

Abstract
Structural biology of membrane proteins has rapidly evolved into a new frontier of science. Although solving the structure of a membrane protein with atomic-level resolution is still a major challenge, separated local field (SLF) NMR spectroscopy has become an invaluable tool in obtaining structural images of membrane proteins under physiological conditions. Recent studies have demonstrated the use of rotating-frame SLF techniques to accurately measure strong heteronuclear dipolar couplings between directly bonded nuclei. However, in these experiments, all weak dipolar couplings are suppressed. On the other hand, weak heteronuclear dipolar couplings can be measured using laboratory-frame SLF experiments, but only at the expense of spectral resolution for strongly dipolar coupled spins. In the present study, we implemented 2D PELF (proton-evolved local-field) pulse sequences using either COMPOZER-CP (composite zero degree cross-polarization) or WIM (windowless isotropic mixing) for magnetization transfer. These PELF sequences can be used for the measurement of a broad range of heteronuclear dipolar couplings, allowing for a complete mapping of protein dynamics in a lipid bilayer environment. Experimental results from magnetically-aligned bicelles containing uniformly 15N-labeled cytochrome b5 are presented and theoretical analyses of the new PELF sequences are reported. Our results suggest that the PELF based experimental approaches will have a profound impact on solid-state NMR spectroscopy of membrane proteins and other membrane-associated molecules in magnetically-aligned bicelles.
Keywords: Bicelles, Lipids, Dipolar coupling, order parameter
INTRODUCTION
Membrane proteins, which constitute a third of all proteins in nature, regulate and direct a host of important cellular processes, ranging from transport of nutrients to generation of energy. Membrane proteins exhibit molecular motions on a wide range of time-scales under physiological conditions and insight into their motions is the key to understanding their functions. Molecular motions on the order of milliseconds are frequently observed in many essential and fascinating biological processes ranging from ligand binding to enzyme catalysis.1–4 Fast timescale side chain dynamics can provide clues into the mechanism by which ions migrate through membrane protein channels. Many membrane-associated enzymes, such as microsomal cytochrome P450, exhibit dynamic structural plasticity that enables the binding of substrates with various sizes and stereochemistry.5, 6 Over the years, significant efforts have been devoted to understand the intricate molecular dynamics of soluble proteins; however, complete characterization of membrane protein dynamics continues to be a challenge.7–14
In principle, solid-state NMR spectroscopy is a versatile tool for probing molecular dynamics occurring on timescales that vary from picoseconds (bond libration) to days (protein aggregation). These motions often influence various NMR observables such as chemical shift anisotropy, dipolar coupling and quadrupole coupling. Standard high-resolution solution NMR techniques have provided valuable insights into membrane protein structure-function relationships, but information regarding protein dynamics in lipid bilayers remains elusive.7–11, 15 The slow tumbling rate of most lipid membrane systems leads to various undesirable line broadening effects that make solution NMR techniques not applicable. Therefore, most solution NMR studies of membrane protein are restricted to proteins reconstituted in detergent micelles in cases where they may retain their functions and tumble rapidly enough to average out various undesirable line broadening effects. However, the dynamics observed in these systems can be different from those in a lipid bilayer environment. Solid-state NMR can overcome the undesirable line broadening effects associated with membrane proteins in lipid bilayers. High-resolution solid-state NMR requires the sample to be mechanically aligned or spun at the magic angle (54.7°) relative to the external magnetic field direction to suppress the line-broadening effects associated with nuclear spin interactions and allow for the reconstitution and study of membrane proteins in their native environment.16–19
Separated local field (SLF) spectroscopy of aligned samples holds a great promise as a practical tool for studying membrane proteins in lipid bilayers.20–23 Alignment media used for membrane protein structural studies can be made to closely mimic the native membrane and experiments can be carried out under physiological conditions.13, 24–26 Indeed, SLF has been successful in providing detailed structural information of α-helical membrane proteins in well-hydrated bilayer environments. A typical SLF spectrum correlates heteronuclear dipolar couplings between low-γ nuclei and their directly bonded 1H nuclei (such as 15N-1H) with the CSAs of the low-γ nuclei (15N, in this case). Importantly, these NMR observables are sensitive to the whole body motions of a protein, and are therefore capable of providing detailed dynamical and structural information.27–29 In order to extract dynamical and structural information from an SLF spectrum, one must be able to accurately measure a broad range of dipolar couplings between directly bonded spin pairs. However, in most commonly used rotating-frame SLF protocols, such as HIMSELF (Heteronuclear Isotropic Mixing Spin Exchange via Local Field), PISEMA (Polarization Inversion Spin Exchange at the Magic Angle) and SAMMY, various weak heteronuclear dipolar couplings are effectively suppressed in favor of simple spectral line-shape and improved resolution in the indirect dipolar coupling dimension.30, 31 Laboratory-frame SLF experiments such as PELF (proton evolved local field) provide high sensitivity and resolution for loop regions, soluble domains, and mobile parts of membrane proteins such that structural and dynamical information can be extracted from these regions in a SLF spectrum.31, 32 Also, PELF is capable of resolving a broad range of dipolar couplings from directly bonded spin pair, which is absolutely necessary for determining both dynamics and structures of membrane proteins. Despite these unique advantages, applications of PELF in structural biology remain to be established.
In the present work, we demonstrate the use of the PELF method to extract dynamical and structural information from cytochrome b5 (Cytb5), a membrane-anchored protein. Cytochrome b5 consists of a transmembrane domain near the C-terminal, a water-soluble heme-containing domain, and a linker region that connects these two domains. Functional studies have shown that the length of the linker region is crucial for cytochrome b5 to interact with and donate an electron to activate cytochrome P450.33 In the present study, a mutant of cytochrome b5 (mutant-Cytb5), which lacks 8 residues in its linker region, is used to demonstrate the efficiency of the PELF sequence. The amino acid sequence of a wild-type rabbit cytochrome b5 is given in Figure 1. A previous solid-state NMR study revealed the various domains of this protein display motions occurring on a wide range of timescales.23 Since the mutant protein used in this study lacks 8 residues in the linker region, the motion of the soluble domain should be significantly reduced such that small, yet measurable, dipolar couplings can be observed. Therefore, it is an ideal system to firmly establish and demonstrate the effectiveness of using the PELF sequence to study the structure and dynamics of membrane proteins in magnetically-aligned bicelles.
Figure 1. The amino acid sequence of a full-length wild-type rabbit cytochrome b5.

High-resolution structures of the soluble domain of the protein have been reported from solution NMR and X-ray crystallography studies, whereas the structure of the full-length protein is unknown as it has not been amenable for studies using high-resolution methods. Secondary structures such as α-helix (blue) and βsheet (green) are indicated based on previous studies. The 8 amino acids that were deleted from the linker region of the wild-type cytochrome b5 to obtain a mutant are shown in red.
THEORY
2D Proton evolved local field
A typical PELF radio frequency (RF) pulse sequence is given in Figure 2. In the PELF sequence, the transverse magnetizations of protons evolve under heteronuclear dipolar coupling (with an evolution frequency ΩIS) during the incrementable t1 period, while chemical shifts of protons and 1H-1H homonuclear dipolar couplings are suppressed.32, 34, 35 After the t1 period, the transverse 1H magnetization is transferred to the rare nucleus (referred to as X, often either 13C or 15N) for detection as shown in Figure 2.
Figure 2. Radio frequency PELF pulse sequences for 2D SLF spectroscopy of solids.
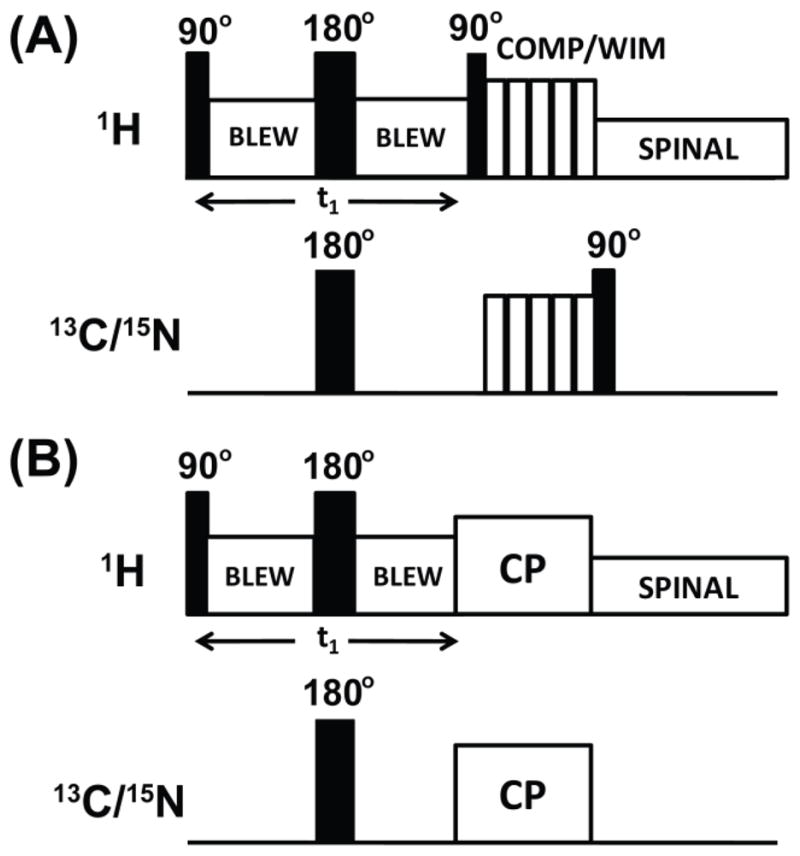
In these pulse sequences, proton homonuclear decoupling in the t1 period is achieved using the BLEW-12 sequence and SPINAL-64 is used to decouple protons during acquisition. Proton transverse magnetization after the t 1 period is transferred to X nuclei using the COMPOZER-CP or WIM pulse sequence. (A) or by the standard cross-polarization sequence (B). A supercycled COMPOZER sequence consists of a series of 360° pulses with phases x, −x, y, −y, −y, y, −x, x. The WIM sequence consists of a series of 90° pulses of phases −x, y, −x, −x, y, −x, x, y, x, x, y, x. Since the z-component of proton magnetization is transferred as the z-component of 15N or 13C spin magnetization under both WIM and COMPOZER-CP sequences, a 90° read pulse on the 15N or 13C channel is used for detection.
Since there is no homonuclear dipolar coupling between rare X nuclei and each 1H spin is coupled to a single X nuclear spin, for every resolvable X resonance, either a single doublet corresponding to a single 1H-X dipolar coupling or a simple superposition of doublets corresponding to different sets of 1H-X pairs is observed in the indirect frequency dimension of a 2D PELF spectrum. The elegant PELF technique thereby avoids the complicated multiplet-type spectral pattern typically observed in the indirect dimension of other SLF sequences where X spin transverse magnetizations are allowed to evolve under the dipolar couplings to many 1H nuclei.31, 32 In other words, after dipolar coupling between a rare X nucleus and the 1H nuclei has evolved for a period of t1, the relevant term of the density operator contributing to the detected signal is given as
| [1] |
Magnetization transfer in a PELF pulse sequence can be accomplished by any of the following means, depending on sample conditions and the spin system dynamics: a standard cross-polarization (CP) scheme, a CP scheme based on the Lee-Goldburg homonuclear dipolar decoupling, WIM (Windowless Isotropic Mixing), COMPOZER-CP (COMPOsite ZERo – Cross Polarization), or INEPT (Insensitive Nuclei Enhanced by Polarization Transfer)/RINEPT (Refocused Insensitive Nuclei Enhanced by Polarization Transfer). The resolution and sensitivity of a 2D PELF spectrum critically depends on the details of the pulse sequence used to transfer the 1H transverse magnetization.31, 32 For example, a short transfer time is required to suppress 1H spin diffusion, which manifests as a broad zero-frequency peak in the indirect dipolar coupling frequency dimension that severely obscures the spectrum.32 Furthermore, for most solid-state systems the rapid rotating-frame relaxation (T1ρ) of protons could hamper the efficiency of magnetization transfer, particularly in the standard CP sequence where the transverse magnetization is transferred from protons to less-sensitive nuclei like 13C or 15N. Since for most systems the proton T1 (spin lattice relaxation time) is much longer than T1ρ, it is advantageous to use a magnetization transfer scheme whereby the transfer of magnetization occurs along the z-axis. Currently, two schemes, WIM and COMPOZER-CP, can render such a magnetization transfer while providing homonuclear decoupling to dampen the 1H-1H spin diffusion process.31, 36 It may be noted that the homonuclear decoupling of WIM sequence is more effective than that of the COMPOZER-CP. Therefore, in this study, the performances of these two schemes (COMPOZER-CP and WIM) in PELF sequences are compared with that of the standard CP. The other two magnetization transfer sequences, namely Lee-Goldburg-CP and INEPT, are excluded from this study. Lee-Goldburg-CP is highly sensitive to the frequency offset and may not be appropriate as a general magnetization transfer protocol.31 Furthermore, magnetization transfer by INEPT sequences only occurs in highly mobile samples and is therefore limited in their applicability to only the mobile regions of membrane proteins.23
MATERIALS AND METHODS
Preparation of magnetically-aligned bicelles
A detailed procedure on the preparation and characterization of magnetically-aligned bicelles containing cytochrome b5 can be found in our previous publication5 while it is outlined very briefly here. In general, the quantity of lipids in bicelles are calculated according to the q = [DMPC]/[DHPC] ratio and in the present study bicelles with a q value of 3.5 was used. Lipid-detergent mixture was hydrated with an appropriate amount of HEPES buffer (10 mM HEPES, pH = 7.0) to obtain the desired wt% of lipids, typically ~ 50 wt%, for the reconstitution of a mutant cytrochrome b5. The solution of lipid-detergent mixture was gently vortexed after every freeze/thaw cycle until an optically clear solution was obtained.
Reconstitution of mutant-cytrochrome b5 in bicelles
Uniformly 15N-labeled mutant-cytochrome b5 expressed and purified according to previous protocols5 was concentrated into a stock solution of 4.3 mM. The amount of protein used in each experiment was calculated according to the desired lipid to protein ratio. A 300:1 DMPC:protein ratio was used in this study. The protein was reconstituted into bicelles by adding an appropriate amount of the protein stock solution to a bicelle solution and the final lipid concentration was diluted to 25 wt% and the final volume was 160 μL. The solution was gently vortexed to ensure proper mixing of protein and lipids and was subsequently transfer to an NMR rotor.
NMR spectroscopy
NMR experiments were carried out on a Varian Infinity-600 MHz solid-state NMR spectrometer using a 4 mm triple-resonance magic angle spinning (MAS) probe under static sample conditions. The 1H and 15N resonance frequencies were 599.8 and 60.78 MHz respectively. About 40 mg of lipids mixed with 3 mg of mutant cytochrome b5 were loaded in a 4 mm NMR glass tube of 4 cm length, and the tube was closed tightly with a Teflon tape to avoid any leakage inside the probe and completely closed with a cap. The sample was then equilibrated for about 30 minutes inside the magnet at the measurement temperature prior to the signal acquisition. The 2D PELF spectra were obtained using the following parameters: 5 μs 90° pulse, 40 kHz 15N sweep width, 28 t1 increments, 3000 scans, a 3 s recycling delay, and a 25 kHz 1H decoupling. A 50 kHz BLEW-12 (Burum, Linder, Ernst Windowless)37 pulse sequence was used for homonuclear decoupling during the t1 period and the SPINAL-6438 pulse sequence was used to decouple protons during signal acquisition in the t2 period.
Simulations and data analysis
Numerical simulations were performed using the program SPINEVOLUTION.39 The simulated spectra and CP buildup curve were plotted using SigmaPlot 9.0. The details of the simulation are described in the results section below. All numerical simulations were performed on a 2.0 GHz Intel Quad Core HP desktop running Window Vista.
RESULTS AND DISCUSSIONS
Simulations of PELF spectra under different magnetization transfer schemes
The influence of magnetization transfer schemes on the resolution and sensitivity of a 2D PELF spectrum, specifically in the heteronuclear dipolar coupling dimension, is investigated through numerical simulations on a hypothetical spin system defined in Figure 3 using the SPINEVOLUTION software.39 In this spin system, an NH group is weakly coupled to an neighboring proton such that it demonstrates the case of a biomolecules embedded in the membranes and/or to reflect motionally averaged N-H dipolar couplings as found in relatively mobile regions of a protein. N-H dipolar coupling slices taken from the indirect dimension of the simulated SLF spectrum are shown in Figures 4. As illustrated by our numerical simulations (Figures 4 and 5), not only the choice of the magnetization transfer scheme but also the duration of the magnetization transfer significantly influence the resolution of a PELF spectrum. With a transfer contact time of approximately 250 μs, all of the magnetization transfer schemes give identical dipolar doublets corresponding to the large dipolar coupling (D1). As the transfer time is increased, significant differences between the spectra are observed, which is a direct consequence of the differences among the magnetization transfer schemes employed (see figure 5). Figure 5 shows that the intensity of the strong dipolar coupling (the outer doublet in figure 4) oscillates strongly as a function of magnetization transfer time when WIM or regular CP was used as a magnetization transfer sequence. The WIM sequence offers the highest homonuclear decoupling and therefore allows for the detection and isolation of the weak dipolar coupling D2 in the presence of the strong coupling D1, which could be suppressed by the choice of an appropriate magnetization transfer time. On the other hand, as the strength of the homonuclear decoupling decreases, the spectral resolution deteriorates because of the obscuring effect of spin diffusion. Spin diffusion manifests as a zero frequency artifact in the indirect dimension (see figures 4(E) and 4(F)). As a result, an intense central peak at the zero frequency is observed, obstructing the measurements of weak dipolar couplings. Also, for a long contact time, D1 is more effectively suppressed when COMPOZER-CP or WIM magnetization transfer schemes are used, which is a consequence of the spin diffusion facilitated by the proton bath. Therefore, the use of these magnetization transfer schemes allow for broader optimal contact time conditions. In summary, from these numerical simulations, we may conclude the following: 1) significant resolution is gained by using a magnetization transfer scheme that suppresses 1H-1H interactions, 2) the main contributors to the central peak observed in the 2D PELF spectrum using the COMPOZER-CP and the regular CP originate from the proton spin diffusion process, and 3) weak 1H-1H dipolar coupling ensures that the amount of 1H magnetization transferred to an X nucleus builds up steadily, which enables a broad range of transfer times to be used. In contrast, when a 1H-1H dipolar coupling is completely eliminated, magnetization is transferred back and forth between insensitive X nuclei and sensitive 1H nuclei, and therefore the transfer time has to be carefully calibrated. This makes PELF-COMPOZER the most generally suitable technique for measuring weak X-1H dipolar couplings (X may be 13C or 15N). Thus, in order to acquire a highly resolved SLF spectrum of an aligned membrane protein using the 2D PELF pulse sequence, one must use a magnetization transfer scheme that suppresses 1H-1H dipolar coupling interactions while using the shortest contact time that permits the observation of the desired range of dipolar coupling frequencies.
Figure 3. A schematic of a hypothetical spin system to evaluate the PELF pulse sequence.
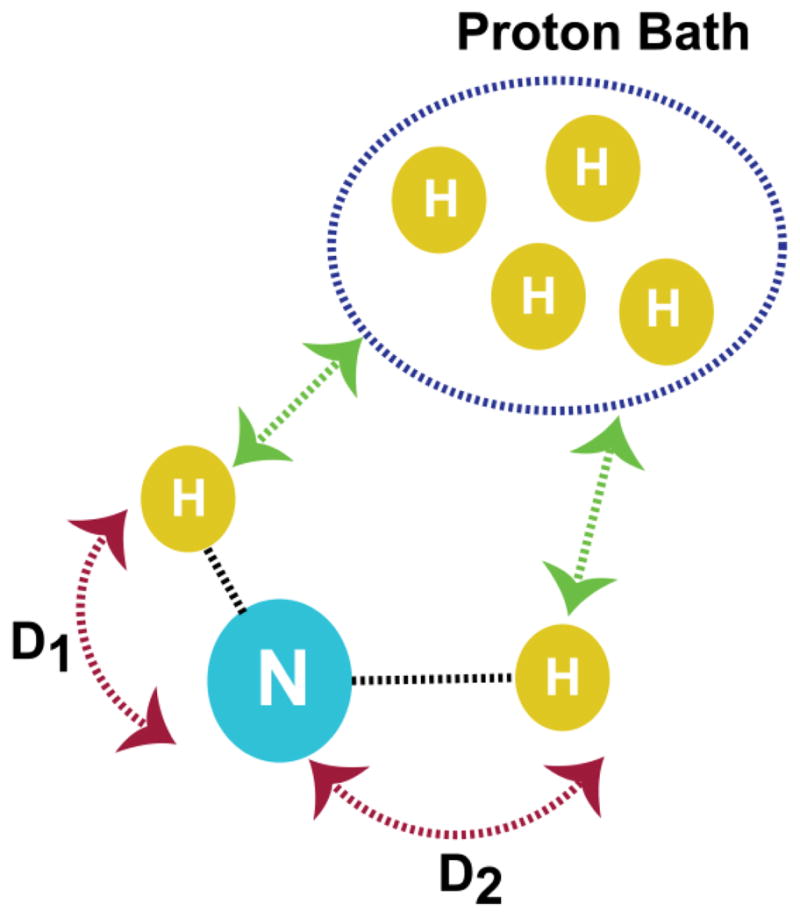
In this system, a 15N nucleus is strongly dipolar coupled to two nearby protons. The dipolar coupling values used in the simulations are D1 = 1750 Hz and D2 = 500 Hz. The 15N-1H dipolar couplings are arranged such that D1 > D2 and each of these protons is weakly dipolar coupled to a proton bath as indicated by the green arrows.
Figure 4. Numerically simulated PELF spectra under different magnetization transfer schemes.

It should be noted that WIM is known for its efficient suppression of 1H-1H dipolar couplings and is better than the COMPOZER-CP sequence. On the other hand, the regular CP sequence does not employ any special sequence to suppress 1H-1H dipolar couplings. Spectra (A–C) were simulated using a contact time of 250 μs and spectra (D–F) were simulated using a contact of 3 ms.
Figure 5.
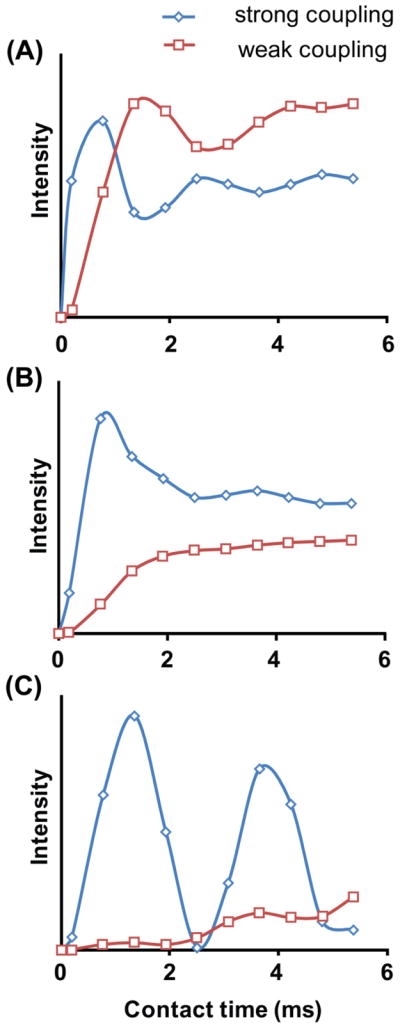
Simulated magnetization transfer efficiency as a function of transfer time by a regular-CP (A), COMPOZER-CP (B), and WIM (C) in a 2D PELF experiment. Details are discussed in the text.
Application of PELF techniques to study the structure and dynamics of 15N labeled mutant-Cyt b5 in magnetically-aligned bicelles
Low resolution and low sensitivity are often the major obstacles in solid-state NMR studies of membrane proteins. The introduction of magnetically-aligned bicelles as an alignment medium has dramatically improved the sample-filling factor of the RF coil, reduced the sample preparation time, and improved the quality of alignment when compared to mechanically aligned glass plate samples.24, 40–42 The use of bicelles has been shown to sufficiently improve the sensitivity and resolution of NMR spectra. Studies have shown that bicelles prepared with different q ratios can be used for solution-like and solid-state-like NMR experiments, enabling the complete characterization of protein structure, protein dynamics and protein-membrane interactions.43,44 Recently, the 2D PELF sequence was demonstrated to be well suited for structural studies of membrane-associated ligands in magnetically aligned bicelles.44,45 In the present work, we demonstrate the unique advantages of combining the PELF sequence with WIM or COMPOZER-CP magnetization transfer scheme for structural studies of membrane proteins in aligned bicelles. To this end, we have acquired a series of 2D PELF spectra of mutant-Cyt b5 in magnetically-aligned bicelles using WIM, COMPOZER-CP or ramp-CP as the magnetization transfer step (figure 6). The contact time used in these experiments was optimized for the maximum signal intensity and spectral resolution.
Figure 6. 2D PELF spectra of magnetically-aligned bicelles containing a uniformly-15N-labeled cytochrome b5.

Spectra were obtained using (A) WIM for 210 μs contact time, (b) COMPOZER-CP with an optimized contact time of 240 μs, and (c) RAMP-CP with an optimized contact time of 800 μs. All PELF spectra were acquired using a 1H RF field strength of 41 kHz and SPINAL proton decoupling67 during acquisition. All spectra are shown at the same contour level for a direct comparison.
In general, the 2D PELF sequence implemented with WIM or COMPOZER-CP offers a substantial improvement in resolution when compared to the one with ramp-CP. The resolution gained using a PELF-COMPOZER or PELF-WIM sequence is due to the ability of COMPOZER-CP and WIM to suppress the 1H-1H dipolar interactions as well as the use of a short contact time, which effectively suppresses the contribution from remote spins due to 1H spin diffusion.31 The central bands (or the zero-frequency peaks) in these experimental spectra are effectively suppressed suggesting a significant reduction in the spin diffusion process as is also illustrated in our simulations. Therefore, clear dipolar coupling doublets are resolved in the indirect frequency dimension. On the other hand, the central bands severely obscure the spectrum when a regular CP sequence is used in the magnetization transfer step, making accurate measurements of the weak dipolar couplings difficult. These experimental results are in good agreement with the numerical simulations for PELF-COMPOZER, PELF-WIM and regular PELF protocols presented in figures 4 and 5.
As shown in figures 6(A) and 6(B), two sets of resonances with vastly different dipolar coupling values are clearly observed. The magnitudes of their dipolar couplings indicate that they correspond to different regions of the proteins.23 The resonances with large dipolar couplings correspond to the membrane-spanning domain while the small dipolar couplings correspond to a more mobile region of the protein, which probably lies near the membrane surface. The spectral resolution of the transmembrane region in the PELF-COMPOZER and PELF-WIM is comparable to that obtained using the rotating frame sequences like HIMSELF and BB-PISEMA (Broad-Band-PISEMA).23 For dipolar couplings originating from the mobile regions of the protein, the gain in spectral resolution when either COMPOZER-CP or WIM is used as the magnetization transfer step (as shown in Figure 7) represents a remarkable improvement over previous PELF studies and demonstrates the effectiveness of this new approach. Thus, these experiments demonstrate the practical application of the 2D PELF pulse sequence with the appropriate magnetization transfer scheme to accurately measure a wide range of heteronuclear dipolar couplings for membrane proteins embedded in magnetically-aligned bicelles.
Figure 7. A 2D PELF-WIM spectrum of aligned bicelles containing a mutant of Cytochrome b5 uniformly labeled with 15N isotope to compare the efficiencies of different PELF sequences.

Sample N-H dipolar coupling slices extracted from 2D PELF spectra, that were obtained using PELF sequences constituted with different magnetization transfer schemes, are included on the right side for a comparison of the efficiency of PELF sequences. In all of these cases, PELF with WIM or COMPOZER-CP offers a significant improvement in resolution particularly in the dipolar coupling dimension. The S/N (signal-to- noise) in the dipolar dimension of WIM, COMPOZER-CP and CP are 7.1, 7.5 and 8, respectively. The line-width taken at half height in the dipolar dimension for WIM, COMPOZER-CP and CP are approximately 150, 140, 400 Hz, respectively.
Wobbling of the transmembrane helix of cytochrome b5 and its influence on the helical wheel pattern of resonances
The proposed average 13° tilt of the mutant-Cytb5 transmembrane domain relative to the membrane normal determined from our PELF experimental data is illustrated in Figure 8A. The resonances corresponding to the transmembrane region are dispersed in a circular pattern, which is unique to proteins with α-helical secondary structure, and the best-fitting helical wheel projection circle is indicated in blue color in Figure 8B.46–48 This is expected for the transmembrane helix of mutant-Cytb5 since both mutant-Cytb5 and the full-length Cytb5 shared identical amino acid sequence in their membrane-spanning domain. Importantly, the degree of dispersion in the resonances of the helical wheel directly reflects the average tilt angle (τ) of the helix with respect to the bilayer normal.46, 47 In Figure 8B, the PISA wheel of an ideal helix was empirically fitted with an average helical tilt angle of 13° and the value of the overall order parameter (S) is estimated to be 0.42. However, no single helical wheel can provide an ideal fit to all the resonances presented in the circular pattern, which indicates non-ideality of the transmembrane helix. A kink due to the Pro residue in the middle of the TM region could be the main reason for the observation of an imperfect PISA (Polarity Index Slant Angle) wheel. Further experimental studies are required to completely assign the spectrum and determinate the origin of this imperfect helix.
Figure 8.

(A) An illustration of the proposed orientation of a mutant-Cytb5 in magnetically-aligned bicelles. (B) A 2D SLF spectrum of a mutant-Cytb5 embedded in aligned bicelles. Blue color represents the best-fit for the helical wheel pattern of resonances from the transmembrane domain of the protein. The resonances in green represent the residues located near the membrane surface.
Over the years, the structure of the membrane-spanning region of Cytb5 has been a source of constant debate.49,50 It was proposed that the transmembrane domain of Cytb5 could adopt two conformations: a helical hairpin conformation or a membrane spanning α-helix. Fluorescence quenching and trypsin digestion assays show that the helical hairpin conformation is a “loose-binding” form of Cytb5, which allows it to exchange between lipid vesicles and doesn’t span the membrane.51 On the other hand, the “tight-binding” form of Cytb5 is believed to span the entire membrane and is firmly anchored to the lipid bilayer.51 Interestingly, the hairpin helical conformation can be converted to the membrane spanning form by increasing the temperature or adding detergents.51 In our case, mutant-Cytb5 most likely incorporates into the bicelle assembly and spans the bilayer due to the presence of DHPC. Also, the value of the overall order parameter is comparable to a single transmembrane helix spanning the whole bilayer as reported in the literature, which provides further evidence that the observed helix of mutant-Cytb5 spans the membrane bilayer in bicelles.25, 42, 52 However, we were unable to resolve all of the resonances in the transmembrane region due to the broadening of resonances in the indirect dimension. The resonances with small N-H dipolar couplings, as shown in green in figure 8B, are probably from the portion of the soluble domain that lies near the membrane surface due to the absence of the 8-residue-linker; however, no satisfactory helical wheel fit can be obtained even though they are dispersed in an apparent circular pattern. Therefore, more experiments using selectively labeled protein samples need to be carried out in order to accurately assign all resonances in the spectrum.
The influence of the whole body motions of the helix can be extracted from careful analysis of the helical wheel even in the absence of spectral assignments.27 In a lipid bilayer system, an alpha helix often exhibits a wide range of motions ranging from long axis rotation to the wobbling of the whole helix. The amplitude of these motions often influences the magnitude of NMR parameters, particularly CSA and dipolar couplings.27, 53 Recently, it was suggested that dynamical information could be extracted from careful analysis of the helical wheel with an appropriate dynamic model.27, 54, 55 Therefore, this will be the focus of the analysis presented in this paper. To model the motion of the whole helix, we invoke the diffusion in a cone model. In this model, the director axis of the helix wobbles freely within a cone described by the semiangle (β) and the wobble is axially symmetric with respect to the azimuthal angle (α). The resulting wobbling motion of the whole protein is shown in figure 9(B) and the best fit of our experimental data to our model is shown in figure 9(A). Notably, the angle β is defined by the tilt angle (τ) of the helix.56 Also, we assumed that this wobbling motion and other collective local fluctuations is fast relative to the measurement time. The order parameter Scone for the diffusion in a cone model is given in Eqn. 2.56
Figure 9.

(A) The fitting of the helical wheel pattern of resonances using the diffusion in a cone model. The open circles (○) denote the position of the observed resonances. The three PISA wheel fits were obtained using an average tilt angle of τ = 13° and a chosen value of σ. In this case, the chosen values of sigma are 1°, 8°, and 16° as indicated in the figure. (B) A diagram showing the wobbling motion of the helix in a lipid bilayer; where τ represents the tilt angle and σ represents the degree of fluctuation in the director axis.
| [2] |
To model the fluctuations in the helical director axis, a normal distribution function, given in Eqn. 3, will be used and the degree of fluctuation is estimated from the spread about the mean. In Eqn. [3], β0 is the average tilt angle of the helix, σ is the standard deviation, and β is the angle that varies between 0 to 90° with respect to the bilayer normal.27
| [3] |
Importantly, the observed helical wheel will be the weighted average of all the probable orientations defined by the degree of fluctuation in the director axis of the helix.
A series of helical wheels, shown in Figure 10, was generated using the proposed model. Our simulations show the position of the helical wheel is sensitive to the degree of helical tilt fluctuation, particularly when the degree of helical tilt is small. In this case, small helical tilt fluctuations result in a significant change in the position of the helical wheel as shown in Figure 10(A). On the other hand, for helix with a large tilt angle, a sizable amplitude of tilt fluctuation is required to change the position of the helical wheel as shown in Figure 10(C). Furthermore, ambiguity can arise when dealing with helical wheels with small dipolar couplings and these cases are illustrated in Figure 10(D–F). As the amplitude of fluctuations increases, helical wheels of different tilt angles will eventually overlap as shown in Figure 10(F). However, if the degree of fluctuation (σ) is much greater than the average tilt angle of the helix (β0), this represents an unrealistic situation and should be ignored. Therefore, from these simulations, our model has shown to be appropriate in extracting dynamical information regarding small amplitude in helical tilt fluctuations (β0 ≥ σ) for helices with tilt angles in the range of 10 to 30°. This range of tilt angles is commonly observed in many transmembrane helices reported in the literature, which makes this model applicable to other helical membrane protein systems.14, 21, 23, 25, 42
Figure 10. A series of helical wheels simulated using the proposed model for cytochrome b5 associated with bicelles.

(A–C) A series of helical wheels simulated with different degree of helical tilt angle fluctuations (σ), while keeping the average tilt (β0) of the helix constant. The average tilt angles (β0) used in the helical wheel simulations in figure 9(A)–9(C) are 10°, 30°, and 45°. (D–F) A series of helical wheels simulated with different degree of helical tilt angles (β0), while keeping the helical fluctuation (σ) constant. The constant helical fluctuation (σ) used in the helical wheel simulations in figures 9(D)–9(F) are 5°, 10°, and 30° and the helical tilt angles (β0) varies from 10°, 30°, and 45° in each of the figures 9(D)–9(F). (Note: If the helical fluctuation is greater than the average helical tilt, it represents an unrealistic situation and was excluded from simulations.)
When our model is incorporated into the fitting of the helical wheel as shown in Figure 9(A), a detailed information regarding the dynamics of the membrane anchor emerges. If the fluctuation of the helical director axis, relative to the bilayer normal, is faster than its diffusion inside the cone, then we will observe only an average cone semiangle, which equals to the average tilt angle of the helix. On the other hand, if the fluctuation of the helical director axis is slow relative to its diffusion inside the cone, then the helix can sample a range of cone semiangles, resulting in a spread about the average helical tilt. These situations are illustrated in Figure 11.
Figure 11. An illustration of the importance of relative timescales in producing a motionally averaged PISA wheel spectrum.

(A) When fluctuations of the helical tilt occur on a faster timescales than diffusion in a cone, only one cone semiangle (β0) has to be considered in the calculation of the motionally averaged helical wheel. (B) When fluctuations of the helical tilt occur on slower timescales than diffusion in the cone, several cone semiangles have to be considered in the calculation of the motionally averaged helical wheel.
The helical wheel that corroborates our experimental data, as illustrated in Figure 9A, requires a spread of approximately 8° about the average tilt angle of the helix, which indicates the diffusion of the helix inside the cone is faster than the fluctuation in the helical tilt. In our proposed model, the helix is wobbling in a cone defined by an average 13° tilt angle of the helix with an approximately 8° fluctuation in the helical tilt (σ). Also, from the magnitude of dipolar couplings, the collective motions of the helix are estimated to be on the order of milliseconds. This 8° fluctuation in the helical tilt defines an upper limit since the collective motions of the bicellar assemblies are incorporated into the current analysis as well. When we extend our current model to the mobile region, no satisfactory helical wheel fit can be obtained. This result demonstrates the dynamics in the mobile region is significantly different from the transmembrane domain. Due to the motional freedom in the mobile region, a greater range of motions can be afforded at various time scales, which dramatically increases the complexity required in our model for a proper description of this region. Thus, despite this shortcoming, our proposed model corroborates well with experimental results for the membrane-spanning domain, providing meaningful insights regarding helical motions in a bilayer environment, which showed the possibility of extracting both structural and dynamical information from the helical wheel. Our current dynamical analysis can be extended, with modifications, to include motions of two tightly bound helices in a lipid bilayer. These systems are commonly observed in nature with profound biological implications. For instance, the association of helices is commonly observed in macromolecular assembly such as viropins or between two cytochrome redox partners.5, 20, 21, 42 Therefore, understanding the dynamics that takes place in these complexes will allow us to gain a better insight into the role of molecular dynamics in facilitating their biological functions.
Further application of PELF technique to other systems and to study molecular dynamics
The resolution afforded using the PELF techniques described here can be extended to other spin systems, particularly the methyl groups. Recently, 13C labeled methyl groups have been demonstrated to be an exquisite NMR probe for investigating dynamics, structure and biological functions of large protein complexes and the fact that Val, Ile, Leu, Ala, Met and Thr, constitute 30 – 45% of all the amino acids in membrane proteins, the methyl group is becoming an important molecular probe for interrogating membrane protein structures and dynamics.57–59 However, the use of methyl 13C-1H dipolar couplings in SLF studies of membrane proteins remains a challenge due to the lack of dispersion in the methyl 13C chemical shifts as well as the complicated multiplet lineshape in the indirect dimension due to the use of SLF sequences such as PISEMA, HIMSELF and SAMMY.57,60 Therefore, the PELF methods implemented in the current studies can be used to simplify the complicated line shape in the indirect dimension, allowing for the accurate measurement of methyl’s 13C-1H dipolar couplings. The methyl 13C-1H dipolar couplings can provide dynamical information on a variety of biological systems. For example, the association of two different helices of a membrane protein often restricts the methyl group rotation at the binding interface, resulting in an increase in the magnitude of methyl’s 13C-1H dipolar couplings. Therefore, through measuring the changes in the 13C-1H dipolar couplings, both the binding interface between two helices and the strength of their interactions at each methyl site can be deduced by comparing the order parameters of methyl groups as long as their resonances are resolvable. Furthermore, methyl 13C-1H dipolar couplings can provide structural constraints regarding the helical tilt similar to the recently proposed GALA (Geometric Analysis of Labeled Alanines) method that exploits the deuterium quadrupolar coupling of a CD3 system.61,62 While the GALA method is successful in providing the tilt angle of a helix in both oriented and unoriented samples, it requires numerous selectively deuterated methyl samples for different Ala residues, which is not practical.62 The advantage of using the CH3 group is that multiple Ala methyl sites can be 13C labeled and their 13C-1H dipolar couplings can be simultaneously measured using the PELF protocols provided their resonances are resolvable. Therefore, the PELF sequence can be extended to CH3 spin systems, providing both side chain dynamics and structural constraints for refining protein secondary structures.57
The ability to measure a broad range of dipolar couplings allows for not only the determination of structures, but also the understanding of protein dynamics at various time scales as illustrated in our studies. Dipolar couplings can provide information on both the orientation of bond vectors and the amplitude of motions up to the millisecond time scale.63–65 Under the commonly used rotating-frame SLF pulse sequences, only motions in the range of 10−4 – 10−5 s are observed, which reflects the rotational diffusion of a transmembrane α-helix.53 However, under the PELF approach, motions up to 10−3 s are revealed; thus, opening up a new avenue for probing protein dynamics via solid-state NMR experiments on aligned samples. In fact, a complete mapping of protein dynamics is now possible, using a combination of relaxation and dipolar coupling measurements.65 Currently, applications of SLF spectroscopy mainly focus on the determination of the transmembrane domain structures of membrane proteins. Yet, in many cases, particularly for the case of membrane-associated receptors, a significant portion of the protein is in the cytoplasmic domain and conformational changes in one region of a protein can be propagated to another region through changes in protein motions such as those observed in dynamically driven allosteric interactions.66 Despite many advances in solid-state NMR methodology, the synergy of these conformational changes has so far not been well characterized. Thus, the ability to produce a complete mapping of protein dynamics and their correlation with each other will allow for a better understanding of the intricate and enigmatic relationships between the structure of a protein and its biological functions.
CONCLUSION
In the present study, we have demonstrated the practicality of using PELF pulse sequence to generate a complete mapping of dynamics, by measuring dipolar couplings of directly bonded N-H spin pairs, of a membrane anchored cytochrome b5 in magnetically-aligned bicelles. The superiority in resolution of a PELF spectrum under WIM-CP or COMPOZER-CP sequence rivals that of conventional rotating frame SLF techniques such as SAMMY, BB-PISEMA and HIMSELF. Importantly, under our approach, an SLF spectrum of both transmembrane and mobile regions is recorded in a single experiment, allowing for a complete correlation of protein dynamics between the two regions. The observed PISA wheel was fit using a dynamical model, which includes the whole body motions of the helix. Under this approach, the helix is modeled as wobbling in a cone with an approximate 8° fluctuation in the tilt of the helix, which is estimated to be 13°. Importantly, this approach represents a first step in extracting the motions of a membrane protein from an aligned sample, allowing for a better understanding of the dynamics of a protein in a bilayer environment. Although this method is demonstrated successfully on a membrane protein, it can also be used for other aligned liquid crystalline materials. Finally, while this method of extracting dynamical information can be extended to system of two binding helixes, MD simulations will be needed to include all the motions pertaining to such a complex in order to provide a proper and insightful description regarding its motions in membranes.
Acknowledgments
This work was supported by the National Institutes of Health (GM084018 and RR023597 to A.R.) and CRIF-NSF funding for the NMR spectrometer.
References
- 1.Loria JP, Berlow RB, Watt ED. Acc Chem Res. 2008;41:214–221. doi: 10.1021/ar700132n. [DOI] [PubMed] [Google Scholar]
- 2.Korzhnev DM, Kloiber K, Kanelis V, Tugarinov V, Kay LE. J Am Chem Soc. 2004;126:3964–3973. doi: 10.1021/ja039587i. [DOI] [PubMed] [Google Scholar]
- 3.Skrynnikov NR, Mulder FA, Hon B, Dahlquist FW, Kay LE. J Am Chem Soc. 2001;123:4556–4566. doi: 10.1021/ja004179p. [DOI] [PubMed] [Google Scholar]
- 4.Zintsmaster JS, Wilson BD, Peng JW. J Am Chem Soc. 2008;130:14060–14061. doi: 10.1021/ja805839y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dürr UHN, Waskell L, Ramamoorthy A. Biochim Biophys Acta. 2007;1768:3235–3259. doi: 10.1016/j.bbamem.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 6.Ellis J, Gutierrez A, Barsukov IL, Huang WC, Grossmann JG, Roberts GC. J Biol Chem. 2009;284:36628–36637. doi: 10.1074/jbc.M109.054304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liang B, Tamm LK. Proc Natl Acad Sci U S A. 2007;104:16140–16145. doi: 10.1073/pnas.0705466104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arora A, Abildgaard F, Bushweller JH, Tamm LK. Nat Struct Biol. 2001;8:334–338. doi: 10.1038/86214. [DOI] [PubMed] [Google Scholar]
- 9.Hwang PM, Bishop RE, Kay LE. Proc Natl Acad Sci U S A. 2004;101:9618–9623. doi: 10.1073/pnas.0402324101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hwang PM, Choy WY, Lo EI, Chen L, Forman-Kay JD, Raetz CR, Prive GG, Bishop RE, Kay LE. Proc Natl Acad Sci U S A. 2002;99:13560–13565. doi: 10.1073/pnas.212344499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Porcelli F, Verardi R, Shi L, Henzler-Wildman KA, Ramamoorth A, Veglia G. Biochemistry. 2008;48:5565–5572. doi: 10.1021/bi702036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Porcelli F, Buck-Koehntop BA, Thennarasu S, Ramamoorthy A, Veglia G. Biochemistry. 2006;45:5793–5799. doi: 10.1021/bi0601813. [DOI] [PubMed] [Google Scholar]
- 13.Traaseth NJ, Buffy JJ, Zamoon J, Veglia G. Biochemistry. 2006;45:13827–13834. doi: 10.1021/bi0607610. [DOI] [PubMed] [Google Scholar]
- 14.Traaseth NJ, Shi L, Verardi R, Mullen DG, Barany G, Veglia G. Proc Natl Acad Sci U S A. 2009;106:10165–10170. doi: 10.1073/pnas.0904290106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Horn WD, Kim HJ, Ellis CD, Hadziselimovic A, Sulistijo ES, Karra MD, Tian C, Sonnichsen FD, Sanders CR. Science. 2009;324:1726–179. doi: 10.1126/science.1171716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ravindranathan KP, Gallicchio E, McDermott AE, Levy RM. J Am Chem Soc. 2007;129:474–475. doi: 10.1021/ja0672371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldbourt A, Gross BJ, Day LA, McDermott AE. J Am Chem Soc. 2007;129:2338–2344. doi: 10.1021/ja066928u. [DOI] [PubMed] [Google Scholar]
- 18.Zhou DH, Shah G, Mullen C, Sandoz D, Rienstra CM. Angew Chem Int Ed Engl. 2009;48:1253–1256. doi: 10.1002/anie.200801029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ader C, Schneider R, Hornig S, Velisetty P, Wilson EM, Lange A, Giller K, Ohmert I, Martin-Eauclaire MF, Trauner D, Becker S, Pongs O, Baldus M. Nat Struct Mol Biol. 2008;15:605–612. doi: 10.1038/nsmb.1430. [DOI] [PubMed] [Google Scholar]
- 20.Traaseth NJ, Verardi R, Torgersen KD, Karim CB, Thomas DD, Veglia G. Proc Natl Acad Sci U S A. 2007;104:14676–14681. doi: 10.1073/pnas.0701016104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramamoorthy A, Kandasamy SK, Lee DK, Kidambi S, Larson RG. Biochemistry. 2007;46:965–975. doi: 10.1021/bi061895g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lambotte S, Jasperse P, Bechinger B. Biochemistry. 1998;37:16–22. doi: 10.1021/bi9724671. [DOI] [PubMed] [Google Scholar]
- 23.Dürr UHN, Yamamoto K, Im SC, Waskell L, Ramamoorthy A. J Am Chem Soc. 2007;129:6670–6671. doi: 10.1021/ja069028m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diller A, Loudet C, Aussenac F, Raffard G, Fournier S, Laguerre M, Grelard A, Opella SJ, Marassi FM, Dufourc EJ. Biochimie. 2009;91:744–751. doi: 10.1016/j.biochi.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Angelis AA, Howell SC, Nevzorov AA, Opella SJ. J Am Chem Soc. 2006;128:12256–12267. doi: 10.1021/ja063640w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buffy JJ, Traaseth NJ, Mascioni A, Gor’kov PL, Chekmenev EY, Brey WW, Veglia G. Biochemistry. 2006;45:10939–10946. doi: 10.1021/bi060728d. [DOI] [PubMed] [Google Scholar]
- 27.Esteban-Martin S, Strandberg E, Fuertes G, Ulrich AS, Salgado J. Biophys J. 2009;96:3233–3241. doi: 10.1016/j.bpj.2008.12.3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Traaseth NJ, Ha KN, Verardi R, Shi L, Buffy JJ, Masterson LR, Veglia G. Biochemistry. 2008;47:3–13. doi: 10.1021/bi701668v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Page RC, Kim S, Cross TA. Structure. 2008;16:787–797. doi: 10.1016/j.str.2008.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramamoorthy A, Wu CH, Opella SJ. J Magn Reson B. 1995;107:88–90. doi: 10.1006/jmrb.1995.1063. [DOI] [PubMed] [Google Scholar]
- 31.Dvinskikh SV, Yamamoto K, Ramamoorthy A. J Chem Phys. 2006;125:34507. doi: 10.1063/1.2212939. [DOI] [PubMed] [Google Scholar]
- 32.Dvinskikh SV, Dürr UHN, Yamamoto K, Ramamoorthy A. J Am Chem Soc. 2006;128:6326–6327. doi: 10.1021/ja061153a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clarke TA, Im SC, Bidwai A, Waskell L. J Biol Chem. 2004;279:36809–36818. doi: 10.1074/jbc.M406055200. [DOI] [PubMed] [Google Scholar]
- 34.Hong M, Pines A, Caldarell S. J phys Chem. 1996;100:14815–14822. [Google Scholar]
- 35.Hong M, Schmidt-Rohr K, Zimmermann H. Biochemistry. 1996;35:8335–8341. doi: 10.1021/bi953083i. [DOI] [PubMed] [Google Scholar]
- 36.Fukuchi M, Ramamoorthy A, Takegoshi K. J Magn Reson. 2009;196:105–109. doi: 10.1016/j.jmr.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burum D, Linder M, Ernst RR. J Magn Reson. 1981;44:173–188. [Google Scholar]
- 38.Fung BM, Ermolaev Y, Yu Y. J Magn Reson. 1999;138:28–35. doi: 10.1006/jmre.1999.1723. [DOI] [PubMed] [Google Scholar]
- 39.Veshtort M, Griffin RG. J Magn Reson. 2006;178:248–282. doi: 10.1016/j.jmr.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto K, Soong R, Ramamoorthy A. Langmuir. 2009;25:7010–7018. doi: 10.1021/la900200s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Angelis AA, Nevzorov AA, Park SH, Howell SC, Mrse AA. J Am Chem Soc. 2004;126:15340–15341. doi: 10.1021/ja045631y. [DOI] [PubMed] [Google Scholar]
- 42.Park SH, De Angelis AA, Nevzorov AA, Wu CH, Opella SJ. Biophys J. 2006;91:3032–3042. doi: 10.1529/biophysj.106.087106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith PES, Brender JR, Ramamoorthy A. J Am Chem Soc. 2009;131:4470–4478. doi: 10.1021/ja809002a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Canlas CG, Ma D, Tang P, Xu Y. J Am Chem Soc. 2008;130:13294–13300. doi: 10.1021/ja802578z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cui T, Canlas CG, Xu Y, Tang P. Biochim Biophys Acta. 2009;1798:161–166. doi: 10.1016/j.bbamem.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang J, Denny J, Tian C, Kim S, Mo Y, Kovacs F, Song Z, Nishimura K, Gan Z, Fu R, Quine JR, Cross TA. J Magn Reson. 2000;144:162–167. doi: 10.1006/jmre.2000.2037. [DOI] [PubMed] [Google Scholar]
- 47.Ramamoorthy A, Wei YF, Lee DK. Ann Rep NMR Spectrosc. 2004;52:1–52. [Google Scholar]
- 48.Denny JK, Wang J, Cross TA, Quine JR. J Magn Reson. 2001;152:217–226. doi: 10.1006/jmre.2001.2405. [DOI] [PubMed] [Google Scholar]
- 49.Everett J, Zlotnick A, Tennyson J, Holloway PW. J Biol Chem. 1986;261:6725–6729. [PubMed] [Google Scholar]
- 50.Vergeres G, Ramsden J, Waskell L. J Biol Chem. 1995;270:3414–3422. doi: 10.1074/jbc.270.7.3414. [DOI] [PubMed] [Google Scholar]
- 51.Ladokhin AS, Wang L, Steggles AW, Malak H, Holloway PW. Biochemistry. 1993;32:6951–6956. doi: 10.1021/bi00078a020. [DOI] [PubMed] [Google Scholar]
- 52.Muller SD, De Angelis AA, Walther TH, Grage SL, Lange C, Opella SJ, Ulrich AS. Biochim Biophys Acta. 2007;1768:3071–3079. doi: 10.1016/j.bbamem.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 53.Park SH, Mrse AA, Nevzorov AA, De Angelis AA, Opella SJ. J Magn Reson. 2006;178:162–165. doi: 10.1016/j.jmr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 54.Shi L, Cembran A, Gao J, Veglia G. Biophys J. 2009;96:3648–3662. doi: 10.1016/j.bpj.2009.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Straus SK, Scott WR, Watts A. J Biomol NMR. 2003;26:283–295. doi: 10.1023/a:1024098123386. [DOI] [PubMed] [Google Scholar]
- 56.Brainard JR, Szabo A. Biochemistry. 1981;16(20):4618–4628. doi: 10.1021/bi00519a016. [DOI] [PubMed] [Google Scholar]
- 57.Wu CH, Das BB, Opella SJ. J Magn Reson. 2009;202:127–134. doi: 10.1016/j.jmr.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sprangers R, Kay LE. Nature. 2007;445:618–622. doi: 10.1038/nature05512. [DOI] [PubMed] [Google Scholar]
- 59.Tugarinov V, Sprangers R, Kay LE. J Am Chem Soc. 2004;126:4921–4925. doi: 10.1021/ja039732s. [DOI] [PubMed] [Google Scholar]
- 60.Dvinskikh SV, Durr UH, Yamamoto K, Ramamoorthy A. J Am Chem Soc. 2007;129:794–802. doi: 10.1021/ja065536k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Strandberg E, Ozdirekcan S, Rijkers DT, van der Wel PC, Koeppe RE, Liskamp RM, Killian JA. Biophys J. 2004;86:3709–3721. doi: 10.1529/biophysj.103.035402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van der Wel PC, Strandberg E, Killian JA, Koeppe RE. Biophys J. 2002;83:1479–1488. doi: 10.1016/S0006-3495(02)73918-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tolman JR, Ruan K. Chem Rev. 2006;106:1720–1736. doi: 10.1021/cr040429z. [DOI] [PubMed] [Google Scholar]
- 64.Zhuang T, Leffler H, Prestegard JH. Protein Sci. 2006;15:1780–1790. doi: 10.1110/ps.051994306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Q, Stelzer AC, Fisher CK, Al-Hashimi HM. Nature. 2007;450:1263–1267. doi: 10.1038/nature06389. [DOI] [PubMed] [Google Scholar]
- 66.Kalodimos CG, Boelens R, Kaptein R. Nat Struct Biol. 2002;9:193–197. doi: 10.1038/nsb763. [DOI] [PubMed] [Google Scholar]