincubated with the Gi inactivator Pertussis toxin (100 ng·mL−1) for 2 h or with the PI3K inhibitor LY294002 (25 µmol·L−1) for 20 min and then stimulated with Cl-IB-MECA (1 µmol·L−1). The preincubation with Pertussis toxin did not modify the Cl-IB-MECA-mediated inhibition of P-p38, NF-κB p50 or p65, TNF-α and IL-8 levels. In contrast, LY294002 incubation completely abolished the inhibition of P-p38, NF-κB p50 or p65, TNF-α and IL-8 levels by Cl-IB-MECA. These data suggest that the A3 adenosine receptors signal through a PI3K pathway (Table 2). In order to verify whether the Gs pathway was involved in A2A receptor responses, human synoviocytes were incubated with a direct activator of adenylyl cyclase activity, forskolin (1 µmol·L−1). This compound was able to reduce P-p38, NF-κB p50 or p65, TNF-α and IL-8 levels suggesting the involvement of cAMP in A2A receptor-mediated responses (Table 2).
Table 2.
Effect of Pertussis toxin, PI3K inhibitor (LY294002) or forskolin on phospho-p38 (P-p38) and NF-κB activation, TNF-α and IL-8 release in human synoviocytes
| LPS (10 µg·mL−1) | LPS(10 µg·mL−1)Cl-IB-MECA(1 µg·mL−1) | LPS(10 µg·mL−1)Pertussis toxin(100 ng·mL−1)Cl-IB-MECA(1 µg·mL−1) | LPS(10 µg·mL−1)LY294002(25 µg·mL−1)Cl-IB-MECA(1 µg·mL−1) | LPS(10 µg·mL−1)Forskolin(1 µg·mL−1) | |
|---|---|---|---|---|---|
| P-p38 | 172 ± 10 | 135 ± 6a | 133 ± 7 | 175 ± 8c | 128 ± 8b |
| NF-κB (p50) | 169 ± 14 | 110 ± 12b | 104 ± 10 | 173 ± 13d | 109 ± 9b |
| NF-κB (p65) | 353 ± 14 | 234 ± 12b | 242 ± 13 | 338 ± 14d | 226 ± 11b |
| TNF-α | 358 ± 21 | 128 ± 13b | 134 ± 12 | 344 ± 19d | 136 ± 14b |
| IL-8 | 455 ± 20 | 262 ± 11b | 278 ± 15 | 437 ± 22d | 287 ± 14b |
The data are expressed as % of control ± SEM.
P < 0.02 versus LPS;
P < 0.01 versus LPS;
P < 0.02 versus Cl-IB-MECA;
P < 0.01 versus Cl-IB-MECA.
IL-8, interleukin-8; TNF-α, tumour necrosis factor α.
Discussion
Chronic inflammatory processes are based on a sustained and tightly regulated communication network among different cells types. It is generally accepted that synoviocytes have a key function in the development of sustained inflammation in joint diseases such as OA. The aetiology of OA is multifactorial and includes the release of both systemic and local biochemical factors (Peat et al., 2001). There is growing evidence that synovial inflammation has an important role in the pathophysiology of OA, contributing to signs and symptoms of the disease (Liu et al., 2009; Sutton et al., 2009). The activation by inflammatory stimuli of synoviocytes mediates the production of different chemokines, cytokines and matrix metalloproteinases (Georganas et al., 2000; Nanki et al., 2001). There is also growing evidence that pro-inflammatory mediators could play critical roles in the development of inflammation and damage in joint tissues (Inoue et al., 2005; Wen et al., 2006). Several studies have indicated that adenosine, via stimulation of its receptors, is involved in the modulation of inflammatory processes (Palmer and Trevethick, 2008; Gessi et al., 2008; Ham and Rees, 2008). In particular, A2A adenosine receptor agonists inhibit cartilage damage when used in the treatment of septic arthritis, by diminishing IL-8 expression and reduce rat adjuvant-induced arthritis (Cohen et al., 2005). In synoviocytes, the selective involvement of A2A and A3 adenosine receptors in the immunomodulatory actions of methotrexate has been studied (Montesinos et al., 2003; Cronstein, 2005). Recently, stimulation of A3 adenosine receptors inhibited human synoviocyte growth and the inflammatory manifestations of arthritis (Ochaion et al., 2008).
The purpose of the present paper was to document the expression and the binding parameters of A1, A2A, A2B and A3 adenosine receptors in human synoviocytes derived from OA patients. Adenosine receptors are coexpressed in these cells and were investigated through mRNA, Western blotting analysis and saturation binding experiments. To exactly quantify the affinity and density of adenosine receptors, saturation binding studies were performed. In human synoviocytes, the adenosine receptor affinities (KD, nmol·L−1) were in the nanomolar range and the receptor densities (Bmax, fmol·mg−1 protein) were from 125 to 287 fmol·mg−1 protein. No data have been published on the binding parameters of adenosine receptors in human synoviocytes, although binding and functional characterization has been performed in bovine synoviocytes. In these cells, adenosine receptor affinity was similar in nanomolar range to those observed in human synoviocytes. Adenosine receptor density was higher in human than bovine synoviocytes (Varani et al., 2008). The competition binding experiments in human synoviocytes were performed to calculate the affinity of adenosine receptor agonists and antagonists which were also studied in functional assays. As expected, competition between [3H] DPCPX and [3H] MRE 3008F20 and increasing concentrations of CHA and Cl-IB-MECA, respectively, revealed two binding sites for these agonists, probably due to the presence of two different high and low receptor affinity states. On the contrary, competition binding curves with antagonists were monophasic (Varani et al., 2000; Merighi et al., 2001). In addition, competition of [3H] ZM 241385 and [3H] MRE 2029F20 by increasing concentrations of CGS 21680 and NECA, respectively, showed simple inhibition curves excluding the involvement of multiple affinity states because of the tight coupling between A2 adenosine receptors and Gs proteins (Varani et al., 1998; Gessi et al., 2005). Affinity values of adenosine receptor agonists and antagonists for human A1, A2A, A2B and A3 adenosine receptors expressed in CHO or HEK 293 cells were closely similar to those obtained in human synoviocytes. CGS 21680 and Cl-IB-MECA were selective for A2A and A3 adenosine receptors, CHA was able to interact with A1 and A3 adenosine receptors while NECA bound all adenosine receptors. The adenosine receptor antagonists, DPCPX, SCH 58261, MRE 2029F20 and MRE 3008F20 chosen in this study are selective for A1, A2A, A2B and A3 adenosine receptors respectively.
Another purpose of the present study was to investigate the functional activities of adenosine receptors in human synoviocytes, where agonists and antagonists were able to modulate cAMP production. As expected, CGS 21680 and NECA stimulated adenylyl cyclase activity, whereas CHA and Cl-IB-MECA decreased cAMP production. These adenosine receptor agonist effects were blocked by selective antagonists which allowed the involvement of specific adenosine receptors to be identified. These data are in agreement with those reported in human or bovine synoviocytes where A2A and A2B adenosine receptors are coupled positively to adenylyl cyclase, whereas A1 and A3 adenosine receptors are linked to Gi proteins and inhibit cAMP production (Boyle et al., 1996; Varani et al., 2008).
There is a large body of evidence suggesting that p38 MAPK represents one key signal transduction pathway crucial for the induction and maintenance of chronic inflammation (Westra and Limburg, 2006). This network comprises the extracellular mediators such as cytokines, chemokines and matrix-degrading proteases which orchestrate the participation of the cells in chronic inflammatory process (Karin, 2005). The mirrors of this outside communication world are intracellular transcription factor pathways such as NF-κB, which shuttle information about inflammatory stimuli to the cell nucleus (Pomerantz and Baltimore, 2002). To address this issue, p38 MAPK activation was studied following adenosine receptors modulation in human synoviocytes. We found that the stimulation of A2A and A3 adenosine receptors by using CGS 21680 and Cl-IB-MECA, respectively, mediated a significant decrease of the phosphorylated, hence activated form of p38 MAPK. There are few papers published on human synoviocytes and the functional response of adenosine receptors (Boyle et al., 1996; Ochaion et al., 2008). On the other hand, these data are in agreement with those previously reported in human pro-monocytic U937 cells where CGS 21680 decreased phospho-p38 protein levels (Fotheringham et al., 2004).
It is well-known that phospho-p38 acts as a kinase implicated in the phosphorylation of the NF-κB inhibitor, IkB, allowing the p50 and p65 subunits to enter the nucleus and promoting the transcription of inflammatory genes. Therefore, we have investigated the effects of adenosine receptor agonists and antagonists on p50 and p65 subunit levels in the nuclear extract of human synoviocytes. Our data demonstrated that adenosine agonists were able to reduce p50 and p65 levels primarily through the involvement of A2A and A3 adenosine receptors as revealed by the use of receptor antagonists. In particular, the effect of CHA, that binds A1 and A3 adenosine receptors with high affinity and A2A adenosine receptors with low affinity (Table 1), was reversed only by the A3 receptor antagonist MRE3008F20 and not by the A1 antagonist DPCPX excluding the involvement of A1 adenosine receptors. The effect of the A2A receptor agonist CGS 21680 was reversed by the selective A2A antagonist SCH 58261, confirming the involvement of A2A adenosine receptors in the reduction in p50 and p65 levels. The inhibitory effect of the pan-adenosine agonist NECA was only blocked by SCH 58261 and MRE 3008F20, but not by the A1 receptor antagonist DPCPX or the A2B antagonist MRE 2029F20, demonstrating the involvement of A2A and A3 adenosine receptors but not of A1 or A2B adenosine receptors. The direct role of A3 adenosine receptors in the down-regulation of nuclear NF-κB levels was further confirmed by the inhibitory effect of the A3 agonist Cl-IB-MECA that was completely abolished by the A3 antagonist MRE 3008F20.
The present data strongly support several pieces of evidence performed in different cellular models such as murine microglial cells, mouse macrophages and leukemic cell line indicating that adenosine and in particular A2A and A3 receptor stimulation mediated the inhibition of NF-κB (Majumdar and Aggarwal, 2003; Lee et al., 2006; Martin et al., 2006).
The transcription factor NF-κB represents a major component of the TNF-α gene activation machinery and its activation is necessary for TNF-α production. In our study we found that the suppression of TNF-α release was mediated by A2A and A3 adenosine receptors. Our data are in agreement with those previously reported by several authors showing that A2A and A3 adenosine receptors mediated a reduction of TNF-α production (Haskòet al., 1996; Szabòet al., 1998; Haskòet al., 2000; Mabley et al., 2003). This reduction of TNF-α levels could be explained as a direct inhibition of NF-κB, at a transcriptional level (Lee et al., 2006). Alternatively, it is also possible that A2A and A3 adenosine receptor-mediated inhibition of TNF-α production is closely associated with p38 MAPK, which has been proposed to be a key regulator of TNF-α mRNA stability and protein translation (Fotheringham et al., 2004).
The cell signalling pathways initiated by pro-inflammatory events converge on the activation of p38 and NF-κB which are also implicated in the regulation of IL-8 expression, a critical mediator of tissue inflammation. As a result of p38 and NF-κB inhibition, stimulation of A2A and A3 adenosine receptors resulted in a decrease of IL-8 production. Previous contrasting studies obtained in different cells have reported that adenosine acts as a positive or negative regulator of IL-8 suggesting that this disparity of the effect could be in part explained by the presence of different target cells or tissues (Murakami et al., 2001; Jijon et al., 2005).
To better investigate the role of G proteins in the function of A2A or A3 adenosine receptors, selective experiments were performed with the aim to evaluate the effect of Cl-IB-MECA in the presence of inhibitors of Gi proteins or PI3K. We found that the A3 receptor-mediated reduction in NF-κB activation and cytokine release was not affected by the block of Gi proteins but only by the inhibition of PI3K suggesting that, for these responses, A3 adenosine receptors signal through a PI3K pathway. In contrast, the A2A receptor-mediated reduction of inflammatory responses was most likely due to the activation of Gs protein and to the increase in cAMP levels, as demonstrated by the direct activator of adenylyl cyclase, forskolin, which induced responses similar to those of the A2A adenosine receptors.
Clearly, further studies will be needed regarding the differential roles of other MAPKs and transcription factors on various inflammatory mediators to delineate the mechanisms underlying cytokine production by adenosine signalling.
In conclusion, the novel findings of this study in human synoviocytes from OA patients are represented by the presence of adenosine receptors that are also quantified by high levels of density. The functional results revealed the direct involvement of A2A and A3 adenosine receptors in the inhibition of inflammatory cascade in human synoviocytes. Taken together, these results suggest that these adenosine receptors could represent potential therapeutic targets in the complex pathways regulating inflammatory processes in joint disease.
Acknowledgments
We thank Dr Agnese Pellati (University of Ferrara, Department of Morphology and Hystology) and Giuliano Marzola (University of Ferrara, Department of Clinical and Experimental Medicine) for helpful advice on the cell culture preparation.
Glossary
Abbreviations:
- IL-8
interleukin-8
- MAPK
mitogen-activated protein kinase
- NF-κB
nuclear factor κB
- OA
osteoarthritis
- PI3K
phosphoinositide 3-kinase
- TNF-α
tumour necrosis factor α
Conflicts of interest
None.
References
- Abeles AM, Pillinger MH. The role of the synovial fibroblast in rheumatoid arthritis: cartilage destruction and the regulation of matrix metalloproteinases. Bull NYU Hosp Jt Dis. 2006;64:20–24. [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman R, Barkin RL. Topical therapy for osteoarthritis: clinical and pharmacologic perspectives. Postgrad Med. 2009;121:139–147. doi: 10.3810/pgm.2009.03.1986. [DOI] [PubMed] [Google Scholar]
- Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- Benito MJ, Veale DJ, FitzGerald O, Van den Berg WB, Bresnihan B. Synovial tissue inflammation in early and late osteoarthritis. Ann Rheum Dis. 2005;64:1263–1267. doi: 10.1136/ard.2004.025270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borea PA, Varani K, Dalpiaz A, Capuzzo A, Fabbri E, Ijzerman AP. Full and partial agonists and thermodynamic binding parameters of adenosine A1 receptor ligands. Eur J Pharmacol. 1994;267:55–61. doi: 10.1016/0922-4106(94)90224-0. [DOI] [PubMed] [Google Scholar]
- Boyle DL, Sajjadi FG, Firestein GS. Inhibition of synoviocyte collagenase gene expression by adenosine receptor stimulation. Arthritis Rheum. 1996;39:923–930. doi: 10.1002/art.1780390608. [DOI] [PubMed] [Google Scholar]
- Bradford M. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein dye-binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Burnstock G. Purinergic signalling and disorders of the central nervous system. Nat Rev Drug Discov. 2008;7:575–590. doi: 10.1038/nrd2605. [DOI] [PubMed] [Google Scholar]
- Cohen SB, Leo BM, Baer GS, Turner MA, Beck G, Diduch DR. An adenosine A2A receptor agonist reduces interleukin-8 expression and glycosaminoglycan loss following septic arthrosis. J Orthop Res. 2005;23:1172–1178. doi: 10.1016/j.orthres.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Cronstein BN. Low-dose methotrexate: a mainstay in the treatment of rheumatoid arthritis. Pharmacol Rev. 2005;57:163–172. doi: 10.1124/pr.57.2.3. [DOI] [PubMed] [Google Scholar]
- De Mattei M, Varani K, Masieri FF, Pellati A, Ongaro A, Fini M, et al. Adenosine analogs and electtromagnetic fields inhibit prostaglandin E2 release in bovine synovial fibroblast. Osteoarthritis Cartilage. 2009;17:252–262. doi: 10.1016/j.joca.2008.06.002. [DOI] [PubMed] [Google Scholar]
- Forrest CM, Harman G, McMillan RB, Stoy N, Stone TW, Darlington LG. Modulation of cytokine release by purine receptors in patients with rheumatoid arthritis. Clin Exp Rheumatol. 2005;23:89–92. [PubMed] [Google Scholar]
- Fotheringham JA, Mayne MB, Grant JA, Geiger JD. Activation of adenosine receptors inhibits tumor necrosis factor-alpha release by decreasing TNF-alpha mRNA stability and p38 activity. Eur Pharmacol. 2004;497:87–95. doi: 10.1016/j.ejphar.2004.06.029. [DOI] [PubMed] [Google Scholar]
- Georganas C, Liu H, Perlman H, Hoffmann A, Thimmapaya B, Pope RM. Regulation of IL-6 and IL-8 expression in rheumatoid arthritis synovial fibroblasts: the dominant role for NF-kappa B but not C/EBP beta or c-Jun. J Immunol. 2000;165:7199–7206. doi: 10.4049/jimmunol.165.12.7199. [DOI] [PubMed] [Google Scholar]
- Gessi S, Cattabriga E, Avitabile A, Gafa' R, Lanza G, Cavazzini L, et al. Elevated expression of A3 adenosine receptors in human colorectal cancer is reflected in peripheral blood cells. Mol Pharmacol. 2004;65:711–719. doi: 10.1158/1078-0432.CCR-1134-03. [DOI] [PubMed] [Google Scholar]
- Gessi S, Merighi S, Varani K, Leung E, Mac Lennan S, Borea PA. The A3 adenosine receptor: an enigmatic player in cell biology. Pharmacol Ther. 2008;117:123–140. doi: 10.1016/j.pharmthera.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Gessi S, Varani K, Merighi S, Cattabriga E, Pancaldi C, Szabadkai Y, et al. Expression, pharmacological profile, and functional coupling of A2B receptors in a recombinant system and in peripheral blood cells using a novel selective antagonist radioligand, [3H]MRE 2029-F20. Mol Pharmacol. 2005;67:2137–2147. doi: 10.1124/mol.104.009225. [DOI] [PubMed] [Google Scholar]
- Goldring MB, Goldring SR. Osteoarthritis. Cell Physiol. 2007;213:626–634. doi: 10.1002/jcp.21258. [DOI] [PubMed] [Google Scholar]
- Gomez Cabrera MC, Martinez A, Santangelo G, Pallardò FV, Sastre J, Vina J. Oxidative stress in marathon runners: interest of antioxidant supplementation. Br J Nutrition. 2006;96:S31–S33. doi: 10.1079/bjn20061696. [DOI] [PubMed] [Google Scholar]
- Ham J, Rees DA. The adenosine a2b receptor: its role in inflammation. Endor Metab Immune Disord Drug Targets. 2008;8:244–254. doi: 10.2174/187153008786848303. [DOI] [PubMed] [Google Scholar]
- Haskò G, Kuhel DG, Chen JF, Schwarzschild A, Deitch EA, Mabley JC, et al. Adenosine inhibits IL-12 and TNF-α production via adenosine A2A receptor-dependent and independent mechanisms. Faseb J. 2000;14:2065–2074. doi: 10.1096/fj.99-0508com. [DOI] [PubMed] [Google Scholar]
- Haskò G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7:759–770. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskò G, Szabò C, Nemeth ZH, Kvetan V, McCarthy Pastores S, Vizi ES. Adenosine receptor agonists differentially regulate IL-10, TNF-α, and nitric oxide production in RAW 264.7 macrophages and in endotoxemic mice. J Immunol. 1996;157:4634–4640. [PubMed] [Google Scholar]
- Inoue T, Hammaker D, Boyle DL, Firestein GS. Regulation of p38 MAPK by MAPK kinases 3 and 6 in fibroblast-like synoviocytes. J Immunol. 2005;174:4301–4306. doi: 10.4049/jimmunol.174.7.4301. [DOI] [PubMed] [Google Scholar]
- Jijon HB, Walker J, Hoentien F, Diaz H, Ewaschuk J, Jobin C, et al. Adenosine is a negative regulator of NF-kappaB and MAPK signaling in human intestinal ephithelial cells. Cell Immunol. 2005;237:86–95. doi: 10.1016/j.cellimm.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Karin M. Inflammation-activated protein kinases as targets for drug development. Proc Am Thorac Soc. 2005;2:386–390. doi: 10.1513/pats.200504-034SR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Jhun BS, Oh YT, Lee JH, Choe W, Baik HH, et al. Activation of adenosine A3 receptor suppresses lipopolysaccharide-induced TNF-alpha production through inhibition of PI 3-kinase/Akt and NF-kappaB activation in murine BV2 microglial cells. Neurosci Lett. 2006;396:1–6. doi: 10.1016/j.neulet.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Liu YZ, Jackson AP, Cosgrove SD. Contribution of calcium-containing crystals to cartilage degradation and synovial inflammation in osteoarthritis. Osteoarthritis Cartilage. 2009;17:1333–1340. doi: 10.1016/j.joca.2009.04.022. [DOI] [PubMed] [Google Scholar]
- Mabley J, Soriano F, Pacher P, Haskò G, Marton A, Wallace R, et al. The adenosine A3 receptor agonist, N6-(3-iodobenzyl)-adenosine-5′-N-methyluronamide, is protective in two murine models of colitis. Eur J Pharmacol. 2003;466:323–329. doi: 10.1016/s0014-2999(03)01570-x. [DOI] [PubMed] [Google Scholar]
- Majumdar S, Aggarwal BB. Adenosine suppresses activation of nuclear factor-kappaB selectively induced by tumor necrosis factor in different cell types. Oncogene. 2003;22:1206–1218. doi: 10.1038/sj.onc.1206184. [DOI] [PubMed] [Google Scholar]
- Martin L, Pingle SC, Hallam DM, Rybak LP, Ramkumar V. Activation of the adenosine A3 receptor in RAW 264.7 cells inhibits lipopolysaccharide-stimulated tumor necrosis factor-alpha release by reducing calcium-dependent activation of nuclear factor-kappaB and extracellular signal-regulated kinase 1/2. J Pharmacol Exp Ther. 2006;316:71–78. doi: 10.1124/jpet.105.091868. [DOI] [PubMed] [Google Scholar]
- Merighi S, Mirandola P, Milani D, Varani K, Gessi S, Klotz K-N, et al. Adenosine receptors as mediators of both cell proliferation and cell death of cultured human melanoma cells. J Invest Dermatol. 2002;119:923–933. doi: 10.1046/j.1523-1747.2002.00111.x. [DOI] [PubMed] [Google Scholar]
- Merighi S, Varani K, Gessi S, Cattabriga E, Iannotta V, Ulouglu C, et al. Pharmacological and biochemical characterization of adenosine receptors in the human malignant melanoma A375 cell line. Br J Pharmacol. 2001;134:1215–1226. doi: 10.1038/sj.bjp.0704352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita T, Kawakami A, Nakashima T, Yamasaki S, Tamai M, Tanaka F, et al. Osteoprotegerin (OPG) acts as an endogenous decoy receptor in tumour necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis of fibroblast-like synovial cells. Clin Exp Immunol. 2004;137:430–436. doi: 10.1111/j.1365-2249.2004.02534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montesinos MC, Desai A, Delano D, Chen JF, Fink JS, Jacobson MA, et al. Adenosine A2A or A3 receptors are required for inhibition of inflammation by methotrexate and its analog MX-68. Arthritis Rheum. 2003;48:240–247. doi: 10.1002/art.10712. [DOI] [PubMed] [Google Scholar]
- Munson PJ, Rodbard D. Ligand: a versatile computerized approach for the characterization of ligand binding systems. Anal Biochem. 1980;107:220–239. doi: 10.1016/0003-2697(80)90515-1. [DOI] [PubMed] [Google Scholar]
- Murakami S, Hashikawa T, Saho T, Takedachi M, Nozaki T, Shimabukuro Y, et al. Adenosine regulates the IL-1 beta-induced cellular functions of human gingival fibroblasts. Int Immunol. 2001;13:1533–1540. doi: 10.1093/intimm/13.12.1533. [DOI] [PubMed] [Google Scholar]
- Nanki T, Nagasaka K, Hayashida K, Saita Y, Miyasaka N. Chemokines regulate IL-6 and IL-8 production by fibroblast-like synoviocytes from patients with rheumatoid arthritis. J Immunol. 2001;167:5381–5385. doi: 10.4049/jimmunol.167.9.5381. [DOI] [PubMed] [Google Scholar]
- Ochaion A, Bar-Yehuda S, Cohen S, Amital H, Jacobson KA, Joshi BV, et al. The A3 adenosine receptor agonist CF502 inhibits the PI3K, PKB/Akt and NF-kappaB signaling pathway in synoviocytes from rheumatoid arthritis patients and in adjuvant-induced arthritis rats. Biochem Pharmacol. 2008;76:482–494. doi: 10.1016/j.bcp.2008.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer TM, Trevethick MA. Suppression of inflammatory and immune responses by the A(2A) adenosine receptor: an introduction. Br J Pharmacol. 2008;153:S27–S34. doi: 10.1038/sj.bjp.0707524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peat G, Croft P, Hay E. Clinical assessment of the osteoarthritis patient. Best Pract Res Clin Rheumatol. 2001;15:527–544. doi: 10.1053/berh.2001.0171. [DOI] [PubMed] [Google Scholar]
- Pomerantz JL, Baltimore D. Two pathways to NF-kappaB. Mol Cell. 2002;10:693–695. doi: 10.1016/s1097-2765(02)00697-4. [DOI] [PubMed] [Google Scholar]
- Schulte G, Fredholm BB. Signalling from adenosine receptors to mitogen-activated protein kinases. Cell Signal. 2003;15:813–827. doi: 10.1016/s0898-6568(03)00058-5. [DOI] [PubMed] [Google Scholar]
- Sutton S, Clutterbuck A, Harris P, Gent T, Freeman S, Foster N, et al. The contribution of the synovium, synovial derived inflammatory cytokines and neuropeptides to the pathogenesis of osteoarthritis. Vet J. 2009;179:10–24. doi: 10.1016/j.tvjl.2007.08.013. [DOI] [PubMed] [Google Scholar]
- Szabò C, Scott GS, Virag L, Egnaczyk G, Salzman AL, Shanley TP, et al. Suppression of macrophage inflammatory protein (MIP)-1α production and collagen-induced arthritis by adenosine receptor agonists. Br J Pharmacol. 1998;125:379–387. doi: 10.1038/sj.bjp.0702040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varani K, De Mattei M, Vincenzi F, Gessi S, Merighi S, Pellati A, et al. Characterization of adenosine receptors in bovine chondrocytes and fibroblast-like synoviocytes exposed to low frequency low energy pulsed electromagnetic fields. Osteoarthritis Cartilage. 2008;16:292–304. doi: 10.1016/j.joca.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Varani K, Gessi S, Dionisotti S, Ongini E, Borea PA. [3H]-SCH 58261 labelling of functional A2A adenosine receptors in human neutrophil membranes. Br J Pharmacol. 1998;123:1723–1731. doi: 10.1038/sj.bjp.0701758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varani K, Gessi S, Merighi S, Vincenzi F, Cattabriga E, Benini A, et al. Pharmacological characterization of novel adenosine ligands in recombinant and native human A2B receptors. Biochem Pharmacol. 2005;70:1601–1612. doi: 10.1016/j.bcp.2005.08.018. [DOI] [PubMed] [Google Scholar]
- Varani K, Merighi S, Gessi S, Klotz KN, Leung E, Baraldi PG, et al. [3H]-MRE 3008-F20: a novel antagonist radioligand for the pharmacological and biochemical characterization of human A3 adenosine receptors. Mol Pharmacol. 2000;57:968–975. [PubMed] [Google Scholar]
- Wen D, Nong Y, Morgan JG, Gangurde P, Bielecki A, Dasilva J, et al. A selective small molecule IkappaB kinase beta inhibitor blocks nuclear factor kappaB-mediated inflammatory responses in human fibroblast-like synoviocytes, chondrocytes and mast cells. J Pharmacol Exp Ther. 2006;317:989–1001. doi: 10.1124/jpet.105.097584. [DOI] [PubMed] [Google Scholar]
- Westra J, Limburg PC. p38 mitogen-activated protein kinase (MAPK) in rheumatoid arthritis. Mini Rev Med Chem. 2006;6:867–874. doi: 10.2174/138955706777934982. [DOI] [PubMed] [Google Scholar]
- Zhang W, Moskowitz RW, Nuki G, Abramson S, Altman RD, Arden N, et al. OARSI recommendations for the management of hip and knee osteoarthritis, Part II: OARSI evidence-based, expert consensus guidelines. Osteoarthritis Cartilage. 2008;16:137–162. doi: 10.1016/j.joca.2007.12.013. [DOI] [PubMed] [Google Scholar]