

Abstract
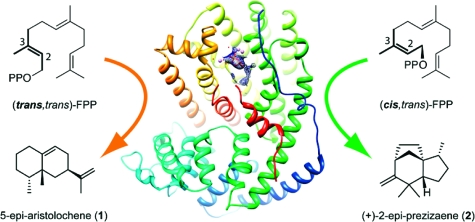
Sesquiterpene skeletal complexity in nature originates from the enzyme-catalyzed ionization of (trans,trans)-farnesyl diphosphate (FPP) (1a) and subsequent cyclization along either 2,3-transoid or 2,3-cisoid farnesyl cation pathways. Tobacco 5-epi-aristolochene synthase (TEAS), a transoid synthase, produces cisoid products as a component of its minor product spectrum. To investigate the cryptic cisoid cyclization pathway in TEAS, we employed (cis,trans)-FPP (1b) as an alternative substrate. Strikingly, TEAS was catalytically robust in the enzymatic conversion of (cis,trans)-FPP (1b) to exclusively (≥99.5%) cisoid products. Further, crystallographic characterization of wild-type TEAS and a catalytically promiscuous mutant (M4 TEAS) with 2-fluoro analogues of both all-trans FPP (1a) and (cis,trans)-FPP (1b) revealed binding modes consistent with preorganization of the farnesyl chain. These results provide a structural glimpse into both cisoid and transoid cyclization pathways efficiently templated by a single enzyme active site, consistent with the recently elucidated stereochemistry of the cisoid products. Further, computational studies using density functional theory calculations reveal concerted, highly asynchronous cyclization pathways leading to the major cisoid cyclization products. The implications of these discoveries for expanded sesquiterpene diversity in nature are discussed.
Terpenes comprise the most structurally diverse class of natural products, playing essential ecological roles by mediating communication between plants and insects, by providing antimicrobial defenses for plants, and likely acting in additional undefined capacities (as reviewed previously (1)). Terpenoids originate from primary isoprenoid metabolism, wherein iterative condensation of 5-carbon isoprene units (isopentyl diphosphate and dimethylallyl diphosphate) catalyzed by prenyltransferases produce polyisoprenoid diphosphate substrates of varying lengths (for a review, see ref (2)). Terpene synthases, in turn, often referred to as cyclases given the cyclic nature of many of their products, transform the polyisoprenoid diphosphate substrates, e.g., geranyl diphosphate C10, farnesyl diphosphate C15, or geranylgeranyl diphosphate C20, into structurally diverse mono-, sesqui-, and diterpene products, respectively.
The structural complexity of these molecules underlies their diverse biological activities. Ruzicka formulated the biogenetic isoprene rule, which predicted the formation of sesquiterpenes arising from the head-to-tail connection of three isoprene units (5 carbons each) where the skeletal complexity can be formally deduced from farnesol(3). Cane later developed a general stereochemical model for sesquiterpene biogenesis involving the idealized fold of the farnesyl chain in the active site posing the reacting carbons to direct a sequence of electrophilic cyclizations and rearrangements following pyrophosphate loss/ionization (4). Moreover, a limited number of conformations of the farnesyl substrate give rise to much greater product diversity. Product specificity or in many cases product diversity arises from a limited number of farnesyl chain conformations, wherein the reacting double bonds reside mutually perpendicular to a common plane. Thus, there is a direct correspondence between the absolute stereochemical configuration of the sesquiterpene product and the inferred conformation of the precursor (4). A central challenge for the structural enzymologist is to define how individual terpene synthases statically or dynamically discriminate between alternative polyprenyl cation conformational modes or selectively favor particular conformations to shepherd reactive intermediates along distinct cyclization cascades.
Structural biology provides a framework for addressing the evolutionary origins of complex terpenoid metabolites and their biosynthetic pathways. Terpene synthases comprise a structurally conserved enzyme family, which adopt a common α-helical architecture termed the class I terpenoid cyclase fold, first revealed from the crystal structures of tobacco 5-epi-aristolochene synthase (TEAS) from Nicotiana tobaccum(5) and pentalene synthase from Streptomyces UC5319(6). The lyase function of these enzymes stems from two conserved metal binding motifs: the “aspartate-rich” DDxxD motif that coordinates two Mg2+ ions and the “NSE/DTE” motif that coordinates a third Mg2+. These static X-ray crystallographic studies show that the binding of (Mg2+)3-PPi stabilizes the active site in a closed conformation that is sequestered from bulk solvent(7). In addition to multiple divalent cation coordination bonds, the PPi anion accepts hydrogen bonds from conserved basic residues when bound to the closed synthase conformation, while a hydrophobic pocket, lined by a number of aromatic residues, cradles the farnesyl chain and most likely templates the cyclization reaction by enforcing particular substrate conformations and stabilizing carbocations through π-stacking interactions.
While the wealth of structural diversity among terpene hydrocarbons arises from bifurcations along multistep cyclization pathways, divergence at the earliest mechanistic step defines two major classes of terpene synthases and hence distinct product families. The “transoid” synthases ionize (trans,trans)-farnesyl diphosphate (FPP) (1a) to generate the 2,3-transoid farnesyl cation (trans along the C2−C3 bond) followed by initial C1 attack on the distal C10−C11 double bond, prior to further downstream cyclizations (1). By contrast, the “cisoid” synthases conduct an initial C2−C3 double bond isomerization prior to cyclization, wherein the nascent farnesyl cation is recaptured at C3 by pyrophosphate to form the neutral, enzyme-bound (3R)- or (3S)-nerolidyl diphosphate (NPP), thus allowing rotation about the C2−C3 bond from trans to cis. Reionization of NPP generates the 2,3-cisoid farnesyl cation, which undergoes further cyclization via initial C1 attack either on the proximal C6−C7 or on the distal C10−C11 double bonds prior to further transformations. This reaction mechanism has been invoked to account for the formation of β-macrocarpene(8), amorpha-4,11-diene(9), trichodiene (10), and cedranes such as isocedrol (11). Moreover, the biosynthesis of epi-isozizaene was recently described in connection with the isolation and functional characterization of epi-isozizaene synthase from Streptomyces coelicolor(12). The cisoid mechanistic class in sesquiterpene cyclases is akin to the majority of monoterpene cyclases that proceed via the cis (neryl) allylic cation to form the corresponding cyclic monoterpene products (for a review, see ref (13)).
Figure 1.

Mechanism of TEAS-catalyzed cyclization of (cis,trans)-FPP to (+)-2-epi-prezizaene (2). a) The structure and semisystematic nomenclature for isomers of the FPP substrate and fluorinated analogues used in this study are indicated. b) Based on biochemical and stereochemical information governing the nature of the cisoid products, a cyclization mechanism is proposed to account for all identified products along this multistep pathway (16). c) The configuration of the terminal isopropenyl tail of the (7R)-β-bisabolyl cation relates to the final cyclization products of the cisoid pathway.
Though most sesquiterpene synthases can be classified as belonging to either the cisoid or transoid classes, some display cryptic activities associated with the other pathway. Notably, TEAS catalyzes the formation of (+)-5-epi-aristolochene (1), the first committed step in the biosynthesis of the phytoalexin capsidiol, the principal component of tobacco’s antifungal chemical defense (14). Aside from its major product, TEAS generates an additional 24 minor products, some of which are derived from the cisoid cyclization pathway (15). The structural variations of these cisoid products result from a multistep mechanism of cyclizations and rearrangements, suggesting that TEAS templates the cisoid cation pathway with fidelity and enables the formation of a distinct set of complex skeletal structures. These unexpected observations give rise to several confounding questions. Does a single parental fold of the farnesyl chain give rise to products along both the cisoid and transoid pathways in TEAS? On a structural level, how are both pathways templated within a single active site? How can the cryptic cisoid pathway in TEAS become activated? Does this “vestigial” activity portend an unanticipated new function for TEAS in tobacco?
To address these questions, we investigated the cisoid cyclization recently discovered in TEAS using synthetically derived (cis,trans)-FPP (1b), a geometrical isomer of the native all-trans substrate (1, panel a). [The descriptors “cis” and “trans” in (cis,trans)-FPP and the fluoroFPP isomers refer to the longest carbon chain about the 2,3 and 6,7 double bonds, respectively, as defined in 1, panel a. For more formalized nomenclature, see refs (116) and (117).] Remarkably, TEAS efficiently converts this alternative substrate into predominantly (+)-2-epi-prezizaene (2), a novel sesquiterpene hydrocarbon related to the naturally occurring alcohol jinkohol(16), along with other cisoid cation-derived products. Large-scale enzyme reactions produced sufficient amounts of hydrocarbon products for stereochemical elucidation and positive identification of nine compounds(16).
In the present investigation, crystallographic analyses of wild-type TEAS and a previously reported promiscuous mutant (TEAS M4) (17), with unreactive 2-fluoro analogues (2a and 2b) of (trans,trans)- and (cis,trans)-FPP (1a and 1b, respectively) revealed catalytically relevant binding modes and distinct farnesyl chain topologies that are consistent with preorganization by the active-site for cisoid or transoid cyclization, and hence, the predicted stereochemical course of the reaction. Further, key transition state geometries calculated using density functional theory revealed concerted, highly asynchronous cyclization pathways. Thus, combining biochemical, computational, and crystallographic analyses with the recently elucidated stereochemistry of the cisoid products, we pictorially reconstruct herein the TEAS-catalyzed transoid and cisoid cyclization pathways. Further, comparison of wild-type and mutant TEAS-analogue complexes provides structural snapshots and insights into product specificity/diversity reflected in the preorganization of the farnesyl chain along 2 major cyclization pathways.
Results and Discussion
TEAS-Directed Cisoid Cyclization with (cis,trans)-FPP
To investigate the cryptic cyclization activity in TEAS, we synthesized the 2,3-cis geometrical isomer of farnesyl diphosphate (cis,trans)-FPP (1b) (16). This substrate analogue is effectively “preisomerized”, and hence its ionization by TEAS would be expected to generate the cisoid farnesyl cation, which in turn should feed directly into the cisoid cyclization pathway (1, panel b). Indeed, our pilot experiments revealed that TEAS generated a near exclusive spectrum of cisoid products when incubated with (cis,trans)-FPP (1b) as substrate, including the previously reported iso-prezizaene ((+)-2-epi-prezizaene, 2) as the dominant reaction product (1 and 2). This result demonstrated the ability of TEAS to template the cisoid cyclization pathway with a high degree of product specificity and catalytic efficiency.
Table 1. Enzymatic products from incubations of TEAS wild-type and the M4 mutant with (cis,trans)- or (trans,trans)-FPP.
| % by mass | % by total ion count (TIC) |
||||
|---|---|---|---|---|---|
| Peak | Product | RT (min) | TEAS wt | TEAS wt | TEAS M4 |
| (cis,trans)-FPP products | |||||
| 3 | (−)-α-cedrene | 15.3 | 18.95 | 18.85 | 18.48 |
| 2 | (+)-2-epi-prezizaene | 15.8 | 37.89 | 46.52 | 30.54 |
| 4 | α-acoradiene | 16 | 3.44 | 6.80 | 13.40 |
| 5 | 4-epi-α-acoradiene | 16.1 | 1.03 | 2.54 | 4.06 |
| 6 | (−)-β-curcumene | 16.5 | 13.78 | 12.30 | 4.65 |
| 7 | nerolidol | 17.1 | 3.61 | 0.90 | 1.81 |
| 8 | α-bisabolol | 18.6 | 1.80 | 0.11 | 0.05 |
| 9 | epi-α-bisabolol | 18.6 | 1.80 | 0.11 | 0.05 |
| 10 | cis-farnesol | 18.8 | 5.69 | 0.29 | 0.50 |
| remaining | n.a. | 11.99 | 11.58 | 26.46 | |
| (trans,trans)-FPP products | |||||
| 11 | germacrene A | 15.1 | 3.65 | 10.98 | |
| 1 | 5-epi-aristolochene | 16.2 | 78.90 | 30.66 | |
| 12 | 4-epi-eremophilene | 16.3 | 6.21 | 27.46 | |
| 13 | premnaspirodiene | 16.48 | 1.66 | 25.47 | |
| remaining | n.a. | 9.58 | 5.43 | ||
Figure 2.

Gas chromatograms of products from incubations of wild-type TEAS and the M4 mutant with (cis,trans)- and (trans,trans)-FPP. TEAS or its M4 mutant were incubated with either (a) (trans,trans)-FPP (1a) or (b) (cis,trans)-FPP (1b) using the vial assay followed with analysis by GC–MS as described in Methods. Major product peaks are labeled according to identified products listed in 1.
As detailed in a concurrent report, the structure, stereochemistry, and enantio-purity was determined for nine cisoid products of TEAS isolated from large-scale enzyme incubations with (cis,trans)-FPP (1b), an achievement enabling the formulation of a mechanistic proposal for their biosynthetic origin (1, panel b) (16). Chromatographic separations or enrichments of five hydrocarbon and three alcohol fractions, together with comparative NMR spectral data, chiral GC analyses, optical rotation measurements, and chemical correlations, allowed assignment of structures for (+)-2-epi-prezizaene (2), (−)-α-cedrene (3), α-acoradiene (4), 4-epi-α-acoradiene (5), (−)-β-curcumene (6), nerolidol (7), the α-bisabalol epimers (8 and 9), and 2,3-cis-farnesol (10) shown in 1, panel b and listed in 1. Importantly, knowledge of the relative and absolute stereochemistry of the cisoid hydrocarbon products provided vital information for guiding computational studies and accurately describing the stereochemical course of the TEAS cisoid cyclization reaction mechanism (1, panel b and below).
To further characterize the product specificity of wild-type TEAS and a previously described promiscuous mutant (A274T V372I Y406L V516I) referred to here as M4 TEAS(17), we performed GC−MS analyses via the vial assay(18) to examine product mass spectra derived from native (1a) and (cis,trans)-FPP (1b) substrates (2 and 1). TEAS generated 18 distinct terpene products from the 2-cis substrate, including (+)-2-epi-prezizaene (2), which constitutes nearly half the product spectrum (46% by TIC). With the native all-trans FPP (1a) substrate, M4 TEAS exhibits relaxed product specificity in the terminal steps of the transoid cyclization pathway, producing roughly equal amounts of 5-epi-aristolochene (1), 4-epi-eremophilene (12), and premnaspirodiene (13). With (cis,trans)-FPP (1b), M4 TEAS produced the same repertoire of hydrocarbons as the wild-type enzyme, again displaying relaxed product specificity with only a third of the turnovers producing (+)-2-epi-prezizaene (2). This result indicates that the catalytic promiscuity of M4 TEAS extends to the cisoid pathway.
To assess the efficiency of the enzymatic conversion of (cis,trans)-FPP (1b) to (+)-2-epi-prezizaene (2) by wild-type TEAS and M4 TEAS, we conducted steady-state kinetic experiments (2). The experiments using (trans,trans)-FPP (1a) reveal that both enzymes display comparable catalytic efficiencies (kcat/Km), with the higher Km of M4 TEAS (13.3 μM) offset by a higher overall turnover number (9.4 min−1). Both enzymes utilize (cis,trans)-FPP (1b) with similar catalytic efficiencies, with the promiscuous TEAS M4 possessing a Km (7.9 μM) lower than that of wild-type but also a slower overall turnover (4.6 min−1), the reverse of the trend observed for (trans,trans)-FPP (1a). Additionally, enzymatic activities were assessed using the 2-fluoro analogues of (trans,trans)-FPP (2a) and (cis,trans)-FPP (2b) (1, panel a). Wild-type TEAS and several of its catalytically active mutants fail to turn over either (2-trans,6-trans)-2-fluorofarnesyl diphosphate (trans-2F-FPP, 2a) or (2-cis,6-trans)-2-fluorofarnesyl diphosphate (cis-2F-FPP, 2b) after 24-h incubation periods. The lack of measurable catalytic activity when using the fluoro-substituted FPPs is most likely due to a strong electron-withdrawing inductive effect due to the presence of the fluoro substituent at position 2 that prevents ionization and pyrophosphate loss. By contrast, the fungal sesquiterpene cyclase aristolochene synthase converted trans-2F-FPP (2a) cleanly to 2-fluorogermacrene A after extended incubation times(19), and trichodiene synthase produced several fluorinated sesquiterpene hydrocarbons of unknown structure (20). Although unreactive fluoro analogues have been useful for crystallography with limonene synthase(21) and more recently 2F-FPP complexes with aristolochene synthase(22), the inertness of the 2-fluoro-FPPs proved useful for our crystallographic experiments.
Table 2. Kinetic parameters of TEAS wild-type and the M4 enzyme determined using either (cis,trans)- or (trans,trans)-FPP.
| (trans,trans)-FPP |
(cis,trans)-FPP |
|||||
|---|---|---|---|---|---|---|
| kcat (min−1) | KM (μM) | kcat/KM (μM−1 min−1) | kcat (min−1) | KM (μM) | kcat/KM (μM−1 min−1) | |
| TEAS wt | 2.5 ± 0.7 | 8.4 ± 0.89 | 0.3 | 5.71 ± 0.08 | 14.03 ± 2.59 | 0.41 |
| TEAS M4 | 9.43 ± 0.21 | 13.31 ± 1.04 | 0.71 | 4.62 ± 0.07 | 7.97 ± 1.26 | 0.58 |
Stereochemical Mechanism of Cyclization
On the basis of the elucidated stereochemistry of the major cisoid products, a reaction mechanism for the TEAS-catalyzed cyclization of the cisoid farnesyl cation is proposed (1, panel b) (16). Catalysis begins with divalent cation-assisted ionization of (cis,trans)-FPP generating the cisoid farnesyl cation. The ensuing 1,6 cyclization involves C1 attack on the re face of C6 of the C6−C7 double bond to produce the (6S)-α-bisabolyl cation. This step is followed by a 120° CW rotation and 6,7 hydride shift to form the (7R)-β-bisabolyl cation. The (7R)-β-bisabolyl cation is a key reaction intermediate, lying at the intersection of the majority of cisoid hydrocarbon products. The orientation of the terminal isoprene unit at this stage directs the subsequent divergence of reaction trajectories at the C6−C10 cyclization step (1, panel c). When the isoprene unit is oriented endo, the C6−C10 cyclization produces the (1R,4R,5S)-α-acorenyl cation. This intermediate undergoes a further C3–C11 cyclization and then a Wagner–Meerwein rearrangement to a tertiary carbocation prior to proton elimination to produce (+)-2-epi-prezizaene (2). Conversely, the exo configuration leads to the (1R 4S,5S)-α-acorenyl cation, possessing the opposite stereochemistry at C4 relative to the aforementioned prezizaene pathway. A C2–C11 cyclization of this cation followed by proton elimination terminates the reaction pathway at (−)-α-cedrene (3).
The remaining stereochemically defined products comprise roughly equal amounts of sesquiterpene hydrocarbons and alcohols, and their formation can be rationalized as branches off the main reaction pathway (1, panel b). Early in the mechanism, water quenching on C1 or C3 of the nascent cis-farnesyl cation accounts for cis-farnesol (10) and nerolidol (7), respectively. Immediately following the initial C1−C6 cyclization to the α-bisabolyl cation, water quenching again intercepts the cyclization path by indiscriminant attack on either face of the cation to produce equal amounts of α- and epi-α-bisabolol (8 and 9), comprising approximately one-third of the alcohol products. Alternative proton eliminations from C5 of the (7R)-β-bisabolyl cation account for the third most abundant product in the TEAS cisoid spectrum of products, namely, (−)-β-curcumene (6), representing 16% of total hydrocarbon product. Finally, α- and 4-epi-α-acoradienes (4 and 5) stem from proton elimination from the terminal isopropenyl tail of the acorenyl cations, representing the remaining products observed at 4% and 1.2% total hydrocarbon, respectively.
Computational Analysis of the TEAS Cisoid Mechanism
The intrinsic reactivity, conformation, and energy of carbocation intermediates define physically allowable cyclization pathways, which ultimately pass through the selectivity filter of active site geometry and electrostatics, most likely modulated by enzyme dynamics. To computationally examine the conformation and intrinsic energetics of the cisoid cyclization pathway, we conducted density functional theory (DFT) calculations. While numerous transition states were identified which connect consecutive intermediates in the proposed reaction mechanism (1), alternative connectivities were discovered that bypass adjacent carbocations, thereby directly linking more distal steps in the cisoid cyclization pathway (3, panel a). For example, a transition structure (14) was found that bypasses the (7R)-β-bisabolyl cation in a concerted, highly asynchronous reaction by directly connecting the (6S)-α-bisabolyl cation to the (4S)-α-acorenyl cation along the pathway to (−)-α-cedrene. Following this, a transition structure was found for the next step, linking the (4S)-α-acorenyl cation to the final carbocation, thereby completing this pathway (via 14) from the (6S)-α-bisabolyl cation to (−)-α-cedrene (3) (3, panel b). Interestingly, the (6S)-α-bisabolyl cation to the (4R)-α-acorenyl connection was not uncovered due to steric occlusion, indicating the importance of the (7R)-β-bisabolyl cation along the pathway to (+)-2-epi-prezizaene (2).
Figure 3.

Computational analysis of the TEAS cisoid cyclization pathway. a) Density functional theory (DFT) calculations were performed on the TEAS cisoid pathway and revealed concerted, highly asynchronous reactions with a transition state (14 or 15) specific to the formation of (−)-α-cedrene (3) or (+)-2-epi-prezizaene (2), respectively (red arrows). Hong and Tantillo (24) previously located an alternative concerted highly asynchronous transition in the cedrene pathway (blue arrow). b) A transition state structure (14) was discovered that connects the (6S)-α-bisabolyl cation to the (4S)-α-acorenyl cation corresponding to point 50 on the intrinsic reaction coordinate plot. The migrating hydrogen (dark blue) and the two carbons (yellow) forming a nascent σ-bond (dashed line) are depicted. The plot shows the change in the dashed bond distance (⧫), the original (◼) and new (▲) C−H bond distances of the migrating hydrogen in 14 during the course of the reaction taken from an IRC calculation. The transition state structure is point 0. c) A transition state structure (15) along the (+)-2-epi-prezizaene (2) pathway is shown. Change in bond distances shown during the course of the IRC calculations bond (⧫, a bond; ●, b bond; ▲, c bond) indicates the highly asynchrous nature of this step. The transition state structure (15) is point 10.
Formally, the formation of (+)-2-epi-prezizaene (2) involves a high-energy secondary carbocation from the anti-Markovnikov C3−C11 cyclization. It has been suggested that such high-energy secondary carbocations in terpene biosynthesis can be avoided in the gas phase via concerted, highly asynchronous mechanisms, in analogy to the formation of the C and D rings in the cyclization of squalene oxide to lanosterol(23). These mechanisms have also been discovered in related computational studies of sesquiterpene cyclization. This was indeed the case here as we located a transition structure (15) and demonstrated with intrinsic reaction coordinate (IRC) calculations that it connected the distal cyclization events leading to (+)-2-epi-prezizaene (2) in a concerted, highly asynchronous step (3, panel c). Hong and Tantillo have independently identified this same transition state (24).
Structure of Wild-Type TEAS and M4 TEAS with 2-Fluoro Analogues
We expected that the three-dimensional structures of TEAS-2F-FPP complexes would be informative regarding the static templating of both cisoid and transoid pathways in the TEAS active site. To investigate the structural basis for substrate preorganization and catalytic promiscuity along the transoid and cisoid cyclization pathways, we carried out crystal soaks with the nonionizable substrate analogues trans-2F-FPP (2a) and cis-2F-FPP (2b), respectively. These experiments yielded protein−small molecule complexes diffracting to resolutions ranging from 2.1 to 2.6 Å (3).
Table 3. Crystallographic data and refinement statisticsa.
| wt TEAS-trans-2F-FPP | wt TEAS-cis-2F-FPP | M4 TEAS-trans-2F-FPP | M4 TEAS-cis-2F-FPP | |
|---|---|---|---|---|
| pdb code | 3M01 | 3M0 | 3LZ9 | 3M00 |
| Space group | P41212 | P41212 | P41212 | P41212 |
| Unit-cell parameters: | ||||
| a(Å) | 125.5 | 125.5 | 126.3 | 126.1 |
| b(Å) | 125.5 | 125.5 | 126.3 | 126.1 |
| c(Å) | 122.7 | 121.3 | 121.9 | 122.4 |
| α-β-γ° | 90 | 90 | 90 | 90 |
| Monomers per Asymm unit | 1 | 1 | 1 | 1 |
| Resolution range (Å) | 500.0−2.6 | 500.0−2.5 | 500.0−2.28 | 500.0−2.1 |
| No. reflections measured | 219537 | 324161 | 377780 | 248702 |
| Merging R-factor | 0.093 (0.325) | 0.077 (0.282) | 0.090 (0.346) | 0.080 (0.358) |
| ⟨I/σ⟩ | 17.28 (4.63) | 23.53 (5.12) | 16.04 (3.59) | 16.45 (3.55) |
| Completeness | 0.963 (0.914) | 0.979 (0.938) | 0.992 (0.903) | 0.956 (0.872) |
| Redundancy | 7.36 (7.48) | 8.54 (8.55) | 7.74 (6.53) | 4.02 (3.92) |
| No. reflections used | 30227 | 30047 | 22460 | 57483 |
| R-factor | 0.2065 | 0.1976 | 0.1935 | 0.2205 |
| Free R-factor | 0.2423 | 0.2257 | 0.2464 | 0.2586 |
| No. amino acid residues | 547 | 547 | 547 | 547 |
| No. water molecules | 150 | 168 | 174 | 412 |
Values in parentheses represent highest resolution shell.
Global comparison of all structures by superpositioning C-α carbons revealed a high degree of similarity with root mean square deviation (rmsd) values ranging from 0.22 to 0.37 Å for all atoms (Supplementary Table S1). Annotating structures according to B-factors suggested a common pattern of dynamic regions across all structures refined (Supplementary Figure S1). In contrast to the originally published TEAS·farnesyl hydroxyphosphonate (FHP) structure, all complexes described here exhibit disorder in a portion of the J−K catalytic loop, a region encompassing amino acids 521−533 that completes the enclosure of the active site during catalysis (5). Several residues were excised from both the wild-type and M4 TEAS models during refinement due to a lack of clearly observable electron density and the attendant poor refinement of these regions (Supplementary Figure S2). As previously noted, the mutations in the M4 TEAS protein reside either in the active site (V516I) or distribute more peripherally around the active site surface (A274T, V372I, and Y406L) with distances from the active site center ranging from 7 to 14 Å (Supplementary Figure S3). While each mutated side chain was readily discernible in the electron density, no significant backbone distortions were evident, strongly hinting at dynamic, not static, modulation of the active site contour for templating transformations of farnesyl cations in TEAS. However, the V516I mutation directly affects the active site contour with implications for substrate binding as discussed below.
Observable electron density is present in the active site regions for all complexes, and the positions of ligand-binding residues were clearly established with the exception of Y527 on the J−K loop. In all the structures, contiguous electron density stretches from the DDxxD motif through the diphosphate moiety into the NSE/DTE motif enshrouding the catalytically essential Mg2+ ions (4, panel c). Although three Mg2+ ions are visible in each complex, a complete octahedral coordination sphere of waters is only discernible in the highest resolution M4 TEAS-cis-2F-FPP complex. Electron density surrounding the diphosphate appendage is the most prominent feature in the calculated electron density (without ligands modeled) with large σ values in the SIGMAA-weighted 2Fo − Fc electron density maps (4, panel b). Clear electron density extends from the diphosphate through the first isoprene unit containing the fluoro substituent in all complexes but trails off through the center of the chain and picks up again at the distal isoprene unit (4, panel e). Despite the waning electron density for the distal isoprene units, the farnesyl chain clearly curls into a U-shape in all complexes, particularly at lower σ where continuous density is apparent in the wild-type complexes (4, panel e). Taken together, these complexes display near complete occupancy (based upon the unmistakable diphosphate and first isoprene unit electron density), and aside from an incomplete J−K loop, Mg2+ ions and ligands are bound with the farnesyl chain folded in a manner consistent with the formation of major cyclization products along both the transoid and cisoid mechanistic pathways.
Figure 4.

Crystallographic analysis of wild-type and M4 TEASs bound to fluoro-FPPs. a) Global structure of TEAS is illustrated as a rainbow-colored ribbon with the active site region boxed. b) Zoomed-in view of the Mg2+-diphosphate coordination complex of the M4 TEAS-cis-2F-FPP complex with the 2Fo − Fc map contoured at 3σ. c) Close-up view of the DDxxD motif (residues 301, 302 (not shown), and 305), neighboring NSE/DTE motif (residues 444, 448, and 452), coordinating Mg2+ and diphosphate in the indicated fluoro-farnesyl diphosphate complexes contoured at 1σ in the 2Fo − Fc SIGMAA-weighted electron density map. d) Close-up of the TEAS-cis-2F-FPP complex active site showing the bound ligand and the neighboring TEAS residues. e) Ligand density for the respective complexes with the SIGMAA-weighted 2Fo − Fc electron density map contoured to either 1σ (dark blue) or 0.6σ (light blue).
Comparison of ligand binding modes between the cis- and trans-2F-FPP complexes reveals important differences relating to catalysis. While the orientation of the C−O bond in both trans-2F-FPP structures is nearly perpendicular to the plane of the C2−C3 double bond as required for maximum activity, the C−O bond adopts a parallel position in the cis-2F-FPP structures and hence represents an inactive conformation. If this conformation were reflective of the (cis,trans)-FPP binding, then rotation of the C1−C2 bond would be required to form a catalytically active complex. This inactive conformation may be promoted by the 2-fluoro moieties in cis-2F-FPP (2b) through its electrostatic interaction with Arg 264 residing 3 Å away (4, panel d).
Spatial Reconstruction of Cisoid and Transoid Reaction Pathways in TEAS
Multiple substrate binding modes discerned during building and refinement for the extended farnesyl chain could potentially satisfy, and likely contribute to, the observed electron densities (Supplementary Figure S4). Despite these ambiguities, the general topology of the farnesyl chain is clear and consistent with the anticipated parental fold inferred from the elucidated stereochemistry of the final products. Importantly, electron density for the cis-2F-FPP complexes reveals that the terminal isoprene unit curls into a helical (endo) fold in accordance with the anticipated conformation (1, panel c). This orients the plane of the C10–C11 double bond parallel to a potentially attacking carbocation at C1. In contrast, the plane of the C10–C11 double bond of the terminal isoprene unit is perpendicular to a nascent C1 carbocation in the trans-2F-FPP complexes, in accord with an initial C1–C10 cyclization along the transoid pathway.
To spatially reconstruct the two major cyclization pathways in TEAS, we manually docked transition state structures and models of the major products into the respective active sites of wild-type trans-2F-FPP and cis-2F-FPP complexes (5, panel a). Restraining the placement of products/intermediates such that cyclization to a specific product most likely proceeds with the minimal amount of conformational distortion for the nascent farnesyl cation en route to the final products results in a mechanistically plausible transition state geometry en route to the observed dominant product.
Figure 5.

Spatial reconstruction of the transoid and cisoid cyclization pathways in TEAS. a) Refined conformations for the trans-2F-FPP or cis-2F-FPP·TEAS complexes are displayed in the binding pocket (clipped surface), and models of indicated reaction intermediates or products were manually positioned relative to the refined conformations. An accompanying schematic of chemical structures designates the 2-fluoro positions in each substrate as H(F). Images were rendered with UCSF Chimera (55). Transition state structures are shown alongside their corresponding rendered figures; dashed lines are used to indicate bond breakage and formation. b) Proposed substrate folds leading to the cisoid and transoid cyclization pathways in TEAS.
Superposition of the transition state structure (15) on the farnesyl chain indicates that substantial contraction of the substrate must occur to produce the compact (+)-2-epi-prezizaene (2) final product. The crucial elements of preorganization are the juxtaposition of C1 and C6 together with the endo orientation of the farnesyl tail, both consistent with the observed electron density of the TEAS·cis-2F-FPP complex. Therefore, the static picture drawn from these observations reveals a catalytically relevant substrate binding conformation and substrate/intermediate preorganization very early along the cisoid pathway catalyzed by TEAS. Based on this model, the pyrophosphate ion would reside close by but suitably sequestered by neighboring interactions to stabilize the developing positive charge of the secondary carbocation while limiting recapture probability prior to the final proton elimination yielding (+)-2-epi-prezizaene (2).
For the transoid pathway, the key transition state structure leading to the major product (+)-5-epi-aristolochene (1) involves a methyl migration atop the decalin ring system of the eremophilyl carbocation (5, panel a, right) as previously reported (17). To achieve this energetically favorable alkyl migration, substrate folding must preorganize an initial electrophilic attack of C1 on C10 of the distal double bond. This requires substantial movement of the chain following ionization, as these atoms are 5 Å apart in the ground state complexes. However, judging by the degree of overlap between the transition state model and the farnesyl chain, this motion can be accommodated largely within the first isoprene unit with minimal conformational adjustments of the more distal isoprene units. Therefore, this conformation of the farnesyl chain is consistent with TEAS preorganizing FPP (or more likely the resultant acyclic farnesyl cation) for cyclization to 5-epi-aristolochene (1), in contrast to the original TEAS·FHP complex(5). The phosphate moiety of FHP overlaps with the β-phosphates of 2F-FPPs, although the farnesyl chain is more extended and folds in essentially the opposite direction (Supplementary Figure S4).
Cisoid Cyclase Activities with (trans,trans)-FPP
On the basis of our spatial reconstruction of the cisoid cyclization pathway, we propose a model describing the “cisoid fold” of (trans,trans)-FPP (1a) that is representative of the preorganization of the farnesyl chain leading to its conversion to cisoid-cation-derived hydrocarbons. Accordingly, an alternative, catalytically productive binding mode of FPP is populated in which the farnesyl chain curls into its typical U-shaped topology, but with the first two isoprenoid units inverted relative to the “transoid fold” configuration (5, panel b). The cisoid binding mode therefore possesses the DU configuration, opposite to that described for germacrene A(25), and importantly, with the distal isoprenoid unit curled below this plane into an alternative binding pocket formed by T401, T402, C440, R441, D444, and the diphosphate moiety (4, panel a). We posit that the positioning and anchoring of the terminal isoprenoid unit is an essential stereochemical feature for triggering ionization, perhaps through both steric and electronic effects. Upon ionization, a kinetically slow initial isomerization occurs as the C10–C11 double bond is rotated out of position for electrophilic attack by C1; in turn, a re face capture by the pyrophosphate ion on C3 of the nascent farnesyl cation generates the neutral (3S)-NPP intermediate. Rotation around the C2–C3 bond followed by reionization generates the 2,3-cis-farnesyl cation and entry into the cisoid cyclization pathway.
Structural Picture of Catalytic Promiscuity
The ligand–protein structures of a promiscuous TEAS mutant offer a glimpse into the structural underpinnings of product specificity or lack thereof. To discern the structural basis for product specificity in both cisoid and transoid cyclization pathways, we conducted a comparative analysis of wild-type TEAS and M4 mutant structures with particular attention focused on the active site contour and farnesyl chain binding modes. The most obvious surface distortion, whether statically or dynamically derived, is contributed by the V516I mutation, which introduces a methyl group into the active site cavity (Supplementary Figure S5). While no drastic distortion is evident in the comparative models of the farnesyl chain, the electron density for the farnesyl chain in the M4 TEAS structures is discontinuous, indicative of increased dynamic motion and/or local disorder (4, panel e). Interestingly, only the M4 TEAS-cis-2F-FPP complex exhibits a significant shift in the position of Y520, which additionally alters the active site surface features. The ligand-dependency for the Y520 shift may reflect the interaction between the farnesyl chain, wherein the central isoprene unit is inverted relative to the corresponding wild type complex, and active site residues in defining the preorganized binding state. Despite the higher resolution of the M4 structures, the density for the ligand is discontinuous, even when the electron density maps are viewed at low σ, in contrast to the wild-type structures (4, panel e). It is probable that a more dynamic farnesyl chain in M4-TEAS-cis/trans-2F-FPP structures explains the lack of electron density for the entire farnesyl chain consistent with this M4 TEAS’s promiscuous catalytic activity along both pathways.
Conclusions
(cis,trans)-FPP proved effective in directing reactions along the cisoid cyclization pathway in TEAS. The isolation and stereochemical elucidation of the products lead to formulation of reasonable reaction pathways to the cisoid-derived sesquiterpene skeletons(16). Traditionally, chemical tools have been a vital part of defining the stereochemical course of terpene biosynthesis, as elegantly exemplified by the use of (1R)-[1-3H]- and (1S)-[1-3H]-geranyl diphosphate to provide direct experimental confirmation that cyclization along the cisoid pathway results in net retention of configuration at C1 of the substrate (26). Fluoro isoprenoid diphosphate substrate analogues also have been instrumental in elucidating mechanistic aspects of terpene biosynthesis, most notably through the interception of reaction intermediates such as 6-fluorogermacrene A(27) and 7-fluoroverticillenes (28) in TEAS and taxadiene synthase enzymes, respectively. Most recently, 2F-FPP and 12,13-difluorofarnesyl diphosphate (DF-FPP) were instrumental in deciphering the probable order of metal-ion binding and conformational changes required for catalysis by aristolochene synthase from Aspergillus terrus(22).
To date, crystallographic analyses of terpene cyclases have yielded important insights into how these enzymes function on the atomic scale. Most notably, anchoring the diphosphate moiety of the substrate and metal coordination by the DDxxD and NSE/DTE motifs shown in crystal structures paints a picture of the fundamental role of these events in terpene synthase catalysis. Structures containing inorganic pyrophosphate or a substrate analogue bound in the active site display ordering of various loops proximal to the active site, consistent with a closed protein conformation that shields reactive carbocation intermediates from solvent5,21,29. Further, alterations in pyrophosphate binding are thought to aid in the modulation of prenyl chain orientation within the active site and most likely modulate the fate of the early intermediates along prescribed mechanistic pathways(30). All structures reported in the current study contain the full complement of Mg2+ ions coordinating the diphosphate of the fluoro-farnesyl analogues. However, despite this clear coordination geometry, elements of the J−K loop remain disordered in both wild-type and mutant structures bound to diphosphate containing ligands with Y527 electron density missing from the active site. This lack of observable density for Y527 stands in contrast to the original TEAS·FHP complex where this residue is clearly discernible (Supplementary Figure S2).
These observations hint at a greater role of dynamics in terpene chain cyclization than evident from the early structural work based upon static crystal structures. The wild-type ligand complexes in the current study revealed density for the farnesyl chain folded in a manner consistent with catalysis, and this can be interpreted in light of the established stereochemistry for all TEAS products. Moreover, these static observations enabled the positing of 2 distinct parental folds each of which gives rise to either cisoid or transoid cyclization pathways for TEAS (5, panel b). The structure of a complex of limonene synthase with 2-fluorolinalyl diphosphate captured another instance where the isoprenoid chain conformation is consistent with the geometry of the final product(21). However, it has been noted that most reported crystal structures of terpene synthases complexed with isoprenoid substrate analogues, including 2F-FPP used here, reveal isoprenoid tail conformations that are not catalytically relevant5,21,22,29. Considering the ideal case, where there is unambiguous density for every atom of the farnesyl chain, a central challenge in the field remains to resolve structural features responsible for product specificity or lack thereof from a static picture alone, given the degeneracy of possible products arising from a single parental substrate fold. Future progress toward defining the origins of sesquiterpene skeletal complexity will undoubtedly benefit from integrating dynamic information from NMR and time-resolved fluorescence (in progress) with computational approaches and protein crystallography to develop a much clearer and time-resolved biophysical picture of terpene synthase directed cyclization.
What possible relevance does the cryptic cisoid cyclization pathway of TEAS have in the natural world? Although (cis,trans)-FPP has not been identified as a metabolite in tobacco or related Solanaceous plants, a (cis,trans)-farnesyl diphosphate synthase has been identified in Mycobacterium tuberculosis involved in bacterial cell wall synthesis (31,32), suggesting the potential relevance of this compound in other biological systems. Moreover, while often observed, the biological significance of small amounts (3–14% of total product) of (cis,trans)-FPP formation by FPP synthases (33) has been ignored to date. Is it possible then that TEAS possesses a “moonlighting” role in vivo by gathering up what we would normally consider biosynthetic “waste” and recycling it into a bioactive product? While TEAS produces cisoid terpenes in vitro, the presence of these metabolites has yet to be confirmed in planta. Nonetheless, TEAS clearly possesses an efficient catalytic potential to access presently unanticipated in vivo chemical diversity from lengthy branches of the cisoid reaction pathway, a property that may have been naturally selected for and that can also be immediately exploited for biotechnological applications starting with (cis,trans)-FPP.
Methods
Organic Synthesis
(cis,trans)-FPP was available from the concurrent investigation(16). (trans, trans)- and (cis,trans)-2-FluoroFPPs were accessed from the corresponding 2-fluorofarnesol isomers (28) by conversion to the respective 2-fluorofarnesyl chlorides and SN2 displacements with (nBu)4N/diphosphate (HOPP)(34) with complete retention of the 2,3-double bond configurations by means of procedures similar to those reported previously (see Supporting Information) (28). (cis,trans)-2-FluoroFPP has not been previously described in the literature. Characterization data for ammonium salt of 2b are as follows: white solid (51 mg, 68%); 1H NMR (CD3OD, 400 MHz) δ 5.17–5.11 (m, 1H, vinyl H), 5.11–5.05 (m, 1H, vinyl H), 4.59 (dd, 2H, J = 23.3, 5.4 Hz, CH2OPP), 2.16–2.11 (m, 4H, CH2), 2.09–2.04 (m, 2H, CH2), 2.00–1.95 (m, 2H, CH2), 1.68 (d, 3H, J = 3.5 Hz, CH3), 1.66 (q, 3H, J = 1.2 Hz, CH3), 1.61 (d, 3H, J = 1.2 Hz, CH3), 1.60 (br d, 3H, J = 0.6 Hz, CH3); 31P NMR (CD3OD, 162 MHz) δ −7.99 (br d, J = 14.9 Hz), −9.30 (br d, J = 14.1 Hz); 19F NMR (CD3OD, 376 MHz) δ −118.9 (td, J = 23.2, 3.5 Hz).
Protein Crystallization and Data Collection
Crystallization of purified proteins was conducted using hanging drops over a 0.5-mL reservoir (15% w/v PEG8K, 200 mM Mg(OAc)2, 100 mM 3-(N-morpholino)-2-hydroxypropanesulfonic acid (MOPSO)-Na+, pH 7.0). Crystal soaks were conducted overnight in reservoir solution containing 10 mM fluoro-farnesyl diphosphate ligand. Crystals were frozen in soak solution also containing 20% v/v ethylene glycol as cryoprotectant. Data collection was performed at the Stanford Synchrotron Radiation Laboratory (SSRL) beamline 1–5 for wild-type TEAS-2F-FPP and cis-2F-FPP complexes, while data for the M4 TEAS mutant structures were collected at the Advanced Light Source (ALS) beamline 8.2.1. All data were processed using XDS software (35). The initial crystallographic structure solutions were obtained through molecular replacement analyses using the TEAS-FHP complex (PDB id 5eat) as the search model with Molrep in Collaborative Computational Project No. 4 (CCP4) (36). Model building was performed using COOT (37) and rounds of refinement were conducted using Crystallography NMR System (CNS) (38). To refine the position of the farnesyl chain, the first isoprene unit containing the 2-flouro group was built and refined, followed by sequential addition, building and refinement of remaining isoprene units. For the wild type cis-2F-FPP complex, additional multi-conformer refinement was undertaken using the program RefMac (39–46) in CCP4(36). The fold inferred from the known stereochemistry of the final products was built into the density followed by a final round of refinement using CNS to produce the current models of the farnesyl chain (Supplementary Figure S5).
Computational Methods
All calculations were performed with GAUSSIAN 98W and GAUSSIAN 09W (47) and the density functional method using B3LYP, Becke’s three-parameter hybrid method (48) with the Lee–Yang–Parr correlation functional (49) and the 6-31G* basis set (50). All stationary points were confirmed with second derivative calculations. Energies reported here include zero-point energy corrections calculated with unscaled B3LYP/6-31G* frequencies obtained analytically with G98W. Intrinsic reaction coordinate calculations (51,52) were used to determine reaction pathways. Single point mpw1pw91/6-311+G(2d,p)//B3LYP/6-31G* calculations as recommended by Matsuda (53) were carried out for all stationary points reported (54).
Product Elucidation
A preparative-scale incubation was carried out using 127 mg (310 mmol) of (cis,trans)-FPP and a total of 18 mg of recombinant wild-type TEAS in order to accumulate sufficient material for chromatographic fractionations, NMR analyses, and optical rotation measurements of the major products as described(16).
Protein expression, purification, and kinetic measurement are detailed in Supporting Information.
Acknowledgments
L.S. acknowledges financial support from the Polish Ministry of Science and Education (research project no. N N 202 187636) This work was supported by the Howard Hughes Medical Institute and the National Science Foundation Grant MCB-0645794 (J.P.N.).
Supporting Information Available
This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Gershenzon J.; Dudareva N. (2007) The function of terpene natural products in the natural world. Nat. Chem. Biol. 3, 408–414. [DOI] [PubMed] [Google Scholar]
- Liang P.; Ko T.; Wang A. (2002) Structure, mechanism and function of prenyltransferases. Eur. J. Biochem. 269, 3339–3354. [DOI] [PubMed] [Google Scholar]
- Ruzicka L.; Eschenmoser A.; Heusser H. (1953) The isoprene rule and the biogenesis of terpenic compounds. Experentia 9, 357–367. [DOI] [PubMed] [Google Scholar]
- Cane D. (1985) Isoprenoid biosynthesis. Stereochemistry of the cyclization of allylic pyrophosphates. Acc. Chem. Res. 18, 220–226. [Google Scholar]
- Starks C.; Back K.; Chappell J.; Noel J. (1997) Structural basis for cyclic terpene biosynthesis by tobacco 5-epi-aristolochene synthase. Science 277, 1815–1820. [DOI] [PubMed] [Google Scholar]
- Lesburg C.; Zhai G.; Cane D.; Christianson D. (1997) Crystal structure of pentalenene synthase: mechanistic insights on terpenoid cyclization reactions in biology. Science 277, 1820–1824. [DOI] [PubMed] [Google Scholar]
- Shishova E.; Di Costanzo L.; Cane D.; Christianson D. (2007) X-ray crystal structure of aristolochene synthase from Aspergillus terreus and evolution of templates for the cyclization of farnesyl diphosphate. Biochemistry 46, 1941–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köllner T.; Schnee C.; Li S.; Svatos A.; Schneider B.; Gershenzon J.; Degenhardt J. (2008) Protonation of a neutral (S)-β-bisabolene intermediate is involved in (S)-β-macrocarpene formation by the maize sesquiterpene synthases TPS6 and TPS11. J. Biol. Chem. 283, 20779–20788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picaud S.; Mercke P.; He X.; Sterner O.; Brodelius M.; Cane D.; Brodelius P. (2006) Amorpha-4,11-diene synthase: mechanism and stereochemistry of the enzymatic cyclization of farnesyl diphosphate. Arch. Biochem. Biophys. 448, 150–155. [DOI] [PubMed] [Google Scholar]
- Cane D.; Ha H. (1988) Trichodiene biosynthesis and the role of nerolidyl pyrophosphate in the enzymatic cyclization of farnesyl pyrophosphate. J. Am. Chem. Soc. 110, 6865–6870. [Google Scholar]
- Mercke P.; Crock J.; Croteau R.; Brodelius P. (1999) Cloning, expression, and characterization of epi-cedrol synthase, a sesquiterpene cyclase from Artemisia annua L. Arch. Biochem. Biophys. 369, 213–222. [DOI] [PubMed] [Google Scholar]
- Lin X.; Cane D. (2009) Biosynthesis of the sesquiterpene antibiotic albaflavenone in Streptomyces coelicolor. Mechanism and stereochemistry of the enzymatic formation of epi-isozizaene. J. Am. Chem. Soc. 131, 6332–6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis E. M.; Croteau R. (2000) Cyclization enzymes in the biosynthesis of monoterpenes, sesquiterpenes, and diterpenes. Top. Curr. Chem. 209, 53–95. [Google Scholar]
- Gordon M.; Stoessl A.; Stothers J. (1973) Post-infectional inhibitors from plants. 4. Structure of capsidiol - antifungal sesquiterpene from sweet peppers. Can. J. Chem. 51, 748–752. [Google Scholar]
- O’Maille P. E.; Chappell J.; Noel J. (2006) Biosynthetic potential of sesquiterpene synthases: alternative products of tobacco 5-epi-aristolochene synthase. Arch. Biochem. Biophys. 448, 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox R. B., and Powell W. H. (2001) Nomenclature of Organic Compounds, 2nd ed., pp 306−308, American Chemical Society and Oxford University Press, Oxford. [Google Scholar]
- Rigaudy J., and Klesney S. P. (1979) IUPAC Nomenclature of Organic Chemistry: Sections A, B, C, D, E, F and H, pp 475−477, Pergamon Press, Oxford. [Google Scholar]
- Faraldos J. A., O’Maille P. E., Dellas N., Noel J., and Coates R. M. (2009) Bisabolyl-derived sesquiterpenes from tobacco 5-epi-aristolochene synthase-catalyzed cyclization of (2Z, 6E)-farnesyl diphosphate. J. Am. Chem. Soc., accepted for publication. [DOI] [PubMed] [Google Scholar]
- O’Maille P. E.; Malone A.; Dellas N.; Andes Hess B. J.; Smentek L.; Sheehan I.; Greenhagen B.; Chappell J.; Manning G.; Noel J. (2008) Quantitative exploration of the catalytic landscape separating divergent plant sesquiterpene synthases. Nat. Chem. Biol. 4, 617–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Maille P. E.; Chappell J.; Noel J. (2004) A single-vial analytical and quantitative gas chromatography-mass spectrometry assay for terpene synthases. Anal. Biochem. 335, 210–217. [DOI] [PubMed] [Google Scholar]
- Miller D.; Yu F.; Allemann R. (2007) Aristolochene synthase-catalyzed cyclization of 2-fluorofarnesyl-diphosphate to 2-fluorogermacrene A. ChemBiochem 8, 1819–1825. [DOI] [PubMed] [Google Scholar]
- Vedula L.; Zhao Y.; Coates R.; Koyama T.; Cane D.; Christianson D. (2007) Exploring biosynthetic diversity with trichodiene synthase. Arch. Biochem. Biophys. 466, 260–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D.; Youn B.; Zhao Y.; Santhamma B.; Coates R.; Croteau R.; Kang C. (2007) Structure of limonene synthase, a simple model for terpenoid cyclase catalysis. Proc. Natl. Acad. Sci. U.S.A. 104, 5360–5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishova E.; Yu F.; Miller D. J.; Faraldos J.; Zhao Y.; Coates R.; Allemann R.; Cane D.; Christianson D. (2008) X-ray crystallographic studies of substrate binding to aristolochene synthase suggest a metal binding sequence for catalysis. J. Biol. Chem. 283, 15431–15439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B. (2002) Concomitant C-ring expansion and D-ring formation in lanosterol biosynthesis from squalene without violation of Markovnikov’s rule. J. Am. Chem. Soc. 124, 10286–10287. [DOI] [PubMed] [Google Scholar]
- Hong Y.; Tantillo D. (2009) Consequences of conformational preorganization in sesquiterpene biosynthesis: theoretical studies on the formation of the bisabolene, curcumene, acoradiene, zizaene, cedrene, duprezianene, and sesquithuriferol sesquiterpenes. J. Am. Chem. Soc. 131, 7999–8015. [DOI] [PubMed] [Google Scholar]
- Faraldos J. A.; Wu S.; Chappell J.; Coates R. M. (2007) Conformational analysis of (+)-germacrene A by variable-temperature NMR and NOE spectroscopy. Tetrahedron 63, 7733–7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croteau R.; Felton N.; Wheeler C. (1985) Stereochemistry at C-1 of geranyl pyrophosphate and neryl pyrophosphate in the cyclization to (−)-bornyl pyrophosphate. J. Biol. Chem. 260, 5956–5962. [PubMed] [Google Scholar]
- Faraldos J. A.; Zhao Y.; O’Maille P. E.; Noel J.; Coates R. M. (2007) Interception of the enzymatic conversion of farnesyl diphosphate to 5-epi-aristolochene by using a fluoro substrate analogue: 1-fluorogermacrene A from (2E,6Z)-6-fluorofarnesyl diphosphate. ChemBioChem 8, 1826–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y. H.; Williams D.; Croteau R.; Coates R. M. (2005) Taxadiene synthase-catalyzed cyclization of 6-fluorogeranylgeranyl diphosphate to 7-fluoroverticillenes. J. Am. Chem. Soc. 127, 7834–7842. [DOI] [PubMed] [Google Scholar]
- Whittington D.; Wise M.; Urbansky M.; Coates R.; Croteau R.; Christianson D. (2002) Bornyl diphosphate synthase: structure and strategy for carbocation manipulation by a terpenoid cyclase. Proc. Natl. Acad. Sci. U.S.A. 99, 15375–15380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedula L.; Cane D.; Christianson D. (2005) Role of arginine-304 in the diphosphate-triggered active site closure mechanism of trichodiene synthase. Biochemistry 44, 12719–12727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulbach M.; Brennan P.; Crick D. (2000) Identification of a short (C15) chain Z-isoprenyl diphosphate synthase and a homologous long (C50) chain isoprenyl diphosphate synthase in Mycobacterium tuberculosis. J. Biol. Chem. 275, 22876–22881. [DOI] [PubMed] [Google Scholar]
- Schulbach M.; Mahapatra S.; Macchia M.; Barontini S.; Papi C.; Minutolo F.; Bertini S.; Brennan P.; Crick D. (2001) Purification, enzymatic characterization, and inhibition of the Z-farnesyl diphosphate synthase from Mycobacterium tuberculosis. J. Biol. Chem. 276, 11624–11630. [DOI] [PubMed] [Google Scholar]
- Thulasiram H.; Poulter C. D. (2006) Farnesyl diphosphate synthase: the art of compromise between substrate selectivity and stereoselectivity. J. Am. Chem. Soc. 128, 15819–15823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodside A., Huang Z., and Poulter C. D. (1993) Trisammonium geranyl diphosphate, in Organic Synthesis, Collect. Vol. 8, pp 616−620, Wiley, New York. [Google Scholar]
- Kabsch W. (1993) Automated processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 26, 795–800. [Google Scholar]
- (1994) The CCP4 suite: programs for protein crystallography, Acta Crystallogr. D 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowton K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Brunger A.; Adams P.; Clore G.; Delano W.; Gros P.; Grosse-Kunstleve R. W.; Jiang J.; Kuszewski J.; Nilges M.; Pannu N.; Read R.; Rice L.; Simonson T.; Warren G. (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Murshudov G.; Vagin A.; Lebedev A.; Wilson K. S.; Dodson E. J. (1999) Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallog.r D 55, 247–255. [DOI] [PubMed] [Google Scholar]
- Murshudov G.; Vagin A.; Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Pannu N.; Murshudov G.; Dodson E.; Read R. (1998) Incorporation of prior phase information strengthens maximum-likelihood structure refinement. Acta Crystallogr. D 54, 1285–1294. [DOI] [PubMed] [Google Scholar]
- Skubak P.; Murshudov G.; Pannu N. (2004) Direct incorporation of experimental phase information in model refinement. Acta Crystallogr. D 60, 2196–2201. [DOI] [PubMed] [Google Scholar]
- Steiner R.; Lebedev A.; Murshudov G. (2003) Fisher’s information in maximum-likelihood macromolecular crystallographic refinement. Acta Crystallogr. D 59, 2114–2124. [DOI] [PubMed] [Google Scholar]
- Vagin A.; Steiner R.; Lebedev A.; Potterton L.; Mcnicholas S.; Long F.; Murshudov G. (2004) REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D 60, 2184–2195. [DOI] [PubMed] [Google Scholar]
- Winn M.; Isupov M.; Murshudov G. (2001) Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D 57, 122–133. [DOI] [PubMed] [Google Scholar]
- Winn M.; Murshudov G.; Papiz M. (2003) Macromolecular TLS refinement in REFMAC at moderate resolutions. Acta Crystallogr. D 374, 300–321. [DOI] [PubMed] [Google Scholar]
- Frisch M.et al. (1998) Gaussian, Inc., Pittsburgh, PA.
- Becke A. (1993) Density-functional thermochemistry 3. The role of exact exchange. J. Chem. Phys. 98, 5648–5652. [Google Scholar]
- Lee C.; Yang W.; Parr R. (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785. [DOI] [PubMed] [Google Scholar]
- Hariharan P.; Pople J. (1973) The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 28, 213. [Google Scholar]
- Gonzalez C.; Schlegel H. (1989) An improved algorithm for reaction path following. J. Chem. Phys. 90, 2154. [Google Scholar]
- Gonzalez C.; Schlegel H. (1990) Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 94, 5523. [Google Scholar]
- Matsuda S.; Wilson W. (2006) Mechanistic insights into triterpene synthesis from quantum mechanical calculations. Detection of systematic errors in B3LYP cyclization energies. Org. Biomol. Chem. 4, 530. [DOI] [PubMed] [Google Scholar]
- Adamo C.; Barone V. (1998) Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models. J. Chem. Phys. 108, 664. [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.