

Abstract
Intracellular Ca2+ ([Ca2+]i) can trigger dual-mode regulation of the voltage gated cardiac sodium channel (NaV1.5). The channel components of the Ca2+ regulatory system are the calmodulin (CaM)-binding IQ motif and the Ca2+ sensing EF hand–like (EFL) motif in the carboxyl terminus of the channel. Mutations in either motif have been associated with arrhythmogenic changes in expressed NaV1.5 currents. Increases in [Ca2+]i shift the steady-state inactivation of NaV1.5 in the depolarizing direction and slow entry into inactivated states. Mutation of the EFL (NaV1.54X) shifts inactivation in the hyperpolarizing direction compared with the wild-type channel and eliminates the Ca2+ sensitivity of inactivation gating. Modulation of the steady-state availability of NaV1.5 by [Ca2+]i is more pronounced after the truncation of the carboxyl terminus proximal to the IQ motif (NaV1.5Δ1885), which retains the EFL. Mutating the EFL (NaV1.54X) unmasks CaM-mediated regulation of the kinetics and voltage dependence of inactivation. This latent CaM modulation of inactivation is eliminated by mutation of the IQ motif (NaV1.54X-IQ/AA). The LQT3 EFL mutant channel NaV1.5D1790G exhibits Ca2+ insensitivity and unmasking of CaM regulation of inactivation gating. The enhanced effect of CaM on NaV1.54X gating is associated with significantly greater fluorescence resonance energy transfer between enhanced cyan fluorescent protein–CaM and NaV1.54X channels than is observed with wild-type NaV1.5. Unlike other isoforms of the Na channel, the IQ-CaM interaction in the carboxyl terminus of NaV1.5 is latent under physiological conditions but may become manifest in the presence of disease causing mutations in the CT of NaV1.5 (particularly in the EFL), contributing to the production of potentially lethal ventricular arrhythmias.
Keywords: voltage-gated sodium channel, EF hand motif, IQ motif, calmodulin, FRET
Sodium channels play a central role in the electrophysiology of the heart, being essential for rapid impulse conduction and regulation of the action potential duration. Congenital or acquired defects in inactivation gating of the Na channel have important implications for maintenance of normal rhythmicity in the heart.1–3 The carboxyl terminus (CT) of NaV1.5 has been shown to play an important role in inactivation.4–6 Ca2+, CaM, CaM kinase are known to modulate inactivation though interaction with structural motifs in the CT.6,7 Ca2+ binding to an EF hand–like (EFL) sequence in the CT of NaV1.5 has been shown to regulate channel gating.8,9 The CT of NaV1.5 contains a CaM binding motif of the IQ type; however, CaM apparently has minimal functional effect on the expressed current, unlike other voltage gated sodium channels.6,10 The functional relationship between the EFL and IQ motifs remains uncertain but it has been suggested that a Ca2+ load displaces CaM from the IQ motif, freeing the IQ to interact with the EFL stabilizing inactivation.8
Mutations near the IQ and EFL motifs (Figure 1A) have been linked to the heritable cardiac rhythm disturbances long QT and Brugada syndromes.11–13 A number of acquired pathological conditions (cardiac hypertrophy and heart failure) are associated with dysregulation of Ca2+ homeostasis, which may generate arrhythmias involving the cardiac Na channel through functional alterations mediated by the IQ and EFL motifs. To better understand the functional relationship between Ca2+, CaM, and Ca2+ binding directly to the channel via the EFL, we mutated the key Ca2+-chelating and IQ residues in the CT of NaV1.5 and studied the interaction of Ca2+ and CaM-based signaling on channel gating.
Figure 1.

Ca2+-mediated modulation of gating. A, Schematic of the structured part of the CT of NaV1.5. The predicted helices are labeled H1 through H6. The EFL is in the loop between H1 and H2 and harbors the 4× mutations (E1788A, D1790A, D1792A, E1799A) or the LQT3 mutation D1790G. The IQ is in H6, and the location of the truncation in NaV1.5Δ1885 is shown. B, Representative families of currents (protocol shown in inset) through NaV1.5 and NaV1.54x channels with Ca2+. Ca2+ does not alter the voltage of the peak IV relationships (C) or the activation curves (dotted lines in D) in the absence or presence of Ca2+. The V1/2 of steady-state inactivation (solid lines in D) of NaV1.5 is modestly shifted in the absence of Ca2+. The half-maximal inactivation of NaV1.54x is shifted by nearly ≈15 mV in the hyperpolarizing direction compared with NaV1.5 (Table). Currents were recorded at a test pulse of −20 mV after a 500-ms conditioning pulse from −140 to +30 mV. For NaV1.5, increasing [Ca2+]i decreases τrec (E) and increases τentry (F) into inactivated states compared with the Ca2+-free condition. Recovery of NaV1.54X is slower than wild-type, and Ca2+ further slows recovery. In 10 μmol/L Ca2+, the entry of NaV1.54X into inactivated states is faster and more extensive than NaV1.5. The entry rates converge in the absence of Ca2+, but NaV1.54X undergoes more extensive inactivation compared with NaV1.5. Unless otherwise specified, the data in this and all other figures are in means±SE, and activation and steady-state inactivation data are fit with Boltzmann functions. The symbols are the same in C through F.
Materials and Methods
An expanded Materials and Methods section describing plasmid construction, solutions, and electrophysiology and fluorescence resonance energy transfer (FRET) measurements is available in the online data supplement at http://circres.ahajournals.org. These procedures were similar to those that we have published previously.10 The bath solution contained (in mmol/L): 145 NaCl, 4 KCl, 1.8 CaCl2, 1 MgCl2, 10 glucose, and 10 Na-HEPES (pH 7.4). The patch pipette solution for the Ca2+-free condition contained (in mmol/L): 10 NaF, 100 CsF, 20 CsCl2, 20 BAPTA, and 10 HEPES, pH adjusted to 7.35 with CsOH. The 10 μmol/L Ca2+ pipette solution was similar to the Ca2+-free solution except that BAPTA was reduced to 1 mmol/L and 1 mmol/L CaCl2 was added. Similarly, the 0.5 μmol/L Ca2+ solution contained 5 mmol/L BAPTA and 4 mmol/L CaCl2. The osmolarity of the bath and pipette solutions were equilibrated with glucose.
Results
Ca2+ Directly Interacts With NaV1.5
We first examined the effect of altering intracellular Ca2+ on gating of wild-type NaV1.5 and channels with the Ca2+-sensing residues of the EFL mutated (NaV1.54X). The pipette solution was either Ca2+-free or contained 10 μmol/L Ca2+ (Figure 1B). The peak current–voltage (IV) relationships and V1/2 of the activation curves of NaV1.5 were unchanged in absence or in presence of 10 μmol/L Ca2+ (Figure 1C and 1D and the Table). The voltage dependence of the steady-state availability of wild-type NaV1.5 is shifted in the depolarizing direction by increases in [Ca2+]i (Figure 1D). To determine whether the observed shift is attributable to direct Ca2+ interaction with the channel, we mutated the predicted Ca2+-sensing residues in the CT of NaV1.5 EFL motif (E1788A, D1790A, D1792A, E1799A). Mutations in the EFL eliminate Ca2+ sensitivity of inactivation gating of NaV1.5 and shift the V1/2 almost ≈15 mV in the hyperpolarizing direction (Figure 1D) without affecting the IV relationship (Figure 1C) and V1/2 of activation (Figure 1D). However in Ca2+-free conditions the reversal potential of NaV1.54X is modestly shifted in the hyperpolarizing direction.
Table.
Effects of Intracellular Ca2+, CaM, and CaM1234 on NaV1.5 Variant Channels
| Activation (mV) |
||||||
|---|---|---|---|---|---|---|
| Channel/Mutant | V1/2 Steady State Inactivation (mV) | V1/2 | IPeak | EReversal | τRec (ms) | τEntry (ms) |
| NaV1.5 | ||||||
| +Ca2+ | −82.6±0.2 (12) | −39.7±1.7 | −22.5±2 | 60±1.1 (10) | 3.9±0.1 (10) | 4523±184 (5) |
| −Ca2+ | −87.5±0.2 (14) | −38±2 | −23±3 | 45±3 (10) | 5.5±0.1 (9) | 2783±109 (5) |
| NaV1.54X | ||||||
| +Ca2+ | −98.6±0.4 (15) | −41±2 | −23.7±1.5 | 56±2 (11) | 14.5±1.2 (7) | 2594±209 (5) |
| −Ca2+ | −99.3±0.1 (11) | −39±2.5 | −21±3.5 | 46±3.5 (9) | 21.3±3 (8) | 3061±16 (5) |
| NaV1.5Δ1885 | ||||||
| +Ca2+ | −81.8±0.1 (10) | −36.8±1.7 | −21.6±1.8 | 55.5±1.5 (9) | 2.7±0.1 (5) | 6173±1037 (5) |
| −Ca2+ | −93.3±0.1 (18) | −34.1±0.8 | −18.3±0.7 | 50±2.6 (18) | 5.3±0.1 (9) | 2318±231 (7) |
| NaV1.5IQ/AA | ||||||
| +Ca2+ | −73.9±0.1 (7) | −31.9±1.3 | −20±1.6 | 64±2 (5) | 3.7±0.1 (6) | 5416±568 (5) |
| −Ca2+ | −82.5±0.4 (8) | −32.7±2.6 | −21.3±1.3 | 49±5.2 (8) | 7.8±1.3 (7) | 2807±90 (6) |
| NaV1.5D1790G | ||||||
| +Ca2+ | −98.2±0.1 (8) | −38.9±0.4 | −20.8±1 | 56.7±1.8 (11) | 5.4±0.07 (5) | 3195±150 (6) |
| −Ca2+ | −99.9±0.1 (9) | −37.6±0.4 | −21.4±1 | 53.8±1.2 (13) | 5.1±0.08 (6) | 3051±144 (6) |
| NaV1.5 (Ca2+-Free) | ||||||
| −CaM | −87.5±0.2 (14) | −38±2 | −23±3 | 45±3 (10) | 5.5±0.1 (9) | 2783±109 (5) |
| +CaM | −82.3±0.6 (6) | −35.1±1.1 | −21.7±1.1 | 53±5 (6) | 6±0.9 (5) | 4309±89 (5) |
| +CaM1234 | −77.4±0.2 (7) | −35±2 | −20±2.5 | 58±5.3 (8) | 7±2.3 (5) | 3452±369 (6) |
| NaV1.5Δ1885 (Ca2+-Free) | ||||||
| −CaM | −93.3±0.1 (18) | −34.1±0.8 | −18.3±0.7 | 50±2.6 (18) | 5.3±0.1 (9) | 2318±231 (7) |
| +CaM | −90.4±0.3 (8) | −32.4±2.7 | −18±1.5 | 51±3.6 (7) | 5.3±0.8 (7) | 2994±315 (6) |
| +CaM1234 | −89.3±0.2 (6) | −28.2±0.8 | −18±1.3 | 62±3 (5) | 4±0.7 (5) | 3112±221 (5) |
| NaV1.5 (0.5 μmol/L Ca2+) | ||||||
| −CaM | −82.6±0.1 (5) | −36±2 | −20.7±0.7 | 55±2.2 (7) | 3.8±0.4 (5) | 3413±470 (5) |
| +CaM | −80±0.08 (12) | −34.6±0.3 | −20±2.2 | 53±2.2 (7) | 3.3±0.6 (5) | 3659±46 (5) |
| +CaM1234 | −75.4±0.04 (14) | −37.8±2.7 | −22.9±20.6 | 45±4.3 (7) | 2.3±0.4 (5 | 3291±203 (5) |
| NaV1.54X (0.5 μmol/L Ca2+) | ||||||
| −CaM | −99.9±0.1 (6) | −37.4±1.7 | −20.8±1.5 | 54.2±4 (6) | 14.9±0.2 (7) | 1704±195 (5) |
| +CaM | −106.8±0.1 (8) | −36±4.3 | −21±4.9 | 54±6.2 (5) | 15.5±0.3 (5) | 1745±120 (5) |
| +CaM1234 | −94.9±0.2 (15) | −35±1.1 | −18.7±1.5 | 55±3.6 (11) | 15.3±0.2 (8) | 1734±142 (7) |
| NaV1.54X+AIP (0.5 μmol/L Ca2+) | ||||||
| −CaM | −101.6±0.5 (5) | −34.4±1.3 | −18.8±2.4 | 52.5±6 (5) | 15.8±0.3 (5) | 1685±133 (5) |
| +CaM | −102±0.2 (11) | −34±0.7 | −18±1.5 | 56.3±1.3 (5) | 14.8±0.2 (5) | 1509±150 (5) |
| +CaM1234 | −103.7±0.1 (15) | −33±3.1 | −20±3.1 | 52±2.8 (10) | 18.1±0.3 (9) | 1587±142 (7) |
| NaV1.54X-IQ/AA (0.5 μmol/L Ca2+) | ||||||
| −CaM | −100.8±0.3 (9) | −32.3±2 | −18.4±1.7 | 55.8±3 (6) | 18.2±0.5 (6) | 1951±111 (5) |
| +CaM | −100.3±0.1 (10) | −32±3.9 | −18±2.6 | 52±3 (5) | 14.3±0.2 (5) | 1942±129 (5) |
| +CaM1234 | −103.1±0.2 (5) | −34±3 | −17±1.2 | 52±5.9 (5) | 15.8±0.2 (5) | 2159±196 (5) |
| NaV1.5D1790G (0.5 μmol/L Ca2+) | ||||||
| −CaM | −96.5±0.1 (6) | −38.9±0.5 | −22.5±1.3 | 62.5±1.3 (6) | 3.5±0.3 (5) | 3565±202 (6) |
| +CaM | −102±0.2 (6) | −39.7±0.4 | −20.7±2 | 62.1±2.4 (6) | 4.9±0.5 (5) | 3122±198 (5) |
| +CaM1234 | −92.3±0.2 (7) | −37.9±0.4 | −21.5±1.5 | 68.5±2.6 (6) | 4.5±0.3 (7) | 3379±109 (7) |
Values are means±SE (n). +Ca2+ indicates 10 μmol/L Ca2+; −Ca2+, Ca2+-free; −CaM, without CaM/CaM1234 overexpression.
Alterations in [Ca2+]i affect both entry into and recovery from inactivated states. Increasing the [Ca2+]i significantly shortens the fast recovery time constant (τrec) from 5.5±0.1 to 3.9±0.1 ms in wild-type NaV1.5 (Figure 1E). However, recovery from slower inactivated states after a prepulse of 1000 ms is not affected by the [Ca2+]i (Figure I in the online data supplement). The EFL mutant, NaV1.54X exhibits slower recovery independent of the [Ca2+]i with a fast τrec that is ≈3- to 4-fold larger than wild-type NaV1.5. Reducing the [Ca2+]i does not affect the current available immediately after the conditioning pulse (P1) but significantly shortens the τrec of NaV1.54X (Figure 1E). Conversely, increasing [Ca2+]i significantly slows entry into inactivation of wild-type NaV1.5 with τentry increasing to 4523±184 ms in the presence of 10 μmol/L Ca2+ from 2783±109 ms in Ca2+-free conditions (Figure 1F). NaV1.54X hastens the rate of entry into inactivated states compared to wild-type NaV1.5 in 10 μmol/L Ca2+ and is not associated with any Ca2+-dependent changes in entry into inactivated states (Figure 1F). Changes in [Ca2+]i have no significant effect on the persistent current through NaV1.5 or NaV1.54X channels but in the absence of Ca2+, the persistent current through NaV1.54X is greater than NaV1.5 (supplemental Figure II).
IQ-CaM Interaction Does Not Modulate Ca2+-Dependent Channel Kinetics
The role of the Ca2+-binding protein, calmodulin (CaM) in mediating Ca2+-dependent regulation of cardiac Na channel gating remains controversial. On the one hand, CaM is a ubiquitous Ca2+ buffer; however, CaM or CAM without bound calcium (apo)-CaM directly binds to the IQ motif in the CT of NaV1.5; thus, the IQ motif binds an extrinsic Ca2+ sensor (CaM) that regulates channel function. We deleted the IQ motif by truncation of the channel after amino acid1885 (NaV1.5Δ1885) and assessed the sensitivity of the truncation mutant to the [Ca2+]i level (Figure 2A). Similar to wild-type NaV1.5and NaV1.54X the IV relationship and voltage dependence of activation of NaV1.5Δ1885 are not altered by changes in [Ca2+]i (Figure 2A and 2B). Previous reports suggested a significant increase in late current with the Δ1885 truncation and the IQ/AA mutation5,14; however, a change in [Ca2+]i had no effect on the magnitude of the late current through IQ/AA mutation.7 The persistent current through NaV1.5Δ1885 was not influenced by [Ca2+]i, although the magnitude of the persistent current was greater than that of wild-type NaV1.5 (supplemental Figure II). However, the voltage dependence of steady-state inactivation of NaV1.5Δ1885 remained sensitive to [Ca2+]i. In the presence of 10 μmol/L Ca2+, the V1/2 of steady-state inactivation is −81.8±0.1mV, similar to wild-type NaV1.5, removal of Ca2+ shifts the V1/2 of NaV1.5Δ1885 approximately −11 mV (Figure 2C). The kinetics of inactivation gating of NaV1.5Δ1885 exhibited changes comparable to wild-type NaV1.5 in response to changes in [Ca2+]i. Increasing the [Ca2+]i significantly shortens τrec of NaV1.5Δ1885 from 5.3±0.1 ms in Ca2+-free conditions to 2.7±0.1 ms (Figure 2D). In contrast, 10 μmol/L Ca2+ significantly increases the τentry of NaV1.5Δ1885 compared to the Ca2+-free condition, an effect similar to wild-type NaV1.5 (Figure 2E). Point mutations in IQ region (NaV1.5IQ/AA) of the CT of NaV1.5 that disable CaM binding exhibit comparable Ca2+-induced changes in inactivation gating (Figure 3 and Table). The preservation of the Ca2+ sensitivity of inactivation by the deletion mutant NaV1.5Δ1885 and NaV1.5IQ/AA suggests that the IQ motif is not required for Ca2+ dependent modulation of inactivation voltage dependence or kinetics.
Figure 2.

Ca2+ regulation of truncated NaV1.5 channels. A, Representative family of NaV1.5Δ1885 currents in absence and presence of 10 μmol/L Ca2+. The normalized IV relationships (B) and the V1/2 of activation (dotted lines in C) are not affected by changes in [Ca2+]i. The steady-state inactivation is shifted in the hyperpolarizing direction in the absence of [Ca2+]i (C). The τrec (D) is shorter and τentry (E) is longer in presence of Ca2+ compared to the Ca2+-free condition. NaV1.5 in 10 μmol/L Ca2+ (Figure 1C) is shown for comparison. The symbols are the same in B through E.
Figure 3.

IQ/AA retains Ca2+ sensitivity of inactivation gating. A, Families of representative NaV1.5IQ/AA currents with and without Ca2+. The IV relationship (B) and the kinetics of activation gating of NaV1.5IQ/AA (dotted lines C) of NaV1.5IQ/AA are not altered by changes in [Ca2+]i. The voltage dependence of steady-state inactivation of NaV1.5IQ/AA remains sensitive to [Ca2+]i. τrec (D) and τentry (E) respond similarly to NaV1.5 and NaV1.5Δ1885 with changes in [Ca2+]i. The symbols are the same in B through E.
Unmasking of Modulation of Inactivation by CaM
The IQ motif is not required for Ca2+ modulation of NaV1.5; however, the existence of direct IQ-CaM regulation of gating of other Na channel isoforms6,10 with homologous carboxyl termini has led to the speculation that there could be latent tuning of normal channel function by CaM binding to the IQ motif in NaV1.5. To test the idea of latent CaM-mediated modulation of NaV1.5 gating, we examined the effects of CaM overexpression in the absence of Ca2+ on wild-type channel function. Coexpression of CaM with wild-type NaV1.5 significantly shifts channel availability in the depolarizing direction compared to NaV1.5 without CaM coexpression in the Ca2+-free condition (Figure 4A). Interestingly mutant CaM1234 also shifts the steady-state V1/2 of the inactivation curve in the Ca2+-free condition (Figure 4A). Neither CaM nor CaM1234 affects the activation curve, recovery from or entry into inactivation of NaV1.5 (Table). However, in physiological [Ca2+]i (0.5 μmol/L), the effect of CaM overexpression on NaV1.5 inactivation gating is not significantly different from that observed in the absence of CaM overexpression. Notably, overexpression of CaM1234 produces a significant depolarizing shift in inactivation (Table and Figure 4A and 4B), consistent with retained binding to the CT and modulation of gating. To verify the involvement of the IQ motif in latent CaM-mediated regulation of the cardiac Na channel, we studied the effect of CaM overexpression on inactivation of the IQ-deficient truncation mutant NaV1.5Δ1885 in Ca2+-free conditions. The steady-state inactivation curve of NaV1.5Δ1885 is not affected by overexpression of CaM or CaM1234 compared to the channel alone and neither CaM nor CaM1234 effects activation of NaV1.5Δ1885 (Table). The data suggest that CaM/CaM1234-mediated modulation of NaV1.5 in Ca2+-free conditions is through binding to the IQ motif.
Figure 4.

CaM modulation of gating. A, The steady-state inactivation curves of NaV1.5 in Ca2+-free conditions are shifted in the depolarizing direction with overexpression of both CaM or CaM1234 compared with NaV1.5 expressed alone. B, In 0.5 μmol/L Ca2+, only CaM1234 overexpression significantly shifts the steady-state inactivation curve. Neither CaM nor CaM1234 overexpression affects the current through NaV1.54X (C) or IV relationships (D) in 0.5 μmol/L Ca2+. E, CaM and CaM1234 shifts the steady-state inactivation curves in the hyperpolarizing and depolarizing direction, respectively, compared to NaV1.54X expressed alone. In contrast, CaM or CaM1234 overexpression does not alter recovery from inactivation τrec (F) or entry into inactivation τentry (G). The symbols are the same in D through G.
We then tested whether CaM modulation of NaV1.5 is unmasked in the EFL mutant channel in physiological [Ca2+]i. Neither CaM nor CaM1234 overexpression alters the Na current or IV relationship (Figure 4C and 4D). In contrast to wild-type NaV1.5, coexpression of CaM with NaV1.54X significantly shifts channel availability in the hyperpolarizing direction (≈7 mV with CaM) compared to NaV1.54X expressed alone in 0.5 μmol/L [Ca2+]i (Figure 4E). Conversely, coexpression of apo-CaM (CaM1234) significantly shifts the V1/2 in the depolarizing direction (≈5 mV with CaM1234; Figure 4E). Compared to NaV1.54X alone, recovery from (Figure 4F), or entry into, inactivation (Figure 4G) of NaV1.54X was not significantly affected by CaM/CaM1234 overexpression. Thus, elimination of Ca2+ sensing by the EFL mutation unmasks a CaM-mediated effect on the voltage dependence of steady-state inactivation.
There are 2 lines of evidence that suggest that the CaM or apo-CaM effects on NaV1.54X occur through an IQ-CaM interaction. First, substitution of the IQ motif (IQ/AA) in NaV1.54X (NaV1.54X-IQ/AA) abolishes the CaM/CaM1234 shift of the inactivation curve without altering the Na current or the IV relationship (Figure 5A through 5C). The kinetics of recovery from or entry into inactivation of NaV1.54x-IQ/AA remains unaffected by CaM or CaM1234 (Table). Thus, NaV1.54X-IQ/AA loses its sensitivity to coexpressed CaM and CaM1234.
Figure 5.

IQ motif mediates the CaM regulation of NaV1.54X. A, Representative current families through NaV1.54X-IQ/AA in the presence of CaM and CaM1234 overexpression. Neither CaM nor CaM1234 overexpression alters the IV relationship (B) or steady-state inactivation curves (C) of NaV1.54X-IQ/AA. D, Representative raw current records of NaV1.54X with overexpression of CaM or CaM1234 and intracellular application of AIP290-309. The IV relationship (E) and steady-state inactivation (F) are not affected by CaM or CaM1234 in the presence of AIP290-309. The symbols are the same in B and C and in E and F.
The second line of evidence that the IQ motif is involved in CaM/CaM1234 regulation of NaV1.54x is the effect of the CaM anti-peptide AIP290–309 on NaV1.54x gating. In the presence of AIP290-309, overexpression of CaM or CaM1234 does not affect the Na current, IV relationship (Figure 5D and 5E), or V1/2 of steady-state inactivation of NaV1.54x (Figure 5F). Similarly, recovery from and entry into inactivation of NaV1.54x are unaffected by CaM or CaM1234 in the presence of AIP290-309 (Table). These data suggest that AIP290-309 competes for CaM binding; displaces CaM from the channel, preventing CaM-induced modulation of the voltage dependence of steady-state inactivation; or binds to an unknown alternate binding site(s) to counteract the effect of CaM on inactivation. The AIP290-309 data and the effect of the IQ/AA mutation in NaV1.54x are consistent with the IQ motif mediating latent CaM/CaM1234 modulation of the cardiac Na channel.
Physical Association of CaM With NaV1.5 and NaV1.54X
The electrophysiological data suggest that CaM/CaM1234 interacts with the EFL mutant channel NaV1.54X and produces a shift in the voltage dependence of gating. Wild-type NaV1.5 channels remain insensitive to CaM in physiological (0.5 μmol/L) [Ca2+]i (Table and Figure 4B), but steady-state inactivation is regulated by CaM and CaM1234 in the Ca2+-free condition (Figure 4A). In the skeletal muscle channel, NaV1.4, CaM is tethered to the channel CT and modulates inactivation gating through the IQ motif.10 The CT of NaV1.4 and NaV1.5 are highly homologous, both containing EFL and IQ motifs. We hypothesize that CaM interacts with NaV1.5, but the functional effects of the interaction on inactivation gating are concealed at physiological [Ca2+]i and that the EFL mutant channel, NaV1.54X, reveals latent regulation of inactivation by CaM binding to the CT of NaV1.5 (Figure 4A and 4E). To test this hypothesis, we looked for direct interaction of CaM with the CT of NaV1.5 and NaV1.54X by coexpressing NaV1.5–enhanced yellow fluorescent protein (EYFP) or NaV1.54X-EYFP with ECFP-CaM, ECFP-CaM1234, or ECFP alone. We used donor dequenching FRET in live cells to study the proximity of ECFP-CaM to the CT of NaV1.5 or NaV1.54X. The channel fusions were designed with the fluorescent proteins linked to the carboxyl terminus, which is close in the linear amino acid sequence to the IQ motif, optimizing the possibility of FRET detection on CaM binding. Both the NaV1.5 and NaV1.54X fusion constructs expressed in HEK293 cells display a distinct fluorescent enrichment at the cell perimeter consistent with surface membrane expression in contrast to the uniform cytosolic expression of ECFP-CaM and ECFP-CaM1234 (Figure 6A).
Figure 6.
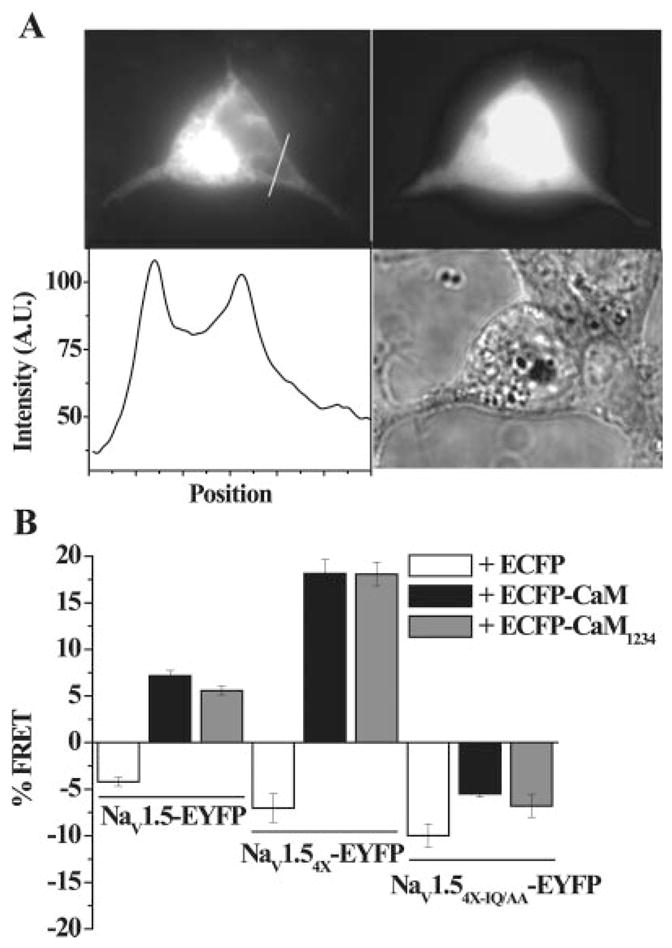
Physical association of CaM and CaM1234 with NaV1.5 variants. A, Epifluorescence image of NaV1.54X-EYFP showing expression of the tagged channel protein (top left panel) and the bright field image of the same cell (bottom right panel). The line scan intensity plot shows channel expression in the cell interior as well as at the perimeter indicating membrane targeting (bottom left panel). To avoid contamination of the FRET signal from cytoplasmic EYFP, we measured FRET by focusing on a region of interest (ROI) only at the membrane (see the online data supplement). In contrast to channel expression, ECFP-CaM is expressed more homogenously in the cytosol (top right). Only cells expressing both fluorophores were chosen for donor dequenching FRET. B, Donor dequenching FRET in HEK293 cells expressing the indicated constructs. The NaV1.54X-EYFP coexpressed with ECFP-CaM or ECFP-CaM1234 yielded significantly more FRET compared to NaV1.5.
The FRET efficiency (percentage) using donor dequenching was computed as previously described.10 A mean FRET efficiency of 7.2±0.4% (Figure 6B) was observed when ECFP-CaM was coexpressed with EYFP-tagged wild-type NaV1.5 channels, indicating that the fluorophores in NaV1.5-EYFP and ECFP-CaM were separated by less than 100 Å. Mutant CaM1234 also associated with the resting channel and exhibited a mean FRET efficiency 5.6±0.5% (Figure 6B) when ECFP-CaM1234 was coexpressed with EYFP-tagged NaV1.5 channels. These data support a physical association between CaM/CaM1234 and NaV1.5, with no apparent effects of CaM overexpression on channel gating in physiological [Ca2+]i.
We then assessed the direct CaM interaction with NaV1.54X channels to study the role of the IQ motif in CaM-mediated regulation of gating of the EFL mutant channel (Figure 4E). The NaV1.54X-EYFP interaction with ECFP-CaM and ECFP-CaM1234 assessed by donor dequenching FRET showed enhanced mean FRET efficiency between NaV1.54X-EYFP and both donors, ECFP-CaM (18.1±1.5%), and ECFP-CaM1234 (18.0±1.3%; Figure 6B). Coexpression of free cytosolic ECFP with NaV1.5-EYFP or NaV1.54X-EYFP and, co expression of ECFP, ECFP-CaM or ECFP-CaM1234 with mutant NaV1.54XIQ/AA-EYFP does not yield FRET (Figure 6B). The absence of FRET between the NaV1.5or NaV1.54X and free cytosolic ECFP, excludes possibility of FRET because of generalized enrichment of CaM/CaM1234 at the cell membrane, and nonspecific binding of CaM to the channel. In addition, the absence of FRET between the EFL and IQ compound mutant NaV1.54X-IQ/AA indicates a specific IQ-CaM/CaM1234 interaction in the CT of NaV1.5. Thus, the enhanced interaction between NaV1.54X channels and CaM/CaM1234 compared to NaV1.5 channels is consistent with unmasking of the latent regulation of NaV1.54X by CaM/CaM1234.
Gating Defects in an LQT3 EFL Mutant Channel
A clinically relevant defect in Ca2+-mediated regulation of NaV1.5 was explored by studying an LQT3 mutation in the EFL, D1790G.15 NaV1.5D1790G expresses robust Na-selective current; the IV relationship (Figure 7A and 7B) and activation voltage dependence are insensitive to changes in [Ca2+]i (Figure 7C). Similar to NaV1.5 4x, NaV1.5D1790G significantly shifts V1/2 of inactivation by ≈15 mV in the hyperpolarizing direction compared to NaV1.5, an effect that was unperturbed by changes in [Ca2+]i (Figure 7C). Changes in [Ca2+]i do not alter recovery from (Figure 7D) or entry into inactivation (Figure 7E and Table). However, D1790G significantly enhances entry into inactivation compared to wild-type channel in high [Ca2+]i (Figure 7E and Table).
Figure 7.

Ca2+/CaM regulation of the LQT3 mutant NaV1.5D1790G. A, Representative families of NaV1.5D1790G currents in presence and absence of intracellular Ca2+. Changes in [Ca2+]i do not alter the normalized IV relationships (B), the activation curves (dotted lines in panel C) or the steady-state inactivation (solid lines in C) of NaV1.5D1790G. Similar to NaV1.54X, the V1/2 of steady-state inactivation of NaV1.5D1790G is significantly shifted in the hyperpolarizing direction compared to NaV1.5. Increasing [Ca2+]i does not alter τrec (D) or τentry (E) into inactivated states compared with the Ca2+-free condition. In 0.5 μmol/L [Ca2+]i, neither CaM nor CaM1234 overexpression alters currents (F) or the IV relationship (G) of NaV1.5D1790G. H, CaM significantly shifts V1/2 of inactivation in hyperpolarizing direction, whereas CaM1234 shifts V1/2 in depolarizing direction compared with NaV1.5D1790G expressed alone. The symbols are the same in panels B–E and, panels G and H.
We tested NaV1.5D1790G for CaM/CaM1234 regulation. Similar to NaV1.54X, the current kinetics and the IV relationship were not affected by overexpression of CaM or CaM1234 (Figure 7F and 7G), but CaM significantly shifts the channel availability in the hyperpolarizing direction in physiological 0.5 μmol/L [Ca2+]i (Figure 7H). Conversely, coexpression of CaM1234 significantly shifts the V1/2 in the depolarizing direction (Figure 7H), and neither CaM nor CaM1234 alters the recovery from or entry into inactivation (Table) of NaV1.5D1790G. Changes in [Ca2+]i have no significant effect on the persistent current of NaV1.5D1790G channel (supplemental Figure II). This single point mutation in the EFL eliminates Ca2+ sensitivity of the mutant channel and unmasks a CaM-mediated effect on the voltage dependence of steady-state inactivation similar to NaV1.54X.
Discussion
Our study demonstrates the capacity for regulation of cardiac Na channels by the Ca2+ regulatory protein CaM, through an IQ motif-CaM interaction at physiological [Ca2+]i. Surprisingly, low [Ca2+]i or mutations in a predicted EFL, upstream of the IQ motif, unmask latent regulation of the NaV1.5 channel by CaM. Furthermore, NaV1.5 is directly and manifestly influenced by [Ca2+]i through the EFL independent of the CaM-IQ interaction. Thus, 2 processes of Ca2+-mediated regulation of NaV1.5 gating exist8 and could be interdependent, particularly when EFL-mediated Ca2+ sensing is compromised by mutations (eg, NaV1.5D1790G or NaV1.54X) or extremely low local [Ca2+]i.
Latent CaM Regulation of Nav1.5 Is Through the IQ Motif
The possibility of latent regulation of NaV1.5 is based on the physical association of CaM/CaM1234 with the wild-type channel CT in vitro6 and in the intact channel in live cells (Figure 6B). Mutations in the EFL shift inactivation in the hyperpolarizing direction (Figure 1D and 7C) simulating low [Ca2+]i, which is in part counteracted by CaM and CaM1234 in the Ca2+-free solution (Figure 4A) and CaM1234 in 0.5 μmol/L Ca2+ (Figure 4B). These data are consistent with an interaction between the EFL and IQ motifs in the CT of NaV1.5. The EFL and IQ motifs are at the N- and C-terminal ends of the proximal structured portion of the CT of NaV1.5, respectively. In the presence of CaM and Ca2+, it is easy to imagine either competitive or synergistic regulation of the channel by these 2 motifs. The existence of manifest regulation of other isoforms of voltage-gated Na channels10 by CaM binding to the IQ suggests the possibility of latent tuning of NaV1.5 channel function by CaM. Our FRET data conclusively demonstrate tethering of CaM to the CT of intact NaV1.5 channels in live cells (Figure 6B) without any apparent effect on channel function at physiological [Ca2+]i, which we interpret as a latent CaM regulation of the wild-type channel. The FRET signal between CaM/CaM1234 and NaV1.5 is more pronounced in the EFL mutant channel (Figure 6B) and is eliminated by the IQ/AA mutation, NaV1.54X-IQ/AA, further supporting the role of the IQ motif in CaM binding and latent and manifest modulation of NaV1.5 inactivation gating. The CaM anti-peptide AIP290–304 abolishes CaM/CaM1234 regulation of inactivation, consistent with CaM or apo-CaM acting on NaV1.54X by binding to the IQ motif.
Remarkably increasing [Ca2+]i shifts the inactivation V1/2 of NaV1.5 in the depolarizing direction, opposite to the effect of CaM on the voltage dependence of NaV1.54X inactivation gating. In addition, mutant CaM1234, which does not bind Ca2+, shifts the NaV1.54X inactivation V1/2 in depolarizing direction in contrast to hyperpolarizing shift of the V1/2 of NaV1.5 observed in the Ca2+-free condition. It is possible that CaM1234 is displacing endogenous CaM from the IQ in NaV1.54X or binding to an alternate binding site(s) accounting for the depolarizing shift in inactivation. The CaM1234-mediated depolarizing shift of NaV1.54X is consistent with the depolarizing shift in inactivation mediated by CaM in NaV1.5 channels in the absence of Ca2+, where CaM may be predominantly in its apo form, similar to CaM1234. In any case, these data confirm the importance of the IQ motif for CaM binding, which could be an evolutionarily conserved process, with concealed modulation of some isoforms and manifest modulation of others (eg, NaV1.4).
Previous studies have suggested that a conformational change in the CT is associated with an interaction involving the III–IV linker that stabilizes the inactivated state of the channel and promotes CaM unbinding from the IQ.4,8 Our current data on NaV1.5 and previous work with NaV1.410 suggest that an interaction between NaV1 channels and CaM is independent of voltage and possibly channel conformation. This suggests that stabilization of inactivation by the interaction of the CT and III–IV is not associated with complete dissociation of CaM from the CT, although we cannot rule out unbinding of a lobe of CaM from the IQ during inactivation.
IQ-Independent Ca2+ Modulation of NaV1.5 Channel Kinetics
The amino acid sequences of the CT of all NaV isoforms, especially the IQ motif regions, are highly homologous, yet regulation of the voltage dependence and kinetics of channel gating mediated through IQ-CaM interactions differ among isoforms.6,7,10,16,17 NaV1.5 channels may be regulated by Ca2+ in 2 ways, acting through the CT of the channel: by direct interaction with the EFL domain and indirectly by binding to CaM and then the IQ domain. Our data show that mutation (NaV1.5IQ/AA) or deletion of the IQ motif of NaV1.5 (NaV1.5Δ1885) does not eliminate the regulation of inactivation gating by intracellular Ca2+. NaV1.5Δ1885 channels exhibited a greater rightward shift of the steady-state inactivation curve in the presence of Ca2+ than NaV1.5 channels; thus, the deletion mutant without the IQ domain remains sensitive to [Ca2+]i. The IQ motif does influence Ca2+-mediated channel modulation, but its absence does not render the channel insensitive to [Ca2+]i, indicating EFL-mediated regulation of NaV1.5 by Ca2+ may be independent of IQ-mediated regulation of the channel (Figures 2C and 3C).
Structural Implications of EFL-IQ–Mediated Inactivation
Inactivation of cardiac Na channels is thought to involve an interaction of the III–IV linker loop and CT.4 Our data suggest removal of Ca2+ exposes negatively charged acidic residues of EFL, leading to conformational modification and disruption of allosteric coupling with other components of the inactivation gating mechanism (eg, III–IV linker loop or inactivation gate receptors formed by IIIS4-S5, IVS4-S5, and IVS6),4 leading to a stabilization of inactivation reflected in the negative shift of fast inactivation of NaV1.5 at low [Ca2+]i (Figure 1D). However, stabilization of channel closure during inactivation is compromised, resulting in an increase in persistent current and may be the result of a change in the interaction with the III–IV linker induced by mutations in EFL. Similar gating alterations are observed with the LQT3 point mutation D1790G, consistent with a congruous disruption of the EFL structure altering the Ca2+-sensing capacity of the EFL loop.18
The EFL and IQ motifs are not immediately contiguous in the linear amino acid sequence but after folding of the CT are predicted to be adjacent.14 In our study, although NaV1.54X is known to disable Ca2+ sensing by the EFL, it is not known whether or not this mutation alters the interaction between the IQ and the EFL. It is possible that CaM access to the IQ domain is enhanced in NaV1.54X compared with wild-type channels, consistent with the enhanced FRET in the mutant (Figure 6B). However, this is speculation and our experiments do not address the structural details of the IQ-EFL interaction.
Conclusion
Ca2+ interaction with the EFL motif plays a critical role in controlling NaV1.5 availability and Ca2+ regulation of gating is independent of the CaM-IQ interaction. Disease-associated mutations in the EFL region of NaV1.5 may be associated with an exaggerated, possibly arrhythmogenic, CaM-induced shift in gating; however, in the intact cardiac Na channel, the concerted action of the 2 motifs finely tune channel inactivation.
Supplementary Material
Acknowledgments
We thank Dr Jeffrey Froehlich for reviewing the manuscript.
Sources of Funding
This work was supported by NIH grant R01HL50411. G.F.T. holds the Michel Mirowski MD Professorship in Cardiology at Johns Hopkins University. S.B. is supported, in part, by a grant from CV Therapeutics.
Footnotes
This manuscript was sent to Harry A. Fozzard, Consulting Editor, for review by expert referees, editorial decision, and final disposition.
Disclosures
None.
References
- 1.Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 2001;104:569–580. doi: 10.1016/s0092-8674(01)00243-4. [DOI] [PubMed] [Google Scholar]
- 2.Deschênes I, Baroudi G, Berthet M, Barde I, Chalvidan T, Denjoy I, Guicheney P, Chahine M. Electrophysiological characterization of SCN5A mutations causing long QT (E1784K) and Brugada (R1512W and R1432G) syndromes. Cardiovasc Res. 2000;46:55–65. doi: 10.1016/s0008-6363(00)00006-7. [DOI] [PubMed] [Google Scholar]
- 3.Deschênes I, Trottier E, Chahine M. Implication of the C-terminal region of the alpha-subunit of voltage-gated sodium channels in fast inactivation. J Membr Biol. 2001;183:103–114. doi: 10.1007/s00232-001-0058-5. [DOI] [PubMed] [Google Scholar]
- 4.Mantegazza M, Yu FH, Catterall WA, Scheuer T. Role of the C-terminal domain in inactivation of brain and cardiac sodium channels. Proc Natl Acad Sci U S A. 2001;98:15348–15353. doi: 10.1073/pnas.211563298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Motoike HK, Liu H, Glaaser IW, Yang AS, Tateyama M, Kass RS. The Na+ channel inactivation gate is a molecular complex: a novel role of the COOH-terminal domain. J Gen Physiol. 2004;123:155–165. doi: 10.1085/jgp.200308929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deschênes I, Neyroud N, DiSilvestre D, Marbán E, Yue DT, Tomaselli GF. Isoform-specific modulation of voltage-gated Na+ channels by cal-modulin. Circ Res. 2002;90:E49–57. doi: 10.1161/01.res.0000012502.92751.e6. [DOI] [PubMed] [Google Scholar]
- 7.Kim J, Ghosh S, Liu H, Tateyama M, Kass RS, Pitt GS. Calmodulin mediates Ca2+ sensitivity of sodium channels. J Biol Chem. 2004;279:45004–45012. doi: 10.1074/jbc.M407286200. [DOI] [PubMed] [Google Scholar]
- 8.Shah VN, Wingo TL, Weiss KL, Williams CK, Balser JR, Chazin WJ. Calcium-dependent regulation of the voltage-gated sodium channel hH1: intrinsic and extrinsic sensors use a common molecular switch. Proc Natl Acad Sci U S A. 2006;103:3592–3597. doi: 10.1073/pnas.0507397103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wingo TL, Shah VN, Anderson ME, Lybrand TP, Chazin WJ, Balser JR. An EF-hand in the sodium channel couples intracellular calcium to cardiac excitability. Nat Struct Mol Biol. 2004;11:219–225. doi: 10.1038/nsmb737. [DOI] [PubMed] [Google Scholar]
- 10.Biswas S, Deschênes I, DiSilvestre D, Tian Y, Halperin VL, Tomaselli GF. Calmodulin regulation of NaV1.4 current: role of binding to the carboxyl terminus. J Gen Physiol. 2008;131:197–209. doi: 10.1085/jgp.200709863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Veldkamp MW, Viswanathan PC, Bezzina C, Baartscheer A, Wilde AA, Balser JR. Two distinct congenital arrhythmias evoked by a multi dysfunctional Na(+) channel. Circ Res. 2000;86:e91–e97. doi: 10.1161/01.res.86.9.e91. [DOI] [PubMed] [Google Scholar]
- 12.Bezzina C, Veldkamp MW, van Den Berg MP, Postma AV, Rook MB, Viersma JW, van Langen IM, Tan-Sindhunata G, Bink-Boelkens MT, van Der Hout AH, Mannens MM, Wilde AA. A single Na(+) channel mutation causing both long-QT and Brugada syndromes. Circ Res. 1999;85:1206–1213. doi: 10.1161/01.res.85.12.1206. [DOI] [PubMed] [Google Scholar]
- 13.Rivolta I, Abriel H, Tateyama M, Liu H, Memmi M, Vardas P, Napolitano C, Priori SG, Kass RS. Inherited Brugada and long QT-3 syndrome mutations of a single residue of the cardiac sodium channel confer distinct channel and clinical phenotypes. J Biol Chem. 2001;276:30623–30630. doi: 10.1074/jbc.M104471200. [DOI] [PubMed] [Google Scholar]
- 14.Cormier JW, Rivolta I, Tateyama M, Yang AS, Kass RS. Secondary structure of the human cardiac Na+ channel C terminus: evidence for a role of helical structures in modulation of channel inactivation. J Biol Chem. 2002;277:9233–9241. doi: 10.1074/jbc.M110204200. [DOI] [PubMed] [Google Scholar]
- 15.An RH, Wang XL, Kerem B, Benhorin J, Medina A, Goldmit M, Kass RS. Novel LQT-3 mutation affects Na+ channel activity through interactions between alpha- and beta1-subunits. Circ Res. 1998;83:141–146. doi: 10.1161/01.res.83.2.141. [DOI] [PubMed] [Google Scholar]
- 16.Herzog RI, Liu C, Waxman SG, Cummins TR. Calmodulin binds to the C terminus of sodium channels Nav1.4 and Nav1.6 and differentially modulates their functional properties. J Neurosci. 2003;23:8261–8270. doi: 10.1523/JNEUROSCI.23-23-08261.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan HL, Kupershmidt S, Zhang R, Stepanovic S, Roden DM, Wilde AA, Anderson ME, Balser JR. A calcium sensor in the sodium channel modulates cardiac excitability. Nature. 2002;415:442–447. doi: 10.1038/415442a. [DOI] [PubMed] [Google Scholar]
- 18.Drake SK, Zimmer MA, Miller CL, Falke JJ. Optimizing the metal binding parameters of an EF-hand-like calcium chelation loop: coordinating side chains play a more important tuning role than chelation loop flexibility. Biochemistry. 1997;36:9917–9926. doi: 10.1021/bi9703913. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.