

Abstract
Sequencing a specific DNA element within a genome or a complex mixture of DNA by the Sanger sequencing method generally involves PCR-mediated amplification of target DNA with forward and reverse primers, followed by a sequencing reaction directed from a single primer. To minimize the contribution of fluorescent signal due to the extension products originating from primers carried over from the amplification step, an intermediate step is routinely incorporated to remove the excess primers before proceeding to the sequencing reaction. We have developed a method called SeqSharp that removes noise in the sequencing data by enzymatically removing chain termination products originating from one or both of the amplification primers. This method substantially improves the quality of sequence information even without an intermediate primer removal step. Importantly, we show that SeqSharp significantly improves the sequence quality from a combined (one-step) amplification/sequencing protocol and provides a more robust method that, unlike previously described one-step sequencing methods, yields high quality sequence data from a single reaction by using equimolar primer concentrations. One-step SeqSharp is generally applicable and produced excellent sequence data from bacterial, fungal, and human DNA.
Despite the recent advent of high-throughput DNA sequencing methods, Sanger's chain termination sequencing method is still widely used for obtaining targeted sequence information of a specific DNA fragment such as a particular gene or part of a gene that can be used for diagnosing an inherited or an infectious disease.1,2 For optimal sensitivity and specificity of the sequencing reaction, the target DNA is generally first amplified by using a pair of specific primers. The amplified product is then sequenced by using a single primer over 25 to 30 temperature cycles with fluorescently labeled dideoxynucleotides and Taq polymerase.1,3,4 A crucial step between amplification and cycle sequencing is the removal of unused primers at the end of the amplification reaction.4 Incomplete removal of primers can lead to noisy sequencing data that are not suitable for making diagnostic decisions. Although there are enzymatic, chromatographic, and ultrafiltrational methods for removing excess primers,5,6 these methods are not always adequate to reduce the noise caused by the extension products from the oppositely oriented primer carried over from the amplification step.
Several attempts have also been made to develop protocols that would combine amplification and sequencing steps and bypass the purification of PCR products, eg, combined amplification and sequencing,7 direct exponential amplification and sequencing,8 and Ampliseq.9 To differentiate signal from noise, combined amplification and sequencing and direct exponential amplification and sequencing used radio-labeled or fluorescently labeled sequencing primers. These methods rely on four separate reactions for specific chain termination by unlabeled dideoxynucleotides. Ampliseq uses the BigDye (Applied Biosystems, Foster City, CA) cycle sequencing kit and requires deoxynucleotide triphosphates (dNTPs) as the only additional reagent and accomplishes amplification and sequencing in a single reaction. This method, however, relies on carefully optimizing unequal forward and reverse primer concentrations to achieve improved signal to noise ratio.
Here we describe a novel method called SeqSharp that can be generally used to remove chain termination products from a specific primer after the sequencing reaction is completed. Because this method does not require any intermediate PCR product purification step, we reasoned that this method would be particularly useful for removing unwanted chain termination products after a one-step process combining both amplification and sequencing. Indeed, SeqSharp substantially improved sequencing data for both regular amplification followed by cycle sequencing, and one-step amplification/cycle sequencing. This method provides superior quality sequencing data by using inexpensive and readily available reagents.
Materials and Methods
Bacterial and fungal genomic DNA were isolated from single colonies by using the Ultraclean DNA extraction kit (MoBio Lab, Inc., Carlsbad, CA) following the procedure recommended by the manufacturer. All primers, phosphorylated and unphosphorylated, were purchased from Invitrogen (Carlsbad, CA). Total human DNA was extracted from a bronchoalveolar lavage specimen by using the QiaAmp Midi kit (Qiagen, Valencia, CA) following the manufacturer's recommendations.
SeqSharp for Sequencing PCR Amplified DNA Fragment
PCR for amplifying the first 500 bp of 16S of bacterial ribosomal gene was performed in 50 μl with 1X AmpliTaq Buffer, 3 mmol/L MgCl2, 200 μmol/L dNTPs, 1 μmol/L primers (5′OH phosphorylated 16SF and 16SR), 1 to 10 ng of bacterial genomic DNA, and 1.25 U of Amplitaq (Applied Biosystems). The sequence of the primers, 16SF and 16SR, were 5′-GAGTTTGATCCTGGCTCAG-3′ and 5′-TTACCGCGGCTGCTGGCA-3′, respectively. The thermal cycler conditions for the amplification was initial denaturation at 95°C for 10 minutes, 30 cycles of 95°C for 30 seconds, 68°C for 30 seconds, 72°C for 45 seconds, and a final extension at 72°C for 10 minutes. Amplification of the 16S gene fragment was confirmed by agarose gel electrophoresis, and 2 μl of the PCR product was used in a 10-μl sequencing reaction using ABI BigDye (version 3.1) reagent (Applied Biosystems). Final concentration of primers (unphosphorylated 16SF or 16SR) in the sequencing reaction was 1 μmol/L. The thermal cycler conditions for cycle sequencing were 25 cycles of 96°C at 10 seconds, 50°C at 5 seconds, and 60°C at 4 minutes. After the sequencing reaction was completed, we treated the products with 2.5 U of lambda exonuclease (New England Biolabs, Ipswich, MA) at 37°C for 30 minutes. BigDye reaction products were purified by using the X-terminator kit (Applied Biosystems) before loading onto Applied Biosystems' 3130 Genetic Analyzer.
SeqSharp for Combined Amplification/Sequencing
A DNA element within the protein A gene (spa) of Staphylococcus aureus was sequenced by combined amplification/sequencing in a 10-μl reaction using ABI BigDye (version 3.1) reagent (Applied Biosystems). The amplification/sequencing reaction for the forward strand contained 1 μmol/L primer 1095F and 0.1 μmol/L 5′-OH phosphorylated primer 1517R.10 Amplification/sequencing of the spa reverse strand was accomplished by using 1 μmol/L 1517R and 0.1 μmol/L 5′ OH phosphorylated 1095F. Extra dNTPs to a final concentration of 125 μmol/L was added to favor amplification over chain termination during initial temperature cycles.11 S. aureus genomic DNA (∼10 ng) was added to the reaction mix. The amplification/sequencing was performed with the following cycling parameters: initial denaturation at 94°C for 30 seconds, followed by 40 cycles consisting of 95°C for 5 seconds, 50°C for 20 seconds, and 60°C for 4 minutes, followed by a hold at 4°C. The single-step amplification/sequencing products were treated with 2.5 U of lambda exonuclease (New England Biolabs) for 30 minutes at 37°C and purified with X-terminator kit (Applied Biosystems) before loading on to Applied Biosystems' 3130 Genetic Analyzer as described for 16S sequencing. For sequencing 16S ribosomal RNA (rRNA) gene, 16SF and 16SR primers were used instead of spa primers. When deoxyinosine triphosphate (dITP) was used, the final dNTP concentrations for deoxyadenosine triphosphate (dATP), deoxycytidine triphosphate (dCTP), deoxythymidine triphosphate (dTTP), dITP, and deoxyguanosine triphosphate (dGTP) were 125 μmol/L, 125 μmol/L, 125 μmol/L, 100 μmol/L, and 25 μmol/L, respectively. When using equimolar concentrations of primers for one-step combined amplification and sequencing, 1 μmol/L final concentration of each primer was used.
SeqSharp for Fungal 26S rRNA Gene and Human β-Globin Gene
Primers NL1 and NL4 were used to sequence the D1 and D2 region within the 26S rRNA gene of Candida famata.12 Bg1 (5′-GGGCTGGGCATAAAAGTCA-3′) and Bg2 (5′-AATAGACCAATAGGCAGA-3′) primers were used to sequence human β-globin gene from total DNA extracted from a bronchoalveolar lavage sample. Both phosphorylated and unphosphorylated primers were at 1 μmol/L concentration (equimolar), and dNTP mix containing dITP were added.
Sequencing by Conventional Methods
Sequencing of PCR amplified DNA fragment (16S gene) and one-step combined amplification and sequencing (spa repeats) were done following an identical procedure as described above for conventional and one-step SeqSharp, except that for conventional methods unphosphorylated primers were used and the sequencing reaction products were not subjected to lambda exonuclease digestion.
Sequence Visualization and Comparison
Applied Biosystems' sequence scanner version 1.2 was used to visualize electropherograms. Vertical colored bars on top of bases indicate the confidence in base-calling by the Applied Biosystems' KB basecaller (QV). The height of the bar is relatively proportional to the score. The blue bar indicates a QV > 20 or <1% predicted error rate for the basecall at that position. Yellow and red bars represent QV < 20.
Results
Presence of primers carried over from the amplification step leads to extension products from both forward and reverse primers (Figure 1A, upper panel). We hypothesized that specifically removing the fluorescent chain termination products originating from the excess amplification primers after the sequencing step would reduce noise in the sequence. Based on this hypothesis, we developed a novel method called SeqSharp that substantially improves the quality of cycle-sequencing data by using phosphorylated primers whose extension products are enzymatically degraded by lambda exonuclease before capillary gel electrophoresis (Figure 1A, lower panel).
Figure 1.

SeqSharp improves sequence quality from a two-step sequencing reaction. A: Schematic comparison of chain termination products from a two-step sequencing reaction and predicted electropherogram without (upper panel) and with SeqSharp (lower panel). B: Comparison of actual electropherograms obtained for S. aureus 16S rRNA gene sequence without (upper panel) or with SeqSharp (lower panel). The region of 16S sequence shown is between positions 239 and 271 within a 530-bp amplicon. Black arrowheads highlight a stretch of four bases where basecalls were affected within the section of electropherogram shown. Blue bars on top of bases indicate QV > 20.
SeqSharp was used to sequence the PCR amplified product from the first 500 bp of bacterial 16S rRNA gene, a common target for amplification and cycle sequencing for identification of bacteria at the species level.13,14,15 Figure 1B shows a section of the electropherogram of the 16S rRNA gene sequence of S. aureus obtained after amplification followed directly by sequencing (upper panel) and the same procedure with SeqSharp (lower panel). Electropherogram quality was markedly improved by the SeqSharp method resulting in highly reliable basecalls (QV > 20). Indeed, multiple base positions were miscalled by the KB Basecaller software (Applied Biosystems) without SeqSharp (flagged by black triangles). This result clearly indicates that a postsequencing treatment with lambda exonuclease,16,17 an enzyme that processively cleaves the 5′ phosphorylated strand in a duplex, is sufficient to remove background signals due to the extension of the oppositely oriented phosphorylated amplification primer, which thereby increases the reliability of basecalls.
Recently a method called Ampliseq has been described for combined single-step amplification and sequencing.9,11 In this method, two primers in unequal concentrations (at 5:1 or 10:1 ratio) are added to BigDye reaction mix along with genomic DNA. Additionally, extra dNTPs are added to help in the amplification process. During the initial PCR cycles, amplification of the template is favored because the concentration of chain terminators is not sufficient to cause appreciable termination in the presence of added extra dNTPs. However, as amplification consumes deoxynucleotides, chain termination by dideoxynucleotides dominates over amplification in later cycles.9 This process leads to a dominance of fluorescence signals in the electropherogram by the primer that was present in higher concentration.
We tested the Ampliseq method to determine the sequence of the repeated DNA sequence element within the S. aureus protein A (spa) gene.10 Because this sequence information is used to distinguish S. aureus strains isolated during hospital outbreaks, it is very important to have high quality sequence information for spa repeats. A single nucleotide difference can potentially change the final conclusion and misdirect an epidemiological investigation. Although the Ampliseq method seemed to yield good quality sequence for some S. aureus strains, in our hands it failed for many other strains under identical conditions (data not shown). It is possible that this method is sensitive to the concentration of starting template or other factors such as presence of PCR inhibitors that can otherwise affect a PCR reaction. Because no intermediate step for removal of unused primers can be used in a one-step combined amplification/sequencing protocol, we predicted that a postsequencing step that specifically removes noise produced by the opposite strand could significantly improve the outcome. On the basis of our results with 16S rRNA gene sequencing by SeqSharp described above, we modified the Ampliseq protocol by using a 5′OH-phosphorylated version of the primer that is present at the lower concentration (minor primer). We hypothesized that a postsequencing treatment with lambda exonuclease could improve the sequence quality by specifically cleaving the extension products originating from the minor primer (Figure 2A, upper versus lower panel). Indeed, when a spa repeat element of ∼350-bp length was sequenced by a combined amplification/sequencing protocol that also incorporated SeqSharp, the quality of the electropherogram obtained was markedly improved (Figure 2B, upper versus lower panel). Without SeqSharp, the predicted error rates for basecalls at almost all base positions were higher with multiple actual wrong basecalls (flagged by black triangles; Figure 2B, upper panel) compared with lower predicted error rates and correct basecalls when SeqSharp was incorporated (Figure 2B, lower panel). These results indicate that SeqSharp can be valuable for improving the quality of data obtained by a one-step combined amplification/sequencing protocol.
Figure 2.
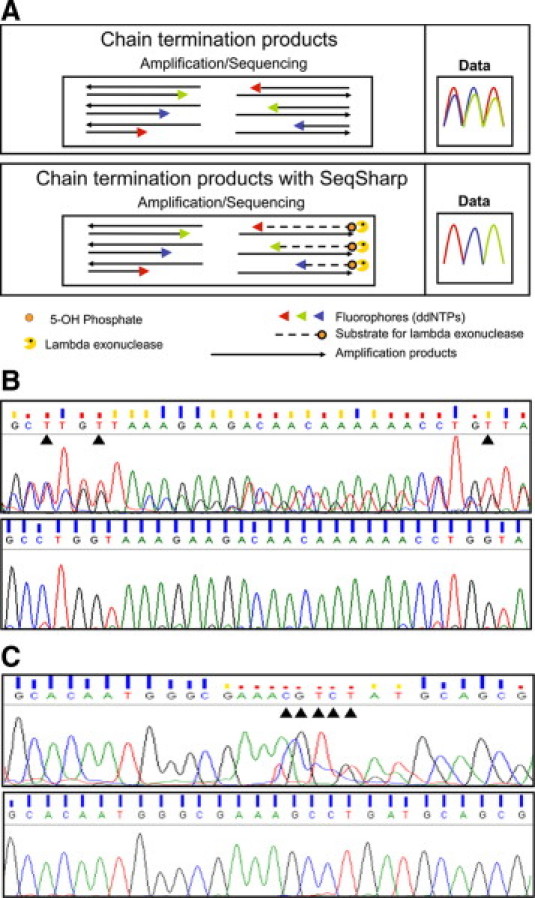
SeqSharp improves sequence quality from a one-step sequencing reaction. A: Schematic comparison of chain termination products from a one-step sequencing reaction and predicted electropherogram without (upper panel) and with SeqSharp (lower panel). B: Comparison of actual electropherograms obtained for spa sequence without (upper panel) or with SeqSharp (lower panel). The region of spa sequence shown is between positions 184 and 214 within a 367-bp amplicon. Black arrowheads highlight base positions where basecalls were wrong. C: One-step SeqSharp of N. farcinica 16S rRNA gene using dNTPs without dITP (upper panel) and with dITP (lower panel). The region of 16S sequence shown is between positions 354 and 381 within a 500-bp amplicon. Incorrect basecalls are highlighted by Black arrowheads. Blue bars on top of bases indicate QV > 20.
Most methods used to purify PCR products from primers also eliminate excess nucleotides. Removal of excess nucleotides does not seem to be crucial for chain termination, but the presence of excess dGTP in a one-step sequencing reaction such as one-step SeqSharp or Ampliseq could lead to incorporation of dG in the chain termination products. The presence of dG in the chain termination product can lead to an aberrant capillary electrophoretic mobility called band-compression, and this can be avoided by including a 4:1 ratio of dITP to dGTP in the assay.18 A sequence motif within the 16S sequence of acid fast bacilli such as Nocardia farcinica resulted in aberrant mobility and incorrect base calls when sequenced by one-step SeqSharp (Figure 2C, upper panel). In contrast, including dITP dramatically improved the electrophoretic mobility (Figure 2C, lower panel) and consequently the confidence in basecalls, and we conclude that adding dITP to the dNTP mix improves the fidelity of sequencing dG containing motifs in a one-step SeqSharp method.
One of the requirements for one-step amplification and cycle sequencing using methods published so far, such as Ampliseq, is the use of unequal forward and reverse primer concentration.11 This is to ensure that signal due to extension of one of the primers (sequencing primer) predominates the fluorescent signals compared with the other primer (opposing primer). However, an equal primer concentration is optimal for the amplification step and may be crucial when attempting to amplify template DNA in low concentrations or from complex biological samples. We hypothesized that the SeqSharp strategy would provide high quality sequencing data from a one-step sequencing protocol even when the opposing primer was present at concentration equal to the sequencing primer. Figure 3A (upper and lower panels) shows a section of forward and reverse sequences we obtained for the16S rRNA gene from N. farcinica by using equimolar primers in a one-step SeqSharp reaction. The high confidence in base calls without any interfering background clearly indicates that unequal primer concentrations are not crucial for a successful one-step SeqSharp sequencing reaction.
Figure 3.

One-step sequencing by SeqSharp using equimolar primer concentrations of forward and reverse primers. A: Complimentary strand sequences between positions 319 and 362 within a 500-bp amplicon from the N. farcinica 16S rRNA gene. B: Complimentary strand sequences between positions 360 and 405 within a 612-bp amplicon from the C. famata 26S rRNA gene. C: Complimentary strand sequences between positions 174 and 222 within a 310-bp amplicon from the human β-globin gene. Upper and lower panels show forward and reverse strands, respectively. Blue bars on top of bases indicate QV > 20.
In order for the one-step SeqSharp to be useful for a wider array of applications, it was important to investigate if eukaryotic genes could be reliably sequenced by this method. Figure 3, B and C, shows sections of electropherograms corresponding to the forward (upper panel) and reverse strand (lower panel) sequences obtained from one-step SeqSharp sequencing of a 500-bp fragment of 26S rRNA gene from C. famata and a 300-bp fragment of the human β-globin gene, respectively. Electropherograms obtained for both of these eukaryotic genes using the one-step SeqSharp were similar in quality to bacterial genes such as 16S rRNA or protein A (spa; Figures 1B and 2B). This indicates SeqSharp provides a robust one-step DNA sequencing protocol that can provide direct sequencing data of single copy human genes.
Discussion
Cycle sequencing from PCR products is a fast and convenient method with a variety of practical applications. At present there are many methods to reduce noise due to carry over primers. These methods assume excess primers are present in free, single-stranded form and can be easily purified or digested away from a PCR product that is double-stranded and substantially higher in molecular weight. However, primers can potentially form both inter- and intramolecular structures, depending on the primer sequences and salt concentrations, which could significantly affect the ability to separate primers from the PCR products using these methods. We have shown that the background noise in the sequencing data from the amplification primers can be easily and efficiently removed by SeqSharp, which entails use of 5′ phosphorylated primers for amplification and nonphosphorylated primers for sequencing and a brief incubation of sequencing reaction products with lambda exonuclease. Lambda exonuclease has been shown to processively cleave the 5′ phosphorylated strand in duplex DNA16 and has been used previously to produce specific single strands from double-stranded DNA.17 This suggests that a significant portion of the extension products from the phosphorylated primer remain hybridized to the complementary strand at the end of the cycle sequencing, and are therefore efficiently cleaved by lambda exonuclease, resulting in an improvement in the quality of sequencing data by SeqSharp.
Sequence quality obtained from a single-step protocol such as Ampliseq is dependent on successful transition from an amplification phase to a sequencing phase in the same reaction mixture. Efficiency of PCR amplification is known to be sensitive to a variety of factors such as template purity, template concentration, and well to well variation within a single PCR run. These variations can prove to be a major impediment to the high-throughput use of a one-step sequencing protocol especially at nonequimolar primer concentrations. Our results indicate selectively digesting the 5′ phosphorylated strands using SeqSharp significantly improves the quality of data generated by a one-step protocol even when both phosphorylated and unphosphorylated primers were present in equimolar concentrations. Using an equimolar primer concentration, SeqSharp lessens the burden of optimizing primer concentration ratios. Our results also indicate that partial substitution of dGTP with dITP improves sequence quality in a one-step sequencing protocol that relies on extra dNTPs to drive amplification. We have used the one-step SeqSharp for identifying more than 500 bacterial isolates belonging to many different genera with consistently good quality sequences comparable with those obtained by conventional cycle sequencing method. We also noted that doubling the concentration of dITP and dGTP to 200 μmol/L and 50 μmol/L had beneficial effect on the sequence quality (data not shown). Furthermore, using equimolar SeqSharp, we successfully sequenced 16S rRNA gene directly from bacterial colonies (data not shown), indicating that high template concentration or purity is not crucial for one-step SeqSharp. Thus the one-step SeqSharp sequencing procedure is both robust and highly user-friendly.
We tested the ability of lambda exonuclease to remove chain terminating products originating from phosphorylated primers in reactions incubated from 0 to 1 hour, and found that 30 minutes was optimal for sequences from 300 to 700 bp in length (data not shown). Incubation for <10 minutes did not show significant improvement in sequence quality especially at the 3′ ends.
In terms of ease of use (reagent addition with incubation), lambda exonuclease treatment in SeqSharp is most similar to digestion of single-stranded primers by exonuclease I, an enzyme widely used for removing excess primers after amplification.19 Our data did not directly compare SeqSharp to commercial products such as ExoSAP-IT (USB Corporation, Cleveland, OH), which contains exonuclease I. Because single-stranded DNA is needed at 37°C to serve as a substrate for exonuclease I, it will be interesting to compare data for these two methods especially with primers that have a propensity for forming homo- or hetero-dimers at lower temperatures.
The SeqSharp protocol for one-step sequencing is effective for targets in microbial and human genomes. Using the one-step protocol, we have successfully sequenced targets that ranged between 300 and 700 bp in length. SeqSharp employs readily available reagents such as phosphorylated primers and lambda exonuclease. Given that DNA sequencing is one of the crucial steps in molecular biological research and diagnostic applications, our technique offers an alternative to the current methods used to remove excess amplification primers. At current market rates, our method will cost only 10 to 15 cents per sequencing reaction to remove background fluorescent signals. Additionally, for laboratories that are interested in further cost and time saving by using a combined amplification and sequencing protocol, our method can help achieve high quality sequence results while substantially reducing personnel and reagent costs.
Acknowledgements
We thank Tia Meuret and Daniel Hoogestraat for excellent technical assistance.
Footnotes
A patent application for the SeqSharp method is pending.
Contributor Information
Dhruba J. SenGupta, Email: dsengup@u.washington.edu.
Brad T. Cookson, Email: cookson@u.washington.edu.
References
- 1.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.ten Bosch JR, Grody WW. Keeping up with the next generation: massively parallel sequencing in clinical diagnostics. J Mol Diagn. 2008;10:484–492. doi: 10.2353/jmoldx.2008.080027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Innis MS, Myambo KB, Gelfand DH, Brown MA. DNA sequencing with Thermus acquaticus DNA polymerase and direct sequencing of polymerase chain reaction-amplified DNA. 1988. Biotechnology. 1992;24:6–10. [PubMed] [Google Scholar]
- 4.Rapley R, editor. PCR Sequencing Protocols: Methods in Molecular Biology. vol 65. Humana Press; New York: 1996. [Google Scholar]
- 5.Dugan KA, Lawrence HS, Hares DR, Fisher CL, Budowle B. An improved method for post-PCR purification for mtDNA sequence analysis. J Forensic Sci. 2002;47:811–818. [PubMed] [Google Scholar]
- 6.Smith PJ, Ballantyne J. Simplified low-copy-number DNA analysis by post-PCR purification. J Forensic Sci. 2007;52:820–829. doi: 10.1111/j.1556-4029.2007.00470.x. [DOI] [PubMed] [Google Scholar]
- 7.Ruano G, Kidd KK. Coupled amplification and sequencing of genomic DNA. Proc Natl Acad Sci USA. 1991;88:2815–2819. doi: 10.1073/pnas.88.7.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kilger C, Paabo S. Direct exponential amplification and sequencing (DEXAS) of genomic DNA. Biol Chem. 1997;378:99–105. doi: 10.1515/bchm.1997.378.2.99. [DOI] [PubMed] [Google Scholar]
- 9.Murphy KM, Eshleman JR. Simultaneous sequencing of multiple polymerase chain reaction products and combined polymerase chain reaction with cycle sequencing in single reactions. Am J Pathol. 2002;161:27–33. doi: 10.1016/S0002-9440(10)64153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shopsin B, Gomez M, Montgomery SO, Smith DH, Waddington M, Dodge DE, Bost DA, Riehman M, Naidich S, Kreiswirth BN. Evaluation of protein A gene polymorphic region DNA sequencing for typing of Staphylococcus aureus strains. J Clin Microbiol. 1999;37:3556–3563. doi: 10.1128/jcm.37.11.3556-3563.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murphy KM, Berg KD, Eshleman JR. Sequencing of genomic DNA by combined amplification and cycle sequencing reaction. Clin Chem. 2005;51:35–39. doi: 10.1373/clinchem.2004.039164. [DOI] [PubMed] [Google Scholar]
- 12.Kurtzman CP, Robnett CJ. Identification and phylogeny of ascomycetous yeasts from analysis of nuclear large subunit (26S) ribosomal DNA partial sequences. Antonie Van Leeuwenhoek. 1998;73:331–371. doi: 10.1023/a:1001761008817. [DOI] [PubMed] [Google Scholar]
- 13.Stackebrandt E, Goebel BM. Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriolog. Int J Syst Bacteriology. 1994;44:846–849. [Google Scholar]
- 14.Drancourt M, Bollet C, Carlioz A, Martelin R, Gayral JP, Raoult D. 16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. J Clin Microbiol. 2000;38:3623–3630. doi: 10.1128/jcm.38.10.3623-3630.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clarridge JE., 3rd Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin Microbiol Rev. 2004;17:840–862. doi: 10.1128/CMR.17.4.840-862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mitsis PG, Kwagh JG. Characterization of the interaction of lambda exonuclease with the ends of DNA. Nucleic Acids Res. 1999;27:3057–3063. doi: 10.1093/nar/27.15.3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boissinot K, Huletsky A, Peytavi R, Turcotte S, Veillette V, Boissinot M, Picard FJ, Martel EA, Bergeron MG. Rapid exonuclease digestion of PCR-amplified targets for improved microarray hybridization. Clin Chem. 2007;53:2020–2023. doi: 10.1373/clinchem.2007.091157. [DOI] [PubMed] [Google Scholar]
- 18.Motz M, Paabo S, Kilger C. Improved cycle sequencing of GC-rich templates by a combination of nucleotide analogs. Biotechniques. 2000;29:268–270. doi: 10.2144/00292st01. [DOI] [PubMed] [Google Scholar]
- 19.Werle E, Schneider C, Renner M, Volker M, Fiehn W. Convenient single-step, one tube purification of PCR products for direct sequencing. Nucleic Acids Res. 1994;22:4354–4355. doi: 10.1093/nar/22.20.4354. [DOI] [PMC free article] [PubMed] [Google Scholar]