

Abstract
Homeostasis of the vertebrate digestive tract requires interactions between an endodermal epithelium and mesenchymal cells derived from the splanchnic mesoderm. Signaling between these two tissue layers is also crucial for patterning and growth of the developing gut. From early developmental stages, sonic hedgehog (Shh) and indian hedgehog (Ihh) are secreted by the endoderm of the mammalian gut, indicative of a developmental role. Further, misregulated hedgehog (Hh) signaling is implicated in both congenital defects and cancers arising from the gastrointestinal tract. In the mouse, only limited gastrointestinal anomalies arise following removal of either Shh or Ihh. However, given the considerable overlap in their endodermal expression domains, a functional redundancy between these signals might mask a more extensive role for Hh signaling in development of the mammalian gut. To address this possibility, we adopted a conditional approach to remove both Shh and Ihh functions from early mouse gut endoderm. Analysis of compound mutants indicates that continuous Hh signaling is dispensable for regional patterning of the gut tube, but is essential for growth of the underlying mesenchyme. Additional in vitro analysis, together with genetic gain-of-function studies, further demonstrate that Hh proteins act as paracrine mitogens to promote the expansion of adjacent mesenchymal progenitors, including those of the smooth muscle compartment. Together, these studies provide new insights into tissue interactions underlying mammalian gastrointestinal organogenesis and disease.
Keywords: Hedgehog, Intestine, Stomach, Endoderm, Mesoderm, Mouse
INTRODUCTION
In vertebrate embryos, the visceral endoderm recruits adjacent splanchnic mesoderm to form a primitive gut tube that is regionally patterned along its length into distinct anatomic and functional domains. From anterior to posterior these are: the esophagus, stomach, small intestine and large intestine. Epithelial differentiation generates distinct self-renewing epithelia; for example, the characteristic villi of the prospective small bowel and glands of the stomach. In addition, the splanchnic mesoderm-derived mesenchyme that underlies the gut is organized along a radial axis of symmetry into distinct cell layers establishing oriented smooth muscle bands that control local movement of the gut tube. Organogenesis and tissue homeostasis within the digestive tract require continuous interaction between endodermal epithelium and subjacent mesenchyme (Haffen et al., 1987; Kedinger et al., 1998), and disruption of these interactions is implicated in human gastrointestinal (GI) birth defects and cancers (Bhowmick et al., 2004; de Santa Barbara et al., 2002; Yauch et al., 2008). Despite their importance, the molecular mediators of epithelial-mesenchymal interchange are poorly characterized.
The mammalian family of lipid-modified hedgehog (Hh) signals comprises three members: sonic (Shh), indian (Ihh) and desert (Dhh) hedgehogs. Each is thought to elicit signaling through a common mechanism (Hooper and Scott, 2005; Ingham and McMahon, 2001). Hh ligands bind to a multi-pass transmembrane receptor, patched 1 (Ptch1), and in so doing counter Ptch1-mediated inhibition of the seven-pass membrane protein, smoothened (Smo). Smo is essential for all Hh signaling (Zhang et al., 2001). Smo-mediated signal transduction controls the activities of zinc-finger transcription factors of the Gli family, and thereby the transcriptional response to a given Hh input within a particular target tissue. Among the generic transcriptional responses observed in all tissues is the upregulation of Ptch1, Gli1 and Hhip, feedback and feed-forward components in the Hh pathway (Ingham and McMahon, 2001; Lum and Beachy, 2004).
Hh proteins direct proliferation, patterning and differentiation of many tissues (McMahon et al., 2003). In the mammalian gut, Shh and Ihh are co-expressed in endodermal epithelium of the primitive gut tube from as early as embryonic day 8.5 (E8.5) (Bitgood and McMahon, 1995; Echelard et al., 1993). Between E10 and E13, Shh expression is more prominent in endoderm of the esophagus and anterior stomach, and Ihh expression is more prominent in endoderm of the posterior stomach and proximal duodenum (Aubin et al., 2002; Bitgood and McMahon, 1995). Both genes are highly expressed in the intestinal epithelium and, after E13.5, Shh and Ihh expression overlaps extensively in the prospective stomach (Ramalho-Santos et al., 2000).
The activation of Hh target genes such as the Hh receptor Ptch1, and a downstream signal, Bmp4, provide an insight into the cellular targets of Hh signals in the gut (Aubin et al., 2002; Bitgood and McMahon, 1995; Madison et al., 2005; Ramalho-Santos et al., 2000; Roberts et al., 1995). For the most part, Shh and Ihh appear to target underlying mesenchyme in the gut tube. Although autocrine signaling within the endodermal epithelium of the hindgut was suggested on the basis of a Ptch1 antibody analysis (van den Brink et al., 2004), a subsequent study concluded that the antibodies did not recognize Ptch1 (van den Brink, 2007). Indeed, the bulk of evidence indicates that paracrine signaling within the gut mesenchyme is the major mode of action shared by mammalian Hh signals from esophagus to colon (Kolterud et al., 2009; Madison et al., 2005; Ramalho-Santos et al., 2000; Sukegawa et al., 2000; Wang et al., 2002; Yauch et al., 2008).
Current understanding of Hh function in early gut development rests on limited studies in chick and mouse embryos (Ramalho-Santos et al., 2000; Roberts et al., 1995; Roberts et al., 1998; Sukegawa et al., 2000; van den Brink, 2007). In a chicken GI explant culture, Shh was shown to regulate mesenchymal patterning and inhibit smooth muscle differentiation (Sukegawa et al., 2000). Proliferation of intestinal intervillus epithelium is reduced in Ihh−/− mice (Ramalho-Santos et al., 2000), whereas Shh−/− mice show multiple gut anomalies, including tracheo-esophageal fistula and anorectal atresia (Litingtung et al., 1998; Ramalho-Santos et al., 2000), among the common congenital GI anomalies observed in humans with mutations in Hh pathway genes (Arsic et al., 2002; de Santa Barbara et al., 2002; Kang et al., 1997; Kim et al., 2001; Nanni et al., 1999). However, the modest phenotypes of individual Shh or Ihh mutants and the extensive overlap in their expression domains are indicative of a potential redundancy in their actions, a conclusion supported by the enhancing action of reducing gene dosage in a background sensitized by the loss of one these proteins (Ramalho-Santos et al., 2000). To broadly attenuate Hh signaling, investigators have overexpressed a secreted form of the Hh inhibitor Hhip1 (Chuang and McMahon, 1999) in the intestine or administered blocking antibodies that bind both ligands (Madison et al., 2005; Wang et al., 2002). These treatments disrupt mouse intestinal villus morphogenesis and the organization of underlying subepithelial myofibroblasts. However, Hh inhibition was probably incomplete and initiated after E12.5, several days after onset of Shh and Ihh expression in gut endoderm. Thus, these studies did not address the early actions of the Hh pathway in the gut. Further, analysis was restricted to the intestine.
Here, we have used conditional gene targeting and complementary approaches to address the overlapping activities of Shh and Ihh. We find that Hh signaling is essential for embryonic gut development, promoting survival and proliferation of mesenchymal progenitors. Surprisingly, activation of Hh signaling in gut mesenchyme does not block differentiation of smooth muscle progenitors but rather promotes expansion of this cell population. The mitogenic actions of Shh and Ihh are crucial for appropriate elaboration of the radial axis of the gut tube and consequently enable a normal endodermal-mesenchymal interplay in mammalian gut development.
MATERIALS AND METHODS
Experimental animals and sampling
ShhCre/Fl;Ihh−/Fl, ShhCre/Fl;Ihh+/Fl, Shh+/Fl;Ihh+/Fl and Shh+/Fl;Ihh−/Fl embryos were obtained from crosses of ShhCre/+;Ihh+/− males and ShhFl/Fl;IhhFl/Fl females. The Cre allele is an insertion of a GFP-Cre fusion cDNA into the Shh locus, inactivating Shh production from that allele (Harfe et al., 2004). ‘Fl’ describes previously documented Cre-dependent conditional alleles of Shh (Lewis et al., 2001) and Ihh (Razzaque et al., 2005). Ihh− encodes a null allele (St-Jacques et al., 1999). Bapx1-Cre;R26-SmoM2 embryos were generated by crossing Bapx1-Cre+/− (Verzi et al., 2009) male and R26-SmoM2+/+ (Mao et al., 2006) female mice. Gli1-CreER;R26R embryos were generated by crossing Gli1-CreER (Ahn and Joyner, 2004) male to R26R (Soriano, 1999) female mice, and a single dose of tamoxifen (5 mg/40 g body weight) was injected intraperitoneally into the pregnant dams at E9.5. The morning of identification of a vaginal plug was regarded as day 0.5 of gestation. Embryos or organs were fixed overnight in 4% paraformaldehyde at 4°C or in Bouin's solution at room temperature, then dehydrated, embedded in paraffin and sectioned (6 μm). Animals were housed and handled according to protocols approved by institutional committees.
Immunohistochemistry, in situ hybridization, β-galactosidase activity and histological analysis
Hematoxylin/Eosin, Periodic Acid Schiff (PAS) and Alcian Blue histological staining of sections, in situ hybridization and immunohistochemistry used conventional approaches described previously (Kim et al., 2005; Wilkinson and Nieto, 1993). Digoxigenin-labeled Shh, Ihh, Gli1 and Apo1a riboprobes were detected by incubating sections overnight at 4°C with alkaline phosphatase-conjugated sheep digoxigenin antibody diluted 1:2000 in blocking solution; color was developed with nitroblue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate (Roche). The following reagents were used for immunofluorescent and immunohistochemical analysis: a mouse or rat monoclonal antibody against Cdx2 (1:20, Biogenex), H/K-ATPase (2B6, 1:1000, MBL), smooth muscle actin (1:2000, Sigma), Muc5AC (45M1, 1:150, Novocastra), BrdU (G3G4, 1:100, Developmental Studies Hybridoma Bank; developed under the auspices of the NICHD and maintained by the University of Iowa, USA) and p63 (1:2000); rabbit antisera against Pdx1 (1:6000, a generous gift from C. Wright, Vanderbilt University, TN, USA), CD31 (1:1000, Santa Cruz), Fli1 (1:500, Labvision), Tuj1 (1:2500, Covance), cleaved caspase 3 (1:200, Cell Signaling), GFP (1:1000, Abcam) and Ki67 (1:2000, Vector Lab). Samples were washed and incubated with biotinylated goat anti-mouse, anti-rabbit or anti-rat IgG, followed by avidin-biotin-peroxidase complex (Vector). Reactions were developed with diaminobenzidine (DAB) hydrochloride (Sigma). For β-gal analysis, ShhCre and Gli1-CreER mice were intercrossed with R26R mice and progeny analyzed for β-gal activity as described previously (Whiting et al., 1991).
Reverse-transcription PCR
Total RNA was extracted using Trizol (Invitrogen), treated with RNase-free DNase (Ambion), and reverse transcribed using oligo-(dT) primers. Relative transcript levels were determined by conventional and SYBR Green real-time PCR (Applied Biosystems) using the primers for Shh (AATGCCTTGGCCATCTCTGT and GCTCGACCCTCATAGTGTAGAGACT), Ihh (GAGCTCACCCCCAACTACAA and TGACAGAGATGGCCAGTGAG), Ptch1 (CTCTGGAGCAGATTTCCAAGG and TGCCGCAGTTCTTTTGAATG), Hhip1 (CCTGTCGAGGCTACTTTTCG and TCCATTGTGAGTCTGGGTCA), Gli1 (GGAAGTCCTATTCACGCCTTGA and CAACCTTCTTGCTCACACATGTAAG), Gli2 (CCTTCTCCAATGCCTCAGAC and GGGGTCTGTGTACCTCTTGG) and GAPDH (AGCCTCGTCCCGTAGACAAAAT and CCGTGAGTGGAGTCATACTGG).
Western blot analysis
Embryonic mouse gastrointestinal tract (stomach and intestine) was isolated, minced and lysed in SDS lysis buffer before loading on SDS-PAGE gels. The following antibodies were used in western blot analysis: a mouse monoclonal antibody against smooth muscle actin (1:2000, Sigma), collagen I (1:2000, Santa Cruz) and an antibody against beta-actin (1:1000, Sigma).
Ex vivo primary mesenchymal culture
E12.5 mouse stomach and intestine mesenchymal cells were isolated as described previously (Kim et al., 2005) and seeded into 12- or 96-well tissue culture dishes. Cells were cultured in alpha-MEM (Mediatech Inc, Herndon, VA, USA) without further serum supplementation. N-terminal hydrophobically modified recombinant N-Shh (Taylor et al., 2001) was added to the cultures at different final concentrations. Recombinant mouse bone morphogenetic protein (Bmp) 4, basic fibroblast growth factor (Fgf) and rat platelet-derived growth factor (Pdgf)-AA were purchased from R&D Systems (Minneapolis, MN, USA) and used according to the manufacturer's instructions. Wnt3a- and N-Shh-conditioned medium were produced by transient transfection of Cos7 cells for 3 days. Cell proliferation was assessed using the CellTiter 96 Non-Radioactive Cell Proliferation Assay kit (Promega, Madison, WI) following the manufacturer's instructions, and confirmed by BrdU staining after exposing cells to 10 μM BrdU (Sigma) for 4 hours.
RESULTS
Removal of Shh and Ihh activities in the early mammalian gut
Shh−/−;Ihh−/− double-mutant embryos arrest around E8.5 (Zhang et al., 2001), precluding an analysis of gastrointestinal (GI) organogenesis. To overcome this limitation, we used the Cre/LoxP system to remove expression of both Shh and Ihh in the early gut endoderm. In this system, Cre recombinase driven from the Shh locus, an allele that is also null for Shh function (Harfe et al., 2004), was used to remove remaining activity from conditional Shh and Ihh alleles. By E10.5, Shh and Ihh were broadly expressed in embryonic intestinal endoderm (Fig. 1). Ihh mRNA extended into the distal stomach but was absent from the proximal stomach and lung buds (Fig. 1D,E), where Shh expression was high (Fig. 1A,B). Although Shh mRNA levels were low in the distal stomach and proximal duodenum (Fig. 1A-C), analysis of intercross progeny between ShhCre/+ and R26R Cre reporter mice (Soriano, 1999) demonstrated that Cre levels are adequate to drive efficient recombination throughout the GI tract endoderm before E10.5 (Fig. 1H,I). ShhCre/+;Ihh+/− mice were intercrossed with a ShhFl/Fl;IhhFl/Fl strain to produce ShhCre/Fl;Ihh−/Fl embryos, potentially removing both Shh and Ihh gut-derived signals. Indeed, a diagnostic PCR assay revealed that both Hh transcripts were absent within the gut tube at E12.5 (see Fig. S1 in the supplementary material). Moreover, no upregulation was observed in several of their target genes, including Ptch1, Gli1 and Hhip, in ShhCre/Fl;Ihh−/Fl embryos (see Fig. S1 in the supplementary material). Importantly, as removal of Shh and Ihh activities requires active transcription of Cre from the Shh locus, a transient pulse of early Hh signaling is probable, and relevant to the resultant phenotype.
Fig. 1.

Removal of Shh and Ihh activities in the developing mouse gut. (A-G) Dynamic Shh and Ihh expression in the gut. Wholemount (A,B,D-F) and tissue-section (C,G) in situ hybridization for Shh (A-C) and Ihh (D-G) in wild-type mouse embryos from E10.5 to E12.5, showing coexpression of Hh ligands and partial overlap in the embryonic stomach (St) and intestine (Int), including small intestine (Sm Int) and large intestine (La Int). A, B, D and F show frontal views of the isolated digestive tract; lung buds (Lu) are included in A, D and E. Inset in E highlights the lack of Ihh expression in the lung buds, esophagus (Es) and rostral stomach. Distinctive fore-stomach and hind-stomach expression is shown in the photomicrographs in C and G. (H,I) Wholemount staining for β-gal in progeny of ShhCre/+ and ROSA26R mice, illustrating efficient recombination throughout the aerodigestive tract at E10.5 (H) and E11.5 (I). Trachea and lung buds are present at the top of both wholemount specimens and blue signal is visible throughout the stomach and intestine. (J-Q) Gross gastrointestinal phenotypes in control and ShhCre/Fl;Ihh−/Fl mutant embryos. Wholemount images of the isolated digestive tract, including stomach and intestine, at E11.5, E12.5, E14.5 and E16.5. Lung buds and esophagus are shown at E11.5 (J,K). As embryos age, the stomach lumen stretches markedly, most likely as a result of an absence of mesenchyme-derived muscular support. The spleen (arrows) is reduced in size at E16.5 (P,Q).
Epithelial and mesenchymal defects in ShhCre/Fl;Ihh−/Fl mutant gut
As the digestive tract of Shh+/Fl;Ihh+/Fl and Shh+/Fl;Ihh−/Fl mice retained Hh signaling and appeared normal, these embryos were used as phenotypically wild-type littermate controls in this study. Gut defects in ShhCre/Fl;IhhFl/+ embryos resembled those previously reported in Shh−/− mice (Ramalho-Santos et al., 2000), whereas absence of Hh signals in ShhCre/Fl;Ihh−/Fl embryos dramatically affected GI development. At E11.5, the digestive tract of ShhCre/Fl;Ihh−/Fl mutant embryos was normal in shape, orientation and location within the embryo, but significantly reduced in size relative to littermate controls (Fig. 1J,K). By E12.5, the digestive tract was reduced to approximately 10% of the length and 33% of the diameter of control littermates, a more profound decrease than overall reduction in embryo size (Fig. 1L,M; data not shown). The deficiency in intestinal growth was evident from the outset, whereas the stomach showed an approximate 1-day delay regional difference that might reflect different periods of transient Hh ligand production before complete removal of signaling activities. In contrast to the significant increase in digestive tract dimensions in control embryos, Shh- and Ihh-null stomach and intestine completely failed to expand and remained nearly as stunted at the end of gestation as they appeared at E12.5 (Fig. 1L-Q). Thus, the effect of endodermal Hh loss was overtly most apparent on organ size.
In spite of markedly reduced dimensions, the histology of Shh- and Ihh-null and control stomachs as late as E11.5 was nearly identical (Fig. 2A,B). The nascent endoderm-derived epithelium comprised viable cells arranged in a well-organized layer resting over an ostensibly healthy mesenchyme of similar thickness to control embryos (Fig. 2A,B). Thereafter, the dramatic expansion of mesenchyme that highlights gut development was notably absent in ShhCre/Fl;Ihh−/Fl stomach. At E12.5, the depth of mesenchyme was reduced (Fig. 2A,B) and this phenotype increased in severity such that little mesenchyme remained between the thin layers of mesentery and epithelium at E16.5 (Fig. 2A,B). Probably as a result of a corresponding loss in mesenchymal integrity, the gut wall stretched significantly between E14.5 and E16.5 (Fig. 1P,Q; Fig. 2A,B) and was surrounded by islands of pancreatic tissue between E16.5 and E18.5 (Fig. 1Q; Fig. 3H). The spleen was hypoplastic (Fig. 1P,Q), a frequent occurrence when stomach morphogenesis is abnormal (Brendolan et al., 2005). In summary, absence of both Hh ligands causes substantial, rapid and progressive mesenchymal loss in the developing GI tract. By contrast, mutants lacking either Ihh or Shh display a milder phenotype indicative of compensatory actions between endoderm-derived Shh and Ihh signals.
Fig. 2.

Digestive tract growth and differentiation in ShhCre/Fl;Ihh−/Fl mutant embryos. (A) Histology of the developing stomach (St) and adjoining spleno (Sp)-pancreatic (Pa) primordium in Shh;Ihh double mutant (right) and control (compound Shh+/Fl;Ihh+/Fl or Shh+/Fl;Ihh−/Fl heterozygote littermates, left) embryos from E11.5 to E16.5. (B) High-magnification micrographs from Shh;Ihh double-mutant (right) and control (left) embryos showing mesenchymal expansion and smooth muscle differentiation in the latter. Progressive attrition of the mesenchymal compartment in mutants is the probable basis for failure of organ growth and lack of epithelial differentiation. Dotted white lines separate gut endodermal epithelium (Ep) from the underlying mesenchyme (Me). Scale bars: 150 μm in A (E16.5, 500 μm); 75 μm in B (E16.5, 500 μm).
Fig. 3.

Epithelial differentiation of ShhCre/Fl;Ihh−/Fl mutant gut. (A) Wholemount of the ShhCre/Fl;Ihh−/Fl digestive tract at E18.5, showing inflation of the stomach as a result of wall thinning. The spleen is reduced in size but liver dimensions are unchanged. (B-D) Histology of ShhCre/Fl;Ihh−/Fl stomach at E18.5, emphasizing loss of wall thickness and regions of limited squamous and glandular differentiation, shown at higher resolution in C and D, respectively. (E-G) The rudimentary stomach mucosa shows isolated areas of foveolar cell differentiation at E18.5, indicated by staining with Periodic Acid Schiff (PAS; E) and Muc5AC (F), largely confined to regions where a few mesenchymal cells remain. H/K-ATPase (H+K+), a parietal cell marker, is expressed in most mucosal cells (G), even when underlying mesenchyme is absent at E18.5. (H-J) Primitive villous differentiation in Shh;Ihh double-mutant intestine at E18.5. Very few intestinal villi (arrows in H) form in the absence of Hh signaling, mainly near residual mesenchymal cells; most of the intestinal epithelium remains flat (arrowheads in H). These few villi carry goblet cells, which stain with Alcian Blue (Alc Bl; I), and express mRNA for the enterocyte marker Apo1a (J). Pa, pancreatic tissue. (K-M) Delineation of gut anteroposterior (A-P) pattern in E12.5 ShhCre/Fl;Ihh−/Fl mutant mouse embryos, as judged by expression of regional epithelial markers. (K) p63, a squamous cell product, reveals a sharp boundary between stomach squamous and glandular mucosae. (L,M) Staining for Pdx1, which marks the stomach (St) antrum, duodenum (Duo) and pancreas (Pa), and the intestine-specific marker Cdx2 in consecutive E12.5 tissue sections further demonstrates correct regional gene expression in absence of Hh signaling.
Despite the evident deficiency in underlying mesenchyme, the endodermal epithelium was largely intact at E11.5 and E12.5 (Fig. 2B). However, epithelial cell numbers declined over time in relation to control embryos and the expected epithelial expansion that normally occurs in mid- and late-gestational stages was not observed in the compound mutant. Furthermore, although squamous, primitive glandular and rudimentary villous epithelia were apparent in the forestomach, distal stomach and intestine, respectively (Fig. 3D-H), mucosal maturation at E18.5, the latest time-point we could evaluate in mutant embryos, was notably impaired.
Several lines of evidence indicate that mesenchymal cell attrition is likely to be the primary defect in Shh and Ihh compund mutants, and that the endodermal anomalies probably follow as a consequence. First, mesenchymal cell loss preceded an overt endodermal phenotype by at least 1 day and probably longer (Fig. 2). Second, differentiation products such as foveolar-cell neutral mucins, reflected in staining with Periodic Acid Schiff (PAS) or Muc5AC antibody, were expressed only in isolated stomach patches adjoining scattered deposits of residual mesenchyme (Fig. 3E,F); only the parietal cell marker H/K-ATPase was expressed in regions devoid of mesenchyme (Fig. 3G). Similarly, the intestine displayed few, rudimentary villi and non-villous endoderm lacked features of maturity. Villi clustered near residual mesenchyme, expressed the enterocyte marker Apo1a and stained with the goblet cell dye Alcian Blue (Fig. 3H-J). Thus, features of normal epithelial development appeared to be restricted to regions that potentially retained the possibility of continued epithelial-mesenchymal interaction. Third, the absence of Ptch1 activity and specific readouts of Hh signaling indicated that the endodermal epithelium was mostly non-responsive to Hh ligands (Kolterud et al., 2009; Madison et al., 2005; Ramalho-Santos et al., 2000; Wang et al., 2002). Thus, gut endoderm-derived Hh ligands signal to underlying mesenchyme (Madison et al., 2005; Ramalho-Santos et al., 2000; Roberts et al., 1995; Sukegawa et al., 2000), which in turn influences endodermal differentiation. Our findings emphasize mutual interdependence of these tissues and the supporting role of Hh ligands in these crucial interactions.
Anterior-posterior gut differentiation in the absence of both Shh and Ihh activities
Hh-mediated regulation of the nuclear receptor COUP-TFII (Nr2f2 – Mouse Genome Informatics) is believed to control anteroposterior (A-P) patterning in the stomach (Takamoto et al., 2005). Surprisingly, despite aberrant GI morphogenesis in ShhCre/Fl;Ihh−/Fl embryos at E12.5, epithelial boundaries were sharply preserved along the A-P axis. Expression of p63 demarcates the fore- and hind-stomach (Fig. 3K), whereas Pdx1 expression distinguishes the antral stomach from the corpus (Fig. 3L) and Cdx2 the presumptive intestine (Fig. 3M). Our genetic strategy requiring transcription from the Shh locus to then eliminate Hh signaling through the activation of Cre inserted at the Shh locus permits transient ligand expression. Thus, the results do not allow an unequivocal assessment of a requirement for Hh signaling in initiating A-P patterning of the gut. However, the sharp preservation of pattern after significant mesenchymal loss, especially in the intestine, suggests that Hh-dependent functions are dispensable for maintaining A-P patterning of specialized gut regions. Unfortunately, the small size of intestines from mutants (Fig. 1K,M,O,Q), coupled with an absence of markers that distinguish small from large intestine in young mouse embryos, precluded critical examination of Hh functions in patterning the distal intestine.
Gut mesenchymal differentiation in the absence of Hh signaling
Despite the importance of mesenchyme in gut morphogenesis, the underlying molecular mechanism governing mesenchymal development is not well understood. To assess the role of Hh signaling in early mesenchyme development, we first examined enteric neurons in the gut of Shh;Ihh double-mutants at E12.5. Enteric neurons are derived from neural crest cells (NCCs) that migrate and colonize the gut mesenchyme during early GI development (Newgreen and Young, 2002). Tissue sections from control and mutant embryos were stained to visualize neuron-specific β-tubulin, Tuj1, a marker of postmitotic neurons. Tuj1-positive cells were present in ShhCre/Fl;Ihh−/Fl mutant gut at E12.5 and E16.5, but their numbers were greatly reduced when compared with control littermates (Fig. 4A,B; see Fig. S2A,B in the supplementary material), consistent with previous reports linking Hh signaling to the regulation of enteric neuron development (Fu et al., 2004; Ramalho-Santos et al., 2000; Sukegawa et al., 2000). Hh signals have been shown to promote vasculogenesis in embryos (Stenman et al., 2008; Vokes et al., 2004). To address this possibility in embryonic gut, we examined blood vessels in ShhCre/Fl;Ihh−/Fl mutants at E12.5. Staining for the vascular endothelial markers CD31 (Fig. 4C,D) and Fli1 (Fig. 4E,F) was similar as in control embryos, and the mutant GI tract had many intact blood vessels containing abundant erythrocytes, without extravasation (Fig. 4G,H). Thus, an early loss of vascular integrity is unlikely to explain the marked mesenchymal growth defect. We also examined the production of extracellular matrix proteins in control and mutant gut at E12.5, and did not detect significant changes in the levels of the extracellular matrix protein collagen I (see Fig. S2F in the supplementary material). The roles for Hh signaling on gut mesenchymal smooth muscle development are discussed later.
Fig. 4.

Mesenchymal differentiation of ShhCre/Fl;Ihh−/Fl mutant gut. (A,B) Neuron-specific β-tubulin, Tuj1, expression in control and ShhCre/Fl;Ihh−/Fl mutant stomachs at E12.5. (C,D) Endothelial marker CD31 immunostaining at E12.5. (E,F) Endothelial marker Fli1 immunostaining in control and ShhCre/Fl;Ihh−/Fl mutant stomachs at E12.5. Dashed white lines separate gut endodermal epithelium (Ep) from the underlying mesenchyme (Me). (G,H) Erythrocytes containing blood vessels are present in the mesenchyme of ShhCre/Fl;Ihh−/Fl mutant stomachs at E12.5 (G) and E16.5 (H).
Hh signaling promotes proliferation of mesenchymal progenitors in the gut
Besides Hh proteins, several other secreted factors (Fgf, Pdgf, Wnt) and transcriptional regulators (Barx1, Foxf1, Foxf2, COUP-TFII, Nkx3.2) have been linked to aspects of gut mesenchyme differentiation (Apelqvist et al., 1997; De Santa Barbara et al., 2005; Geske et al., 2008; Karlsson et al., 2000; Kim et al., 2005; Ormestad et al., 2006; Takamoto et al., 2005; Verzi et al., 2009). However, the major signals governing mesenchymal expansion and organ growth are unclear. The dramatic loss of gut mesenchyme and failure of organ enlargement in Hh double-mutant embryos prompted us to examine Hh requirements in mesenchymal cell growth. When compared at E12.5, stomach mesenchyme in ShhCre/Fl;Ihh−/Fl embryos showed a dramatic reduction in the proliferation marker Ki67 relative to Shh+/Fl;Ihh+/Fl littermates (Fig. 5A,B,E). Thus, Hh signals might act directly as mitogens, as for example in the Shh-mediated expansion of cerebellar granule cells (Rowitch et al., 1999; Wechsler-Reya and Scott, 1999) and Ihh-driven growth of the mammalian skeleton (Long et al., 2001; St-Jacques et al., 1999). Consistent with the presence of an intact vasculature (Fig. 4C-H), we did not observe necrosis in ShhCre/Fl;Ihh−/Fl gut mesenchyme. We also used caspase 3 immunostaining to investigate the possibility of apoptotic cell death in the absence of Hh signaling. We did not detect a significant increase in caspase 3-positive cells or cell fragments in mutant gut mesenchyme at E12.5 (Fig. 5C,D), suggesting that cell apoptosis is not the major contributor to the GI phenotype of ShhCre/Fl;Ihh−/Fl embryos.
Fig. 5.
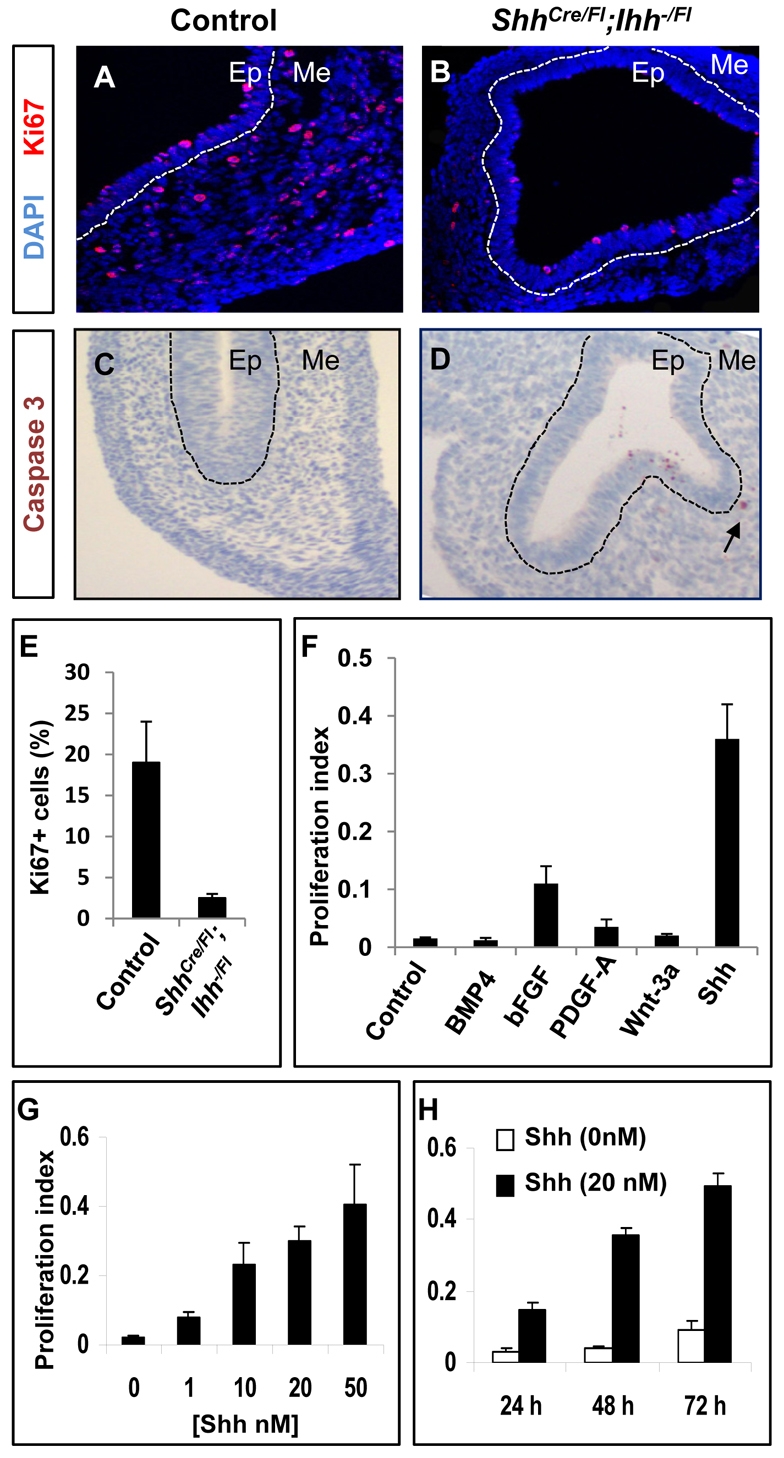
Hh ligands promote proliferation of fetal GI mesenchymal cells. (A,B) Ki67 immunostaining in E12.5 ShhCre/Fl;Ihh−/Fl mutant stomach shows reduced proliferative activity in mesenchymal cells relative to controls. Dashed white lines delineate epithelium (Ep) and mesenchyme (Me). (C,D) cleaved caspase 3 immunostaining in control and Hh mutant stomachs at E12.5. Arrow in D shows the positive caspase 3 staining in Hh mutant mesenchyme. (E) Graph of the percentage of Ki67-expressing mesenchymal cells per 40× field in control and ShhCre/Fl;Ihh−/Fl mutant E12.5 stomachs. Two mice were analyzed per genotype counting a minimum of eight fields per mouse. (F) Proliferative effects of different signaling molecules on fetal stomach mesenchymal cells. Isolated E12.5 stomach mesenchymal cells were cultured for 72 hours as indicated in the presence of N-Shh or Wnt3a conditioned medium, or recombinant Bmp4 (100 ng/ml), bFGF (25 ng/ml) or Pdgf-AA (25 ng/ml). (G,H) Stomach mesenchymal cells were isolated from wild-type E12.5 embryos and cultured in the presence of increasing concentrations (G) of recombinant N-Shh for 3 days, or for different periods in 20 mM N-Shh (H). Proliferative activity was measured by the CellTiter 96 Non-Radioactive Cell Proliferation Assay kit (F-H).
To directly examine the proliferative effects of Hh signaling on mesenchymal cells, we cultured primary E12.5 mouse stomach (Fig. 5F-H) or intestinal (see Fig. S3 in the supplementary material) mesenchymal progenitors in vitro in the presence of Shh or other signals implicated in gut development: Wnt, Fgf, Bmp-4 and Pdgf. In the absence of exogenous recombinant N-terminal hydrophobically modified Shh (N-Shh) (Taylor et al., 2001), primary bulk mesenchymal cells proliferated little but survived for several days. Exposure to N-Shh at increasing concentrations or lengths of time increased cell proliferation substantially, as judged by incorporation of MTT (Fig. 5G,H; see Fig. S3 in the supplementary material) or BrdU (data not shown). By contrast, Wnt3a, Bmp4 and Pdgf-AA showed no significant stimulatory response (Fig. 5F). Basic bFGF (Fgf2) induced a modest but significant mesenchymal proliferation (Fig. 5F), consistent with recent genetic evidence linking Fgf9 signaling through Fgfr1 and 2 to the development of mouse fetal gut mesenchyme (Geske et al., 2008). In summary, both in vivo and in vitro analyses support the conclusion that Hh signaling is a major and necessary mitogenic factor in expansion of embryonic gut mesenchymal progenitors.
Hh pathway activation in gut mesenchyme and smooth muscle development
Gut smooth muscle cells derived from local mesenchymal progenitors are first detected by smooth muscle α-actin (SMA or Acta2) expression around E12 in mouse gut (McHugh, 1995). However, the exact involvement of Hh signaling in GI smooth muscle development has been controversial. Interestingly, we found that, in contrast to wild-type littermates, ShhCre/Fl;Ihh−/Fl mutants do not express SMA at E12.5 (Fig. 6A,B) or later stages (see Fig. S2C-E in the supplementary material), suggesting that Hh signaling is essential for smooth muscle development. However, this view contrasts with a study in the chick which concluded that epithelial-derived Shh inhibits mesenchymal smooth muscle differentiation (Sukegawa et al., 2000).
Fig. 6.

Hh pathway activation and smooth muscle development in GI mesenchyme. (A,B) The absence of smooth muscle actin (SMA) expression in ShhCre/Fl;Ihh−/Fl mutant stomachs at E12.5. (C-G) Hh-responding early progenitor cells give rise to smooth muscle cells and myofibrolasts in gut mesenchyme. β-gal staining (C,D) of E11 and E16.5 Gli1-CreER;R26R gastric mesenchyme following Cre activation by tamoxifen (TM) at E9.5. β-gal and SMA immunostaining (E-G) of Gli1-CreER;R26R gastric mesenchyme at E16.5. (H,I) Wholemount images of E14.5 GI tracts, including stomach (St) and intestine (Int), from R26-SmoM2 and Bapx1-Cre;R26-SmoM2 embryos. (J,K) Smooth muscle actin (SMA) expression in R26-SmoM2 and Bapx1-Cre;R26-SmoM2 embryonic gut at E14.5. SMA immunostaining shows marked expansion of the mesenchymal compartment, including SMA-expressing smooth muscle cells in the Bapx1-Cre;R26-Smom2 stomach mesenchyme. Dashed white lines delineate epithelium (Ep) and mesenchyme (Me).
To address this question further in mice, we first examined the cellular targets of Hh signaling in the gut. Recent studies have established that Hh activity is both necessary and sufficient for Gli1 transcription (Ahn and Joyner, 2004; Lee et al., 1997). Therefore, Gli1 expression provides a faithful and sensitive readout of the Hh signaling activity. In the Gli1-CreER mouse line (Ahn and Joyner, 2004), a tamoxifen-inducible Cre recombinase (CreERT2) was targeted into the Gli1 locus under the control of the endogenous Gli1 promoter. When crossed with a ROSA26 reporter strain (R26R) (Soriano, 1999), administration of tamoxifen indelibly marks Hh-responding cells at a given developmental stage and enables subsequent tracing of the fates of descendant progeny. We examined the cellular fate of Gli1-expressing cells at E11, and later in GI development at E16.5, following a single administration of tamoxifen at E9.5, prior to smooth muscle development (see Fig. S4 in the supplementary material). Early mesenchymal progenitor cells that receive active Hh signals are broadly distributed in the gut mesenchyme at E11 (Fig. 6C). A subset of these early progenitors gave rise at later stages to smooth muscle cells in the outer layers of the gut mesenchyme, and to subepithelial myofibroblasts expressing SMA (Fig. 6D-G). Thus, the mitogenic role of Hh is probably directed, at least in part, at regulating the proliferation and expansion of a pre-smooth muscle progenitor population.
To test this hypothesis, we used a Bapx1-Cre mouse line (Verzi et al., 2009), which drives Cre expression in embryonic gut mesenchyme as early as E9.5, to activate expression of a YFP-tagged, ligand-independent, constitutively active form of Smo (SmoM2-YFP) (Mao et al., 2006; Xie et al., 1998). Although Bapx1-Cre;R26-SmoM2 embryos died around E14.5, the size of the GI tract in these mutants was significantly larger than that of the R26-SmoM2 controls (Fig. 6H,I). More importantly, in agreement with our mesenchymal-mitogen model, the SmoM2-expressing mesenchyme was dramatically expanded (Fig. 6J,K; see Fig. S5 in the supplementary material). In addition, the SMA population was markedly increased (Fig. 6J,K; see Fig. S5 in the supplementary material), although the proper organization of the smooth muscle layer was disrupted. Further, the continued presence of activated SmoM2 in differentiating cells (see Fig. S5J-L in the supplementary material) indicates that Hh pathway activity does not inhibit the progression of mesenchymal progenitors to smooth muscle.
DISCUSSION
Endoderm-derived Hh signals have long been thought to play a crucial role in embryonic gut development. Abnormal Hh signaling is also implicated in the molecular etiology of most common congenital GI malformations (Arsic et al., 2002; de Santa Barbara et al., 2002; Kim et al., 2001). However, the precise function of Hh proteins in GI organogenesis has remained elusive, partly owing to the overlapping distribution and functional redundancy between Shh and Ihh, and partly because of changing requirements at different developmental stages. The phenotypes of single mutant mice might reflect distinct requirements for Shh and Ihh in particular aspects of gut development (Ramalho-Santos et al., 2000) and each might regulate other factors including Bmp4, Foxf1 and 2, Foxl1 and COUP-TFII (Madison et al., 2009; Ormestad et al., 2006; Roberts et al., 1995; Sukegawa et al., 2000). Here, we performed genetic studies in mutant mouse embryos either lacking both Shh and Ihh ligands from early gut endoderm or with constitutive Hh pathway activation in adjacent mesenchyme. Together, these studies provide clear evidence that Shh and Ihh signaling promote the growth of mesenchymal progenitors of the gut. Further, they indicate that primary patterning of the gut into distinct organ segments along the A-P axis is unlikely to rely upon any substantial Hh input. Different experimental approaches will be necessary to unambiguously reconcile whether transient signaling plays any patterning role but we consider this possibility unlikely from the kinetics of our analysis. In regulating growth and differentiation of mesenchyme along the radial axis, Hh signaling facilitates expansion and maturation of the overlying endodermal epithelium through secondary interactions that depend on the mesodermal layer rather than through direct Hh signaling to the epithelium.
The role for Hh signaling in the differentiation of gut mesenchymal progenitors to smooth muscle has been controversial. Although smooth muscle differentiation appears relatively normal in Shh−/− mice, the number of smooth muscle cells was reduced in Ihh−/− mouse intestine, indicative of a positive role (Ramalho-Santos et al., 2000). By contrast, Shh was reported to inhibit SMA expression and smooth muscle differentiation in a chicken GI explant culture (Sukegawa et al., 2000). Our data demonstrate that, in the mouse, Hh signaling is required for, and does not inhibit, smooth muscle differentiation. Hh signaling might directly stimulate muscle differentiation but also probably supports this process indirectly. In this capacity, Hh-mediated signaling might expand a population of Hh-responsive early progenitors to a critical mass that is essential for differentiation, the rate of differentiation being determined by the expansion of this progenitor pool. Mesenchymal progenitors are dramatically reduced on removal of Hh signaling and smooth muscle differentiation is absent in the Hh-depleted gut. By contrast, mesenchymal progenitors are markedly increased on activation of Smo and increased numbers of SMA-positive cells are observed. In this scenario, other cell interactions would more directly regulate differentiation and proper organization of muscle layers. Interestingly, mesenchymal tumors arising in the pancreas from SmoM2-mediated activation show extensive activation of SMA (Tian et al., 2009).
Hh signaling continues within the mesenchyme of the adult GI tract (van den Brink, 2007; van den Brink et al., 2004; Yauch et al., 2008). Although our analysis highlights the early major role of Hh signals in organizing radial growth of the mesodermal compartment, it is reasonable to envision a similar function in postnatal gut homeostasis. With growing evidence for Hh pathway activity in digestive tract tumorigenesis (Olive et al., 2009; Rubin and de Sauvage, 2006; Taipale and Beachy, 2001; Tian et al., 2009; Yauch et al., 2008), paracrine Hh signals also serve to maintain or promote tumor stroma (Olive et al., 2009; Tian et al., 2009; Yauch et al., 2008), as they do for the normal cellular counterpart in the mammalian gut. Thus, a more detailed mechanistic insight into the interactions we report here might help in the future design of tailored approaches to tumor therapy.
Supplementary Material
Acknowledgements
This work was supported by an NIH Javits award (R37 NS033642) to A.P.M. from NINDS and R01 DK081113 to R.A.S. from NIDDK. Work in J.M.'s laboratory is supported by the Charles H. Hood Foundation and J.M. is a member of the UMass DERC (DK32520). Core resources supported by the Diabetes Endocrinology Research Center grant DK32520 were also used. We thank Dr Alex Joyner and Warren Zimmer for providing the Gli1-CreER and Bapx1-Cre mouse lines. Hydrophobically modified N-Shh protein was a generous gift from Dr R. Blake Pepinsky (Biogen). We also thank Jill McMahon and Julie Brooks for their technical support and members of the Mao, Shivdasani and McMahon groups for helpful discussions. J.M., B.-M.K. and M.R. performed the research; all authors designed and interpreted the studies, analyzed the data and drafted the manuscript. Deposited in PMC for release after 12 months.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.044586/-/DC1
References
- Ahn S., Joyner A. L. (2004). Dynamic changes in the response of cells to positive hedgehog signaling during mouse limb patterning. Cell 118, 505-516 [DOI] [PubMed] [Google Scholar]
- Apelqvist A., Ahlgren U., Edlund H. (1997). Sonic hedgehog directs specialised mesoderm differentiation in the intestine and pancreas. Curr. Biol. 7, 801-804 [DOI] [PubMed] [Google Scholar]
- Arsic D., Qi B. Q., Beasley S. W. (2002). Hedgehog in the human: a possible explanation for the VATER association. J. Paediatr. Child Health 38, 117-121 [DOI] [PubMed] [Google Scholar]
- Aubin J., Dery U., Lemieux M., Chailler P., Jeannotte L. (2002). Stomach regional specification requires Hoxa5-driven mesenchymal-epithelial signaling. Development 129, 4075-4087 [DOI] [PubMed] [Google Scholar]
- Bhowmick N. A., Neilson E. G., Moses H. L. (2004). Stromal fibroblasts in cancer initiation and progression. Nature 432, 332-337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitgood M. J., McMahon A. P. (1995). Hedgehog and Bmp genes are coexpressed at many diverse sites of cell-cell interaction in the mouse embryo. Dev. Biol. 172, 126-138 [DOI] [PubMed] [Google Scholar]
- Brendolan A., Ferretti E., Salsi V., Moses K., Quaggin S., Blasi F., Cleary M. L., Selleri L. (2005). A Pbx1-dependent genetic and transcriptional network regulates spleen ontogeny. Development 132, 3113-3126 [DOI] [PubMed] [Google Scholar]
- Chuang P. T., McMahon A. P. (1999). Vertebrate Hedgehog signalling modulated by induction of a Hedgehog-binding protein. Nature 397, 617-621 [DOI] [PubMed] [Google Scholar]
- de Santa Barbara P., van den Brink G. R., Roberts D. J. (2002). Molecular etiology of gut malformations and diseases. Am. J. Med. Genet. 115, 221-230 [DOI] [PubMed] [Google Scholar]
- de Santa Barbara P., Williams J., Goldstein A. M., Doyle A. M., Nielsen C., Winfield S., Faure S., Roberts D. J. (2005). Bone morphogenetic protein signaling pathway plays multiple roles during gastrointestinal tract development. Dev. Dyn. 234, 312-322 [DOI] [PubMed] [Google Scholar]
- Echelard Y., Epstein D. J., St-Jacques B., Shen L., Mohler J., McMahon J. A., McMahon A. P. (1993). Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 75, 1417-1430 [DOI] [PubMed] [Google Scholar]
- Fu M., Lui V. C., Sham M. H., Pachnis V., Tam P. K. (2004). Sonic hedgehog regulates the proliferation, differentiation, and migration of enteric neural crest cells in gut. J. Cell Biol. 166, 673-684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geske M. J., Zhang X., Patel K. K., Ornitz D. M., Stappenbeck T. S. (2008). Fgf9 signaling regulates small intestinal elongation and mesenchymal development. Development 135, 2959-2968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffen K., Kedinger M., Simon-Assmann P. (1987). Mesenchyme-dependent differentiation of epithelial progenitor cells in the gut. J. Pediatr. Gastroenterol. Nutr. 6, 14-23 [DOI] [PubMed] [Google Scholar]
- Harfe B. D., Scherz P. J., Nissim S., Tian H., McMahon A. P., Tabin C. J. (2004). Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell 118, 517-528 [DOI] [PubMed] [Google Scholar]
- Hooper J. E., Scott M. P. (2005). Communicating with Hedgehogs. Nat. Rev. Mol. Cell Biol. 6, 306-317 [DOI] [PubMed] [Google Scholar]
- Ingham P. W., McMahon A. P. (2001). Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 15, 3059-3087 [DOI] [PubMed] [Google Scholar]
- Kang S., Graham J. M., Jr Olney, A. H. and Biesecker L. G. (1997). GLI3 frameshift mutations cause autosomal dominant Pallister-Hall syndrome. Nat. Genet. 15, 266-268 [DOI] [PubMed] [Google Scholar]
- Karlsson L., Lindahl P., Heath J. K., Betsholtz C. (2000). Abnormal gastrointestinal development in PDGF-A and PDGFR-(alpha) deficient mice implicates a novel mesenchymal structure with putative instructive properties in villus morphogenesis. Development 127, 3457-3466 [DOI] [PubMed] [Google Scholar]
- Kedinger M., Duluc I., Fritsch C., Lorentz O., Plateroti M., Freund J. N. (1998). Intestinal epithelial-mesenchymal cell interactions. Ann. New York Acad. Sci. 859, 1-17 [DOI] [PubMed] [Google Scholar]
- Kim B. M., Buchner G., Miletich I., Sharpe P. T., Shivdasani R. A. (2005). The stomach mesenchymal transcription factor Barx1 specifies gastric epithelial identity through inhibition of transient Wnt signaling. Dev. Cell 8, 611-622 [DOI] [PubMed] [Google Scholar]
- Kim J., Kim P., Hui C. C. (2001). The VACTERL association: lessons from the Sonic hedgehog pathway. Clin. Genet. 59, 306-315 [DOI] [PubMed] [Google Scholar]
- Kolterud A., Grosse A. S., Zacharias W. J., Walton K. D., Kretovich K. E., Madison B. B., Waghray M., Ferris J. E., Hu C., Merchant J. L., et al. (2009). Paracrine Hedgehog signaling in stomach and intestine: new roles for hedgehog in gastrointestinal patterning. Gastroenterology 137, 618-628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Platt K. A., Censullo P., Ruiz i Altaba A. (1997). Gli1 is a target of Sonic hedgehog that induces ventral neural tube development. Development 124, 2537-2552 [DOI] [PubMed] [Google Scholar]
- Lewis P. M., Dunn M. P., McMahon J. A., Logan M., Martin J. F., St-Jacques B., McMahon A. P. (2001). Cholesterol modification of sonic hedgehog is required for long-range signaling activity and effective modulation of signaling by Ptc1. Cell 105, 599-612 [DOI] [PubMed] [Google Scholar]
- Litingtung Y., Lei L., Westphal H., Chiang C. (1998). Sonic hedgehog is essential to foregut development. Nat. Genet. 20, 58-61 [DOI] [PubMed] [Google Scholar]
- Long F., Zhang X. M., Karp S., Yang Y., McMahon A. P. (2001). Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development 128, 5099-5108 [DOI] [PubMed] [Google Scholar]
- Lum L., Beachy P. A. (2004). The Hedgehog response network: sensors, switches, and routers. Science 304, 1755-1759 [DOI] [PubMed] [Google Scholar]
- Madison B. B., Braunstein K., Kuizon E., Portman K., Qiao X. T., Gumucio D. L. (2005). Epithelial hedgehog signals pattern the intestinal crypt-villus axis. Development 132, 279-289 [DOI] [PubMed] [Google Scholar]
- Madison B. B., McKenna L. B., Dolson D., Epstein D. J., Kaestner K. H. (2009). FoxF1 and FoxL1 link hedgehog signaling and the control of epithelial proliferation in the developing stomach and intestine. J. Biol. Chem. 284, 5936-5944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J., Login K. L., Rakhlin E. Y., Thayer S. P., Bronson R. T., Rowitch D., McMahon A. P. (2006). A novel somatic mouse model to survey tomorigenic potential applied to the Hedgehog pathway. Cancer Res. 66, 10171-10178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh K. M. (1995). Molecular analysis of smooth muscle development in the mouse. Dev. Dyn. 204, 278-290 [DOI] [PubMed] [Google Scholar]
- McMahon A. P., Ingham P. W., Tabin C. J. (2003). Developmental roles and clinical significance of hedgehog signaling. Curr. Top Dev. Biol. 53, 1-114 [DOI] [PubMed] [Google Scholar]
- Nanni L., Ming J. E., Bocian M., Steinhaus K., Bianchi D. W., Die-Smulders C., Giannotti A., Imaizumi K., Jones K. L., Campo M. D., et al. (1999). The mutational spectrum of the sonic hedgehog gene in holoprosencephaly: SHH mutations cause a significant proportion of autosomal dominant holoprosencephaly. Hum. Mol. Genet. 8, 2479-2488 [DOI] [PubMed] [Google Scholar]
- Newgreen D., Young H. M. (2002). Enteric nervous system: development and developmental disturbances-part 1. Pediatr. Dev. Pathol. 5, 224-247 [DOI] [PubMed] [Google Scholar]
- Olive K. P., Jacobetz M. A., Davidson C. J., Gopinathan A., McIntyre D., Honess D., Madhu B., Goldgraben M. A., Caldwell M. E., Allard D., et al. (2009). Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457-1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormestad M., Astorga J., Landgren H., Wang T., Johansson B. R., Miura N., Carlsson P. (2006). Foxf1 and Foxf2 control murine gut development by limiting mesenchymal Wnt signaling and promoting extracellular matrix production. Development 133, 833-843 [DOI] [PubMed] [Google Scholar]
- Ramalho-Santos M., Melton D. A., McMahon A. P. (2000). Hedgehog signals regulate multiple aspects of gastrointestinal development. Development 127, 2763-2772 [DOI] [PubMed] [Google Scholar]
- Razzaque M. S., Soegiarto D. W., Chang D., Long F., Lanske B. (2005). Conditional deletion of Indian hedgehog from collagen type 2alpha1-expressing cells results in abnormal endochondral bone formation. J. Pathol. 207, 453-461 [DOI] [PubMed] [Google Scholar]
- Roberts D. J., Johnson R. L., Burke A. C., Nelson C. E., Morgan B. A., Tabin C. (1995). Sonic hedgehog is an endodermal signal inducing Bmp-4 and Hox genes during induction and regionalization of the chick hindgut. Development 121, 3163-3174 [DOI] [PubMed] [Google Scholar]
- Roberts D. J., Smith D. M., Goff D. J., Tabin C. J. (1998). Epithelial-mesenchymal signaling during the regionalization of the chick gut. Development 125, 2791-2801 [DOI] [PubMed] [Google Scholar]
- Rowitch D. H., St-Jacques B., Lee S. M., Flax J. D., Snyder E. Y., McMahon A. P. (1999). Sonic hedgehog regulates proliferation and inhibits differentiation of CNS precursor cells. J. Neurosci. 19, 8954-8965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin L. L., de Sauvage F. J. (2006). Targeting the Hedgehog pathway in cancer. Nat. Rev. Drug Discov. 5, 1026-1033 [DOI] [PubMed] [Google Scholar]
- Soriano P. (1999). Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21, 70-71 [DOI] [PubMed] [Google Scholar]
- St-Jacques B., Hammerschmidt M., McMahon A. P. (1999). Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 13, 2072-2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenman J. M., Rajagopal J., Carroll T. J., Ishibashi M., McMahon J., McMahon A. P. (2008). Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science 322, 1247-1250 [DOI] [PubMed] [Google Scholar]
- Sukegawa A., Narita T., Kameda T., Saitoh K., Nohno T., Iba H., Yasugi S., Fukuda K. (2000). The concentric structure of the developing gut is regulated by Sonic hedgehog derived from endodermal epithelium. Development 127, 1971-1980 [DOI] [PubMed] [Google Scholar]
- Taipale J., Beachy P. A. (2001). The Hedgehog and Wnt signalling pathways in cancer. Nature 411, 349-354 [DOI] [PubMed] [Google Scholar]
- Takamoto N., You L. R., Moses K., Chiang C., Zimmer W. E., Schwartz R. J., DeMayo F. J., Tsai M. J., Tsai S. Y. (2005). COUP-TFII is essential for radial and anteroposterior patterning of the stomach. Development 132, 2179-2189 [DOI] [PubMed] [Google Scholar]
- Taylor F. R., Wen D., Garber E. A., Carmillo A. N., Baker D. P., Arduini R. M., Williams K. P., Weinreb P. H., Rayhorn P., Hronowski X., et al. (2001). Enhanced potency of human Sonic hedgehog by hydrophobic modification. Biochemistry 40, 4359-4371 [DOI] [PubMed] [Google Scholar]
- Tian H., Callahan C. A., DuPree K. J., Darbonne W. C., Ahn C. P., Scales S. J., de Sauvage F. J. (2009). Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. USA 106, 4254-4259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Brink G. R. (2007). Hedgehog signaling in development and homeostasis of the gastrointestinal tract. Physiol. Rev. 87, 1343-1375 [DOI] [PubMed] [Google Scholar]
- van den Brink G. R., Bleuming S. A., Hardwick J. C., Schepman B. L., Offerhaus G. J., Keller J. J., Nielsen C., Gaffield W., van Deventer S. J., Roberts D. J., et al. (2004). Indian Hedgehog is an antagonist of Wnt signaling in colonic epithelial cell differentiation. Nat. Genet. 36, 277-282 [DOI] [PubMed] [Google Scholar]
- Verzi M. P., Stanfel M. N., Moses K. A., Kim B. M., Zhang Y., Schwartz R. J., Shivdasani R. A., Zimmer W. E. (2009). Role of the homeodomain transcription factor Bapx1 in mouse distal stomach development. Gastroenterology 136, 1701-1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vokes S. A., Yatskievych T. A., Heimark R. L., McMahon J., McMahon A. P., Antin P. B., Krieg P. A. (2004). Hedgehog signaling is essential for endothelial tube formation during vasculogenesis. Development 131, 4371-4380 [DOI] [PubMed] [Google Scholar]
- Wang L. C., Nassir F., Liu Z. Y., Ling L., Kuo F., Crowell T., Olson D., Davidson N. O., Burkly L. C. (2002). Disruption of hedgehog signaling reveals a novel role in intestinal morphogenesis and intestinal-specific lipid metabolism in mice. Gastroenterology 122, 469-482 [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya R. J., Scott M. P. (1999). Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 22, 103-114 [DOI] [PubMed] [Google Scholar]
- Whiting J., Marshall H., Cook M., Krumlauf R., Rigby P. W., Stott D., Allemann R. K. (1991). Multiple spatially specific enhancers are required to reconstruct the pattern of Hox-2.6 gene expression. Genes Dev. 5, 2048-2059 [DOI] [PubMed] [Google Scholar]
- Wilkinson D. G., Nieto M. A. (1993). Detection of messenger RNA by in situ hybridization to tissue sections and whole mounts. Methods Enzymol. 225, 361-373 [DOI] [PubMed] [Google Scholar]
- Xie J., Murone M., Luoh S. M., Ryan A., Gu Q., Zhang C., Bonifas J. M., Lam C. W., Hynes M., Goddard A., et al. (1998). Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 391, 90-92 [DOI] [PubMed] [Google Scholar]
- Yauch R. L., Gould S. E., Scales S. J., Tang T., Tian H., Ahn C. P., Marshall D., Fu L., Januario T., Kallop D., et al. (2008). A paracrine requirement for hedgehog signalling in cancer. Nature 455, 406-410 [DOI] [PubMed] [Google Scholar]
- Zhang X. M., Ramalho-Santos M., McMahon A. P. (2001). Smoothened mutants reveal redundant roles for Shh and Ihh signaling including regulation of L/R asymmetry by the mouse node. Cell 105, 781-792 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.