

Abstract
Hypoglycosylation of α‐dystroglycan underpins a subgroup of muscular dystrophies ranging from congenital onset of weakness, severe brain malformations and death in the perinatal period to mild weakness in adulthood without brain involvement. Mutations in six genes have been identified in a proportion of patients. POMT1, POMT2 and POMGnT1 encode for glycosyltransferases involved in the mannosylation of α‐dystroglycan but the function of fukutin, FKRP and LARGE is less clear. The pathological hallmark is reduced immunolabeling of skeletal muscle with antibodies recognizing glycosylated epitopes on α‐dystroglycan. If the common pathway of these conditions is the hypoglycosyation of α‐dystroglycan, one would expect a correlation between clinical severity and the extent of hypoglycosylation. By studying 24 patients with mutations in these genes, we found a good correlation between reduced α‐dystroglycan staining and clinical course in patients with mutations in POMT1, POMT2 and POMGnT1. However, this was not always the case in patients with defects in fukutin and FKRP, as we identified patients with mild limb–girdle phenotypes without brain involvement with profound depletion of α‐dystroglycan. These data indicate that it is not always possible to correlate clinical course and α‐dystroglycan labeling and suggest that there might be differences in α‐dystroglycan processing in these disorders.
Keywords: α‐dystroglycan, congenital muscular dystrophy, glycosylation, immunohistochemistry, limb–girdle muscular dystrophy
INTRODUCTION
α‐Dystroglycan is a component of the dystrophin‐associated protein (DAP) complex, which binds to various ligands in the extracellular matrix of muscle and other tissues, including laminin‐α2, perlecan, neurexin and agrin 21, 35, 46. At the muscle cell surface, α‐dystroglycan binds to β‐dystroglycan, thus linking the extracellular matrix to dystrophin and the actin cytoskeleton 9, 16, 17, 46. Dystroglycan plays an important role in the organization of laminins during the development of basement membranes in muscle and nonmuscle tissues 13, 45, 47. In skeletal muscle, dystroglycan and integrin α7B1D function as the receptors for laminin‐α2, and this interaction is necessary for the organization of laminin and the reorganization of the actin cytoskeleton. In turn, the presence of functional laminin‐α2 is necessary to organize dystroglycan, integrin, dystrophin and spectrin at the sarcolemma and costameres 13, 47.
Laminin‐α2 binding to α‐dystroglycan is mediated by the C‐terminal globular LG domains (LG 4‐5) in the laminin molecule and the O‐linked carbohydrate moieties in the mucin‐like domain of dystroglycan 13, 46. Differential glycosylation of the α‐dystroglycan domain confers tissue‐specific functional variability and results in different dystroglycan glycoforms (33). A number of different glycans have been demonstrated on α‐dystroglycan, of which O‐linked mannose represents one of the best characterized ones (10). Its formation involves the action of several enzymes (collectively referred to as glycosyltransferases) that add different monosaccharides in a stepwise manner. These carbohydrate groups also mediate binding with the LG domains of other ligands such as perlecan, agrin and neurexin (2).
Disruption of the interaction between α‐dystroglycan and its ligands has severe consequences for muscle and brain function and structure and results in a group of muscular dystrophies with and without central nervous system involvement 11, 22, 35, 36. These disorders are now collectively referred to as “dystroglycanopathies”2, 24, 30, 38 and are caused by gene defects in fukutin (OMIM 607440), fukutin‐related protein (FKRP; OMIM 606596), protein‐O‐mannose 1,2‐N‐acetylglucosaminyltransferase 1 (POMGnT1; OMIM 606822), protein‐O‐mannosyl transferases (POMT1 and POMT2; OMIM 607423 and 607439, respectively) and LARGE (OMIM 603590). However, mutations in these six genes account for only a proportion of patients with dystroglycan glycosylation defects (19), indicating that other unknown genes are involved.
Dystroglycanopathies constitute a heterogeneous and complex group of disorders as mutations in any of these genes can result in various phenotypes ranging from severe forms of congenital muscular dystrophy (CMD) to milder limb–girdle muscular dystrophy (LGMD) forms 4, 8, 18, 19, 30, 38, 42. Conversely, similar phenotypes can be caused by mutations in the different genes. This has led to the concept that it is the specific mutation that determines the severity of the phenotype rather than the individual gene. To reflect this complexity, a different classification/categorization has been suggested based on the age at onset of weakness (CMD if within the 6 months of life or LGMD if after acquiring ambulation), presence of mental retardation and/or structural brain abnormalities, and eye involvement, rather than on the specific gene defect and the original clinical entity (19).
At the pathological level, these conditions have in common a reduction in the glycosylated epitopes on α‐dystroglycan. The degree of reduction is variable, and while in some patients with severe Walker–Warburg syndrome (WWS) there is virtual absence of these epitopes in muscle, other patients with a mild phenotype only show a minimal reduction 12, 24.
Genotype–phenotype correlations have been described in only a small number of patients with defects in the individual genes 1, 4, 6, 14, 26, 44. Early studies of patients with FKRP mutations showed a broad correlation between the levels of α‐dystroglycan immunolabeling and the clinical severity, such that severely affected patients with the MDC1C phenotype had a more marked reduction than milder LGMD2I patients (6). More recently, the number of genes involved, the spectrum of clinical severity and our understanding of these conditions have significantly expanded. The aims of this study were to (i) determine if the previous correlation noted in patients with defects in FKRP could also be applied to the remaining dystroglycanopathies, and (ii) investigate whether the status α‐dystroglycan glycosylation and/or other pathological features could be used to differentiate between the different primary gene defects.
For this purpose, we have correlated clinical and pathological data in a cohort of 24 patients with confirmed mutations in POMT1, POMT2, POMGnT1, fukutin, LARGE or FKRP from whom muscle biopsies were available.
MATERIALS AND METHODS
Patients
All biopsies were obtained with informed written consent and studied under ethical approval. Details of the 24 patients in this study, the clinical categories and mutations are listed in Table 1. Four cases had mutations in the gene encoding POMT1, five in POMT2, two in POMGnT1, two in fukutin, 10 in FKRP and one in LARGE. Some of these cases have been reported previously as indicated in Table 1. The phenotypic categorization in order of severity and as described in Godfrey et al (19) is as follows:
Table 1.
Summary of genetic and clinical features. Abbreviations: CK = creatine kinase; N/A = not available; WWS = Walter–Warburg syndrome; MEB = muscle–eye–brain disease; FCMD = Fukuyama congenital muscular dystrophy; CMD‐CRB = congenital muscular dystrophy with cerebellar involvement; CMD‐MR = congenital muscular dystrophy with mental retardation; LGMD‐MR = limb–girdle muscular dystrophy with mental retardation; LGMD‐No MR = limb–girdle muscular dystrophy without mental retardation; het = heterozygous.
| Patient | Gene | Inheritance | Type of mutation | Reference | CK | Maximum motor ability | Phenotypic category |
|---|---|---|---|---|---|---|---|
| P1 | POMT1 | Homozygous | Frameshift | (19) | 3 400 | Never sat | MEB‐FCMD |
| P2 | POMT1 | Homozygous | Frameshift | (19) | 4 000 | Never sat | WWS |
| P3 | POMT1 | Compound het | Insertion/deletion | (19) | 8 000 | Walking | LGMD‐MR |
| Missense | |||||||
| P4 | POMT1 | Compound het | Nonsense | (19) | 19 000 | Walking | CMD‐MR |
| Missense | |||||||
| P5 | POMT2 | Compound het | Missense | (19) | 5 500 | Sitting with support | MEB‐FCMD |
| Missense | |||||||
| P6 | POMT2 | Compound het | Missense | 19, 34 | 2 000 | Sitting with support | MEB‐FCMD |
| Missense | |||||||
| P7 | POMT2 | Compound het | Missense | (19) | 6 000 | Never sat | MEB‐FCMD |
| Missense | |||||||
| P8 | POMT2 | Compound het | Missense | (19) | 3 000 | Sitting | CMD‐CRB |
| Frameshift | |||||||
| P9 | POMT2 | Compound het | Missense | 3 000 | Walking | CMD‐MR | |
| P10 | POMGnT1 | Homozygous | Splice site | 1 000 | Sitting | MEB‐FCMD | |
| P11 | POMGnT1 | Homozygous | Missense | (12) | 5 000–12 000 | Walking | LGMD‐No MR |
| P12 | LARGE | Compound het | Missense | (28) | 500–4 500 | Walking | MEB‐FCMD |
| Frameshift | |||||||
| P13 | fukutin | Compound het | Frameshift | 18, 19 | 13 000 | Walking | LGMD‐No MR |
| Frameshift | |||||||
| P14 | fukutin | Compound het | Missense | 18, 19 | 60 000 | Walking | LGMD‐No MR |
| Frameshift | |||||||
| P15 | FKRP | Homozygous | Missense | 8 000 | Walking | LGMD‐No MR (2I‐DMD‐like) | |
| P16 | FKRP | Compound het | Missense | 24 000 | Sitting | CMD‐No MR | |
| P17 | FKRP | Homozygous | Missense | 41, 43 | 6 500 | Sitting | CMD‐CRB |
| P18 | FKRP | Homozygous | Missense* | 25 000 | Walking | LGMD‐MR (2I‐BMD‐like) | |
| P19 | FKRP | Compound het | Missense* | 4 900 | Walking | LGMD‐No MR (2I‐BMD‐like) | |
| Nonsense | |||||||
| P20 | FKRP | Compound het | Missense* | N/A | Walking | LGMD‐No MR (2I‐BMD‐like) | |
| Missense | |||||||
| P21 | FKRP | Compound het | Missense | 11 814 | Walking | LGMD‐No MR (2I‐DMD‐like) | |
| Frameshift | |||||||
| P22 | FKRP | Compound het | Missense* | 3 607 | Walking | LGMD‐No MR (2I‐mild) | |
| Missense | |||||||
| P23 | FKRP | Compound het | Missense* | 5 500 | Walking | LGMD‐No MR (2I‐mild) | |
| Missense | |||||||
| P24 | FKRP | Homozygous | Missense* | N/A | Walking | LGMD‐No MR (2I‐mild) |
Common mutation p.Leu276Ile.
-
(i)
WWS (and WWS‐like): onset prenatally or at birth; severe structural brain abnormalities (complete agyria or severe lissencephaly), severe cerebellar involvement, and complete or partial absence of the corpus callosum; eye abnormalities are common; motor development is absent.
-
(ii)
Muscle–eye–brain disease/Fukuyama congenital muscular dystrophy‐like (MEB/FCMD‐like): CMD with brain abnormality less severe than that seen with WWS (pachygyria with preferential frontoparietal involvement and polymicrogyria), cerebellar hypoplasia and dysplasia, and frequent flattening of the pons and brainstem; eye abnormalities are frequent; individuals may, rarely, acquire the ability to walk, although this is delayed; marked speech delay.
-
(iii)
CMD with cerebellar involvement (CMD‐CRB): CMD with mental retardation and cerebellar involvement on magnetic resonance imaging (MRI) scan as the only structural abnormality (including cysts, hypoplasia or dysplasia).
-
(iv)
CMD with mental retardation (CMD‐MR): CMD with mental retardation and structurally normal brain; patients with isolated microcephaly or minor white matter changes on MRI are included in this group.
-
(v)
CMD with no mental retardation (CMD‐No MR): patients with CMD and normal intellectual function.
-
(vi)
LGMD with mental retardation (LGMD‐MR): LGMD with mental retardation and structurally normal brain; patients with minor white matter abnormalities and microcephaly were included in this group; this category would include patients with LGMD2I 4, 6 and LGMD2K (1).
-
(vii)
LGMD with no mental retardation (LGMD‐No MR): LGMD with no mental retardation; this category would include the LGMD phenotypes such as LGMD2I and 2L (18).
Muscle biopsy
Needle or open muscle biopsies taken from the quadriceps, except patient P13, who had a biopsy taken from a foot muscle, were transversely orientated, mounted in OCT (Tissue‐Tek, Sakura Finetek, Zoeterwoude, The Netherlands) and frozen in isopentane cooled in liquid nitrogen. Standard histological and histochemical techniques were applied to cryostat sections, including hematoxylin and eosin, Gomori trichrome, periodic acid Schiff, nicotinamide adenine dinucleotide dehydrogenase‐tetrazolium reductase (NADH‐TR), succinic dehydrogenase and cytochrome oxidase (15).
Immunohistochemistry
Dystroglycan antibodies were β‐dystroglycan (NCL‐b‐DAG, 1/50; Novocastra, Leica‐microcystems, Wetzlar, Germany), α‐dystroglycan mouse monoclonal IIH6, 1/200 (a gift from K. Campbell) and α‐dystroglycan goat polyclonal GT20ADG, 1/50 25, 27, 35. Antibody GT20ADG was raised against the entire dystroglycan glycoprotein complex and purified against a hypoglycosylated full‐length α‐dystroglycan human fusion protein expressed in HEK‐293 cells. Anti‐α‐dystroglycan sheep polyclonal (a kind gift of S. Kröger) was raised against a synthetic peptide corresponding to the last 20 amino acids of chick α‐dystroglycan 1/500 (20).
Other antibodies used were laminin‐α2 (MAB1922 Chemicon (Millipore, Billerica, MA, USA) to the 80‐kDa C‐terminal fragment, 1/4000, and 4H8 Alexis Corporation (Lausen, Switzerland) to the 300‐kDa N‐terminal fragment, 1/100), spectrin (NCL‐SPEC1, 1/20), dystrophin (NCL‐DYS1, 1/3, NCL‐DYS2, 1/20, and NCL‐DYS3, 1/10), neonatal isoform of myosin heavy chain (NCL‐MHCn, 1/15), developmental isoform of myosin (NCL‐MHCd, 1/20) and utrophin (NCL‐DRP2, 1/5), all from Novocastra. Caveolin‐3 was from Becton Dickinson (Franklin Lakes, NJ, USA) (C38320, 1/400), and the HLA (Major Histocompatibility Complex I) was from Dako (Glostrup, Denmark) (1/400).
Cryostat sections (8 µm) were incubated with primary antibodies for 1 h at room temperature, followed by incubation with an appropriate biotinylated secondary antibody [anti‐mouse IgM (Dako), 1/200 for IIH6; anti‐mouse IgG (Amersham, plc, Little Chalfont, Buckinghamshire, UK), 1/200; anti‐goat or anti‐sheep IgG (Jackson Immunoresearch, West Grove, PA, USA), 1/500] for 30 minutes, and visualized by streptavidin conjugated to Alexa 594 (Molecular Probes, Invitrogen, Carlsbad, CA, USA, 1/1000) for 15 minutes. All dilutions and washings were made in phosphate buffered saline. Sections were mounted in aqueous mountant (Hydromount; National Diagnostics, Atlanta, GA, USA) and viewed with epifluorescence using a Leica DMR microscope (Leica Microsystems, Wetzlar, Germany) linked to Metamorph Molecular Devices (Sunnyvale, CA, USA). Control sections were labeled without primary antibodies, and all sections were compared with age‐matched control samples from other neuromuscular disorders and with normal muscle.
The level of dystroglycan labeling was scored using a scale based on the proportion of positive fibers and intensity relative to the controls used in each sample set. In order to obtain an objective indication of intensity, we used control sections to set the exposure time and scale settings for the digital capturing system. Two independent observers scored the results blindly (C.J.M. and S.T.). The scale is as follows:
-
1
++++ = normal (based on the control sample in the same sample set)
-
2
+++/++++ = minimal reduction
-
3
+++ = mild reduction
-
4
++ = significant reduction
-
5
+ = very marked reduction
-
6
0/+ = weak traces
-
7
0 = negative
RESULTS
Pathology
All samples had features compatible with a muscular dystrophy, but these were variable in type and degree (Table 2). There were variable degrees of variation in fiber size, with atrophy and hypertrophy of fibers, fibrosis, adipose tissue, internal nuclei, necrosis and regeneration (basophilic fibers and expression of neonatal and/or developmental myosins). It was not possible to exclude the influence of factors such as sampling and size of biopsy on some of the features. Some of the biopsies (P1, P2, P5, P7 and P6) contained only a small number of fibers surrounded by connective tissue and fat. There was no correlation between the degree of pathology and age, nor with the defective gene. P1, aged 2 months, and P2, aged 5 years, both had marked connective tissue and fat proliferation (Figure 1A,B). On the other hand, patients with mutations in the same gene and of similar age at the time of biopsy showed different degrees of pathology (P5 and P9, both with POMT2 mutations) (Figure 1C,H). As reported for other neuromuscular disorders (15), there was no correlation between the degree of pathology and clinical severity (compare P10 and P9; Figure 1E,H).
Table 2.
Muscle pathology. Abbreviations: N/A = not available; NMY + ve = fibers positive for neonatal myosin.
| Patient | Gene | Age at biopsy | Fibrosis | Adipose tissue | Internal nuclei | Regeneration | Necrosis |
|---|---|---|---|---|---|---|---|
| P1 | POMT1 | 2 months | Marked | No | A few | No basophilia, many NMY + ve | No |
| P2 | POMT1 | 5 years | Marked | Marked | A few | No basophilia, a few NMY + ve | No |
| P3 | POMT1 | 3 years | Marked | Mild | Yes | Basophilia, many NMY + ve | Yes |
| P4 | POMT1 | 2 years | Yes | Yes | Yes | Basophilia, several NMY + ve | Yes |
| P5 | POMT2 | 2 years | Yes | Yes | Yes | No basophilia, some NMY + ve | No |
| P6 | POMT2 | 1 year | Yes | Marked | Yes | Basophilia | No |
| P7 | POMT2 | 10 years | Yes | Yes | Yes | Several NMY + ve | No |
| P8 | POMT2 | 15 months | Yes | Some | Yes | Basophilia, many NMY + ve | Yes |
| P9 | POMT2 | 2 years | Yes | Yes | Yes | Basophilia, some NMY + ve | No |
| P10 | POMGnT1 | 2 years | No | No | A few | Basophilia, many NMY + ve | No |
| P11 | POMGnT1 | 14 years | Some | Yes | Some | Basophilia, many NMY + ve | Yes |
| P12 | LARGE | 17 years | Traces | No | Yes | Basophilia, few NMY + ve | Yes |
| P13 | fukutin | 4 years | Yes | Yes | Yes | Basophilia, few NMY + ve | Yes |
| P14 | fukutin | 7 years | Yes | Yes | Yes | Basophilia, many NMY + ve | Yes |
| P15 | FKRP | 2 years | Yes | No | Yes | No basophilia, several NMY + ve | Yes |
| P16 | FKRP | 1 year | Yes | Yes | Yes | Basophilia, many NMY + ve | Yes |
| P17 | FKRP | 4 years | Marked | Mild | Few | Basophilia | Yes |
| P18 | FKRP | 14 years | Yes | Yes | Yes | Yes, NMY + ve | Yes |
| P19 | FKRP | 11 years | Yes | Yes | Yes | Yes, basophilia, many NMY + ve | Yes |
| P20 | FKRP | 11 years | Yes | No | No | No basophilia, many NMY + ve | Yes |
| P21 | FKRP | 8 years | N/A | N/A | N/A | Many NMY + ve | N/A |
| P22 | FKRP | 11 months | Yes | No | Yes | Basophilia, several NMY + ve | No |
| P23 | FKRP | 13 years | Yes | No | Yes | Basophilia, several NMY + ve | Yes |
| P24 | FKRP | 10 years | Yes | No | Yes | Basophilia, a few NMY + ve | Yes |
Figure 1.
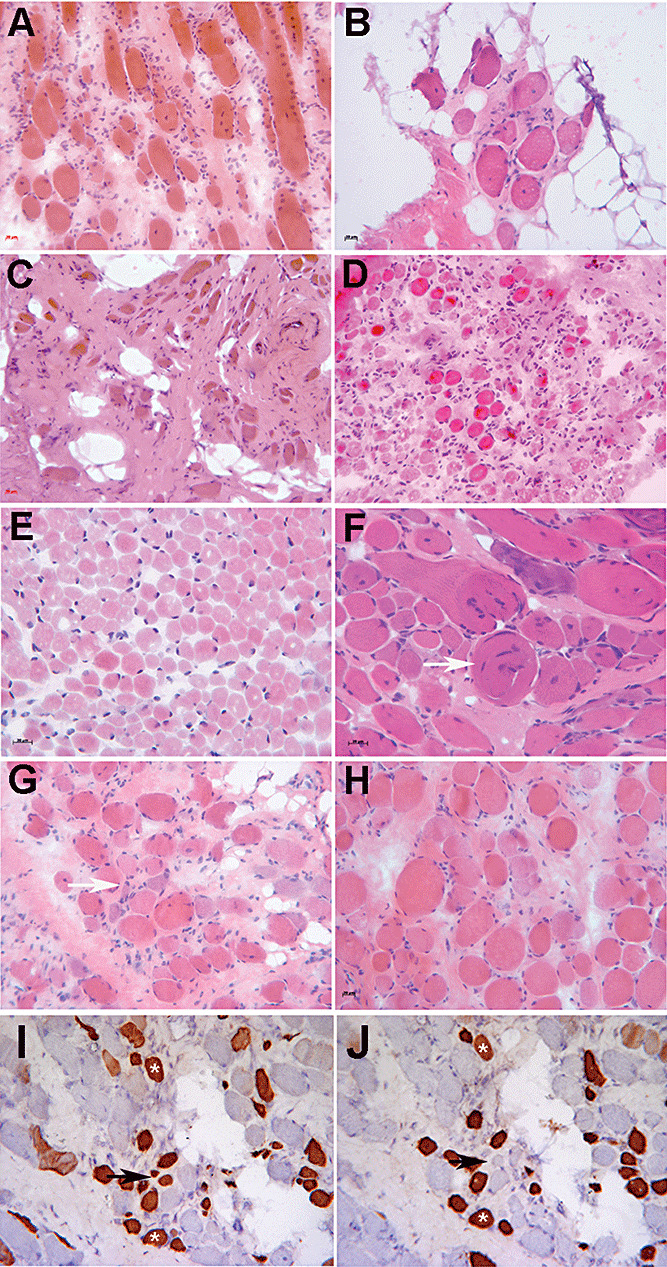
Illustrative images of the spectrum of pathology seen in this cohort of patients. Hematoxylin and eosin: A. P1. B. P2. C. P5. D. P8. E. P10. F. P3. G. P16. H. P9. Immunohistochemistry: I. P16, neonatal isoform of myosin heavy chain. J. P16, developmental isoform of myosin heavy chain. Scale bars represent 20 µm. White arrows in F and G point to whorled and basophilic fibers, respectively. Black arrows in I and J point to fibers that are positive for neonatal myosin but negative for developmental myosin, whereas other fibers are positive for both antibodies (asterisks).
Proliferation of endomysial connective tissue was seen in all cases except P10 (Figure 1E) and was often extensive even in the clinically milder cases. Basophilic fibers were present in some cases, and all biopsies contained a proportion of fibers expressing markers associated with regeneration such as neonatal and developmental myosins (Figure 1I,J), utrophin and MHC‐class I (15). In P10 and P11, the basophilic fibers were often granular in appearance and stained more darkly with NADH‐TR, suggesting a possible mitochondrial proliferation in these fibers. Hypercontracted fibers were present but were not as abundant as in other muscular dystrophies such as Duchenne muscular dystrophy (DMD). Other features included split fibers and whorled fibers (Figure 1F). Fiber typing was often indistinct with oxidative enzyme staining. Fibers expressing slow myosin were predominant in most cases, and some fibers coexpressed more than one isoform of myosin (a common myopathic feature).
Glycosylation of α‐dystroglycan
The glycosylation of α‐dystroglycan was assessed with the monoclonal antibody clone IIH6, which is known to bind to a carbohydrate moiety that mediates and functionally blocks the interaction between α‐dystroglycan and laminin‐α2 2, 7, 16. This was evaluated in parallel with β‐dystroglycan labeling as previously described 6, 23.
The reduction of IIH6 labeling ranged from an absence on most fibers to a very mild reduction. There was no correlation between the extent of the reduction and the particular gene affected; cases with absent or very weak traces of IIH6 were observed in patients with mutations in POMT1, POMT2, POMGnT1, fukutin and FKRP (Table 3). An example for each gene group is shown in Figure 2.
Table 3.
Results of immunohistochemistry with dystroglycan and laminin‐α2 antibodies. Abbreviations: β‐DG = β‐dystroglycan; IIH6, GT20ADG, core sheep = α‐dystroglycan antibodies (see Materials and Methods); N/A = not available; WWS = Walter–Warburg syndrome; MEB = muscle–eye–brain disease; FCMD = Fukuyama congenital muscular dystrophy; CMD‐CRB = congenital muscular dystrophy with cerebellar involvement; CMD‐MR = congenital muscular dystrophy with mental retardation; LGMD‐MR = limb–girdle muscular dystrophy with mental retardation; LGMD‐No MR = limb–girdle muscular dystrophy without mental retardation.
| Patient | Gene | Phenotype | β‐DG | IIH6 | GT20ADG | Core sheep | Laminin‐α2 |
|---|---|---|---|---|---|---|---|
| P1 | POMT1 | MEB‐FCMD | +++ (small fibers) | 0 | +++ | 0 | +++ |
| P2 | POMT1 | WWS | +++/++++ | 0 | N/A | 0/+ | +++ |
| P3 | POMT1 | LGMD‐MR | +++/++++ | ++ | +++/++++ | + | +++/++++ |
| P4 | POMT1 | CMD‐MR | ++++ | +/++ | +++ (small fibers) | + | +++/++++ |
| P5 | POMT2 | MEB‐FCMD | ++ | 0 | ++ | 0/+ | ++/+++ |
| P6 | POMT2 | MEB‐FCMD | +++/++++ (small fibers) | + | ++++ | ++ | +++ |
| P7 | POMT2 | MEB‐FCMD | +++/++++ | 0 | N/A | 0/+ | +++ |
| P8 | POMT2 | CMD‐CRB | ++++ | ++ | ++/+++ | ++ | +++/++++ |
| P9 | POMT2 | CMD‐MR | ++++ | ++ | ++++ | N/A | +++ |
| P10 | POMGnT1 | MEB‐FCMD | +++/++++ | 0/+ | N/A | +/++ | ++++ |
| P11 | POMGnT1 | LGMD‐No MR | +++/++++ | +++/++++ | ++++ | +++/++++ | ++++ |
| P12 | LARGE | MEB‐FCMD | ++++ | ++/+++ | ++/+++ | +++/++++ | ++++ |
| P13 | fukutin | LGMD‐No MR | ++++ | 0 | +++/++++ | 0/+ | +++ |
| P14 | fukutin | LGMD‐No MR | ++++ | 0/+ | +++/++++ | 0/+ | +++/++++ |
| P15 | FKRP | LGMD‐No MR | +++/++++ | 0 | +++/++++ | + | +++ |
| P16 | FKRP | CMD‐No MR | +++/++++ | 0 | +++ | +/++ | ++++ |
| P17 | FKRP | CMD‐CRB | ++++ | 0 | ++++ | N/A | ++/+++ |
| P18 | FKRP | LGMD‐MR (mild) | ++++ | ++ | ++++ | +++ | +++ |
| P19 | FKRP | LGMD‐No MR (intermediate) | +++/++++ (small fibers) | ++ | +++/++++ (small fibers) | +++ | +++/++++ |
| P20 | FKRP | LGMD‐No MR (intermediate) | ++++ | + | ++++ | N/A | ++++ |
| P21 | FKRP | LGMD‐No MR (severe) | ++++ | + | ++++ | N/A | ++++ |
| P22 | FKRP | LGMD‐No MR (2I‐mild) | +++/++++ (small fibers) | +++ | N/A | ++/+++ | ++++ |
| P23 | FKRP | LGMD‐No MR (2I‐mild) | ++++ | ++/+++ | N/A | ++ | +++/++++ |
| P24 | FKRP | LGMD‐No MR (2I‐mild) | ++++ | +++ | N/A | ++/+++ | +++ |
Scoring scale: ++++ = normal (based on the control sample in the same sample set); +++/++++ = minimal reduction; +++ = mild reduction; ++ = significant reduction; + = very marked reduction; 0/+ = weak traces; 0 = negative.
Figure 2.
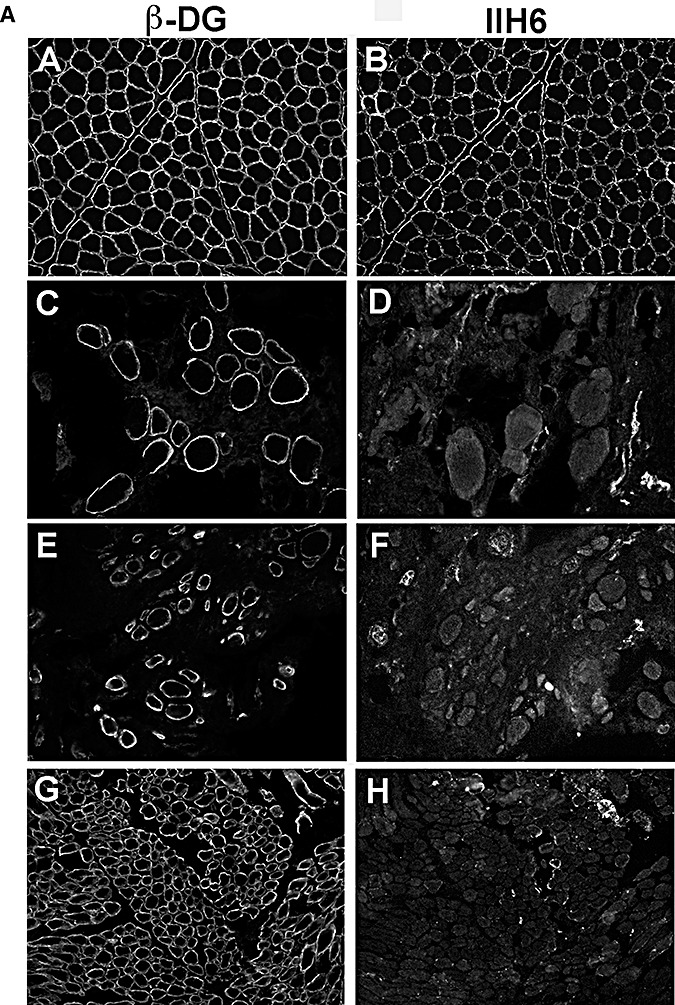
Representative images of the group of cases with virtually absent or very weak traces of glycosylated α‐dystroglycan labeling (0 or 0/+). Images shown were captured with autoscale to allow visualization of the tissue. A. A,C,E,G: β‐dystroglycan (β‐DG). B,D,F,H: α‐dystroglycan using IIH6 antibody. A,B: control; C,D: P2 (POMT1); E,F: P5 (POMT2); G,H: P10 (POMGnT1). B. A,C,E: β‐DG. B,D,F: α‐dystroglycan using IIH6 antibody. A,B: P14 (fukutin); C,D: P16 (FKRP); E,F: P17 (FKRP with cerebellar cysts).
Some biopsies showed a marked variability in the labeling of individual fibers across the section, with some fibers appearing negative while others were brightly labeled. The reason for these intensely labeled fibers is unclear but in most cases, these fibers did not express the neonatal isoform of myosin (as shown in the biopsy of P4; asterisks in Figure 3), suggesting that they are unlikely to be regenerating fibers. They also showed good labeling of caveolin‐3 and other membrane proteins, including β‐dystroglycan (Figure 3), indicating that the sarcolemma was well preserved.
Figure 3.
Muscle biopsy from P4 showing that the fibers that are brightly labeled with IIH6 (B, asterisks) do not express neonatal myosin (D) and their sarcolemma is well preserved [as seen with antibodies to β‐dystrolgycan (β‐DG) in A and caveolin‐3 (cav‐3) in C].
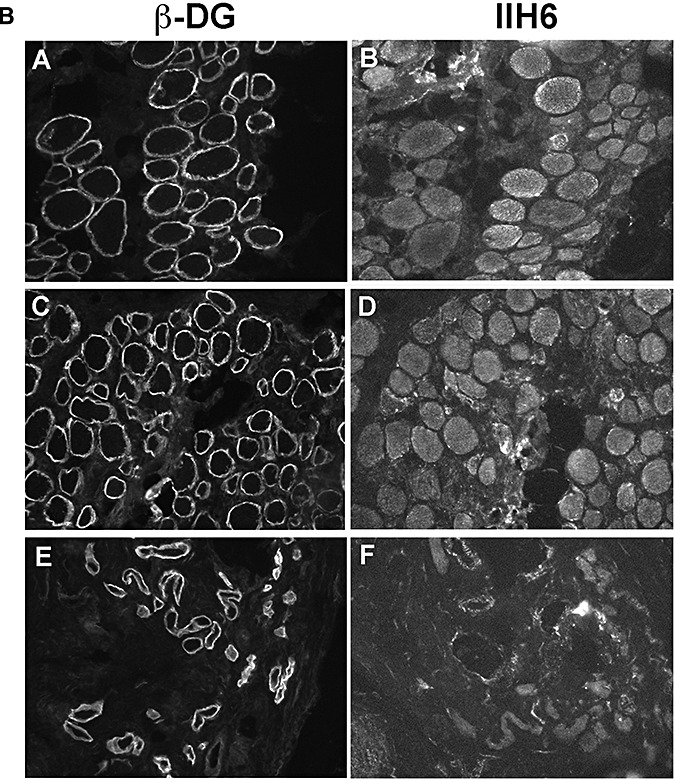
Patients with mutations in either POMT1 or POMT2 genes and a relatively milder phenotype (LGMD‐MR, CMD‐MR) showed more immunolabeling of IIH6 (Table 3; P3 and P9 are shown in Figure 4) than more severe POMT1 and POMT2 cases with WWS or MEB‐FCMD phenotypes (P1, P2 and P6).
Figure 4.
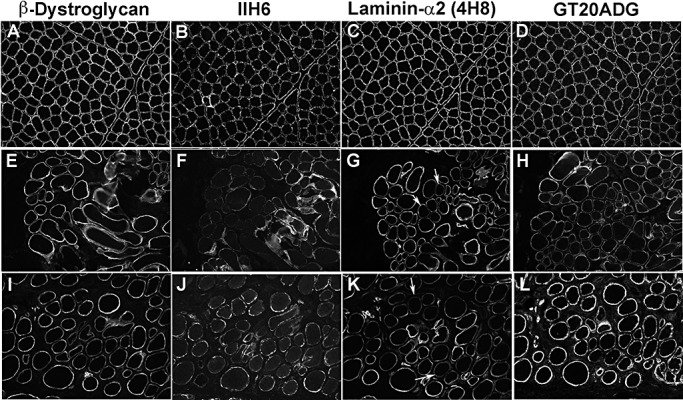
Muscle biopsy of two patients with mutations in POMT1 and POMT2 with a significant reduction of IIH6 labeling (++). Laminin‐α2 was mildly reduced in some fibers in both patients. A,E,I.β‐Dystroglycan. B,F,J. IIH6. C,G,K. Laminin‐α2 (4H8 antibody to the N‐terminal 300 kDa fragment). D,H,L. GT20ADG α‐dystroglycan antibody. A–D: control; E–H: P3; I–L: P9. Arrows indicate some of the fibers with reduced laminin‐α2 labeling.
Labeling of glycosylated α‐dystroglycan was reduced in all the patients with FKRP mutations. The spectrum of IIH6 reduction was very variable and ranged from an absence (P15, P16 and P17; Figure 2B) to a mild reduction (Figure 5, Table 3). Some of these cases showed a mosaic pattern of positive and negative fibers as described previously in other LGMD2I patients (6).
Figure 5.
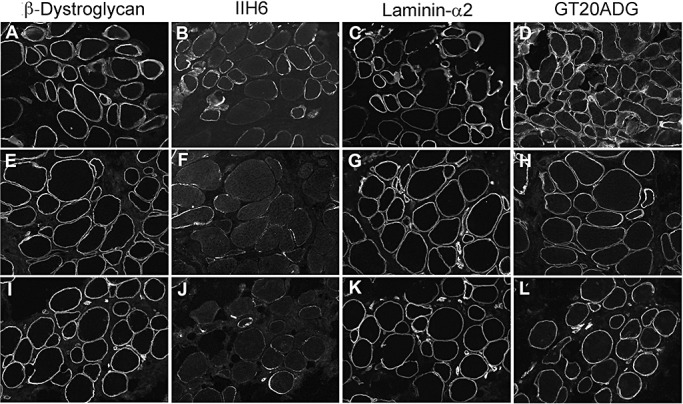
Muscle biopsy of patients with mutations in FKRP. A,E,I.β‐Dystroglycan. B,F,J. IIH6. C,G,K. Laminin‐α2 (4H8 antibody to the N‐terminal 300 kDa fragment). D,H,L. GT20ADG α‐dystroglycan antibody. A–D: P18; E–H: P20; I–L: P21.
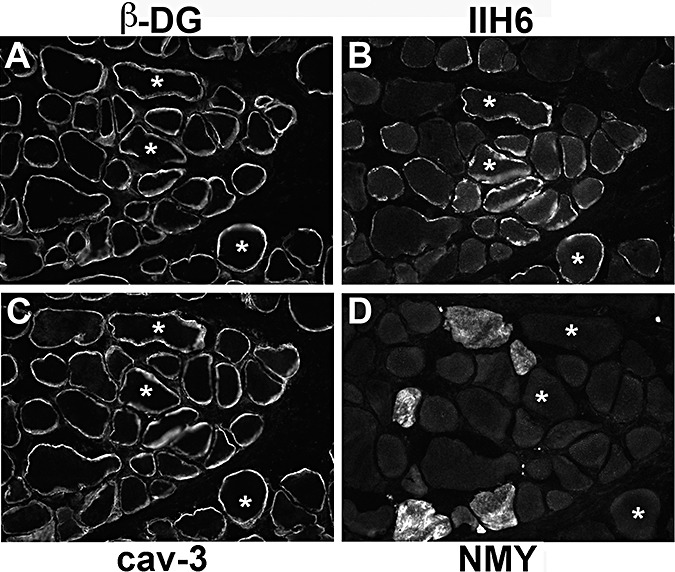
P11 [LGMD‐No MR (12)] had a mild phenotype and only very mild reduction of IIH6 labeling, compared with P10 who also had mutations in POMGnT1 with a more severe phenotype (MEB‐FCMD) and a pronounced reduction of IIH6 immunolabeling (Figure 6). In P11, there was a population of fibers with brighter IIH6 and β‐dystroglycan on the cell surface and intracellular labeling of caveolin‐3 but no neonatal myosin labeling (Figure 6).
Figure 6.
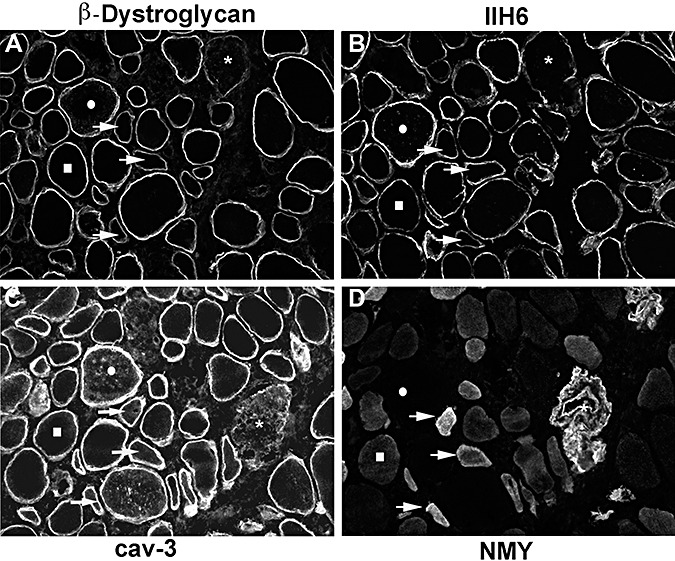
The biopsy of P11 showed the mildest reduction in IIH6 labeling. The labeling of individual fibers with antibodies to β‐dystroglycan (A), IIH6 (B), caveolin‐3 (cav‐3; C) and neonatal myosin (NMY; D) is compared. A few fibers with reduced IIH6 labeling and normal β‐dystroglycan immunoreactivity were seen (squares). Some fibers showed reduced IIH6 and β‐dystroglycan and were positive for the neonatal isoform of myosin so they may represent regenerating fibers (arrows). Fibers with low β‐dystroglycan, IIH6 and reduced sarcolemmal cav‐3 labeling may reflect sarcolemmal damage (asterisks). A population of fibers has marked IIH6 and β‐dystroglycan labeling on the cell surface and intracellular labeling of cav‐3 but no neonatal myosin labeling (circles).
Laminin‐α2 immunolabeling was also variable between and within cases, and was only reduced in 17 out of 24 cases (Table 3). Even in cases with mutations in the same gene, the degree of reduction varied and there was no correlation with either clinical severity or the degree of reduction of α‐dystroglycan labeling. This secondary reduction in laminin‐α2 was sometimes subtle and only detectable with the antibody to the 300‐kDa fragment, as seen in cases of MDC1A with a partial primary reduction of laminin‐α2. However, and in contrast to primary laminin‐α2 deficiency, laminin‐α2 labeling of peripheral nerves was not affected in the secondary dystroglycanopathies (not shown), suggesting that the secondary reduction in laminin‐α2 is a result of the defect in α‐dystroglycan glycosylation specific to the muscle fiber basement membrane rather than that of the peripheral nerve.
Antibodies to the core protein of α‐dystroglycan
Some of the α‐dystroglycan antibodies recognize the primary amino acid sequence in the protein, in theory, independently of whether it is glycosylated or not. Using some of these antibodies [sheep core (20), and IB7 (37)], we and others have previously described a reduction in sarcolemmal labeling that varied from a complete absence to a mild reduction 6, 23, 28, 29.
In the present study, we used a sheep polyclonal antibody raised against the last 20 amino acids of α‐dystroglycan (20). In all cases studied, the staining was abnormal, ranging from an absence to a mild reduction (5, 7). This reduction often correlated with the level of IIH6 labeling (Table 3). It is not clear if the reduction in labeling with this core antibody is a direct consequence of the hypoglycosylation of α‐dystroglycan or the result of the inability of the antibody to bind to the epitope because of masking. A third possibility is that it is a combination of both these possibilities.
Figure 7.
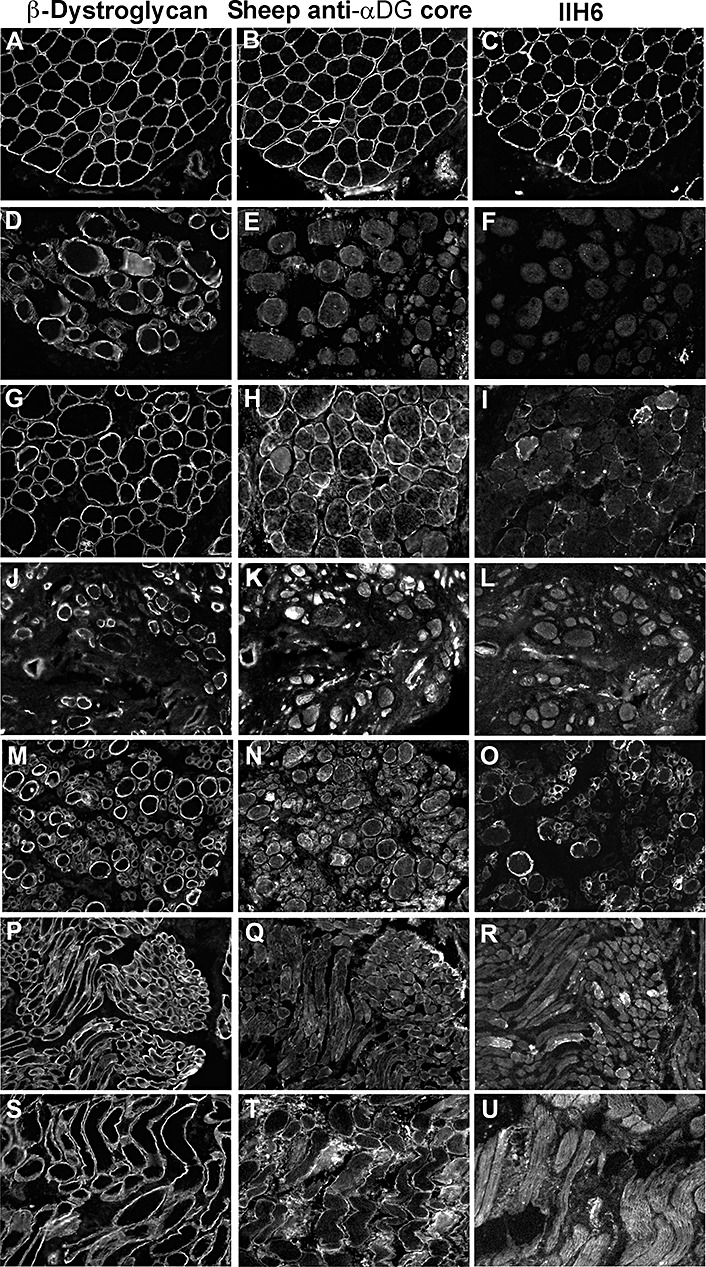
Spectrum of the reduction in α‐dystroglycan labeling using a sheep polyclonal “core” antibody. A,D,G,J,M,P,S.β‐Dystroglycan. B,E,H,K,N,Q,T. Sheep anti‐α‐dystroglycan (α‐DG) core. C,F,I,L,O,R,U. IIH6 antibody. A–C: disease control (Becker muscular dystrophy patient showing a group of regenerating fibers with low labeling of both α‐ and β‐dystroglycan; arrow); D–F: P1 (POMT1); G–I: P4 (POMT1); J–L: P5 (POMT2); M–O: P8 (POMT2); P–R: P10 (POMGnT1); S–U: P15 (FKRP).
In support of the second hypothesis is our observation that studies of cases with either a total or partial primary laminin‐α2 deficiency and mutations in the LAMA2 gene (MDC1A) showed a severe reduction of sarcolemmal labeling with this sheep core antibody (Figure 8). In these MDC1A cases, IIH6 labeling was variable, with only a mild reduction in some fibers but never reduced to the extent observed with the sheep core antibody or to that seen in the dystroglycanopathy cases. Interestingly, in MDC1A patients, the reduction of labeling with the sheep core was independent from the levels of laminin‐α2. For example, both the patient shown in Figure 8A–D, who had complete laminin‐α2 deficiency, and the patient shown in Figure 8E–H, who had partial laminin‐α2 deficiency, had a similar reduction of the sheep core.
Figure 8.
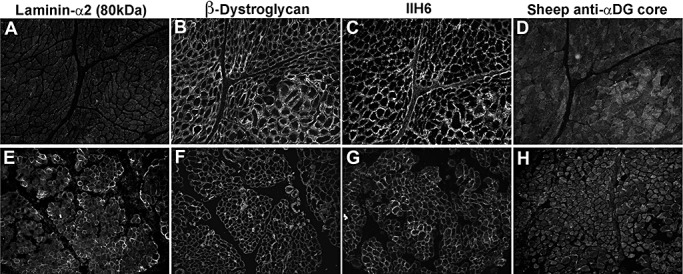
Cases with confirmed primary laminin‐α2‐deficient congenital muscular dystrophy. A,E. Laminin‐α2 (MAB1922 antibody to the 80 kDa C‐terminal fragment). B,F.β‐Dystroglycan. C,G.α‐Dystroglycan IIH6. D,H.α‐Dystroglycan sheep polyclonal core. A–D: MDC1A (total absence of laminin‐α2); E–H: MDC1A (partial laminin‐α2 deficiency).
In contrast to those findings, the labeling obtained using the core antibody GT20ADG was usually well preserved in dystroglycanopathy patients, compared with IIH6 and sheep core. Only rarely was there some variability with the GT20DAG antibody on fibers with well‐preserved β‐dystroglycan (4, 9). This was most apparent in P8 (POMT2; Figure 9A, panel H) and in P12 (LARGE; Figure 9B, panel B). In the latter, labeling with GT20DAG was markedly reduced despite preserved β‐dystroglycan labeling, suggesting this was not a result of sarcolemmal damage.
Figure 9.
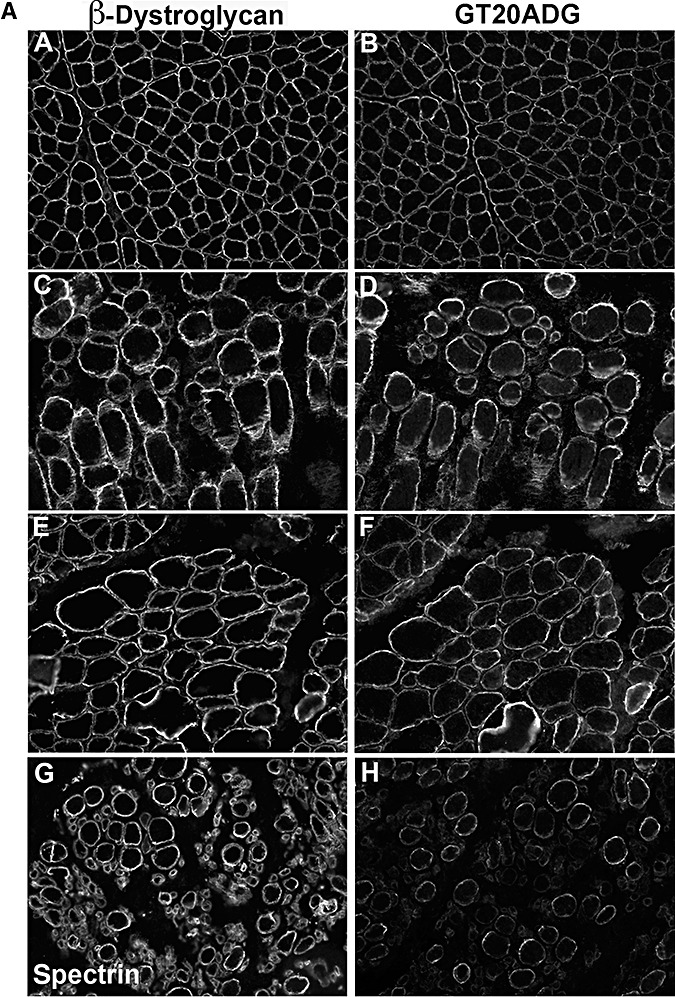
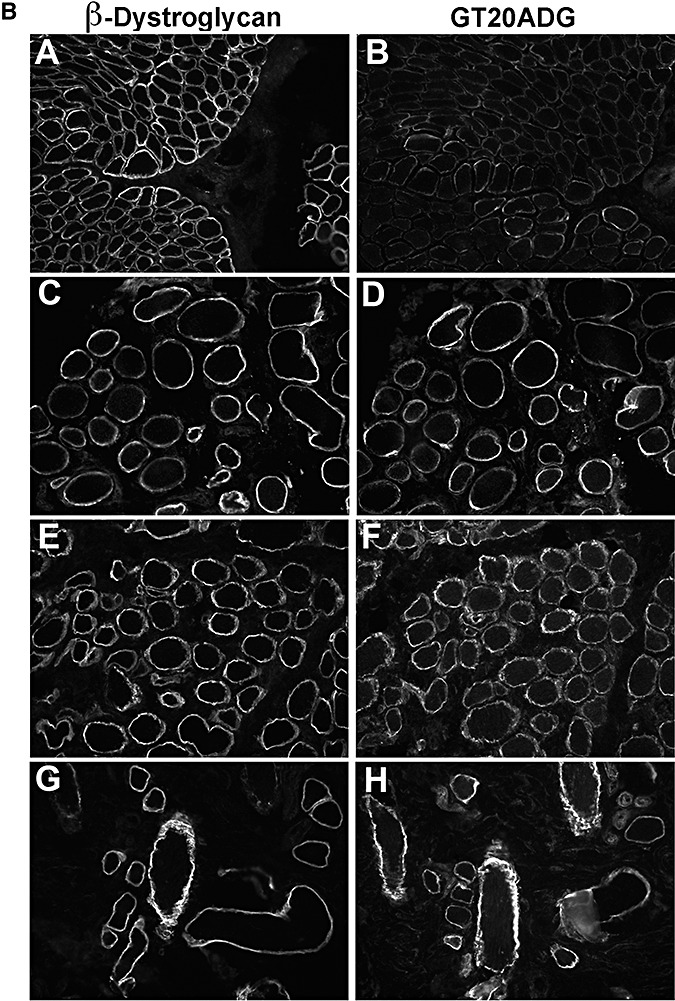
Immunodetection of α‐dystroglycan using GT20ADG antibody. A. A,C,E: β‐dystroglycan. G: β‐spectrin (as a control of sarcolemma preservation as an adjacent image of β‐dystroglycan labeling was not available). B,D,F,H: GT20ADG. A,B: control; C,D: P1 (POMT1); E,F: P4 (POMT1); G,H: P8 (POMT2). B. A,C,E,G: β‐dystroglycan. B,D,F,H: GT20ADG. A,B: P12 (LARGE); C,D: P14 (fukutin); E,F: P16 (FKRP); G,H: P17 (FKRP).
Similarly, in the MDC1A cases, the GT20ADG was well preserved, despite the reduction of the sheep core. Unfortunately, we did not have sufficient tissue to perform the wheat germ agglutinin (WGA) purification that is required prior to using this antibody in Western blots.
We found a reduction in β‐dystroglycan labeling on the sarcolemma of a proportion of fibers (Table 3). In most cases, these were small and expressed neonatal myosin, suggesting that these were regenerating. It cannot be completely excluded, however, that the whole of the dystrophin‐associated glycoprotein complex may be affected in these conditions. As a control for basement membrane preservation, we used antibodies to the laminin‐γ1 and laminin‐β1 chains. We found that while laminin‐γ1 was normal in all cases, laminin‐β1 was variably reduced in 10 of the 24 cases. A reduction in laminin‐β1 has been reported in other neuromuscular disorders (40).
DISCUSSION
The main conclusions from this study are that irrespective of the primary gene defect, the overall pathology and extent of α‐dystroglycan glycosylation in muscle (as determined by the level of IIH6 immunolabeling) is variable. Nevertheless, in this cohort of patients, there was a broad correlation between the amount of glycosylated α‐dystroglycan and the severity of the phenotype in patients with mutations in POMT1, POMT2 and POMGnT1. The severity of the phenotype in these patients was assigned following the clinical categories described in Godfrey et al (19), in which the age of onset of muscle weakness and the presence and severity of brain abnormalities were taken into consideration. It is important to highlight that the limitation of this approach relates to the fact that these clinical categories are based on the combined severity of the skeletal muscle and brain involvement, while in this manuscript we have only studied the glycosylation of α‐dystroglycan in the skeletal muscle. In spite of this, there was also a broad correlation between the reduction of glycosylated α‐dystroglycan and the maximum motor ability within these gene groups (see Table 1). Patients with mutations in POMT1, POMT2 and POMGnT1 that achieved independent ambulation showed more α‐dystroglycan labeling than those who never walked, while the most severe cases who never acquired the independent sitting position had absent labeling.
In contrast to patients with mutations in POMT1, POMT2 and POMGnT1, one patient with FKRP mutations (P15) and two patients with mutations in fukutin (P13 and P14) and an LGMD phenotype with absent brain involvement, had a complete absence of IIH6 labeling. The mutation in P15 (homozygous p.Pro89Leu) has not previously been reported but it affects a conserved amino acid in FKRP, which has been found to be mutated to arginine in a MDC1C patient in combination with a deletion leading to premature termination of translation (31). This patient presented in early infancy following a severe Duchenne‐like course. He lost ambulation before 10 years of age. His severity is comparable with that of P21 (FKRP mutation; see Table 1), who also lost ambulation at the age of 11. These two patients had, on the whole, a severe depletion of IIH6 immunoreactivity, similar to what we have previously described in nonambulant children with MDC1C 5, 6, despite the fact that they did acquire ambulation. The pattern of IIH6 expression in these two patients and in MDC1C cases is, however, substantially different from the one observed in the majority of the LGMD2I cases who follow a much milder disease course and in whom there appears to be a better correlation between IIH6 expression and clinical severity. For example, P18, P19 and P20, who followed a “typical Becker Muscular Dystrophy (BMD)” course had more IIH6 expression compared with the two DMD‐like patients described above but nonetheless showed a significant reduction. Three more LGMD2I patients (P22, P23 and P24), at the milder end of the LGMD phenotype, had well‐preserved IIH6 labeling (mosaic pattern) relative to the other LGMD2I cases discussed above (DMD and BMD‐like).
We also studied two patients with fukutin mutations and an LGMD phenotype. These two patients have been reported previously (18), have no brain involvement and followed a clinical course similar to DMD. As in the FKRP‐related DMD‐like condition, these patients (P14 and P15) had virtual absence of IIH6 labeling. A similar severe depletion of IIH6 has also been observed in other mild fukutin patients, as in the cases with dilated cardiomyopathy and minimal muscle weakness described by Murakami et al (39). The severity of the IIH6 depletion in these patients contrasts with their disease course. The reason for the relatively limited pathology and the mild phenotypes is unclear but it could relate to the presence of additional laminin‐α2‐binding epitope(s) on α‐dystroglycan, which are not recognized by the IIH6 antibody (32).
Although this cohort of patients encompassed a wide range of phenotypes, it did not include the mildest end of the cases with POMT2 spectrum, as recently described by Biancheri et al (3), who displayed an early‐onset LGMD without mental retardation and severely reduced α‐dystroglycan labeling with another antibody directed against the glycosylated epitope VIA4‐1. Likewise, we have not been able to assess any correlations in cases with mutations in LARGE as too few have been identified. The patient described here showed a moderate reduction of IIH6 labeling (28), although we have identified mutations in LARGE in a patient with a severe WWS phenotype in whom absence of IIH6 was demonstrated in another center (19). Other cases with mutations in LARGE have recently been reported but no immunohistochemical studies were reported (44).
In P11, the variant p.Asp556Asn was detected in POMGnT1 along with an altered kinetic profile (12). Very recently, we have become aware that this sequence alteration has been detected in a French family, heterozygously in a patient with MEB yet homozygously in the apparently unaffected sibling. In addition, the variant was detected heterozygously in eight out of 218 alleles from healthy French controls (Céline Bouchet, unpub. obs.). We are carrying out further work to characterize this finding that, however, may point to a secondary reduction in POMGnT1 activity.
The present studies draw attention to the significantly different patterns of immunolabeling obtained with the so‐called “core”α‐dystroglycan antibodies and indicate that more studies are needed to understand what they exactly recognize and their significance in the context of dystroglycanopathies. The absence of one of these epitopes in primary laminin‐α2 deficiency may provide important clues. As previous work has shown that the glycosylation of α‐dystroglycan is extremely complex and that a number of different glycoforms are known to exist in muscle (33), which may be differentially affected by different gene mutations. The generation of new antibodies to known glycosylated epitopes will undoubtedly help clarify some of these issues.
In summary, the present study highlights the limitations of interpreting the extent of α‐dystroglycan functionality by evaluating a single epitope, albeit one, that identifies the laminin‐binding site. Despite these limitations, we found that α‐dystroglycan labeling broadly correlated with clinical severity in cases with mutations in POMT1, POMT2 and POMGnT1 mutations and also within each gene group. However, this conclusion could not always be extended to patients with FKRP or fukutin mutations. Although the function of FKRP and fukutin are still unknown, our findings indicate that their role in α‐dystroglycan glycosylation may be different or broader than that of POMT1, POMT2 and POMGnT1, whose enzymatic activity has been demonstrated. The identification of patients with FKRP and fukutin mutations and a relatively mild muscle phenotype despite absent IIH6 labeling suggests that disruption of the laminin–α‐dystroglycan interaction in muscle, as recognized by the IIH6 antibody, might not be the sole pathological mechanism of disease and that in patients with a severe phenotype, other mechanisms may also operate.
ACKNOWLEDGMENTS
We are grateful to the National Commissioning Group (NCG) Department of Health UK for their financial support. C.G. and E.C. are funded by the Muscular Dystrophy Campaign (RA3/734 and PO0916 respectively). B.T. is funded by Hacettepe University Research Grant 02G129.
REFERENCES
- 1. Balci B, Uyanik G, Dincer P, Gross C, Willer T, Talim B et al (2005) An autosomal recessive limb girdle muscular dystrophy (lgmd2) with mild mental retardation is allelic to Walker‐Warburg syndrome (WWS) caused by a mutation in the pomt1 gene. Neuromuscul Disord 15:271–275. [DOI] [PubMed] [Google Scholar]
- 2. Barresi R, Campbell KP (2006) Dystroglycan: from biosynthesis to pathogenesis of human disease. J Cell Sci 119:199–207. [DOI] [PubMed] [Google Scholar]
- 3. Biancheri R, Falace A, Tessa A, Pedemonte M, Scapolan S, Cassandrini D et al (2007) POMT2 gene mutation in limb‐girdle muscular dystrophy with inflammatory changes. Biochem Biophys Res Commun 363:1033–1037. [DOI] [PubMed] [Google Scholar]
- 4. Brockington M, Blake DJ, Prandini P, Brown SC, Torelli S, Benson MA et al (2001) Mutations in the fukutin‐related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha‐dystroglycan. Am J Hum Genet 69:1198–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brockington M, Yuva Y, Prandini P, Brown SC, Torelli S, Benson MA et al (2001) Mutations in the fukutin‐related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum Mol Genet 10:2851–2859. [DOI] [PubMed] [Google Scholar]
- 6. Brown SC, Fassati A, Popplewell L, Page AM, Henry MD, Campbell KP, Dickson G (1999) Dystrophic phenotype induced in vitro by antibody blockade of muscle alpha‐dystroglycan‐laminin interaction. J Cell Sci 112:209–216. [DOI] [PubMed] [Google Scholar]
- 7. Brown SC, Torelli S, Brockington M, Yuva Y, Jimenez C, Feng L et al (2004) Abnormalities in alpha‐dystroglycan expression in MDC1C and LGMD2I muscular dystrophies. Am J Pathol 164:727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bushby K, Anderson LV, Pollitt C, Naom I, Muntoni F, Bindoff L (1998) Abnormal merosin in adults. A new form of late onset muscular dystrophy not linked to chromosome 6q2. Brain 121:581–588. [DOI] [PubMed] [Google Scholar]
- 9. Chen YJ, Spence HJ, Cameron JM, Jess T, Ilsley JL, Winder SJ (2003) Direct interaction of beta‐dystroglycan with F‐actin. Biochem J 375:329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chiba A, Matsumura K, Yamada H, Inazu T, Shimizu T, Kusunoki S et al (1997) Structures of sialylated O‐linked oligosaccharides of bovine peripheral nerve alpha‐dystroglycan. The role of a novel O‐mannosyl‐type oligosaccharide in the binding of alpha‐dystroglycan with laminin. J Biol Chem 272: 2156–2162. [DOI] [PubMed] [Google Scholar]
- 11. Chiyonobu T, Sasaki J, Nagai Y, Takeda S, Funakoshi H, Nakamura T et al (2005) Effects of fukutin deficiency in the developing mouse brain. Neuromuscul Disord 15:416–426. [DOI] [PubMed] [Google Scholar]
- 12. Clement EM, Godfrey C, Tan J, Brockington M, Torelli S, Feng L et al (2008) Mild POMGnT1 mutations underlie a novel limb‐girdle muscular dystrophy variant. Arch Neurol 65:137–141. [DOI] [PubMed] [Google Scholar]
- 13. Colognato H, Yurchenco PD (2000) Form and function: the laminin family of heterotrimers. Dev Dyn 218:213–234. [DOI] [PubMed] [Google Scholar]
- 14. D'Amico A, Tessa A, Bruno C, Petrini S, Biancheri R, Pane M et al (2006) Expanding the clinical spectrum of POMT1 phenotype. Neurology 66:1564–1567; discussion 461. [DOI] [PubMed] [Google Scholar]
- 15. Dubowitz VSC (2007) Muscle Biopsy: A Practical Approach, 3rd edn. Saunders Elsevier: London. [Google Scholar]
- 16. Ervasti JM, Campbell KP (1993) A role for the dystrophin‐glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol 122:809–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Esapa CT, Bentham GR, Schroder JE, Kroger S, Blake DJ (2003) The effects of post‐translational processing on dystroglycan synthesis and trafficking. FEBS Lett 555:209–216. [DOI] [PubMed] [Google Scholar]
- 18. Godfrey C, Escolar D, Brockington M, Clement EM, Mein R, Jimenez‐Mallebrera C et al (2006) Fukutin gene mutations in steroid‐responsive limb girdle muscular dystrophy. Ann Neurol 60:603–610. [DOI] [PubMed] [Google Scholar]
- 19. Godfrey C, Clement E, Mein R, Brockington M, Smith J, Talim B et al (2007) Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain 130:2725–2735. [DOI] [PubMed] [Google Scholar]
- 20. Herrmann R, Straub V, Blank M, Kutzick C, Franke N, Jacob EN et al (2000) Dissociation of the dystroglycan complex in caveolin‐3‐deficient limb girdle muscular dystrophy. Hum Mol Genet 9:2335–2340. [DOI] [PubMed] [Google Scholar]
- 21. Holt KH, Crosbie RH, Venzke DP, Campbell KP (2000) Biosynthesis of dystroglycan: processing of a precursor propeptide. FEBS Lett 468:79–83. [DOI] [PubMed] [Google Scholar]
- 22. Jayasinha V, Nguyen HH, Xia B, Kammesheidt A, Hoyte K, Martin PT (2003) Inhibition of dystroglycan cleavage causes muscular dystrophy in transgenic mice. Neuromuscul Disord 13:365–375. [DOI] [PubMed] [Google Scholar]
- 23. Jimenez‐Mallebrera C, Torelli S, Brown SC, Feng L, Brockington M, Sewry CA et al (2003) Profound skeletal muscle depletion of alpha‐dystroglycan in Walker‐Warburg syndrome. Eur J Paediatr Neurol 7:129–137. [DOI] [PubMed] [Google Scholar]
- 24. Jimenez‐Mallebrera C, Brown SC, Sewry CA, Muntoni F (2005) Congenital muscular dystrophy: molecular and cellular aspects. Cell Mol Life Sci 62:809–823. [DOI] [PubMed] [Google Scholar]
- 25. Jung D, Yang B, Meyer J, Chamberlain JS, Campbell KP (1995) Identification and characterization of the dystrophin anchoring site on beta‐dystroglycan. J Biol Chem 270:27305–27310. [DOI] [PubMed] [Google Scholar]
- 26. Kondo‐Iida E, Kobayashi K, Watanabe M, Sasaki J, Kumagai T, Koide H et al (1999) Novel mutations and genotype‐phenotype relationships in 107 families with Fukuyama‐type congenital muscular dystrophy (FCMD). Hum Mol Genet 8:2303–2309. [DOI] [PubMed] [Google Scholar]
- 27. Kunz S, Sevilla N, McGavern DB, Campbell KP, Oldstone MB (2001) Molecular analysis of the interaction of LCMV with its cellular receptor [alpha]‐dystroglycan. J Cell Biol 155:301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Longman C, Brockington M, Torelli S, Jimenez‐Mallebrera C, Kennedy C, Khalil N et al (2003) Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of alpha‐dystroglycan. Hum Mol Genet 12:2853–2861. [DOI] [PubMed] [Google Scholar]
- 29. Longman C, Mercuri E, Cowan F, Allsop J, Brockington M, Jimenez‐Mallebrera C et al (2004) Antenatal and postnatal brain magnetic resonance imaging in muscle‐eye‐brain disease. Arch Neurol 61:1301–1306. [DOI] [PubMed] [Google Scholar]
- 30. Martin PT (2006) Mechanisms of disease: congenital muscular dystrophies‐glycosylation takes center stage. Nat Clin Pract Neurol 2:222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Matsumoto H, Hayashi YK, Kim DS, Ogawa M, Murakami T, Noguchi S et al (2005) Congenital muscular dystrophy with glycosylation defects of alpha‐dystroglycan in Japan. Neuromuscul Disord 15:342–348. [DOI] [PubMed] [Google Scholar]
- 32. McDearmon EL, Combs AC, Ervasti JM (2001) Differential Vicia villosa agglutinin reactivity identifies three distinct dystroglycan complexes in skeletal muscle. J Biol Chem 276:35078–35086. [DOI] [PubMed] [Google Scholar]
- 33. McDearmon EL, Combs AC, Sekiguchi K, Fujiwara H, Ervasti JM (2006) Brain alpha‐dystroglycan displays unique glycoepitopes and preferential binding to laminin‐10/11. FEBS Lett 580:3381–3385. [DOI] [PubMed] [Google Scholar]
- 34. Mercuri E, D'Amico A, Tessa A, Berardinelli A, Pane M, Messina S et al (2006) POMT2 mutation in a patient with “MEB‐like” phenotype. Neuromuscul Disord 16:446–448. [DOI] [PubMed] [Google Scholar]
- 35. Michele DE, Barresi R, Kanagawa M, Saito F, Cohn RD, Satz JS et al (2002) Post‐translational disruption of dystroglycan‐ligand interactions in congenital muscular dystrophies. Nature 418:417–422. [DOI] [PubMed] [Google Scholar]
- 36. Moore SA, Saito F, Chen J, Michele DE, Henry MD, Messing A et al (2002) Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature 418:422–425. [DOI] [PubMed] [Google Scholar]
- 37. Moukhles H, Roque R, Carbonetto S (2000) Alpha‐dystroglycan isoforms are differentially distributed in adult rat retina. J Comp Neurol 420:182–194. [DOI] [PubMed] [Google Scholar]
- 38. Muntoni F, Brockington M, Blake DJ, Torelli S, Brown SC (2002) Defective glycosylation in muscular dystrophy. Lancet 360:1419–1421. [DOI] [PubMed] [Google Scholar]
- 39. Murakami T, Hayashi YK, Noguchi S, Ogawa M, Nonaka I, Tanabe Y et al (2006) Fukutin gene mutations cause dilated cardiomyopathy with minimal muscle weakness. Ann Neurol 60:597–602. [DOI] [PubMed] [Google Scholar]
- 40. Sewry CA, Brown SC, Mercuri E, Bonne G, Feng L, Camici G et al (2001) Skeletal muscle pathology in autosomal dominant Emery‐Dreifuss muscular dystrophy with lamin A/C mutations. Neuropathol Appl Neurobiol 27:281–290. [DOI] [PubMed] [Google Scholar]
- 41. Talim B, Ferreiro A, Cormand B, Vignier N, Oto A, Gogus S et al (2000) Merosin‐deficient congenital muscular dystrophy with mental retardation and cerebellar cysts unlinked to the LAMA2, FCMD and MEB loci. Neuromuscul Disord 10:548–552. [DOI] [PubMed] [Google Scholar]
- 42. Toda T, Chiyonobu T, Xiong H, Tachikawa M, Kobayashi K, Manya H et al (2005) Fukutin and alpha‐dystroglycanopathies. Acta Myol 24:60–63. [PubMed] [Google Scholar]
- 43. Topaloglu H, Brockington M, Yuva Y, Talim B, Haliloglu G, Blake D et al (2003) FKRP gene mutations cause congenital muscular dystrophy, mental retardation, and cerebellar cysts. Neurology 60:988–992. [DOI] [PubMed] [Google Scholar]
- 44. Van Reeuwijk J, Grewal PK, Salih MA, Beltran‐Valero de Bernabe D, McLaughlan JM, Michielse CB et al (2007) Intragenic deletion in the LARGE gene causes Walker‐Warburg syndrome. Hum Genet 121:685–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Williamson RA, Henry MD, Daniels KJ, Hrstka RF, Lee JC, Sunada Y et al (1997) Dystroglycan is essential for early embryonic development: disruption of Reichert's membrane in Dag1‐null mice. Hum Mol Genet 6:831–841. [DOI] [PubMed] [Google Scholar]
- 46. Winder SJ (2001) The complexities of dystroglycan. Trends Biochem Sci 26:118–124. [DOI] [PubMed] [Google Scholar]
- 47. Yurchenco PD, Cheng YS, Campbell K, Li S (2004) Loss of basement membrane, receptor and cytoskeletal lattices in a laminin‐deficient muscular dystrophy. J Cell Sci 117:735–742. [DOI] [PubMed] [Google Scholar]