

Abstract
KRAS mutation testing has become a standard procedure in the management of patients with carcinomas. The most frequently used method for KRAS testing is direct sequencing of PCR products. The development of commercial real-time quantitative PCR kits offers a useful alternative since they are in theory much more sensitive than direct sequencing and they avoid post- PCR handling. We present our experience as a reference center for the study of KRAS mutations, comparing direct sequencing and the use of a commercial real-time quantitative PCR kit, as well as determining the sensitivity of both procedures in clinical practice. The TheraScreen K-RAS Mutation Kit identified mutations in 75 (44%) of the 170 tumors. Three cases were tested positive using TheraScreen K-RAS Mutation Kit and negative by direct sequencing. We then compared the sensitivity of the kit and that of direct sequencing using 74 mutant tumors. The kit was able to detect the presence of a mutation in a 1% dilution of the total DNA in 13.5% of the tumors and, in 84%, KRAS mutation was identified at a dilution of 5%. Sequencing was able to detect KRAS mutations when the mutant DNA represented 10% of the total DNA in 20/74 (27%) of the tumors. When the mutant DNA represented 30% of the total DNA, sequencing could detect mutations in 56/74 (76%).
KRAS mutation testing has become a standard procedure in the management of patients with carcinomas. Patients with colorectal carcinoma (CRC) that carry mutations in KRAS gene do not benefit from the administration of anti-epidermal growth factor receptor (anti-EGFR) monoclonal antibodies, as a primary, secondary or third line treatment.1 Therefore, testing KRAS gene mutations should be taken into account before therapy selection for all patients with CRC in the near future.1 Likewise, there is also an increasing need to study the mutations in the KRAS gene in patients with pulmonary adenocarcinomas, as this is a primary marker of resistance to tyrosine kinase inhibitors of EGFR.2
This necessity is creating some important logistical problems worldwide, since there is not currently a standardized test approved by the U.S. Food and Drug Administration.1 In accordance with a recent review, two CE-marked KRAS mutation test kits currently exist in Europe for diagnostic use: TheraScreen (DxS Ltd.) and KRAS LightMix (by TIB MolBiol).3
At present, the most frequently used method to test KRAS mutation is the direct sequencing of PCR products.1,4,5 This method has two key disadvantages: its low sensitivity (20 to 50%) and the important risk of contamination when handling the products of the PCR reaction.6 The development of commercial real-time quantitative PCR kits may offer a useful alternative since they are in theory much more sensitive than direct sequencing and they avoid the post-PCR handling.7 Interestingly, one of these assays was used in one of the metastatic CRC phase III trial, which led to the approval of panitumumab in patients with wild-type KRAS tumors by the European Medicines Agency.8 Indeed, using this kit in the context of a clinical trial was so successful it has received public recognition in a high-impact journal.9
The development of biomarker-assessing kits opens a new era, not only in CRC, but also in the future approval of other drugs, and/or indications. However, despite this approval of a drug associated to a biomarker which was studied by a specific method, it is noteworthy that the same approach has not been universally accepted in clinical practice.4
In this article we present our experience as a reference center for the study of KRAS mutations, comparing direct sequencing and the use of a commercial real-time PCR kit, as well as determining the true sensitivity of both procedures by serial dilution.
Materials and Methods
As a reference laboratory for the centralized determination of KRAS mutation, we collected 170 formalin-fixed, paraffin embedded (FFPE) tumors from patients diagnosed with metastatic colorectal cancer from 15 hospitals throughout Spain in the context of a clinical trial. Informed consent was obtained from each patient. The material available for two of these tumors was unstained slides, while for the remaining 168 tumors, tissue blocks were available. Of all of the samples analyzed, 21 were endoscopic biopsies and 149 was tissue from surgical resections. Before DNA extraction, representative sections from tissues were stained with H&E and tumors were reviewed by two pathologists (E.G.-G. and F.L.-R.) to assess the percentage of tumor cells; and then, whether there was a relevant amount of extracellular mucin in the tumors (>50% of the tumor) or lymphocyte inflammation (more than 10% of lymphocytes at ×20 magnification). In the samples with a small proportion of tumor tissue, relevant lymphocyte inflammation or extracellular mucin, macrodissection of the tumor from the paraffin block was performed to enrich the final amount of tumor DNA and at the same time to eliminate the nonmutated DNA coming from the nonneoplastic cells, to avoid competition in the amplification of the final PCR product. The macrodissection procedure consisted of melting the paraffin block at 65°C for one hour to liberate the tissue from the surrounding paraffin and then to facilitate the separation of the tumor tissue from the nontumor area with a scalpel, followed by reconstruction in two different paraffin blocks, one with the tumor area, from which DNA was extracted, and a second block with the nonneoplastic tissue. To determine how to make the macrodissection, it was absolutely necessary to have an H&E-stained section alongside which had the tumor area previously marked with a permanent marker by a pathologist.
The DNA extraction was performed in duplicate. Fifteen freshly cut sections of FFPE tissue were collected from each tumor in two separate tubes, each with a thickness up to 15 μm for surgical samples, or 15 to 20 μm for diagnostic biopsies. To prevent cross-contamination, disposable microtome blades were used. Moreover, the microtome was cleaned with bleach (10%) and ethanol (70%) after the sections from each sample were collected, and the technician changed gloves after each cut. Before proceeding to the extraction, tissue sections were deparaffinized using two washes with xylol and a final wash in absolute ethanol. The sections were washed with 1 ml of xylol or ethanol, vortexed and centrifuged at maximum speed for 10 minutes, and the supernatant was then removed. Samples were incubated in 180 μl ATL buffer and 20 μl proteinase K at 56°C with shaking and left overnight to ensure that they were completely lysed. DNA extraction was performed with the QIAamp DNA FFPE Tissue kit and automated on the QIAcube robot (QIAGEN, Valencia, CA) following the manufacturer's instructions. The elution volume was 50 μl to ensure that highly concentrated DNA was obtained. That way, a minimal concentration of 25 ng/μL was obtained.
A small aliquot of DNA (1 μl) was separated for quantification in a NanoDrop spectrophotometer. A260/A280 and A260/A230 ratios were recorded to assess the purity and quality of the extracted DNA, with ratios of A260/A280 >1.8 and A260/A230 ∼2 as reference values to accept the sample. Extracted DNA was stored at −20°C.
A detailed description of the pre-analytic phase of the procedure can be found elsewhere.10
The presence of KRAS mutations was determined by two methods. One method used the TheraScreen K-RAS Mutation Kit (CE-IVD) (DxS Ltd, Manchester, UK), which combines two technologies (Amplification Refractory Mutation System, Astrazeneca, and Scorpions, DxS) to detect the most commonly reported KRAS mutations (G12D, G12A, G12R, G12C, G12S, G12V, and G13D) by real-time quantitative PCR.11,12,13 Allele specific amplification was achieved with the Amplification Refractory Mutation System and Scorpions technology was used as a fluorescent signaling system to detect the PCR products. The analysis was performed in duplicate according to the manufacturer's instructions using an ABI PRISM 7300 (Applied Biosystems Inc, Foster City, CA). In addition, mutation screening of exon 2 of the KRAS gene was performed using PCR and automatic direct sequencing as previously described.14,15 Exon 2 of the KRAS gene was amplified in duplicate and all variants were confirmed by resequencing independent PCR products.
Using all KRAS positive tumors, we assessed the sensitivity of the TheraScreen K-RAS Mutation Kit to detect KRAS mutations in a routine diagnostic laboratory. For this purpose, we selected pairs of mutant and wild-type tumors that had a similar percentage of tumor cells and that were equivalent in terms of the quantity and quality of the DNA extracted. Dilutions for the sensitivity study were performed by mixing DNA extracted from a mutant tumor into DNA extracted from a wild-type tumor. Each mixture contained a final proportion of mutant DNA relative to wild-type DNA of 1% (the limit for detection of the TheraScreen K-RAS Mutation Kit provided by the manufacturer), 2%, 3%, 4%, and 5%. Following the same approach, we tested the sensitivity of PCR and direct sequencing to detect KRAS mutations, taking into account that traditional Sanger sequencing can detect 10 to 25% mutant DNA in a background of wild-type DNA. When the KRAS mutation was detected in the presence of 10% mutant DNA, we also tested a proportion of 5%. On the other hand, in cases where a proportion of 25% was not enough to detect the KRAS mutation, a proportion of 30% was assessed.
Finally, we wanted to determine whether the mutational status of the KRAS gene was associated with any of the histological parameters evaluated (presence of extracellular mucin or lymphocyte inflammation, as defined previously), and if any of these histological characteristics influenced the experimental study of sensitivity. Accordingly, comparisons were made using the parametric χ2 test or the Fisher's exact test depending on the number of occurrences in each category of analysis (Fisher's exact test for cases with less than five occurrences). The level of statistical significance was established at 5%.
Results
To ensure an appropriate ratio of tumor cell DNA to nontumor cell DNA, macrodissection was performed on 107 of the 170 tumors analyzed (63%). All macrodissected tumors were surgical specimens. The median tumor cell percentage was 48% (range, 10 to 80%) for nonmacrodissected samples and 70% (range, 10 to 95%) for macrodissected cases.
After digestion, at least 100 ng/μL of DNA was obtained from 90% of the samples and the 10% of samples that yielded a lower concentration of DNA were all derived from endoscopic biopsies. We obtained sufficient DNA to perform a mutational analysis from all samples (mean values of approximately 8 μg for the biopsies and of 23 μg for the surgically resected material).
KRAS mutations were successfully analyzed in each of the 170 tumors by real-time quantitative PCR. Two of these tumors could not be evaluated by direct sequencing of the PCR product. Although the absorbance ratios were adequate, it is possible that the extracted DNA from these FFPE tumors was fully fragmented, so adequate PCR products were not obtained for direct sequencing. It is tempting to speculate that the product amplified by the kit may be smaller (product size not provided by the manufacturer) than that amplified by conventional PCR (product size of 164 bp according to our protocol14,15). Interestingly, it should be noted that of the two cases that were uninformative by direct sequencing one also performed poorly in the subsequent sensitivity analysis with TheraScreen K-RAS Mutation test kit (5%); interestingly, this tumor was characterized by a relevant lymphocyte inflammation and that probably influenced the sensitivity analysis (see below).
The TheraScreen K-RAS Mutation Kit identified mutations in 75 (44%) of the 170 tumors, an incidence of KRAS mutations in colorectal cancer consistent with previous studies. The frequencies of the different types of KRAS mutations were as follows: 23/75 (30.7%) G12D, 16/75 (21.3%) G12V, 13/75 (17.3%) G13D, 12/75 (16%) G12S, 6/75 (8%) G12C, and 5/75 (6.7%) G12A. We observed a clear prevalence of the G>A transition characteristic of colorectal cancer. No G12R mutations were detected in this study. In another series analyzed in our laboratory, we found a frequency of 5.9% (11/187) for this mutation (data not shown).
Several significant findings were evident when the detection of KRAS mutations with the TheraScreen K-RAS Mutation Kit was compared with those identified by direct sequencing. Three cases were tested positive using TheraScreen K-RAS Mutation Kit and negative by direct sequencing. One of these tumors corresponded to the sample with unstained slides as material available and all three samples had relevant lymphocyte inflammation. For two samples, the sequence analysis was not clear and experience in sequence interpretation was required to interpret the data. One of these two cases had 10% tumor cells. Overall, direct sequencing could have produced a false negative result in 1.8% (3/168) of the tumors analyzed. Direct sequencing did not identify any mutation not targeted by the TheraScreen K-RAS Mutation Kit.
We then compared the sensitivity of the kit and of direct sequencing using the 74 mutant tumors for which adequate DNA quality and quantity was available (Table 1). The kit was able to detect the presence of a KRAS mutation in a 1% dilution of the total DNA in 10/74 (13.5%) of the tumors analyzed and, in most cases (almost 84%), KRAS mutation was identified at a dilution of 5% (Figure 1). However, it is important to note that we could not establish the limit of detection with the kit for 12 cases when mutant DNA relative to wild-type DNA represented ≤5% of the tested DNA. Among these 12 cases, it is noteworthy that one was the tumor that was analyzed from unstained slides; in one case macrodissection was not performed due to an error in the work-flow, and in three cases the contribution of the tumor cells was relatively low (10%, 20%, and 40%), below the median percentage of tumor cells for both macrodissected and nonmacrodissected cases.
Table 1.
Sensitivity of KRAS Mutation Testing: A Comparison of Two Methods Using Serial Dilutions
| % mutant DNA relative to wild-type DNA | TheraScreen KRAS mutation Kit (n = 74) | % mutant DNA relative to wild-type DNA | Direct sequencing (n = 74) |
|---|---|---|---|
| 1% | 10/74 (13.5%) | ≤5% | 14/74 (18.9%) |
| 2% | 19/74 (26%) | 10% | 20/74 (27%) |
| 3% | 31/74 (41.9%) | 20% | 41/74 (55.4%) |
| 4% | 49/74 (66.2%) | 25% | 52/74 (70.3%) |
| 5% | 62/74 (83.8%) | 30% | 56/74 (75.7%) |
| N.D. | 12/74 (16.2%) | N.D. | 18/74 (24.3%) |
N.D., not determined.
Figure 1.
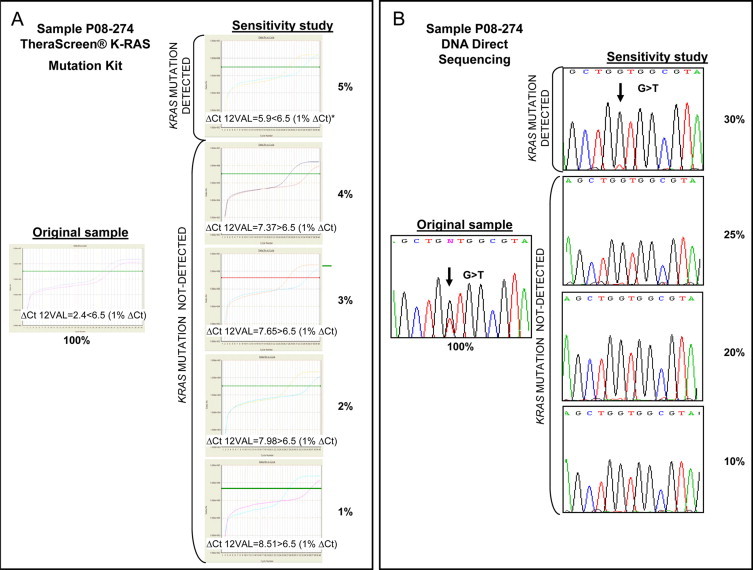
The sensitivity of the TheraScreen K-RAS Mutation Kit in comparison with direct sequencing. Mixtures of DNA from KRAS mutant and wild-type FFPE tumors were used to compare the sensitivity of the two methods. A: For sample P08-274, the TheraScreen K-RAS Mutation Kit was able to detect the KRAS mutation G12V when the DNA from a mutant tumor represented 5% of the total DNA. The 1%ΔCt value is the cut-off level provided by the manufacturer to detect the presence of a G12V KRAS mutation and it is derived from cell lines and synthetic constructs. B: For the same sample, sequencing was unable to detect the KRAS mutation G12V when mutant DNA was present as <30% of the total DNA. Percentages indicate the proportion of DNA from a mutant tumor relative to DNA from a wild-type tumor.
For direct sequencing analysis, we commenced with proportions of mutant DNA relative to wild-type DNA of 10% and 20%, given the general accepted sensitivity of this method (Table 1). Sequencing was able to detect KRAS mutations when the mutant DNA represented 10% of the total tested DNA in 20/74 (27%) of the tumors analyzed. When the mutant DNA represented 20% of the total DNA, sequencing could detect mutations in 41/74 (55.4%) of the clinical samples. Moreover, when the mutant DNA comprised 30% of the total DNA, sequencing could detect mutations in 56/74 (76%) of the clinical samples (Figure 1). We could not establish the sensitivity for direct sequencing when mutant DNA represented ≤30% of the total DNA mixture in 18 cases. Interestingly, 10 of these were also samples that performed poorly when the mutations were detected with the kit.
If we consider the results of the sensitivity study in function of the type of mutation, it is important to note that for detecting the G12A alteration a greater proportion of mutant DNA relative to wild-type DNA was required in both the procedures used. None of the G12A mutations was detected at a dilution <4% of the total DNA with the kit, and at a dilution of <20% of the total DNA with direct sequencing.
Finally, a relevant presence of lymphocytes (Figure 2), as defined in the Materials and Methods, was associated with the presence of mutations in KRAS (P = 0.0207, Table 2). Moreover, we also found evidence that this histological parameter influenced the sensitivity study we performed, significantly diminishing the number of samples with relevant lymphocyte inflammation for which KRAS mutation could be detected at 3% by real-time quantitative PCR (P = 0.0481) or at 20% by direct sequencing (P = 0.0122) (Table 3). The small number of samples may explain the lack of statistical significance (P ≥ 0.05) observed in some categories (1% and 2%, for real-time quantitative PCR, and 5% and 10%, for direct sequencing). The presence of extracellular mucin (Table 3) also appeared to affect the sensitivity of direct sequencing (P = 0.0297 at 30% dilution).
Figure 2.
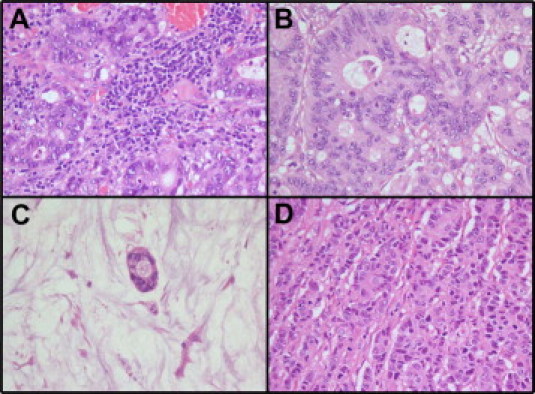
Morphological parameters that might interfere in the sensitivity of the KRAS mutational study. The samples were evaluated to establish the percentage of relevant lymphocytes (>10% in a ×20 field), (A) versus no lymphocytes (B); and relevant extracellular mucin (>50% of the tumor) (C) versus no mucin (D).
Table 2.
Association Between Certain Histological Parameters and KRAS Mutation Status
|
KRAS |
KRAS |
||||
|---|---|---|---|---|---|
| % of extracellular mucin in the tumor | WT (n = 95) | MUT (n = 75) | % of lymphocytes in the tumor | WT (n = 95) | MUT (n = 75) |
| ≤50% | 83 (87.4%) | 57 (76%) | ≤10%* | 14 (14.7%) | 22 (29.3%) |
| >50% | 12 (12.6%) | 18 (24%) | >10%* | 81 (85.3%) | 53 (70.7%) |
| (P = 0.1932) | (P = 0.0207) | ||||
WT: wild-type; MUT: mutant.
Percentage of lymphocyte inflammation was defined in a ×20 field.
Table 3.
Influence of Certain Histological Parameters on Sensitivity when Studied by Serial Dilutions, either Using the TheraScreen KRAS Mutation Kit or by Direct Sequencing
| TheraScreen K-RAS mutation test kit | ||||||
|---|---|---|---|---|---|---|
| % of extracellular mucin in the tumor |
% of lymphocytes in the tumor |
|||||
| Dilution | >50% (n = 13) | ≤50% (n = 61) | P value | >10%* (n = 24) | ≤10%* (n = 50) | P value |
| 1% | 0 | 10 | 0.1928 | 2 | 8 | 0.4836 |
| 2% | 4 | 15 | 0.7292 | 4 | 15 | 0.2664 |
| 3% | 7 | 24 | 0.3683 | 6 | 25 | 0.0481 |
| 4% | 10 | 39 | 0.5227 | 14 | 35 | 0.4316 |
| 5% | 11 | 51 | 1.0000 | 20 | 42 | 1.0000 |
| Direct sequencing | ||||||
|---|---|---|---|---|---|---|
| % of extracellular mucin in the tumor |
% of lymphocytes in the tumor |
|||||
| Dilution | >50% (n = 13) | ≤50% (n = 61) | P value | >10%* (n = 24) | ≤10%* (n = 50) | P value |
| ≤5% | 3 | 11 | 0.7020 | 2 | 12 | 0.1270 |
| 10% | 5 | 15 | 0.3194 | 3 | 17 | 0.0572 |
| 20% | 10 | 31 | 0.1255 | 8 | 33 | 0.0122 |
| 25% | 12 | 40 | 0.0920 | 16 | 36 | 0.7865 |
| 30% | 13 | 43 | 0.0297 | 17 | 39 | 0.5676 |
Percentage of lymphocyte inflammation was defined in a ×20 field. Statistically significant results are shown in bold.
Discussion
The need to know the mutational status of an increasing number of genes (KIT, EGFR, BRAF, KRAS, PI3KCA, etc.) in patients with solid tumors to determine their response to a given treatment is already a clinical reality.16,17,18,19,20 However, despite the clinical importance of these predictive markers of response, there are two issues that must be addressed.
First, there is a lack of standardization and quality control when assaying the mutational status of a gene, especially when compared with the molecular diagnosis of hematological or infectious diseases.21,22,23 For example, 5 years after having shown the importance of determining the mutational status of EGFR in lung cancer, there is still no standardized method to perform the mutational analysis. At present, there are a variety of laboratory developed tests and direct sequencing is still considered the gold standard for gene mutation analysis, despite its low sensitivity. These methods are subject to great inter- and intralaboratory variability and are not always prone to adequate Quality Control schemes that ensure reproducibility of results.24 If we compared the current situation of the standardization of the DNA-based assays with that of fluorescence in situ hybridization, it would be as if we were still using laboratory-developed bacterial artificial chromosome clones to diagnose the amplification of HER2 gene.
Second, there are a large number of methods that could be used to study somatic mutations, but there are few comparative studies and analyses of the sensitivity of these assays in the clinical setting. Paradoxically, given the low sensitivity of direct sequencing, none of these methods has replaced it as the gold standard for the characterization of somatic mutations in tumors.4,8,19,25,26 This is particularly relevant if we bear in mind the events associated with the methods to determine other predictive markers. For example, 20 years after the discovery of HER2 in breast carcinomas, the best way to select the patients that might benefit from trastuzumab therapy is still under debate. Indeed, fluorescence in situ hybridization was recently proposed as the best alternative, considering that immunohistochemistry is not sufficiently reproducible.27,28
On the basis of the above, we proposed to perform a study of KRAS mutations, comparing the true sensitivity of direct sequencing and a commercial kit in a clinical setting. The prevalence of KRAS mutations we found was in agreement with previous studies.1,5 Interestingly, we found an association between relevant inflammatory infiltration and the existence of KRAS mutations, two features that have been etiologically linked in mouse pancreatic ductal adenocarcinoma.29
The experimental sensitivity of the TheraScreen K-RAS Mutation Kit was 5% for the majority of the samples (84%), as long as strict morphological control of the procedure was maintained. As we have demonstrated, this sensitivity is much higher than that obtained by direct sequencing (76% of the mutations were detected when DNA from a mutant tumor reached a proportion of 30% relative to wild-type DNA). In the similar experimental studies that we are aware of, the serial dilution studies were performed using DNA extracted from cell lines.19,30 However, we believe that our procedure is closer to the clinical situation as the sensitivity was tested in a series of mutated tumors performing mixing experiments with DNA from mutant and wild-type tumors.
To the best of our knowledge, there are few studies that have compared different methods to study the mutational status of KRAS. An unpublished analysis that compared five different KRAS tests, four of which are commercially available, concluded that the DxS kit and the Allele-Specific PCR supplied by the Genzyme Corporation yielded the best performance.7 Another poster presentation compared direct sequencing, pyrosequencing and the DxS kit, with the latter showing the highest sensitivity.30 However, despite the higher sensitivity of the TheraScreen K-RAS Mutation Kit, it is important to note one of its limitations. This kit is only able to detect mutations targeted by the designed primers. Although the frequency of mutations not targeted by the TheraScreen K-RAS Mutation Kit is relatively low (1.6% in another series studied in our laboratory, data not shown), the kit would provide a false negative result in these situations. For direct sequencing, the false negative rate identified in a recent publication was very similar to that identified here (1.8%) and, although a study of sensitivity using serial dilutions was not performed, these authors indicated that mutations in KRAS might not be detected if the proportion of tumor tissue is below 30%.31 In our series, the principal reason for the failure of direct sequencing to identify mutants detected with the TheraScreen K-RAS Mutation Kit was the presence of relevant lymphocyte inflammation, followed by difficulties in the postanalytical phase (interpretation). Application of co-amplification-at-lower denaturation-temperature PCR (COLD-PCR) may improve the detection sensitivity of sequencing without additional costs.32
Practically all of the mutational analyses in CRC are performed on paraffin-embedded tissue samples. As such, there are two main types of samples: small biopsies and surgical specimens. Our study demonstrates that it is possible to obtain DNA of sufficient quality to perform mutational studies of the KRAS gene, even when the parameters used to process the samples are unknown. However, it must be recognized that less DNA was recovered from endoscopic biopsies. It is important to note that, although the most popular fixative in most countries is 10% neutral buffered formalin, the results obtained could vary when other fixatives are used, eg, alcoholic fixatives, mercury based fixatives, etc.33
In light of the above, it is evident that a collaborative effort between clinicians and pathologists is critical to ensure the quality of KRAS testing, even when the DNA extraction and the analysis (PCR) are to be performed in another Department. Although important advances in this field are being made with the increasing use of CE marked kits on platforms that should ensure the quality of the analytical and postanalytical phases of the process, we must not forget that the pre-analytical part should be standardized.10
All of the tissue that is to be subjected to mutational analysis must be examined by a pathologist to select the appropriate area from which the DNA should be extracted, and to determine the proportion of tumor cells. As shown, the minimum percentage of mutant DNA should be >30% if direct sequencing is to be used, and >5% when using real-time quantitative PCR. Thus, to ensure sufficient sensitivity of the mutational analysis, macrodissection of the tissue should be performed, a precaution that is not often followed.34 In our series, it was necessary to perform macrodissection in 63% of the samples (107/170), and we must emphasize that this is a procedure that is not time-consuming and that does not interfere with the work-flow of a surgical pathology laboratory. If the sample available is a surgical specimen, the pathologist responsible for selecting the block should choose one that has the greatest percentage of neoplastic cells, avoiding the zones with many lymphocytes or extracellular mucin. All these situations, as well as providing cut slides rather than the original block, can diminish the sensitivity of the technique, above all if direct sequencing is used.34 In fact, all of the cases considered positive with the kit but that were identified as wild-type by direct sequencing had a relevant number of lymphocytes. As we demonstrated in our study of sensitivity, this histological parameter affects the detection of mutations, even when real-time quantitative PCR is used. The significant presence of extracellular mucin can also interfere with the study of KRAS mutations by direct sequencing. This scenario is not relevant for in-house cases, but it could be critical for referral cases or in a clinical trial setting. As such, it is important to specify how the tissue should be selected when designing a clinical trial, not only focusing on the collection of the tissue (which is usually the case).
In summary, we have presented a comparison and a study of the sensitivity of KRAS testing using direct sequencing and an ingenious commercial real-time quantitative PCR platform. The conclusions could also be valid for the study of other mutations in CRC or in other solid tumors (ie, for the mutational study of KRAS in lung adenocarcinomas where specimens are often very small). The publication of consensus guidelines, the determination of the sensitivity and the limitations of the tests and their quality control are all issues that we should consider to be urgent when carrying out mutational studies for predictive oncology. Otherwise, we may have the right drug and the right biomarker, but without the adequate method we will not target the right patient.
Acknowledgements
We thank Rosalind Franklin for her contribution to this work. We also thank the Tumor Bank at the Laboratorio de Dianas Terapéuticas, Hospital Universitario Madrid Sanchinarro, for handling the samples used in this study. We are most grateful to the Sequencing service of the Genomic Unit at the Spanish National Cancer Centre (CNIO) for performing the direct sequencing assays.
Footnotes
Supported by Amgen, (provided the TheraScreen K-RAS Mutation Kits) and DxS Ltd. and Roche Diagnostics (technical assistance).
The ‘Laboratorio de Dianas Terapéuticas’ participates in the European Quality Assurance Programe for KRAS testing organized by the European Society of Pathology.
References
- 1.Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, McAllister PK, Morton RF, Schilsky RL. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol. 2009;27:2091–2096. doi: 10.1200/JCO.2009.21.9170. [DOI] [PubMed] [Google Scholar]
- 2.Riely GJ, Ladanyi M. KRAS mutations: an old oncogene becomes a new predictive biomarker. J Mol Diagn. 2008;10:493–495. doi: 10.2353/jmoldx.2008.080105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Krieken JH, Jung A, Kirchner T, Carneiro F, Seruca R, Bosman FT, Quirke P, Fléjou JF, Plato Hansen T, de Hertogh G, Jares P, Langner C, Hoefler G, Ligtenberg M, Tiniakos D, Tejpar S, Bevilacqua G, Ensari A. KRAS mutation testing for predicting response to anti-EGFR therapy for colorectal carcinoma: proposal for an European quality assurance program. Virchows Arch. 2008;453:417–431. doi: 10.1007/s00428-008-0665-y. [DOI] [PubMed] [Google Scholar]
- 4.Jimeno A, Messersmith WA, Hirsch FR, Franklin WA, Eckhardt SG. KRAS mutations and sensitivity to epidermal growth factor receptor inhibitors in colorectal cancer: practical application of patient selection. J Clin Oncol. 2009;27:1130–1136. doi: 10.1200/JCO.2008.19.8168. [DOI] [PubMed] [Google Scholar]
- 5.Linardou H, Dahabreh IJ, Kanaloupiti D, Siannis F, Bafaloukos D, Kosmidis P, Papadimitriou CA, Murray S. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008;9:962–972. doi: 10.1016/S1470-2045(08)70206-7. [DOI] [PubMed] [Google Scholar]
- 6.Lyon E, Wittwer CT. LightCycler technology in molecular diagnostics. J Mol Diagn. 2009;11:93–101. doi: 10.2353/jmoldx.2009.080094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Juan T, Suggs S, Wolf M, Sarosi I, Freeman D, Oliner K, Bakkar A, Patterson SD: A comparatibility study of 4 commercial KRAS tests. American Association for Cancer Res (AACR) Annual Meeting, April 12–16, 2008, Abstract #1811
- 8.Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, Patterson SD, Chang DD. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 9.Roses AD. Pharmacogenetics in drug discovery and development: a translational perspective. Nat Rev Drug Discov. 2008;7:807–817. doi: 10.1038/nrd2593. [DOI] [PubMed] [Google Scholar]
- 10.García-García E, Angulo B, Martínez R, Suárez-Gauthier A, Conde E, Hidalgo M, López-Ríos F. The pre-analytical phase in the study of KRAS mutations in colorectal carcinoma: a proposal to standardize the pre-PCR procedure through a morphology-based approach. APJOH. 2009;1:1–5. [Google Scholar]
- 11.Clayton SJ, Scott FM, Walker J, Callaghan K, Haque K, Liloglou T, Xinarianos G, Shawcross S, Ceuppens P, Field JK, Fox JC. K-ras point mutation detection in lung cancer: comparison of two approaches to somatic mutation detection using ARMS allele-specific amplification. Clin Chem. 2000;46:1929–1938. [PubMed] [Google Scholar]
- 12.Thelwell N, Millington S, Solinas A, Booth J, Brown T. Mode of action and application of Scorpion primers to mutation detection. Nucleic Acids Res. 2000;28:3752–3761. doi: 10.1093/nar/28.19.3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whitcombe D, Theaker J, Guy SP, Brown T, Little S. Detection of PCR products using self-probing amplicons and fluorescence. Nature Biotechnol. 1999;17:804–807. doi: 10.1038/11751. [DOI] [PubMed] [Google Scholar]
- 14.Conde E, Angulo B, Tang M, Morente M, Torres-Lanzas J, Lopez-Encuentra A, Lopez-Rios F, Sanchez-Cespedes M. Molecular context of the epidermal growth factor receptor mutations: evidence for the activation of mTOR/S6K signaling. Clin Cancer Res. 2006;12:710–717. doi: 10.1158/1078-0432.CCR-05-1362. [DOI] [PubMed] [Google Scholar]
- 15.Fernandez P, Carretero J, Medina PP, Jimenez AI, Rodriguez-Perales S, Paz MF, Cigudosa JC, Esteller M, Lombardia L, Morente M, Sanchez-Verde L, Sotelo T, Sanchez-Cespedes M. Distinctive gene expression of human lung adenocarcinomas carrying LKB1 mutations. Oncogene. 2004;23:5084–5091. doi: 10.1038/sj.onc.1207665. [DOI] [PubMed] [Google Scholar]
- 16.Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, Bardelli A. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26:5705–5712. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- 17.Heinrich MC, Owzar K, Corless CL, Hollis D, Borden EC, Fletcher CD, Ryan CW, von Mehren M, Blanke CD, Rankin C, Benjamin RS, Bramwell VH, Demetri GD, Bertagnolli MM, Fletcher JA. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: cALGB 150105 study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol. 2008;26:5360–5367. doi: 10.1200/JCO.2008.17.4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirsch FR, Bunn PA., Jr EGFR testing in lung cancer is ready for prime time. Lancet Oncol. 2009;10:432–433. doi: 10.1016/S1470-2045(09)70110-X. [DOI] [PubMed] [Google Scholar]
- 19.Lièvre A, Bachet JB, Boige V, Cayre A, Le Corre D, Buc E, Ychou M, Bouché O, Landi B, Louvet C, André T, Bibeau F, Diebold MD, Rougier P, Ducreux M, Tomasic G, Emile JF, Penault-Llorca F, Laurent-Puig P. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008;26:374–379. doi: 10.1200/JCO.2007.12.5906. [DOI] [PubMed] [Google Scholar]
- 20.Sartore-Bianchi A, Martini M, Molinari F, Veronese S, Nichelatti M, Artale S, Di Nicolantonio F, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, Bardelli A. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009;69:1851–1857. doi: 10.1158/0008-5472.CAN-08-2466. [DOI] [PubMed] [Google Scholar]
- 21.Leonard DGB. In: Molecular pathology in clinical practice: infectious diseases. Leonard DGB, editor. Springer Science; New York: 2009. [Google Scholar]
- 22.Leonard DGB. In: Molecular pathology in clinical practice: genetics. Leonard DGB, editor. Springer Science; New York: 2009. [Google Scholar]
- 23.Leonard DGB. In: Molecular pathology in clinical practice: oncology. Leonard DGB, editor. Springer Science; New York: 2009. [Google Scholar]
- 24.Eberhard DA, Giaccone G, Johnson BE. Biomarkers of response to epidermal growth factor receptor inhibitors in Non-Small-Cell Lung Cancer Working Group: standardization for use in the clinical trial setting. J Clin Oncol. 2008;26:983–994. doi: 10.1200/JCO.2007.12.9858. [DOI] [PubMed] [Google Scholar]
- 25.De Roock W, Piessevaux H, De Schutter J, Janssens M, De Hertogh G, Personeni N, Biesmans B, Van Laethem JL, Peeters M, Humblet Y, Van Cutsem E, Tejpar S. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol. 2008;19:508–515. doi: 10.1093/annonc/mdm496. [DOI] [PubMed] [Google Scholar]
- 26.Van Cutsem E, Köhne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D'Haens G, Pintér T, Lim R, Bodoky G, Roh JK, Folprecht G, Ruff P, Stroh C, Tejpar S, Schlichting M, Nippgen J, Rougier P. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408–1417. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- 27.Sauter G, Lee J, Bartlett JM, Slamon DJ, Press MF. Guidelines for human epidermal growth factor receptor 2 testing: biologic and methodologic considerations. J Clin Oncol. 2009;27:1323–1333. doi: 10.1200/JCO.2007.14.8197. [DOI] [PubMed] [Google Scholar]
- 28.Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, Levin WJ, Stuart SG, Udove J, Ullrich A, Press M. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–712. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- 29.Guerra C, Schuhmacher AJ, Cañamero M, Grippo PJ, Verdaguer L, Pérez-Gallego L, Dubus P, Sandgren EP, Barbacid M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by KRAS oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 30.Lamy PJ, Montels F, Servanton AC, Ychou M, Crapez E: Diagnostic of KRAS gene mutations in colorectal cancer: evaluation of direct sequencing, pyrosequencing and allele specific amplification. ECCO 15 and 34th ESMO Multidisciplinary Congress, September 20–24, 2009. Abstract #1322
- 31.Tol J, Dijkstra JR, Vink-Börger ME, Nagtegaal ID, Punt CJ, van Krieken JH, Ligtenberg MJ. High sensitivity of both sequencing and real-time PCR analysis of KRAS mutations in colorectal cancer tissue. J Cell Mol Med. 2009 doi: 10.1111/j.1582-4934.2009.00788.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zuo Z, Chen SS, Chandra PK, Galbincea JM, Soape M, Doan S, Barkoh BA, Koeppen H, Medeiros LJ, Luthra R. Application of COLD-PCR for improved detection of KRAS mutations in clinical samples. Mod Pathol. 2009;22:1023–1031. doi: 10.1038/modpathol.2009.59. [DOI] [PubMed] [Google Scholar]
- 33.Hewitt SM, Lewis FA, Cao Y, Conrad RC, Cronin M, Danenberg KD, Goralski TJ, Langmore JP, Raja RG, Williams PM, Palma JF, Warrington JA. Tissue handling and specimen preparation in surgical pathology: issues concerning the recovery of nucleic acids from formalin-fixed, paraffin-embedded tissue. Arch Pathol Lab Med. 2008;132:1929–1935. doi: 10.5858/132.12.1929. [DOI] [PubMed] [Google Scholar]
- 34.Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, Price TJ, Shepherd L, Au HJ, Langer C, Moore MJ, Zalcberg JR. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–1765. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]