

Abstract
A panel of amino acid analogs and conformationally-restricted amino acids bearing a sulfonic acid were synthesized and tested for their ability to preferentially inhibit the obligate cysteine-glutamate transporter system xc− versus the vesicular glutamate transporter (VGLUT). Several promising candidate molecules were identified: R/S-4-[4′-carboxyphenyl]-phenylglycine, a biphenyl substituted analog of 4-carboxyphenylglycine and 2-thiopheneglycine-5-sulfonic acid both of which reduced glutamate uptake at system xc− by 70–75% while having modest to no effect on glutamate uptake at VGLUT.
Keywords: glutamate, amino acid analog, inhibitor, VGLUT, system Xc−, sulfonic acid
L-Glutamate (1) is a key neurotransmitter responsible for the vast majority of the fast excitatory synaptic communication in the mammalian CNS. L-Glutamate acts at ionotropic glutamate receptors to mediate ligand gated ion channels and at metabotropic glutamate receptors to couple intracellular second messenger systems via G-proteins.1–5 The importance of L-glutamate as a contributor to higher order processing required in development, plasticity, learning, and memory is well established.4,6,7 However, glutamatergic excitotoxicity can result when an excess of L-glutamate occurs and continually activates glutamate receptors.2,5,8 To maintain the proper titer of L-glutamate there is a network of strategically positioned transporters that shuttle L-glutamate in and out of cells and organelles. Most notable among these transporters are the excitatory amino acid transporters (EAATs) that facilitate the uptake of L-glutamate into neurons.8
In addition to EAATs, other transporters maintain intra- and extra-cellular levels of glutamate including; system xc−, a chloride-dependent, sodium-independent obligate exchanger that couples the export of intracellular L-glutamate with the import of extracellular L-cystine9–12 and the vesicular glutamate transporter (VGLUT) that mediates the uptake of intracellular glutamate into synaptic vesicles.6,13,14
System xc− and VGLUT are structurally and functionally distinct from the EAATs and also from each other. Although system xc− and VGLUT differ pharmacologically,5,8,15,16 both transporters remove L-glutamate from the cytosol. In principle, therefore, intracellular L-glutamate levels could be regulated by modulating one or both of these transporters.16 As such, the development of inhibitors that selectively block system xc− and/or VGLUT represents an interesting pharmacologic challenge. Some inhibitors9,16,17 have been reported for system xc− and for VGLUT6,15,18–26 (Fig. 1). Two interesting features common to several system xc− and VGLUT inhibitors are the use of aromatic rings to conformationally lock27 the acid groups (e.g., CPG) and sulfonic acid isosteres 24,25,28 in place of a carboxylic acid. Seeing these as opportunities to explore similarities and differences in the specificities of system xc− and VGLUT, we prepared a number of conformationally-restricted glutamate analogs bearing a sulfonic acid group in place of the distal (γ) carboxylic acid of glutamate.
Figure 1.

Structures of glutamate, VGLUT and system xc− inhibitors and their corresponding IC50 values.
The target compounds were synthesized as shown in Schemes 1 and 2. Simple amino acid analogs (3a–e) of phenylglycine were synthesized via hydrolysis of the corresponding hydantoin intermediates (2a–e).29–32 The preparation of sulfonic acid analogs 5a–i was carried out by reaction of commercially available amino acids with fuming sulfuric acid to afford monosulfonic acid analogs.33 To explore the relative contribution of the amino acid center to inhibition two additional targets, compounds 7a–b, were synthesized by hydrolysis of the commercially available structures 6a–b using 2N NaOH. Each synthesized compound was characterized by 1H NMR, IR and mass spectral analysis34 prior to testing at the two transporter systems (Table 1). Activity was assessed by quantifying the ability of the compounds to inhibit the specific uptake of 3H-L-gluamate by either system xc- or VGLUT. System xc− mediated uptake of L-glutamate (100 μM) was measured in SNB19 glioma cells under Na-free conditions, corrected for non-specific uptake, and normalized to protein content.9 VGLUT mediated uptake of L-glutamate (250 μM) was measured in synaptic vesicles isolated from rat brain, corrected for non-specific uptake, and normalized to protein content.20
Scheme 1.

Synthesis of amino acid analogs 3a–f. Reagents and conditions: (i) (NH4)2CO3, KCN, 1:1 MeOH, H2O, 50–60 °C, 3 h; (ii) Ba(CO3)2, H2O, 100 °C, 72 h.
Scheme 2.

Preparation of thiophene analogs 7a/7b.
Table 1.
Percent of L-glutamate uptake in system xc− and VGLUT for compounds 3a–f, 5a–i and 7ab. System xc− assay: 100 uM L-glutamate and 500 uM of inhibitor. VGLUT assay: 250 uM L-glutamate and 5 mM of inhibitor.
| Entry | Structure or name | system xc− (% of uptake)9 | VGLUT (% of uptake)22 |
|---|---|---|---|
| 3a | R/S-5-(2-thienyl)-2- thienylglycine | 69±1 | 53±4 |
| 3b | R/S-4,5-dimethyl-2- thienylglycine | 73±3 | 57±10 |
| 3c | R/S-5-imidazoylglycine | 96±6 | 95±4 |
| 3d | R/S-5-bromo-2-thienylglycine | 83±17 | 37±6 |
| 3e | R/S-4-[4′-carboxyphenyl]-phenylglycine | 27±2 | 75±7 |
| 3f | R/S-benzothiophene-3-glycine | 53±2 | 31±4 |
| 5a | 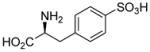 |
90±3 | 131±31 |
| 5b | 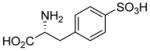 |
101±6 | 102±10 |
| 5c | 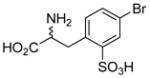 |
83±2 | 77±9 |
| 5d | 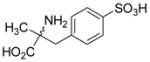 |
106±13 | 98±13 |
| 5e | 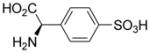 |
98±9 | 108±9 |
| 5f | 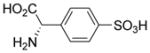 |
55±4 | 105±10 |
| 5g | 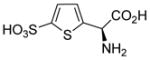 |
32±1 | 92±4 |
| 5h | 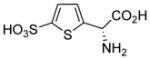 |
55±3 | 104±9 |
| 5i | 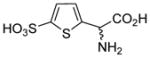 |
26±3 | 81±7 |
| 7a | 3-amino, 4-methylthiophene-2- carboxylic acid | 83±2 | 91±6 |
| 7b | 3-methyl, 5-aminothiophene-2,4-dicarboxylic acid | 57±4 | 44±3 |
| Cntrls | L-cystine Congo Red |
22±3 n/a |
n/a 31±2 |
The rationale for testing compounds 3a–f was based on the fact that a thienylglycine heterocycle contains an embedded cysteine. The imidazole structure was prepared as a control analog. Structure 3e was built as a chain extended homologue of 4-CPG, which was found to be a good inhibitor of system xc− but a poor inhibitor of VGLUT. The activity of 3e also suggests the likelihood that the compounds are interacting with lipophilic domains associated with the transporter, as has also been shown to occur with EAAT inhibitors.12 Interestingly, all the thiophene-containing structures showed inhibition of system xc− and VGLUT with the 5-bromo thienylglycine 3d and benzothienylglycine 3f blocking about 60% and 70% of VGLUT uptake, respectively (Table 1). The imidazo analog 3c was completely inactive indicating the importance of the thiophene ring and/or possibly contribution by the sulfur atom. Most surprising in this initial screen was the finding that the biphenyl analog of CPG 3e blocked 73% of glutamate uptake at system xc− but was a poor inhibitor of VGLUT.
Sulfonic acid analogs of the amino acids phenylglycine, phenylalanine and thienylglycine 5a–i were prepared to determine the role of stereochemistry, isostere contribution and limitations of the γ-carboxylic acid group. We rationalized that CPG and cysteate are inhibitors of system xc− and therefore, CPG analogs bearing a γ-sulfonic acid would be more potent, and potentially highly selective inhibitors when compared as inhibitors of VGLUT. Neither D- or L-4-sulfophenylalanine 5ab nor α-methyl 4-sulfophenylalanine 5d blocked glutamate uptake at either transporter (Table 1). Sulfonation of 4-bromophenylalanine was conducted to afford the phenylalanine-2-sulfonic acid analog 5c to reduce the distance between the two acid groups, and render it a conformationally-restricted analog of homocysteate.
However, compound 5c was a poor inhibitor of both transporters. (R)-4-sulfo-phenylglycine 5e did not block glutamate uptake at either transporter, yet 5f was a selective inhibitor of system xc−. We attribute this selectivity to the fact that system xc− generally requires an S-configured amino acid center for inhibitors whereas VGLUT shows no need for this center and, in fact, does not require a basic amine.
Using (S)-4-sulfophenylglycine as a new lead, we prepared the thiophene analogs that position the sulfonic acid and amino acid groups at a distance midway between 4-sulfophenylglycine and homocysteic acid. Both (R)-5h and (S)-4-sulfothienylglycine 5g blocked uptake of glutamate at system xc−, 45% and 70%, respectively (Table 1). The latter compound proved as potent as the endogenous substrate L-cystine. Unlike system xc− both were less potent at VGLUT.
The last set of analogs we prepared to test specificity differences between system xc− and VGLUT were aminothiophenecarboxylic acids 7ab. Since the thiophene scaffold showed promise in system xc− inhibitors, we next queried whether or not replacement of the α-amino acid group with an aniline-type amine and carboxylic acid would preferentially block VGLUT. In both instances, glutamate uptake was blocked at system xc− and not VGLUT indicating that the presence of an α-amino acid group is not a requirement for system xc− inhibitor structure. This is also consistent with the activity of sulfazaline, an inhibitor of system xc−, which lacks the free α-amino acid head group that typifies the majority of known inhibitors. Sulfasalazine is of particular interest because it suggests that system xc− may represent a viable point of therapeutic intervention in the treatment of glial brain tumors.35
Overall, the only sulfonic acid analog of phenylglycine or phenylalanine that showed activity was 5f that selectively blocked 45% transport at system xc−. Substituted thienyl- and benzthienylglycines blocked VGLUT with marginal selectivity over system xc−, however, the presence of a sulfonic acid group on the thiophene (5fgh) afforded selective system xc− transport inhibitors. We are currently developing a system xc− pharmacophore model using the thiophene template to produce better inhibitors. One inhibitor that effectively reduced the uptake of glutamate at both transporters was R/S-benzothiophene-3-glycine. In summary, we have identified new system xc− inhibitors that we envision will become important pharmacologic tools, but additional work is needed to identify more effective dual inhibitors.
Acknowledgments
The authors thank J. Gerdes and N. Natale for advice. This research was supported in part by NIH NS38248 (CMT), SBIR grants to ATERIS Technologies LLC (R43 ES016392 U44 NS058229), NS30570 (RJB) and NS42077 (RJB), as well as by the Core Laboratory for Neuromolecular Production (NIH P30-NS055022) and the COBRE Center for Structural and Functional Neuroscience (NIH P20-RR015583).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Byrnes KR, Loane DJ, Faden AI. Neurotherapeutics. 2009;6:94. doi: 10.1016/j.nurt.2008.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choi DW, Rothman SM. Annu Rev Neurosci. 1990;13:171. doi: 10.1146/annurev.ne.13.030190.001131. [DOI] [PubMed] [Google Scholar]
- 3.Balazs R, Bridges RJ, Cotman CW. Excitatory amino acid transmission in health and disease. Oxford University Press; New York: 2006. [Google Scholar]
- 4.Jane DE, Lodge D, Collingridge GL. Neuropharmacology. 2009;56:90. doi: 10.1016/j.neuropharm.2008.08.023. [DOI] [PubMed] [Google Scholar]
- 5.Swanson GT, Sakai R. Prog Mol Subcell Biol. 2009;46:123. doi: 10.1007/978-3-540-87895-7_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shigeri Y, Shimamoto K. Nippon Yakurigaku Zasshi. 2003;122:253. doi: 10.1254/fpj.122.253. [DOI] [PubMed] [Google Scholar]
- 7.Reis HJ, Guatimosim C, Paquet M, Santos M, Ribeiro FM, Kummer A, Schenatto G, Vsalgado JV, Vieira LB, Teixeira AL, Palotas A. Curr Med Chem. 2009;16:796. doi: 10.2174/092986709787549271. [DOI] [PubMed] [Google Scholar]
- 8.Bridges RJ, Esslinger CS. Pharmacol Ther. 2005;107:271. doi: 10.1016/j.pharmthera.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 9.Patel SA, Warren BA, Rhoderick JF, Bridges RJ. Neuropharmacology. 2004;46:273. doi: 10.1016/j.neuropharm.2003.08.006. SNB-19 transport assay: Wells were pre-incubated (30°C, 10 min) in HEPES (pH7.4) Hank’s balanced salt solution (HBHS). Uptake was initiated by addition of 3H-L-glutamate (20–500 μM) and incubated for 5 min. For inhibition, 3H-L-glutamate (100 μM) and inhibitor (500 μM) were added. Each well was rinsed with HBHS (0°C, 3x) and the cells treated with 1mL 0.4 M NaOH for 24 h. Acetic acid (5 μl) was added to an aliquot (200 μl) and the radioactivity quantified vs background. [DOI] [PubMed] [Google Scholar]
- 10.Domercq M, Sanchez-Gomez MV, Sherwin C, Etxebarria E, Fern R, Matute C. J Immunol. 2007;178:6549. doi: 10.4049/jimmunol.178.10.6549. [DOI] [PubMed] [Google Scholar]
- 11.Burdo J, Dargusch R, Schubert D. J Histochem Cytochem. 2006;54:549. doi: 10.1369/jhc.5A6840.2006. [DOI] [PubMed] [Google Scholar]
- 12.Bridges RJ, Patel SA. Pharmacology of glutamate transport in the CNS: substrates and inhibitors of excitatory amino acid transporters and the glutamate cystine exchanger system xc- Springer; NY: 2009. [Google Scholar]
- 13.Takamori S. Neurosci Res. 2006;55:343. doi: 10.1016/j.neures.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 14.Moriyama Y, Omote H. Biol Pharm Bull. 2008;31:1844. doi: 10.1248/bpb.31.1844. [DOI] [PubMed] [Google Scholar]
- 15.Carlson MD, Kish PE, Ueda T. J Neurochem. 1989;53:1889. doi: 10.1111/j.1471-4159.1989.tb09258.x. [DOI] [PubMed] [Google Scholar]
- 16.Natale NR, Magnusson KR, Nelson JK. Curr Top Med Chem. 2006;6:823. doi: 10.2174/156802606777057535. [DOI] [PubMed] [Google Scholar]
- 17.Warren BA, Patel SA, Nunn PB, Bridges RJ. Toxicol Appl Pharmacol. 2004;200:83. doi: 10.1016/j.taap.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 18.Bartlett RD. PhD Thesis. The University of Montana; Missoula MT: 1999. [Google Scholar]
- 19.Bartlett RD, Esslinger CS, Thompson CM, Bridges RJ. Neuropharmacology. 1998;37:839. doi: 10.1016/s0028-3908(98)00080-x. [DOI] [PubMed] [Google Scholar]
- 20.Carrigan CN, Bartlett RD, Esslinger CS, Cybulski KA, Tongcharoensirikul P, Bridges RJ, Thompson CM. J Med Chem. 2002;45:2260. doi: 10.1021/jm010261z. [DOI] [PubMed] [Google Scholar]
- 21.Carrigan CN, Esslinger CS, Bartlett RD, Bridges RJ, Thompson CM. Bioorg Med Chem Lett. 1999;9:2607. doi: 10.1016/s0960-894x(99)00444-8. [DOI] [PubMed] [Google Scholar]
- 22.Kish PE, Ueda T. Methods Enzymol. 1989;174:9. doi: 10.1016/0076-6879(89)74005-2. Assays were intiated by the addition of 3H-glutamate± inhibitors (0.01–5 mM) to the synaptic vesicles (approx. 0.1mg protein). Rates of uptake were normalized to protein content. [DOI] [PubMed] [Google Scholar]
- 23.Patel SA, Nagy JO, Bolstad ED, Gerdes JM, Thompson CM. Bioorg Med Chem Lett. 2007;17:5125. doi: 10.1016/j.bmcl.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roseth S, Fykse EM, Fonnum F. J Neurochem. 1995;65:96. doi: 10.1046/j.1471-4159.1995.65010096.x. [DOI] [PubMed] [Google Scholar]
- 25.Roseth S, Fykse EM, Fonnum F. Biochem Pharmacol. 1998;56:1243. doi: 10.1016/s0006-2952(98)00200-7. [DOI] [PubMed] [Google Scholar]
- 26.Thompson CM, Davis E, Carrigan CN, Cox HD, Bridges RJ, Gerdes JM. Curr Med Chem. 2005;12:2041. doi: 10.2174/0929867054637635. [DOI] [PubMed] [Google Scholar]
- 27.Bridges RJ, Stanley MS, Anderson MW, Cotman CW, Chamberlin AR. J Med Chem. 1991;34:717. doi: 10.1021/jm00106a037. [DOI] [PubMed] [Google Scholar]
- 28.Dunlop J, Fear A, Griffiths R. Neuroreport. 1991;2:377. doi: 10.1097/00001756-199107000-00005. [DOI] [PubMed] [Google Scholar]
- 29.Synthesis of hydantoins 2a–e. Compounds 2a–e were synthesized from aldehydes (1a–e). Compounds 1a–e (1.0 g; 5.40 mmol) were dissolved in 1:1 CH3OH/H2O and (NH4)2CO3 (4.5 g; 47.4 mmol) and KCN (1.3 g, 20 mmol) were added. The mixture was heated (58–60°C; 3h), concentrated to two-thirds, and chilled to 0°C. Crystalline products were filtered, washed with water, dried and characterized by 1H-NMR and MS. The resultant hydantoins were hydrolyzed directly.
- 30.Chruma JJ, Liu L, Zhou W, Breslow R. Bioorg Med Chem. 2005;13:5873. doi: 10.1016/j.bmc.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 31.Nenajdenko VG, Zakurdaev EP, Prusov EV, Balenkova ES. Russian Chem Bulletin. 2004;53:2866. [Google Scholar]
- 32.Stalker RA, Munsch TE, Tran JD, Nie X, Warmuth R, Beatty A, Aakeröy CB. Tetrahedron. 2002;58:4837. [Google Scholar]
- 33.Shanbhag VM, Martell AE. Inorganic Chemistry. 2002;29:1023. [Google Scholar]
- 34.Spectral data for selected compounds. Compound 2c: Yield 75%; mp 258–261°C; 1H NMR (400MHz, DMSO- d6): δ 12.02 (br s, 1H), 10.59 (br s, 1H), 8.06 (s, 1H), 7.59 (s, 1H), 7.09 (bs, 1H), 4.99 (s, 1H); ESI MS m/z = 167 (M+1); IR (KBr) (υmax/Cm−1): 3414, 3241, 2700, 1729, 1456. Anal. Calcd for C6H6N4O2: C, 43.38; H, 3.64; N, 33.72. Found: C, 43.33; H, 3.59; N, 33.62. Compound 3c: Yield 55%; mp >300 °C; 1H NMR (400MHz, D2O): δ 7.51 (s, 1H), 6.88 (s, 1H), 4.91 (s, 1H); ESI MS m/z = 142 (M+1); IR (KBr) (υmax/Cm−1): 3250, 2348, 2287, 1593, 1462, 1377. Anal. Calcd for C5H7N3O2: C, 42.55; H, 5.00; N, 29.77. Found: C, 41.88; H, 5.20; N, 29.64. Compound 5h: Yield 52%; mp > 300 °C; 1H NMR (400MHz, D2O): δ 7.32 (d, J = 7.35 Hz, 1H), 7.11 (d, J = 7.35 Hz, 1H), 5.07 (s, 1H); 13C: δ 173.5, 148.3, 141.2, 131.8, 131.5, 55.7; HRMS m/z = 237.9833 (M+1); IR (KBr) (υmax/Cm−1): 2634, 1746, 1613, 1527, 1214, 1165. Anal. Calcd for C6H7NO5S2: C, 30.37, H, 2.97, N, 5.90. Found: C, 30.51, H, 2.88, N, 5.94.
- 35.Sontheimer H. J Neurochem. 2008;105:287. doi: 10.1111/j.1471-4159.2008.05301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]