

Abstract
Transactive response (TAR) DNA-binding protein 43 (TDP-43) is a major protein component within ubiquitin-positive inclusions of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Although TDP-43 is a nuclear DNA/RNA-binding protein, in pathological conditions, TDP-43 has been reported to redistribute to the cytoplasm where it is cleaved and forms insoluble, ubiquitinated, and phosphorylated inclusions. Here we present a cellular model in which full-length human TDP-43 or a splicing isoform (TDP-S6) that lacks the C terminus is overexpressed in a human cell line and mouse primary neurons. Whereas recombinant and endogenous TDP-43 was primarily localized in the nucleus, the shorter TDP-S6 formed highly insoluble cytoplasmic and nuclear inclusions reminiscent of disease-specific pathology. Western blot analysis of detergent-insoluble extracts showed an increase in high molecular weight immunoreactive species for TDP-S6 compared with TDP-43, consistent with ubiquitination or ubiquitin-like modifications. We used a multiplex stable isotope labeling with amino acids in cell culture approach to compare the detergent-insoluble proteome from mock-, TDP-43-, and TDP-S6-transfected cells. TDP-S6 overexpression caused a concomitant increase in both ubiquitin (Ub) and the small Ub-like modifier-2/3 (SUMO-2/3) within the insoluble proteome. Similarly, full-length TDP-43 overexpression also resulted in the elevation of SUMO-2/3. Immunofluorescence showed strong co-localization of endogenous Ub with both cytoplasmic and nuclear TDP-S6 inclusions, whereas SUMO-2/3 was co-localized mainly with the nuclear inclusions. Quantitative mass spectrometry further revealed that mixed Lys-48 and Lys-63 polyUb linkages were associated with the TDP insoluble fractions. Together our data indicate that expression of a TDP-43 splice variant lacking a C terminus recapitulates many of the cellular and biochemical features associated with disease pathology and that the interplay of ubiquitination and SUMOylation may have an important role in TDP-43 regulation.
Frontotemporal lobar degeneration (FTLD)1 is a progressive neurodegenerative disease characterized by prominent behavioral abnormalities, personality changes, and language dysfunction, and it co-occurs with the development of parkinsonism and motor neuron disease in some patients (1, 2). Like other neurodegenerative diseases, FTLD is characterized by the abnormal accumulation of ubiquitinated intracellular protein aggregates (3, 4). In addition to the aggregation of tau in some forms of FTLD, more than half of cases are marked by ubiquitin-positive inclusions and are subclassified as FTLD-U. Transactive response (TAR) DNA-binding protein 43 (TDP-43) has been identified as a major protein component of inclusions in FTLD-U and amyotrophic lateral sclerosis (ALS) (5). TDP-43 aggregation is also observed in hereditary inclusion body myopathy and Paget disease of the bone with frontotemporal dementia (6) as well as in some cases of Alzheimer and Parkinson diseases (7, 8). This indicates that a common underlying mechanism may broadly define a spectrum of neurodegenerative disorders termed “TDP-43 proteinopathies” (9, 10).
TDP-43 is a 414-amino acid protein that contains two RNA recognition motifs (RRM1 and RRM2) and a glycine-rich C-terminal domain. It is highly conserved in human, mouse, fly, and worm and is expressed in all tissues, including the brain (11–13). First characterized to bind and repress the promoter activity of TAR DNA in the human immunodeficiency virus 1 long terminal repeat region (14), TDP-43 was later found to regulate splicing of the transcripts of apolipoprotein A-II (15) and cystic fibrosis transmembrane conductance regulator (16). Therefore, TDP-43 can act both as a transcriptional repressor and as a splicing regulator. Although physiological TDP-43 resides mainly in the nucleus, pathology-relevant TDP-43 redistributes from the nucleus to the cytoplasm where it is cleaved and forms phosphorylated and ubiquitinated inclusions (5, 17–19). The degree to which TDP-43 is post-translationally modified and whether other modifications initiate nuclear to cytoplasmic translocation and aggregation remain unknown.
The primary transcripts of TDP-43 in mouse and human undergo multiple alternative splicing events in which 10 splice variants (S1–S10) have been documented (11). With the exception of full-length TDP-43, all alternatively spliced isoforms are expressed as truncated proteins that lack the C-terminal glycine-rich domain. Isoforms that lack this domain are no longer capable of enhancing the skipping of exon 9 of the cystic fibrosis transmembrane conductance regulator gene via interactions with heterogeneous nuclear ribonucleoprotein A/B (11, 20). To date, the vast majority of sporadic and familial TARDBP gene variants found in ALS cases (21–25) are reported to have missense mutations resulting in single amino acid substitutions located in the glycine-rich domain. This establishes an intriguing relationship between ALS disease etiology and the function of the TDP-43 C-terminal region. Whether TDP-43 splice variants that lack this C terminus contribute to translocation and aggregation requires further investigation.
In this study, we overexpressed human TDP-43 and TDP-S6, a splice variant lacking the glycine-rich C terminus, in human embryonic kidney 293 (HEK-293) cells and mouse hippocampal neurons. When compared with TDP-43, TDP-S6 showed both nuclear and cytoplasmic location and extensive aggregation. Biochemical analysis revealed that TDP-S6 was almost completely Sarkosyl-insoluble. Moreover, both insoluble TDP-43 and TDP-S6 displayed varying levels of post-translational modifications that included ubiquitination and/or ubiquitin-like modifications, phosphorylation, and proteolytic cleavage. To assess protein differences among the detergent-insoluble extracts from mock-, TDP-43-, and TDP-S6-transfected cells, a multiplex stable isotope labeling with amino acids in culture (SILAC) strategy was used in combination with LC-MS/MS (26). This quantitative proteomics approach metabolically introduces a mass difference into proteins by incorporation of heavy isotopic forms of arginine and lysine. We identified and characterized a novel association between the small ubiquitin (Ub)-like modifier-2/3 (SUMO-2/3) and TDP-43. Additional targeted proteomics analysis found multiple polyubiquitin structures in the TDP insoluble fractions. Our data indicate that overexpression of a truncated TDP-43 splice variant recapitulates many of the features associated with disease pathology and that SUMOylation and ubiquitination may play important roles in regulating TDP-43 functions.
EXPERIMENTAL PROCEDURES
DNA Constructs
The full-length TDP-43 coding sequence (nucleotides 1–1245, encoding amino acids 1–414) was amplified by PCR from a TDP-43 expression plasmid (Open Biosystems, Huntsville AL; Clone ID 3038905); the forward and reverse primers were 5′-GGTAGGATCCATGTCTGAATATATTCGGGTAA-3′ and 5′-TTATATAGGGCCCCTACATTCCCCAGCCAGA-3′, respectively. The PCR products were digested and cloned into pcDNA3.1-HA expression vector downstream of the two HA motifs (peptide YPYDVDYA). The TDP-S6 splice variant (nucleotides 1–888 (11), encoding amino acids 1–295) was also cloned into pcDNA3.1-HA. Both N-terminal HA expression vectors (TDP-43 and TDP-S6) were confirmed by DNA sequencing (Agencourt Bioscience Corp., Beverly MA). The SUMO-2 construct was a kind gift from Dr. Keith Wilkinson (Emory University) as described (27). Mature SUMO-2 (93 amino acids with two glycine residues at the C terminus) was subcloned into pEGFP-C3 vector (Clontech).
Primary Neuronal Cultures
Primary hippocampal neuron cultures were prepared from wild type C57BL/6 mice (Charles River Laboratories, Wilmington, MA) at embryonic day E18. The embryos were dissected, and the hippocampus was isolated in dissection buffer (Hanks' balanced salt solution, 10 mm HEPES, 1% penicillin/streptomycin). After mild trypsinization with 0.25% trypsin and 0.01% deoxyribonuclease in dissection buffer for 15 min at 37 °C, the tissue fragments were rinsed twice with dissection buffer and twice with plating medium (buffered minimum Eagle's medium essential medium (Invitrogen), 0.6% glucose (Invitrogen), 2 mm l-glutamine (Mediatech Inc., Manassas, VA), 10% heat-inactivated horse serum (Invitrogen), 1% penicillin/streptomycin). The tissue was then subjected to mechanical dissociation by repeated aspiration through a fire-polished Pasteur pipette in dissociation medium. Viable cells were determined by trypan blue exclusion. Neurons were plated at a density of 50,000 cells/cm2 on poly-l-lysine (Sigma)-coated coverslips (Propper Manufacturing Co., Long Island City, NY) and maintained in Neurobasal medium (Invitrogen) containing B-27 supplement (Invitrogen), 2 mm l-glutamine, and 1% penicillin/streptomycin at 37 °C under 5% CO2.
Cell Culture and Immunocytochemistry
HEK-293 cells were cultured in Dulbecco's modified Eagle's medium (Cambrex, Walkersville, MD) supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin/streptomycin (Cambrex). After plating on Matrigel-coated coverslips, cells were transfected using FuGENE 6 reagent (Roche Applied Science) followed by immunocytochemistry 2 days later as described previously (28). Primary antibodies included rabbit TDP-43 antibody raised against recombinant TDP-43 (ProteinTech Group, Chicago, IL), HA monoclonal antibody (clone 16B12; Covance, Emeryville CA), rabbit SUMO-2/3 antibody (Zymed Laboratories Inc.), rabbit antibody against the neuron-specific marker enolase (Chemicon, Temecula, CA), rabbit Ub antibody (Dako, Carpinteria, CA), and Lys-48 or Lys-63 linkage-specific antibodies, a gift from Dr. V. M. Dixit (29). Briefly, the cells were fixed with 2% paraformaldehyde in PBS, permeabilized and blocked with PBS containing 5% BSA and 0.05% Triton X-100, and incubated in primary antibodies overnight. The cells were then rinsed and incubated with either fluorophore-conjugated or biotinylated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) for 60 min. Cells treated with biotinylated secondary antibodies were subsequently incubated with avidin-biotin-peroxidase complex and tyramide-conjugated fluorophores according to the manufacturer's protocol (PerkinElmer Life Sciences). Following additional rinses, cells were incubated with a Hoechst reagent for nuclear localization, mounted with Vectashield (Vector Laboratories, Burlingame, CA), and imaged on a Zeiss LSM 510 confocal microscope (Zeiss, Thornwood, NY). Images were captured with a 1-μm optical thickness for subsequent analysis.
Subcellular Fractionation
To examine the solubility of TDP-43 and TDP-S6, sequential extractions were performed as described previously (5) with slight modification. Cells (∼1 × 107) were washed twice, collected with ice-cold PBS buffer, and then lysed in Sarkosyl buffer (10 mm Tris, pH 7.5, 5 mm EDTA, 1 mm DTT, 0.5 m NaCl, 10% sucrose, 1% N-lauroylsarcosine, 10 mm β-glycerophosphate, 10 mm sodium orthovanadate, 10 mm tetrasodium pyrophosphate, 50 mm sodium fluoride). Resulting lysates were spun at 180,000 × g for 30 min at 22 °C to generate the detergent-soluble samples. Insoluble pellets were washed three times with additional Sarkosyl buffer, then extracted with urea buffer (30 mm Tris, pH 8.5, 7 m urea, 2 m thiourea, 2% sodium dodecyl sulfate), and centrifuged at 25,000 × g for 30 min at 22 °C. Protease inhibitors (Roche Applied Science) were added to all buffers prior to use. Protein concentration was determined for Sarkosyl fractions by bicinchoninic acid (BCA) protein assay (Pierce) according to the manufacturer's instructions. For urea fractions, protein concentration was determined likewise and/or by estimating Coomassie Blue G-250 staining intensity of a small fraction of the urea extracts following electrophoresis in polyacrylamide gels using titrated BSA as a standard (30).
Biochemical Assays
To assess TDP-43 phosphorylation, total cell lysate was incubated with 0, 0.15, 0.3, or 0.6 mg/ml bovine alkaline phosphatase Type VII-S (3533 units/mg of protein; Sigma-Aldrich) for 2.5 h at 37 °C followed by Western blot analysis. For deSUMOylation, sentrin-specific endopeptidase 2 (SENP2; Boston Biochem, Cambridge, MA) was incubated with 10-μg detergent-soluble or 1-μg insoluble fractions from TDP-43-transfected cells in 20-μl reactions (buffer: 50 mm Tris, 100 mm NaCl, 5 mm DTT, pH 8.0; titrated SENP2 concentration: 0, 1, or 5 μm). All samples were incubated for 3 h at 37 °C and then analyzed by Western blotting.
Western Blotting
Immunoblotting was performed according to standard procedures. Briefly, samples in Laemmli sample buffer were separated by SDS-PAGE and transferred overnight to PVDF Immobilon-P membranes (Millipore, Billerica, MA). To ensure both equal loading and complete transfer of proteins from the gel, membranes were reversibly stained with Ponceau S (Diasys Europe Ltd., Workingham, UK). Blots were blocked for 1 h at room temperature using 1× Blocking Buffer (USB Corp., Cleveland OH), probed with primary antibody in TBS with 0.1% Tween 20 overnight at 4 °C, and incubated for 1 h at room temperature with secondary antibodies (1:20,000) conjugated to fluorophores (Molecular Probes, Eugene OR; Rockland, Gilbertsville PA). Blots were dried, scanned, and quantified with an Odyssey Infrared Imaging System (Li-Cor Biosciences, Lincoln, NE).
SILAC and LC-MS/MS
Cells were cultured in Dulbecco's modified Eagle's medium (deficient in l-lysine and l-arginine) supplemented with 5% dialyzed fetal calf serum (Invitrogen) as described (31). For stable isotopic labeling, arginine and lysine were added in light (Arg0/Lys0; Sigma), medium (Arg6/Lys4), or heavy forms (Arg10/Lys8; Cambridge Isotope Laboratories, Andover, MA) to a final concentration of 0.26 mm. Cells were cultured for seven passages to ensure full labeling and then transfected with pcDNA3.1-HA (mock), HA-TDP-43, and HA-TDP-S6 plasmids, respectively. After 2 days, the cells were harvested, equally mixed, and lysed to prepare the Sarkosyl-insoluble, urea-soluble fraction as described above. The fraction was reduced with 10 mm DTT, alkylated with 50 mm iodoacetamide for 30 min in the dark, and then resolved on a 10% polyacrylamide-SDS gel. After staining with Coomassie Blue, one gel lane was cut into five gel bands, and bands were subjected to in-gel digestion (12.5 μg/ml trypsin). Extracted peptides were loaded onto a C18 column (75-μm inner diameter, 10 cm long, ∼300 nl/min flow rate, 5-μm resin from Michrom Bioresources, Auburn, CA) and eluted during a 10–30% gradient (Buffer A: 0.4% acetic acid, 0.005% heptafluorobutyric acid, 5% ACN; Buffer B: 0.4% acetic acid, 0.005% heptafluorobutyric acid, 95% ACN). The eluted peptides were detected by Orbitrap (350–1500 m/z; 1,000,000 automatic gain control target; 1000-ms maximum ion time; resolution, 60,000 full-width at half-maximum) followed by five data-dependent MS/MS scans in the linear ion trap quadrupole (2 m/z isolation width, 35% collision energy, 5,000 automatic gain control target, 200-ms maximum ion time) on a hybrid mass spectrometer (Thermo Finnigan, San Jose, CA).
Acquired MS/MS spectra were extracted and searched against the human reference database from the National Center for Biotechnology Information (December 13, 2007) using the SEQUEST Sorcerer algorithm (version 2.0, SAGE-N) (32). Searching parameters included mass tolerance of precursor ions (±50 ppm) and product ion (±0.5 m/z); partially tryptic restriction; fixed mass shift for modification of carboxamidomethylated Cys (+57.0215 Da); dynamic mass shifts for oxidized Met (+15.9949), Lys (+4.02511 for 2H2 or +8.01420 for 13C615N2), and Arg (+6.02013 for 13C6 or +10.00827 for 13C615N4); five maximal modification sites; and three maximal missed cleavages. Only b and y ions were considered during the database match. To compare medium and heavy labeled samples, searches were performed with static modifications +4.02511 on Lys and +6.02013 on Arg and dynamic modifications of +3.98909 on Lys and +3.98814 on Arg to account for the mass difference between medium and heavy labeled peptides. To evaluate the false discovery rate (FDR), all original protein sequences were reversed to generate a decoy database that was concatenated to the original database (a total of 53,830 protein entries) (33, 34). The FDR was estimated by the number of decoy matches (nd) and total number of assigned matches (nt). FDR = 2 × nd/nt, assuming that mismatches in the original database were the same as in the decoy database. To remove false positive matches, assigned peptides were grouped by a combination of trypticity (fully, partial, and non-tryptic) and precursor ion charge state (1+, 2+, 3+, and 4+). Each group was first filtered by mass accuracy (10 ppm for high resolution MS) and by dynamically increasing XCorr (minimal 1.8) and ΔCn (minimal 0.05) values to reduce protein FDR to less than 0.2%. The identified proteins/peptides are listed in the supplemental data (supplemental Tables S1–S6 and S8) with accession number, matched peptide number, sequencing coverage, mass shift, and matching scores. Every peptide in the tables was linked to assigned MS/MS spectra, precursor ion mass, and charge state. If peptides were matched to multiple members of a protein family, the matched members were clustered into a single group (supplemental Tables S7 and S9).
SILAC Quantification and Bioinformatics Analysis
Quantitative pairwise comparisons of control, TDP-S6-, and TDP-43-transfected cells were carried out according to reported methods (35, 36) with slight modification. (i) For, ion extraction from MS scans, the ion currents for identified peptides were extracted in MS survey scans of high resolution (60,000) based on the isotopic ion selected for MS/MS sequencing. A number of parameters were defined, including precursor m/z, charge state, retention time, ion peak width, height, area, and noise level. The noise level was derived by averaging signal intensity of all ions in the MS scan after removing outliers that were at least two S.D. away from the mean. The intensity of ions was presented by the peak height and normalized according to the noise intensity under the assumption that the noise level of MS scans reflects, at least partially, variable ionization efficiency. The peaks used in the analysis had a minimal intensity of 2 signal-to-noise ratio. (ii) Ion matching among light, medium, and heavy isotopes was allowed with a tolerance of 10 ppm. If a sequenced peptide could not be matched, we estimated that the maximum ion current for undetected signal was equal to the noise level and used it to derive the peptide ratio. (iii) For data integration, the ratio of every peptide was transformed into logarithmic (log2) values that were averaged over all peptides of a particular protein to determine the protein ratio. If a protein was quantified by both matched peptide ratios and unmatched peptide ratios, only the matched data were averaged. The results are shown in supplemental Table S7 with the number of peptides quantified for every protein. (iv) For data normalization, according to the null hypothesis, the histogram of all protein log2 ratios was fitted to a Gaussian distribution to evaluate systematic bias (according to the mean) and experimental variation (based on S.D.). The data were then normalized by subtracting the mean in every protein ratio. The S.D. values in all SILAC comparisons were less than 0.4. (v) For data filtering, we selected the cutoff of log2 ratios that were outside a 95% confidence interval (∼2 S.D.) from the mean of the Gaussian distribution. Finally, the quantified proteins were manually examined with respect to MS/MS assignment, ion peak matching, and ion intensity.
Semiquantitative Proteomics by Spectral Counts
To compare differences between the detergent-soluble and -insoluble proteome, we compared the identified proteins in both samples based on spectral counts. The spectral counts were first normalized to ensure that average spectral counts per protein was the same in the two data sets (37). A G test was used to judge statistical significance of protein abundance difference (38). Briefly, the G value of each protein was calculated as shown in Equation 1,
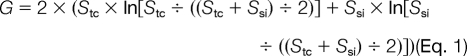 |
where Stc and Ssi are the detected spectral counts of a given protein in the total cell lysate and in the Sarkosyl-insoluble fraction, respectively, and “ln” is the natural logarithm. Although theoretical distribution of the G values is complex, these values approximately fit to the χ2 distribution (1 degree of freedom), allowing the calculation of related p values.
Quantitative Analysis of Polyubiquitin Chains and Other Targeted Proteins by LC/Selective Reaction Monitoring (SRM)
The analysis of polyUb linkages, ubiquitin E1 enzyme, and proteasome subunit Rpn2 was performed with metabolically heavy labeled cells as internal standards (+8.01420 for Lys and +10.00827 for Arg) using a previously reported protocol (36, 39). The labeled cells were spiked into transfected cells followed by protein extraction. Sarkosyl-insoluble fractions were resolved on a one-dimensional SDS gel. The gel regions above 80 kDa, which contained the vast majority of polyUb species, were used for in-gel trypsin digestion that produced a pair of light and heavy GG-linked ubiquitin peptides corresponding to every polyUb linkage. Digested peptides were analyzed by the same LC system as above in which peptide ion pairs of interest were selected for fragmentation and quantified by related product ion pairs, a process termed SRM or multiple reaction monitoring. The detailed LC/SRM parameters are shown in supplemental Table S10.
RESULTS
Expression, Localization, and Biochemical Properties of Recombinant TDP-43 and TDP-S6
We cloned and overexpressed HA-tagged (at the N terminus) human TDP-43 and TDP-S6 in HEK-293 cells (Fig. 1). Human TDP-S6 is generated via an additional splicing event within exon 6 and encodes a 295-amino acid protein in which the first 277 amino acids are identical to those of TDP-43 (Fig. 1A). Although mouse TDP-S6 has been shown to have altered nuclear structure in mammalian cells (40), the human TDP-S6 transcript, identified in HEK-293 cells (11), has not yet been characterized. Western blotting of total cell lysate prepared from the transfected cells indicated that both recombinant proteins were expressed and recognized by the HA antibody. TDP-43 also displayed two higher molecular mass species at ∼50 kDa (Fig. 1, B and C, asterisk). After treatment with an increasing amount of alkaline phosphatase, the upper band disappeared, and the middle band became significantly weaker, whereas the intensity of unmodified TDP-43 remained stable. This supports that TDP-43 is preferentially phosphorylated on the C terminus on at least two different residues (Fig. 1C), consistent with current identification of two phosphorylation sites near the C terminus of TDP-43 in diseased tissues (41, 42).
Fig. 1.

Expression of TDP-43 and shorter alternative splicing isoform, TDP-S6, in HEK-293 cells. A, TDP-43 consists of six exons that encode a protein of 414 amino acids. RRM1 spans exons 3 and 4, RRM2 spans exons 5 and 6, and the glycine-rich C-terminal domain lies within exon 6. TDP-S6 is generated by an additional splicing event resulting in a reading frameshift after amino acid 277. Thus, TDP-S6 contains 18 unique amino acids (278–295) on its C terminus (grey) corresponding to residual intron RNA included during an alternative splicing event. B, Western blotting (WB) analysis of transfected control (mock plasmid), HA-tagged TDP-43, or TDP-S6. C, loss of TDP-43 phosphorylation during phosphatase treatment. The HA-TDP-43-transfected total cell lysate was incubated for 2.5 h with the addition of an increasing concentration of alkaline phosphatase followed by Western analysis. Phosphorylated TDP-43 isoforms are indicated by asterisks.
Subcellular localization of full-length TDP-43 and TDP-S6 was assessed by immunofluorescence confocal microscopy. Overexpressed TDP-43 in HEK-293 cells had primarily nuclear localization (Fig. 2A, upper panel), consistent with that of endogenous TDP-43 in untransfected cells (Fig. 2A, upper panel, left bottom corner). In contrast, TDP-S6 formed aggregates that were mainly localized to the cytoplasm with some inclusions in the nucleus (Fig. 2A, middle and bottom panels). Observed puncta were of variable size, ranging from fine granular deposits to large, aggresome-like inclusions. To assess whether the TDP-S6 phenotype observed in HEK-293 cells could be recapitulated in neurons, we transfected primary mouse hippocampal neurons with TDP-43 or TDP-S6 constructs (Fig. 2B). Consistent with results in HEK-293 cells, TDP-43 in neurons was nuclear, whereas the majority of TDP-S6 formed granular deposits in the cytoplasm of the soma and neurites (Fig. 2, B and C). Enolase was stained as a neuron-specific marker.
Fig. 2.

TDP-S6 translocation and aggregation in HEK-293 cells and primary hippocampal neurons. A, HEK-293 cells were transfected with HA-TDP-43 or HA-TDP-S6 and stained with TDP-43 antibody for both recombinant and endogenous proteins (green), HA antibody for recombinant TDP-43 exclusively (red), and Hoechst stain for the nucleus (blue). The white bars represent a distance of 10 μm. B, neurons were transfected and stained with the neuron-specific marker enolase (green), HA antibody (red), and Hoechst stain (blue). Two different sections of the same neuron, corresponding to separate focal planes, are shown in the middle and bottom panels of TDP-S6 (Soma), indicating cytoplasm and nucleus, respectively. C, TDP-S6 was also aggregated in neuronal processes, shown under higher magnification.
To evaluate the solubility of overexpressed TDP-43 or TDP-S6, HEK-293 cells were transfected with either construct and then mixed equally to eliminate experimental variability in subsequent processing. The mixed cells were sequentially extracted with Sarkosyl-containing buffer and urea and then analyzed by immunoblotting. The differences in molecular mass between HA-TDP-43 (47 kDa), native TDP-43 (43 kDa), and HA-TDP-S6 (37 kDa) allowed analysis of all three proteins in a single lane (Fig. 3A). In addition, phosphorylated TDP-43 isoforms were also resolved. Although almost all of HA-TDP-S6 was insoluble, 60 ± 7% of unmodified HA-TDP-43 and virtually none of the native unmodified TDP-43 were present in the insoluble fraction (Fig. 3, A and B). The degree of TDP-S6 and TDP-43 enrichment in the insoluble fraction was also repeatable using a second preparation of cells (data not shown). Based on these findings, biochemical insolubility of TDP-S6 is consistent with the inclusions observed by immunofluorescence (Fig. 2). The overexpression of HA-TDP-43 may account for the differences in solubility between the recombinant and native proteins as has been recently reported (43). Interestingly, the phosphorylated species of recombinant HA-TDP-43 (91 ± 4%) and endogenous TDP-43 (44 ± 1%) were more enriched in the insoluble fraction than the unmodified forms, respectively (60 ± 7 and <5%), supporting previous findings linking TDP-43 phosphorylation with aggregation and insolubility (5). To ensure that overexpression of TDP-43 or TDP-S6 did not dramatically bias the global distribution of proteins to the insoluble fraction, the percentage of total protein in each fraction (detergent-soluble and -insoluble) was calculated from mock-, HA-TDP-43-, and HA-TDP-S6-transfected cells. Approximately 96% of all cellular proteins were detergent-soluble irrespective of the recombinant protein expressed, indicating that overexpression of our target proteins did not cause a gross increase in the total amount of protein within the insoluble fraction.
Fig. 3.

Characterization of Sarkosyl-insoluble fraction by Western blotting and mass spectrometry. A, Western blot (WB) analysis of sarkosyl-soluble and -insoluble HA-TDP-43, native TDP-43, and HA-TDP-S6 using an anti-TDP-43 antibody. The asterisk (*) indicates phosphorylated forms. B, densitometry analysis of multiple TDP-43 forms. C, large scale profiling of Sarkosyl-soluble and -insoluble fractions by LC-MS/MS. Spectral counts of identified proteins were used as a semiquantitative index to derive probability values. snRNP, small nuclear ribonucleoprotein.
To further characterize proteins enriched in the Sarkosyl-insoluble fraction, we identified proteins in the total cell lysate and in the Sarkosyl-insoluble sample using a label-free LC-MS/MS approach. The relative abundance of proteins in the two samples was compared by spectral counts, and the difference was statistically evaluated by G test (38). Among 1265 proteins profiled (supplemental Table S9), the level of 267 proteins was significantly altered with corresponding p values below 0.01. Listed in Fig. 3C are proteins with large changes, including components associated with the proteasome, caveolae, aggresomes, and a number of nuclear complexes. Specifically, flotillin-1 and -2 are markers for caveolae/lipid rafts that are highly enriched in cholesterol and are thus more resistant to detergent extraction (44). Vimentin is a protein marker of aggresomes that is often associated with proteasome proteins (45). Finally, particular DNA/RNA-interacting proteins were found to be intrinsically detergent-insoluble. Therefore, the protocol of Sarkosyl-based differential extraction is capable of enriching both aggregated proteins and other detergent-insoluble complexes.
Further analysis of detergent-soluble and insoluble fractions from transfected cells revealed that high molecular mass TDP-43-immunoreactive species were more abundant in fractions corresponding to TDP-S6, rather than TDP-43, overexpression. This observation is consistent with ubiquitination, ubiquitin-like modifications, and/or protein polymerization typically associated with protein insolubility (Fig. 4). To this end, immunoblotting of Sarkosyl-insoluble fractions with a ubiquitin-specific antibody demonstrated an increase in global protein ubiquitination with TDP-43 (≥250 kDa) and more dramatically with TDP-S6 overexpression (Fig. 4). TDP-S6 also displayed two short TDP-43-immunoreactive species (∼30 and ∼20 kDa) that were not detected by an HA antibody. In addition, a third fragment (∼37 kDa) was identified in TDP-S6 urea fractions by both TDP-43 and HA antibodies. Although TDP-S6 lacks the extreme C terminus, the proteolytic fragmentation observed here is in agreement with reported C-terminal TDP-43 fragmentation in FTLD-U cases (19). Full-length TDP-43 was also cleaved but at a much lower level (observed on overexposed images; data not shown). These results show that an increase in phosphorylation, ubiquitination, and proteolysis is related to the insolubility of TDP proteins. Taken together, the microscopic and biochemical data for TDP-S6 in cell culture are highly consistent with that of pathologic TDP-43 observed in disease tissue (5, 9, 18, 19).
Fig. 4.

TDP-43 and TDP-S6 display post-translational modifications reminiscent of biochemical signature observed in neurodegeneration. The cells were transfected with mock, HA-TDP-43, or HA-TDP-S6 plasmids and extracted by a Sarkosyl buffer (A), and the pellet was solubilized with urea buffer (B). The sequentially solubilized proteins were analyzed by Western blotting (WB). In detergent-insoluble fractions, modified TDP-43 phosphorylated species (marked by asterisks), proteolytic fragments (marked by arrowheads), and high molecular mass TDP immunoreactivity are observed. An increase in global protein ubiquitination with TDP-43 and more dramatically with TDP-S6 overexpression is observed using a Ub-specific antibody (B, right panel).
Quantitative Analysis of Insoluble TDP-43 and TDP-S6 Proteome Using Multiplex SILAC
As both recombinant TDP-43 and TDP-S6 are highly enriched in detergent-insoluble extracts, we used a multiplex SILAC approach to evaluate proteins that co-enrich in the detergent-insoluble proteome of cells overexpressing TDP-43 or TDP-S6 (46) (Fig. 5A). Human HEK-293 cells were fully labeled with light, medium, or heavy arginine and lysine amino acids. The three labeled cell populations were transfected with HA-TDP-43, HA-TDP-S6, or a mock plasmid. After incubation for 2 days, the cells were harvested, equally mixed, and subjected to sequential protein extraction. Although pooling the labeled cells minimizes experimental variation during protein extraction and subsequent steps in the SILAC analysis, it is possible for biological variation to occur during cell labeling and DNA transfection. Therefore, we performed a second independent analysis as a biological replicate with the labeled isotopes “swapped” in HEK-293 cells prior to transfection (Fig. 5A). The detergent-insoluble extracts were then resolved on an SDS gel, excised into gel bands, digested with trypsin, and analyzed by LC-MS/MS on a high resolution Orbitrap mass spectrometer (Fig. 5B). After database searching and stringent filtering mainly by mass accuracy (≤10 ppm) and SEQUEST scores (XCorr and ΔCn), a total of 2670 proteins that were clustered into 1117 groups were identified and quantified (supplemental Tables S1–S7). The corresponding false discovery rate was calculated to be less than 0.2% according to the target-decoy strategy (33, 34).
Fig. 5.

Quantitative proteomics of Sarkosyl-insoluble fraction by multiplex SILAC. A, diagram for biological replicates of SILAC analysis (experiments 1 and 2). The amino acid labeling order was swapped in the repeated analysis. B, silver-stained SDS gel of the isolated Sarkosyl-insoluble fractions. The gel lanes were excised as indicated. C, a representative fitting of a pairwise comparison in the first experiment, shown as a histogram fitted to a Gaussian curve. D, the experimental variations were similar in multiple comparisons, indicated by the values of S.D. L, light; M, medium; H, heavy; Ctl, control. GeLC-MS/MS is the method of one dimensional SDS gel and LC-MS/MS.
To evaluate the quality of the quantitative data, we analyzed the results based on the null hypothesis as described previously (35). The protein ratios in all pairwise comparisons were converted into log2 values, and the resulting histogram of all values was fitted to a Gaussian distribution (Fig. 5C). The majority of proteins fit the curve well, indicating that they did not change under the conditions analyzed. Although the fitted mean (−0.01) suggested little systematic bias introduced by sample handling (e.g. a slightly different amount of starting cells), the S.D. (0.31) provided a good measure of the magnitude of variations in the analysis. In all six pairwise comparisons of light, medium, and heavy labeling in the two experiments, the value of the S.D. was stable (0.28 ± 0.03; Fig. 5D), indicating that the variations in our SILAC analyses were consistent. Proteins were considered changed if their values fell outside more than 2 S.D. (0.6; ∼95% confidence interval) and showed consistency in experiments 1 and 2. A large number of proteins were removed when comparing results from experiments 1 and 2, suggesting that the biological replicate is essential to reduce false positives in the SILAC assay. The filtered proteins were further validated by manual examination of paired ion currents in raw files, resulting in the acceptance of six proteins with altered expression (Table I).
Table I. List of proteins that were altered in cells overexpressing TDP-43 or TDP-S6.
The log2 ratios summed over all molecular mass regions (gel bands 1–5) are shown as the mean of two biological replicates ±S.E. If the proteins were quantified only in one experiment, the S.E. value was not available. NC, not changed (the measured difference was not statistically significant). The accession numbers are from the National Center for Biotechnology Information Reference Sequence Database.
| Accession no. | Protein names | log2(TDP-43/control) | log2(S6/control) | log2(S6/TDP-43) |
|---|---|---|---|---|
| Proteins affected by TDP-43 and TDP-S6 expression | ||||
| NP_031401.1 | TDP-43 or TDP-S6 | 2.8 ± 0.3 | 3.7 ± 0.3 | 1.3 ± 0.4 |
| NP_002945.1 | Ubiquitin | NC | 1 ± 0.4 | 1 ± 0.4 |
| NP_008868.3 | SUMO-2/3 | 0.9 | 2.4 ± 0.0 | 1.2 ± 0.3 |
| NP_005726.1 | ARP1 | 1.2 ± 0.5 | 2.8 | NC |
| Proteins affected by TDP-S6 expression | ||||
| NP_006169.2 | N-Ethylmaleimide-sensitive factor | NC | −1 | −1.2 ± 0.5 |
| NP_056384.2 | Dynamin 3 | NC | −2.5 | −1.2 ± 0.6 |
Validation and Subcellular Co-localization of SUMO-2/3 and Ubiquitin
The list of proteins with altered levels in the Sarkosyl-insoluble fraction included overexpressed TDP proteins (TDP-43 or TDP-S6), ubiquitin, and SUMO-2/3 (Table I). In gel band 1 (>180 kDa; Fig. 5B), the MS results (Fig. 6, A and B) indicated that TDP-S6-expressing cells had more TDP in the insoluble proteome compared with TDP-43-expressing cells, consistent with the high molecular mass immunoreactivity in Western blotting (Fig. 4B). As expected, native TDP-43 from mock-transfected cells was not observed at this high molecular mass range. SILAC also confirmed our Western blot results for ubiquitin (Fig. 4B), which increased concomitantly with TDP levels within this molecular mass range. However, although TDP-S6 showed significant ubiquitin enrichment compared with mock-transfected cells when summed over all molecular mass regions (gel bands 1–5), no enrichment was observed for ubiquitin in the TDP-43 insoluble fraction (Table I). In contrast, the significant increase in total ubiquitin enrichment for TDP-S6 may be a reflection of its aggregation observed in cell culture. Interestingly, SUMO-2/3 was identified as a novel component enriched in both the TDP-43 and TDP-S6 insoluble proteome (summed over all molecular mass regions), suggesting the presence of both polyubiquitination and SUMOylation in the insoluble (Fig. 6C) proteome. The elevation of SUMO-2/3 modifications was correlated to the degree of TDP protein insolubility (Fig. 3), suggesting that insoluble TDP proteins are direct targets of SUMOylation.
Fig. 6.

SILAC analysis of TDP insoluble proteome and characterization of SUMOylation. Shown are MS spectra of detected peptide ions for MOV10 (a negative control), TDP proteins, Ub, and SUMO-2/3 in one dimensional SDS gel and LC-MS/MS from the top gel band (>180 kDa) in experiments 1 (A) and 2 (B). C, Western blotting of SUMO-2/3 and HA in Sarkosyl-insoluble fractions from cells transfected with mock, TDP-43, and TDP-S6 plasmids. D, both Sarkosyl-soluble (10 μg) and -insoluble (1 μg) fractions were incubated with SENP2 followed by immunoblotting with antibodies against SUMO-2/3, TDP-43, and HA. The three bands indicated by arrows correspond to TDP-43 modified by one, two, or three SUMO-2/3 molecules, respectively. WB, Western blot.
To assess whether TDP-43 is covalently modified by SUMO-2/3, we performed a deSUMOylation assay using SENP2 (47). An increasing amount of SENP2 was added to either detergent-soluble or -insoluble samples from cells overexpressing TDP-43. A Western blot with anti-SUMO-2/3 showed a decrease in high molecular mass SUMO-2/3 immunoreactivity with increasing enzyme concentration (Fig. 6D). In addition, three specific bands (∼55, ∼65, and ∼75 kDa) on the blots were recognized by SUMO-2/3, TDP-43, and HA antibodies. Because unmodified HA-TDP-43 has a molecular mass of ∼45 kDa and SUMO-2/3 is ∼10 kDa in size, these bands likely represent mono-, di-, and triSUMOylated forms of TDP-43. The tri- and diSUMOylated HA-TDP-43 showed decreasing signals when incubated with titrated SENP2. In contrast, the monoSUMOylated band was increased at 1 μm SENP2, possibly due to processing of tri- and diSUMOylated bands. This increase was appropriately reduced with increased SENP2 concentration (5 μm). Thus, HA-TDP-43 is directly modified by SUMO-2/3 in detergent-insoluble fractions.
To assess whether SUMO-2/3 co-localizes with TDP-43 or TDP-S6 in HEK-293 cells, we performed immunofluorescence confocal microscopy (Fig. 7A). Endogenous SUMO-2/3 in untransfected cells was diffusely expressed mainly in the nucleus with some accumulation in nuclear bodies as described previously (48). In cells expressing full-length TDP-43, the majority of endogenous SUMO-2/3 was diffusely localized throughout the nucleus, although co-localization was also observed in certain SUMO-2/3 bodies when TDP-43 expression levels were higher (Fig. 7A, arrowheads). In TDP-S6-expressing cells, SUMO-2/3 was sequestered within TDP-S6 nuclear inclusions. Notably, SUMO-2/3 did not co-localize with cytoplasmic aggregates of TDP-S6. Similar results were obtained in cells co-expressing green fluorescent protein-tagged SUMO-2 and TDP constructs (data not shown). These data strongly support that SUMOylation is associated with TDP protein insolubility and localization in the nuclei but not in the cytoplasm.
Fig. 7.

Localization of SUMO-2/3, polyubiquitin (Lys-48 and Lys-63), and TDP proteins in HEK-293 cells. Cells were transfected with plasmids expressing HA-TDP-43 or HA-TDP-S6 and then co-stained with HA antibodies (red), and other antibodies (green) against SUMO-2/3 (A) and Lys-48 (B) and Lys-63 (C) polyUb linkages. The nuclei were stained by Hoechst (blue). Nuclear SUMO-2/3 and TDP-43 co-localization is indicated by white arrowheads (A).
Ub is attached to protein substrates in monomeric form or as polymers (polyUb) assembled through the N-terminal amino group and the side chains of all seven lysine residues (Lys-6, Lys-11, Lys-27, Lys-29, Lys-33, Lys-48, and Lys-63). These diverse polyUb structures may regulate downstream signaling specificity and determine the consequence of ubiquitination (36, 47). Whereas the Lys-63 polyUb linkage is proposed to mediate nonproteolytic events, including protein trafficking and inclusion formation (49), the Lys-48 linkage and other atypical polyUb linkages mediate proteasomal degradation (36). To elucidate the role of different polyUb linkages with TDP protein aggregates, we examined transfected cells with Lys-48 or Lys-63 polyUb linkage-specific antibodies (29) compared with untransfected cells and found that both linkages were clearly present in the cytoplasmic and nuclear TDP inclusions (Fig. 7, B and C). These results indicate that mixed polyUb chains (Lys-48 and Lys-63) are specifically associated with TDP protein aggregates.
To confirm the presence of Lys-48 and Lys-63 polyUb linkages observed in TDP-S6 cytoplasmic aggregates, we analyzed the levels of polyUb linkages in the Sarkosyl-insoluble proteome by targeted mass spectrometry (36, 39). Metabolically heavy labeled proteins were used as internal standards for their unlabeled counterparts, allowing the quantitation of native proteins in each sample. Trypsin cleaves endogenous polyUb chains and their corresponding internal standards to generate linkage-specific peptides tagged with two Gly residues (51). During the LC/SRM analysis, heavy and light peptides co-eluted and were fragmented to produce product ion pairs (Fig. 8, A and B), which were used as surrogates to provide sensitive measurements of relative protein abundance. Among all polyUb linkages, Lys-63 increased ∼3.3-fold in the insoluble fraction of TDP-S6-expressing cells over that of mock-transfected cells. In addition, Lys-48 linkages (∼2.0-fold), Lys-11 linkages (∼1.5-fold), and Lys-29 linkages (∼1.3-fold) were also measured in this sample (Fig. 8C), whereas the other four potential linkages mediated by three lysines (Lys-6, Lys-27, and Lys-33) and the N terminus were not detected. In the TDP-S6 insoluble fraction, all measured linkages other than Lys-29 were significantly increased. In comparison, within the TDP-43 insoluble fraction, Lys-63 was the only polyUb linkage significantly increased (∼ 2.2-fold) over the level in mock-transfected cells. This is in good agreement with the localization of Lys-63-linked polyUb and nuclear TDP-43 bodies by immunofluorescence in HEK-293 cells expressing a high level of TDP-43 (Fig. 7C). These data further support the involvement of multiple polyUb chains in TDP protein aggregation and indicate that the Lys-63 polyUb linkage may be the most significant form associated with TDP-43 or TDP-S6 overexpression.
Fig. 8.

Multiple polyUb linkages were detected in TDP-S6 and TDP-43 insoluble proteome. Transfected cells were harvested and mixed with metabolically labeled cells as internal standards followed by differential extraction. The Sarkosyl-insoluble samples were used for SRM-based analysis to quantify all polyUb linkages and two other proteins (E1 and Rpn2 as loading control). A, the monitored product ion pair of Lys-48 linkage-specific peptide. B, the co-elution of Lys-48 linkage peptide pair during an LC/SRM run. C, the comparison of measured linkages and proteins in three samples: mock-, HA-TDP-43- or HA-TDP-S6-transfected cells. The data were normalized to values in the mock sample. Error bars indicate S.E., and significance (*) was determined by performing an unpaired two-tailed t test (p < 0.01). WT, wild type (Full length TDP-43).
DISCUSSION
The abnormal accumulation of phosphorylated and ubiquitinated species in protein inclusions is observed in a wide variety of neurodegenerative diseases (3, 4). In Alzheimer and Parkinson diseases, these intracellular inclusions are called neurofibrillary tangles and Lewy bodies and are composed of pathologically altered forms of tau and α-synuclein, respectively (3, 52). In FTLD-U and ALS, TDP-43 has been identified as the major component of tau- and α-synuclein-negative inclusions where it is both phosphorylated and ubiquitinated (5). Although there are 10 known splice variants of human TDP-43, their roles in abnormal TDP-43 translocation and pathologic aggregation have not yet been characterized. In this study, we expressed human recombinant TDP-43 and a truncated splice variant, TDP-S6, in HEK-293 cells and primary neurons. The full-length protein was expressed almost exclusively in the nucleus where it co-localized with endogenous TDP-43. In sharp contrast, the shorter TDP-S6 formed highly insoluble cytoplasmic and nuclear inclusions reminiscent of disease-specific pathology. Moreover, overexpression of both TDP-43 and TDP-S6 resulted in the enrichment of specific post-translational modifications within their respective insoluble protein extracts. These post-translational modifications included ubiquitination, phosphorylation, and TDP-43-specific C-terminal fragmentation. Based on our multiplex SILAC proteomics approach, we identified the significant enrichment of SUMO-2/3 and Ub within the Sarkosyl-insoluble, urea-soluble extracts.
In our cellular model, endogenous Ub showed strong association with the TDP aggregates in both biochemical analysis and immunostaining, supporting the hypothesis that aggregation-prone TDP-43 is a major ubiquitinated species. Although TDP-S6-expressing cells displayed inclusions of various sizes, only a fraction of recombinant TDP-43-expressing cells formed small nuclear aggregates dependent on high level expression (Fig. 7). Quantitative MS analysis revealed that several main polyUb chains were present in the insoluble extracts from these cells with Lys-63 linkages being the most up-regulated. These findings are reminiscent of previous characterization of tau- and SOD1-positve inclusions (49) and are consistent with the idea that Lys-63-linked chains may facilitate the formation of inclusions and direct subsequent clearance by the autophagy pathway.
Recent studies have suggested that ubiquitinated inclusions in FTLD-U brains are primarily composed of C-terminal fragments of TDP-43 and that these fragments may comprise the primary pathologic species of TDP-43 aggregates by serving as a seed for aggregation (53). Moreover, caspase-3 activation has been implicated in the proteolytic processing of TDP-43 into three C-terminal fragments similar to those seen in disease tissues (54). Although TDP-S6 lacks the final 137 amino acids of full-length TDP-43, the splice variant still contains all three purported caspase-3 cleavage sites at amino acids 10–13 (DEND), 86–89 (DETD), and 216–219 (DVMD), corresponding to the 42-, 35-, and 25-kDa fragments, respectively, in the full-length protein (54). Interestingly, the fragmentation of TDP-S6 resulted in three lower molecular mass species that approximate the molecular mass expected of caspase-3 cleavage products in this HA-tagged construct (37, 30, and 20 kDa). These data suggest that TDP-S6 cleavage preferentially occurs in cells and potentially as a result of a caspase-3-mediated mechanism. Alternatively, these fragments may instead be derived from the cleavage of sequestered endogenous TDP-43 in TDP-S6-expressing cells, but further studies using targeted MS analysis will be necessary to validate enzyme-specific TDP-43 cleavage.
Currently, it remains unknown whether aberrant splicing of the TDP-43 transcript results in the production of pathologic TDP isoforms in ALS or FTLD-U. However, two TDP-43 splicing isoforms have been identified previously in human brain and spinal cord tissue (55). One of these isoforms, which closely resembles TDP-S6, was identified in two ALS cases. This novel transcript lacked all of exon 3 and a significant portion of exon 6, resulting in expression of a protein product (∼28 kDa) without a glycine-rich C terminus (55). In this study, TDP-S6, a C-terminal truncated splice variant previously identified in HEK-293 cells (11), was found predominately localized to the cytoplasm and mimicked the aggregation observed in FTLD-U and ALS. It should be noted that although the human TDP-S6 variant formed both cytoplasmic and nuclear inclusions, overexpression of the mouse TDP-S6 in HEK-293 cells resulted only in the formation of nuclear specific speckle-like structures, termed TDP bodies (40). Thus, despite sharing 96% amino acid identity, inconsistency between mouse and human TDP-S6 localization may result from differences in species-specific nuclear/cytoplasmic translocation or different experimental conditions.
SILAC analysis of this cellular model revealed novel components in the formation of protein inclusions. SUMO-2/3 was found to be preferentially enriched within the TDP-43 and TDP-S6 insoluble proteomes and was subsequently validated by Western blotting. Interestingly, immunofluorescence confocal microscopy indicated that SUMO-2/3 co-localized mainly with TDP-S6 nuclear inclusions, thereby implicating a unique nuclear association between SUMO-2/3 and TDP-S6. This nuclear association is not completely surprising because TDP-43 has been previously found to associate with promyelocytic leukemia bodies (40), nuclear structures known to co-localize with SUMO (27, 48). Moreover, sequence analysis of TDP-43 revealed a canonical SUMO conjugation site motif (ΨKX(D/E); where Ψ indicates hydrophobic and χ indicate any residue, amino acids 135–138 of TDP-43) among its potential SUMO-targeted lysines (56). Using an in vitro deSUMOylation assay, we provide experimental evidence that TDP-43 itself is directly SUMOylated within the insoluble fraction. This is also supported by a recent large scale study wherein TDP-43 was identified as a SUMO-2 conjugate that was accumulated over 7-fold in response to heat shock (57). Notably, protein SUMOylation has also been linked to aggregated proteins in Alzheimer disease, Parkinson disease, and poly(Q) disorders (58). In addition, SUMO-1 has been shown to specifically co-localize with ubiquitinated nuclear neuronal inclusions in FTLD-U tissues (59). Although protein SUMOylation functions in regulating transcription and nuclear transport (60), it has also been shown to cross-talk with ubiquitination because the SUMO tag on proteins may serve as a recognition signal for subsequent ubiquitination and proteasome-mediated degradation (61–63). More specifically, the recent identification of SUMO-targeted ubiquitin ligases (STUbLs) (64) raises the intriguing possibility that upon SUMOylation misfolded nuclear TDP-S6 is subjected to sequestration in nuclear aggregates and STUbL-dependent ubiquitination and degradation (65). Interestingly, a STUbL-mediated mechanism for the cellular regulation of insoluble TDP-S6 is supported by the immunochemical co-localization of polyubiquitin chains with both nuclear and cytoplasmic TDP-S6 inclusions (Fig. 7). The mechanism of TDP-43 SUMOylation underlying disease-specific protein translocation, degradation, or aggregation is worth further investigation.
In addition to Ub and SUMO-2/3, actin-related protein 1 (ARP1) was also found to be enriched in the Sarkosyl-insoluble fractions of TDP-43- and TDP-S6-transfected cells. ARP1 is a subunit of dynactin, a macromolecular complex that interacts with both microtubules and cytoplasmic dynein. ARP1 is also involved in protein transport and vesicular trafficking (66). Interestingly, gene mutation in another subunit of dynactin, p150Glued, has been reported in ALS cases (67), and the related mouse model developed motor neuron disease (68). In addition, overexpression of TDP-S6 reduces N-ethylmaleimide sensitive factor, an ATPase involved in intracellular trafficking, and dynamin 3, a microtubule-associated protein involved in vesicle budding (69). These results suggest that protein transportation may be dysregulated in neurodegenerative diseases. Furthermore, these findings, coupled with an increase in Lys-63 polyUb linkages, further support an association between TDP-43 and protein sorting pathways.
In sum, our data show that the C-terminal domain is necessary for TDP-43 nuclear localization and that without this region TDP-43 becomes primarily sequestered within cytoplasmic aggregates, consistent with a previous report showing that the C terminus is essential for the solubility and cellular localization of TDP-43 (50). Thus, it might be plausible that the genetic mutations in TDP-43 found within the C-terminal region (21–25) somehow disrupt critical amino acid residues needed for TDP-43 nuclear localization and proper function. Although further studies are needed to evaluate the presence of TDP-S6 or other similar short isoforms in disease tissue, our data raise the intriguing possibility that dysregulation of human TDP-43 alternative splicing or preferential proteolytic processing to produce TDP N-terminal fragments can contribute to the pathology of FTLD-U and ALS.
Supplementary Material
Acknowledgments
We thank Drs. Jonathan Glass and Ping Xu for discussion and critically reading the manuscript.
* This work was supported, in whole or in part, by the National Institutes of Health through the Emory Alzheimer's Disease Center (Grant P50AG025688), the Emory Neuroscience NINDS Core Facilities (Grant P30NS055077), and Training Grants T32NS007480 (to N. S.) and F30NS057902 (to Y. G.). This work was also supported by Research Scholar Grant RSG-09-181-01 from the American Cancer Society.
 This article contains supplemental Tables S1–S10.
This article contains supplemental Tables S1–S10.
1 The abbreviations used are:
- FTLD
- frontotemporal lobar degeneration
- TDP
- TAR DNA-binding protein
- FTLD-U
- ubiquitin-positive inclusions of frontotemporal lobar degeneration
- ALS
- amyotrophic lateral sclerosis
- SILAC
- stable isotope labeling with amino acids in culture
- Ub
- ubiquitin
- SUMO
- small ubiquitin-like modifier
- RRM
- RNA recognition motif
- TAR
- transactive response
- HEK
- human embryonic kidney
- HA
- hemagglutinin
- SENP2
- sentrin-specific endopeptidase 2
- FDR
- false discovery rate
- SRM
- selective reaction monitoring
- STUbL
- SUMO-targeted ubiquitin ligase
- ARP1
- actin-related protein 1.
REFERENCES
- 1.Kumar-Singh S., Van Broeckhoven C. (2007) Frontotemporal lobar degeneration: current concepts in the light of recent advances. Brain Pathol 17, 104–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neary D., Snowden J., Mann D. (2005) Frontotemporal dementia. Lancet Neurol 4, 771–780 [DOI] [PubMed] [Google Scholar]
- 3.Ross C. A., Poirier M. A. (2004) Protein aggregation and neurodegenerative disease. Nat. Med 10, (suppl.) S10–S17 [DOI] [PubMed] [Google Scholar]
- 4.Taylor J. P., Hardy J., Fischbeck K. H. (2002) Toxic proteins in neurodegenerative disease. Science 296, 1991–1995 [DOI] [PubMed] [Google Scholar]
- 5.Neumann M., Sampathu D. M., Kwong L. K., Truax A. C., Micsenyi M. C., Chou T. T., Bruce J., Schuck T., Grossman M., Clark C. M., McCluskey L. F., Miller B. L., Masliah E., Mackenzie I. R., Feldman H., Feiden W., Kretzschmar H. A., Trojanowski J. Q., Lee V. M. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 [DOI] [PubMed] [Google Scholar]
- 6.Neumann M., Mackenzie I. R., Cairns N. J., Boyer P. J., Markesbery W. R., Smith C. D., Taylor J. P., Kretzschmar H. A., Kimonis V. E., Forman M. S. (2007) TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J. Neuropathol. Exp. Neurol 66, 152–157 [DOI] [PubMed] [Google Scholar]
- 7.Amador-Ortiz C., Lin W. L., Ahmed Z., Personett D., Davies P., Duara R., Graff-Radford N. R., Hutton M. L., Dickson D. W. (2007) TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann. Neurol 61, 435–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakashima-Yasuda H., Uryu K., Robinson J., Xie S. X., Hurtig H., Duda J. E., Arnold S. E., Siderowf A., Grossman M., Leverenz J. B., Woltjer R., Lopez O. L., Hamilton R., Tsuang D. W., Galasko D., Masliah E., Kaye J., Clark C. M., Montine T. J., Lee V. M., Trojanowski J. Q. (2007) Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol 114, 221–229 [DOI] [PubMed] [Google Scholar]
- 9.Cairns N. J., Neumann M., Bigio E. H., Holm I. E., Troost D., Hatanpaa K. J., Foong C., White C. L., 3rd, Schneider J. A., Kretzschmar H. A., Carter D., Taylor-Reinwald L., Paulsmeyer K., Strider J., Gitcho M., Goate A. M., Morris J. C., Mishra M., Kwong L. K., Stieber A., Xu Y., Forman M. S., Trojanowski J. Q., Lee V. M., Mackenzie I. R. (2007) TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am. J. Pathol 171, 227–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Forman M. S., Trojanowski J. Q., Lee V. M. (2007) TDP-43: a novel neurodegenerative proteinopathy. Curr. Opin. Neurobiol 17, 548–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang H. Y., Wang I. F., Bose J., Shen C. K. (2004) Structural diversity and functional implications of the eukaryotic TDP gene family. Genomics 83, 130–139 [DOI] [PubMed] [Google Scholar]
- 12.Ayala Y. M., Pantano S., D'Ambrogio A., Buratti E., Brindisi A., Marchetti C., Romano M., Baralle F. E. (2005) Human, Drosophila, and C. elegans TDP43: nucleic acid binding properties and splicing regulatory function. J. Mol. Biol 348, 575–588 [DOI] [PubMed] [Google Scholar]
- 13.Buratti E., Baralle F. E. (2001) Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J. Biol. Chem 276, 36337–36343 [DOI] [PubMed] [Google Scholar]
- 14.Ou S. H., Wu F., Harrich D., García-Martínez L. F., Gaynor R. B. (1995) Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J. Virol 69, 3584–3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mercado P. A., Ayala Y. M., Romano M., Buratti E., Baralle F. E. (2005) Depletion of TDP 43 overrides the need for exonic and intronic splicing enhancers in the human apoA-II gene. Nucleic Acids Res 33, 6000–6010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buratti E., Dörk T., Zuccato E., Pagani F., Romano M., Baralle F. E. (2001) Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J 20, 1774–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mackenzie I. R., Bigio E. H., Ince P. G., Geser F., Neumann M., Cairns N. J., Kwong L. K., Forman M. S., Ravits J., Stewart H., Eisen A., McClusky L., Kretzschmar H. A., Monoranu C. M., Highley J. R., Kirby J., Siddique T., Shaw P. J., Lee V. M., Trojanowski J. Q. (2007) Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol 61, 427–434 [DOI] [PubMed] [Google Scholar]
- 18.Mackenzie I. R., Rademakers R. (2007) The molecular genetics and neuropathology of frontotemporal lobar degeneration: recent developments. Neurogenetics 8, 237–248 [DOI] [PubMed] [Google Scholar]
- 19.Neumann M., Kwong L. K., Sampathu D. M., Trojanowski J. Q., Lee V. M. (2007) TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis: protein misfolding diseases without amyloidosis. Arch. Neurol 64, 1388–1394 [DOI] [PubMed] [Google Scholar]
- 20.Buratti E., Brindisi A., Giombi M., Tisminetzky S., Ayala Y. M., Baralle F. E. (2005) TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J. Biol. Chem 280, 37572–37584 [DOI] [PubMed] [Google Scholar]
- 21.Van Deerlin V. M., Leverenz J. B., Bekris L. M., Bird T. D., Yuan W., Elman L. B., Clay D., Wood E. M., Chen-Plotkin A. S., Martinez-Lage M., Steinbart E., McCluskey L., Grossman M., Neumann M., Wu I. L., Yang W. S., Kalb R., Galasko D. R., Montine T. J., Trojanowski J. Q., Lee V. M., Schellenberg G. D., Yu C. E. (2008) TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol 7, 409–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sreedharan J., Blair I. P., Tripathi V. B., Hu X., Vance C., Rogelj B., Ackerley S., Durnall J. C., Williams K. L., Buratti E., Baralle F., de Belleroche J., Mitchell J. D., Leigh P. N., Al-Chalabi A., Miller C. C., Nicholson G., Shaw C. E. (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kabashi E., Valdmanis P. N., Dion P., Spiegelman D., McConkey B. J., Vande Velde C., Bouchard J. P., Lacomblez L., Pochigaeva K., Salachas F., Pradat P. F., Camu W., Meininger V., Dupre N., Rouleau G. A. (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet 40, 572–574 [DOI] [PubMed] [Google Scholar]
- 24.Yokoseki A., Shiga A., Tan C. F., Tagawa A., Kaneko H., Koyama A., Eguchi H., Tsujino A., Ikeuchi T., Kakita A., Okamoto K., Nishizawa M., Takahashi H., Onodera O. (2008) TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann. Neurol 63, 538–542 [DOI] [PubMed] [Google Scholar]
- 25.Gitcho M. A., Baloh R. H., Chakraverty S., Mayo K., Norton J. B., Levitch D., Hatanpaa K. J., White C. L., 3rd, Bigio E. H., Caselli R., Baker M., Al-Lozi M. T., Morris J. C., Pestronk A., Rademakers R., Goate A. M., Cairns N. J. (2008) TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol 63, 535–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mann M. (2006) Functional and quantitative proteomics using SILAC. Nat. Rev. Mol. Cell Biol 7, 952–958 [DOI] [PubMed] [Google Scholar]
- 27.Mukhopadhyay D., Ayaydin F., Kolli N., Tan S. H., Anan T., Kametaka A., Azuma Y., Wilkinson K. D., Dasso M. (2006) SUSP1 antagonizes formation of highly SUMO2/3-conjugated species. J. Cell Biol 174, 939–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Volpicelli L. A., Lah J. J., Fang G., Goldenring J. R., Levey A. I. (2002) Rab11a and myosin Vb regulate recycling of the M4 muscarinic acetylcholine receptor. J. Neurosci 22, 9776–9784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newton K., Matsumoto M. L., Wertz I. E., Kirkpatrick D. S., Lill J. R., Tan J., Dugger D., Gordon N., Sidhu S. S., Fellouse F. A., Komuves L., French D. M., Ferrando R. E., Lam C., Compaan D., Yu C., Bosanac I., Hymowitz S. G., Kelley R. F., Dixit V. M. (2008) Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell 134, 668–678 [DOI] [PubMed] [Google Scholar]
- 30.Xu P., Duong D. M., Peng J. (2009) Systematical optimization of reverse-phase chromatography for shotgun proteomics. J. Proteome Res 8, 3944–3950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ong S. E., Mann M. (2006) A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC). Nat. Protoc 1, 2650–2660 [DOI] [PubMed] [Google Scholar]
- 32.Eng J., McCormack A. L., Yates J. R., 3rd. (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 33.Peng J., Elias J. E., Thoreen C. C., Licklider L. J., Gygi S. P. (2003) Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J. Proteome Res 2, 43–50 [DOI] [PubMed] [Google Scholar]
- 34.Elias J. E., Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 35.Cheng D., Hoogenraad C. C., Rush J., Ramm E., Schlager M. A., Duong D. M., Xu P., Wijayawardana S. R., Hanfelt J., Nakagawa T., Sheng M., Peng J. (2006) Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol. Cell. Proteomics 5, 1158–1170 [DOI] [PubMed] [Google Scholar]
- 36.Xu P., Duong D. M., Seyfried N. T., Cheng D., Xie Y., Robert J., Rush J., Hochstrasser M., Finley D., Peng J. (2009) Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 137, 133–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kislinger T., Cox B., Kannan A., Chung C., Hu P., Ignatchenko A., Scott M. S., Gramolini A. O., Morris Q., Hallett M. T., Rossant J., Hughes T. R., Frey B., Emili A. (2006) Global survey of organ and organelle protein expression in mouse: combined proteomic and transcriptomic profiling. Cell 125, 173–186 [DOI] [PubMed] [Google Scholar]
- 38.Xia Q., Liao L., Cheng D., Duong D. M., Gearing M., Lah J. J., Levey A. I., Peng J. (2008) Proteomic identification of novel proteins associated with Lewy bodies. Front. Biosci 13, 3850–3856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kirkpatrick D. S., Hathaway N. A., Hanna J., Elsasser S., Rush J., Finley D., King R. W., Gygi S. P. (2006) Quantitative analysis of in vitro ubiquitinated cyclin B1 reveals complex chain topology. Nat. Cell Biol 8, 700–710 [DOI] [PubMed] [Google Scholar]
- 40.Wang I. F., Reddy N. M., Shen C. K. (2002) Higher order arrangement of the eukaryotic nuclear bodies. Proc. Natl. Acad. Sci. U.S.A 99, 13583–13588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hasegawa M., Arai T., Nonaka T., Kametani F., Yoshida M., Hashizume Y., Beach T. G., Buratti E., Baralle F., Morita M., Nakano I., Oda T., Tsuchiya K., Akiyama H. (2008) Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol 64, 60–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Inukai Y., Nonaka T., Arai T., Yoshida M., Hashizume Y., Beach T. G., Buratti E., Baralle F. E., Akiyama H., Hisanaga S., Hasegawa M. (2008) Abnormal phosphorylation of Ser409/410 of TDP-43 in FTLD-U and ALS. FEBS Lett 582, 2899–2904 [DOI] [PubMed] [Google Scholar]
- 43.Johnson B. S., Snead D., Lee J. J., McCaffery J. M., Shorter J., Gitler A. D. (2009) TDP-43 is intrinsically aggregation-prone and ALS-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem 284, 20329–20339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Browman D. T., Hoegg M. B., Robbins S. M. (2007) The SPFH domain-containing proteins: more than lipid raft markers. Trends Cell Biol 17, 394–402 [DOI] [PubMed] [Google Scholar]
- 45.Kopito R. R. (2000) Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 10, 524–530 [DOI] [PubMed] [Google Scholar]
- 46.Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 47.Kerscher O., Felberbaum R., Hochstrasser M. (2006) Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu. Rev. Cell Dev. Biol 22, 159–180 [DOI] [PubMed] [Google Scholar]
- 48.Vertegaal A. C., Ogg S. C., Jaffray E., Rodriguez M. S., Hay R. T., Andersen J. S., Mann M., Lamond A. I. (2004) A proteomic study of SUMO-2 target proteins. J. Biol. Chem 279, 33791–33798 [DOI] [PubMed] [Google Scholar]
- 49.Tan J. M., Wong E. S., Kirkpatrick D. S., Pletnikova O., Ko H. S., Tay S. P., Ho M. W., Troncoso J., Gygi S. P., Lee M. K., Dawson V. L., Dawson T. M., Lim K. L. (2008) Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet 17, 431–439 [DOI] [PubMed] [Google Scholar]
- 50.Ayala Y. M., Zago P., D'Ambrogio A., Xu Y. F., Petrucelli L., Buratti E., Baralle F. E. (2008) Structural determinants of the cellular localization and shuttling of TDP-43. J. Cell Sci 121, 3778–3785 [DOI] [PubMed] [Google Scholar]
- 51.Peng J., Schwartz D., Elias J. E., Thoreen C. C., Cheng D., Marsischky G., Roelofs J., Finley D., Gygi S. P. (2003) A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol 21, 921–926 [DOI] [PubMed] [Google Scholar]
- 52.Lee V. M., Goedert M., Trojanowski J. Q. (2001) Neurodegenerative tauopathies. Annu. Rev. Neurosci 24, 1121–1159 [DOI] [PubMed] [Google Scholar]
- 53.Igaz L. M., Kwong L. K., Xu Y., Truax A. C., Uryu K., Neumann M., Clark C. M., Elman L. B., Miller B. L., Grossman M., McCluskey L. F., Trojanowski J. Q., Lee V. M. (2008) Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am. J. Pathol 173, 182–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y. J., Xu Y. F., Dickey C. A., Buratti E., Baralle F., Bailey R., Pickering-Brown S., Dickson D., Petrucelli L. (2007) Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J. Neurosci 27, 10530–10534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Strong M. J., Volkening K., Hammond R., Yang W., Strong W., Leystra-Lantz C., Shoesmith C. (2007) TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol. Cell. Neurosci 35, 320–327 [DOI] [PubMed] [Google Scholar]
- 56.Geiss-Friedlander R., Melchior F. (2007) Concepts in sumoylation: a decade on. Nat. Rev. Mol. Cell Biol 8, 947–956 [DOI] [PubMed] [Google Scholar]
- 57.Golebiowski F., Matic I., Tatham M. H., Cole C., Yin Y., Nakamura A., Cox J., Barton G. J., Mann M., Hay R. T. (2009) System-wide changes to SUMO modifications in response to heat shock. Sci. Signal 2, ra24. [DOI] [PubMed] [Google Scholar]
- 58.Martin S., Wilkinson K. A., Nishimune A., Henley J. M. (2007) Emerging extranuclear roles of protein SUMOylation in neuronal function and dysfunction. Nat. Rev. Neurosci 8, 948–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mackenzie I. R., Baker M., West G., Woulfe J., Qadi N., Gass J., Cannon A., Adamson J., Feldman H., Lindholm C., Melquist S., Pettman R., Sadovnick A. D., Dwosh E., Whiteheart S. W., Hutton M., Pickering-Brown S. M. (2006) A family with tau-negative frontotemporal dementia and neuronal intranuclear inclusions linked to chromosome 17. Brain 129, 853–867 [DOI] [PubMed] [Google Scholar]
- 60.Dorval V., Fraser P. E. (2007) SUMO on the road to neurodegeneration. Biochim. Biophys. Acta 1773, 694–706 [DOI] [PubMed] [Google Scholar]
- 61.Lallemand-Breitenbach V., Jeanne M., Benhenda S., Nasr R., Lei M., Peres L., Zhou J., Zhu J., Raught B., de Thé H. (2008) Arsenic degrades PML or PML-RARalpha through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat. Cell Biol 10, 547–555 [DOI] [PubMed] [Google Scholar]
- 62.Schimmel J., Larsen K. M., Matic I., van Hagen M., Cox J., Mann M., Andersen J. S., Vertegaal A. C. (2008) The ubiquitin-proteasome system is a key component of the SUMO-2/3 cycle. Mol. Cell. Proteomics 7, 2107–2122 [DOI] [PubMed] [Google Scholar]
- 63.Tatham M. H., Geoffroy M. C., Shen L., Plechanovova A., Hattersley N., Jaffray E. G., Palvimo J. J., Hay R. T. (2008) RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat. Cell Biol 10, 538–546 [DOI] [PubMed] [Google Scholar]
- 64.Sun H., Leverson J. D., Hunter T. (2007) Conserved function of RNF4 family proteins in eukaryotes: targeting a ubiquitin ligase to SUMOylated proteins. EMBO J 26, 4102–4112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Perry J. J., Tainer J. A., Boddy M. N. (2008) A SIM-ultaneous role for SUMO and ubiquitin. Trends Biochem. Sci 33, 201–208 [DOI] [PubMed] [Google Scholar]
- 66.Schroer T. A., Bingham J. B., Gill S. R. (1996) Actin-related protein 1 and cytoplasmic dynein-based motility—what's the connection? Trends Cell Biol 6, 212–215 [DOI] [PubMed] [Google Scholar]
- 67.Münch C., Sedlmeier R., Meyer T., Homberg V., Sperfeld A. D., Kurt A., Prudlo J., Peraus G., Hanemann C. O., Stumm G., Ludolph A. C. (2004) Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63, 724–726 [DOI] [PubMed] [Google Scholar]
- 68.Laird F. M., Farah M. H., Ackerley S., Hoke A., Maragakis N., Rothstein J. D., Griffin J., Price D. L., Martin L. J., Wong P. C. (2008) Motor neuron disease occurring in a mutant dynactin mouse model is characterized by defects in vesicular trafficking. J. Neurosci 28, 1997–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schekman R., Orci L. (1996) Coat proteins and vesicle budding. Science 271, 1526–1533 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.