

Abstract
High-resolution melting analysis was applied to X-linked chronic granulomatous disease, a rare disorder resulting from mutations in CYBB. Melting curves of the 13 PCR products bracketing CYBB exons were predicted by Poland's algorithm and compared with observed curves from 96 normal individuals. Primer plates were prepared robotically in batches and dried, greatly simplifying the 3- to 6-hour workflow that included DNA isolation, PCR, melting, and cycle sequencing of any positive products. Small point mutations or insertions/deletions were detected by mixing the hemizygous male DNA with normal male DNA to produce artificial heterozygotes, whereas detection of gross deletions was performed on unmixed samples. Eighteen validation samples and 22 clinical kindreds were analyzed for CYBB mutations. All blinded validation samples were correctly identified. The clinical probands were identified after screening for neutrophil oxidase activity. Nineteen different mutations were found, including seven near intron-exon boundaries predicting splicing defects, five substitutions within exons, three small deletions predicting premature termination, and four gross deletions of multiple exons. Ten novel mutations were found, including (c.) two missense (730T>A, 134T>G), one nonsense (90C>A), four splice site defects (45 + 1G>T, 674 + 4A>G, 1461 + 2delT, and 1462-2A>C), two small deletions (636delT, 1661_1662delCT), and one gross deletion of exons 6 to 8. High-resolution melting can provide timely diagnosis at low cost for effective clinical management of rare, genetic primary immunodeficiency disorders.
Chronic granulomatous disease (CGD) is a rare (1 per 250,000) inherited disorder of phagocytes caused by defects in the respiratory burst NADPH oxidase complex.1,2,3,4 Intracellular bactericidal defects in phagocytes lead to recurrent, severe bacterial and fungal infections of the skin, lungs, visceral organs, and bones. Granuloma formation often occurs at the site of infections.5,6,7 The disorder is typically caused by mutations in four phagocyte oxidase (phox) subunits including a 91-kDa glycoprotein (gp91phox) encoded by CYBB on the X chromosome and three autosomal components, p47phox encoded by NCF1, p67phox encoded by NCF2, and p22phox encoded by CYBA.8 Most cases (65 to 70%) are X-linked and arise from mutations in CYBB, whereas ∼25% arise from mutations in NCF1.
Screening for CGD is usually performed by functional assays such as the neutrophil respiratory burst assay performed by flow cytometry.9 Incubation of normal cells with the nonfluorescent compound dihydrorhodamine-1,2,3 followed by stimulation, results in a 100-fold increase in fluorescence by formation of the oxidized metabolite rhodamine-1,2,3. In classic X-linked CGD, phagocytic cells do not express the mutated protein (X0) and no oxidase activity is detected by flow cytometry.
In addition to classic X0 CGD, variant X-linked CGD has been described.8,10,11,12 X-linked variants occur in both X+ and X− forms.13 Fewer than 20 cases of X+ CGD, in which the neutrophils express normal levels of the mutated protein but lack oxidase activity, have been described. Approximately 5% of X-linked CGD is of the X− form, with underexpression of the mutated protein and either absent or decreased oxidase activity. Decreased oxidase activity is observed as an intermediate population of fluorescent cells by flow cytometry.
Although mutations in CYBA usually result in absent oxidase activity and mutations in NCF1 and NCF2 usually show decreased oxidase activity, different mutations in these autosomal recessive genes may result in either absent or decreased oxidase activity.14 Therefore, it is not possible to distinguish autosomal recessive from X-linked disease by flow cytometry analysis of the patients. However, heterozygous mothers and fathers of patients with autosomal recessive disease will have normal oxidase activity. In contrast, carrier females of X-linked CGD usually have both normal and abnormal cell populations in varying proportions. Even so, some female carriers may selectively inactivate their functional X chromosome, resulting in absent oxidase activity and the disease phenotype. Furthermore, 10 to 20% of X-linked CGD occurs from new spontaneous mutations with no detectable abnormalities in the mother.
Additional limitations to the flow cytometry oxidase assay have been described,15 and correct diagnosis continues to be a challenge.16 Pre-activation of neutrophils by infection, inflammation, sample contamination, or harsh conditions of blood transport or processing can deplete cellular respiratory capacity and confound the assay resulting in intermediate populations of fluorescent cells. Genetic analysis is, thus, critical for diagnostic confirmation, genetic counseling, prenatal diagnosis, and appropriate initiation of prophylactic antimicrobial or interferon-γ therapy, up to and including potential bone marrow transplantation or gene therapy.8,17
More than 500 CYBB mutations have been described with 30% missense, 31% nonsense, 15% deletions (including large deletions of multiple exons), 8% insertions, 13% splice site, and 1% promoter mutations, and new mutations continue to be identified.10,13,18,19,20 CYBB variants can be identified by single-strand conformation polymorphism analysis,21 denaturing high-pressure liquid chromatography,22 or sequencing20 of all 13 exons and adjacent splice sites. Because the disease is rare, few centers offer genetic analysis, and turnaround times are typically long.
High-resolution melting analysis is a new gene scanning method with high sensitivity that promises to decrease the cost and turnaround time of genetic analysis.23,24,25 Because CYBB is on the X chromosome, deletions of one or more exons can also be detected. Furthermore, benign polymorphisms are rare in CYBB, suggesting that high-resolution melting analysis, followed by targeted sequencing only when necessary, may be an ideal method of analysis.
Materials and Methods
Neutrophil Oxidative Burst Assay
The neutrophil respiratory burst assay was performed in the ARUP Cellular and Innate Immunology Laboratory as described previously.9,15 In brief, the red blood cells of heparinized whole blood were lysed, and the white blood cells incubated with phorbol-myristate acetate. After addition of catalase and dihydrorhodamine-1,2,3, the production of fluorescent rhodamine-1,2,3 was measured by flow cytometry. Physicians of the patients with abnormal results were contacted and permission, consent, and assent (depending on the patient's age) were obtained for DNA analysis of the patient and family members if available, according to University of Utah Institutional Review Board Protocols 00018916 and 00029386.
DNA Samples
Three sets of DNA samples were analyzed. A random control DNA panel of 96 Caucasian blood donors from the UK provided by the European Collection of Cell Cultures (HRC1, Sigma-Aldrich, St. Louis, MO) was used to screen for common CYBB polymorphisms.
The second set consisted of 18 blinded DNA samples with established CYBB variants selected from prior studies,10,20 provided by the Sanquin Research Institute (Amsterdam, The Netherlands) to the University of Utah for validation of high-resolution melting analysis of CYBB. These samples were whole genome-amplified (illustra GenomiPhi HY DNA Amplification Kit, GE Healthcare, Piscataway, NJ) as directed by the manufacturer.
The third set consisted of DNA from 22 families (37 samples) obtained after defective oxidative burst activity was established on clinical samples submitted to the Associated Regional and University Pathologists laboratories. DNA was extracted from 400 μl of heparinized whole blood with an automated MagNA Pure Compact using the Nucleic Acid Isolation Kit I (Roche, Indianapolis, IN). Extraction required 30 minutes to complete. Template DNA was quantified by spectroscopy (ND-1000, NanoDrop Technologies, Wilmington, DE) and adjusted to 50 ng/μl in 10 mmol/L Tris (pH 8.0)-0.1 mmol/L EDTA.
CYBB Primer Plates
Primers bracketing the 13 exons of CYBB were designed with LightScanner Primer Design software (Idaho Technology, Salt Lake City, UT) to include the exon flanking regions where splice site mutations are common. The primer sequences and the sizes of the resulting 13 PCR products (179 to 401 bp) are shown in Table 1. Batches of 96-well microtiter plates (black shell, white well plates, HSP-9665, Bio-Rad Laboratories, Hercules, CA) with primers dried in each well for CYBB scanning were robotically prepared by Idaho Technology on a BioMek FX system (Beckman Coulter, Fullerton, CA) by dispensing 5 μl of each combined primer solution into the appropriate wells, air drying at room temperature overnight, and vacuum packaging in foil pouches at 150 torr. The primer plates were stored for up to 8 months without noticeable degradation in performance. The 13 CYBB primer pairs were arranged in six replicates, each replicate occupying two columns of the microtiter plate. Five picomoles of each primer was spotted, so that when reconstituted to 10 μl with PCR Master Mix and DNA, their concentration was 0.5 μmol/L.
Table 1.
Primers for CYBB High-Resolution Melting
| Name | Sequence | Product size (bp) |
|---|---|---|
| Ex1F | 5′-AGAAGCATAGTATAGAAGAAAGGC-3′ | 179 |
| Ex1R | 5′-CCCGAGAAGTCAGAGAATTTATAAC-3′ | |
| Ex2F | 5′-CTACTGTGGAAATGCGGA-3′ | 214 |
| Ex2R | 5′-AGCCAATATTGCATGGGAT-3′ | |
| Ex3F | 5′-GGACAGGGCATATTCTGTG-3′ | 249 |
| Ex3R | 5′-GCCTTTGAAAATTAGAGGAACTTAGTA-3′ | |
| Ex4F | 5′-CTTTCCTGTTAACAATTACTATTCCAT-3′ | 202 |
| Ex4R | 5′-TCCCTGGTTCCAAGTTTTCTTTAATA-3′ | |
| Ex5F | 5′-TCATACCCTTCATTCTCTTTGTTT-3′ | 281 |
| Ex5R | 5′-AGTCCTCAATTGTAATGGCCTA-3′ | |
| Ex6F | 5′-TGTGTGTGTGTGTGTTTATATTTTAC-3′ | 309 |
| Ex6R | 5′-CTGCCTAGAAATTGAGGGAC-3′ | |
| Ex7F | 5′-TTAATTTCCTATTACTAAATGATCTGGACTT-3′ | 255 |
| Ex7R | 5′-TGTCAGTAATGAAACTGTAATAACAAC-3′ | |
| Ex8F | 5′-CCTCTGAATATTTTGTTATCTATTACCACTTA-3′ | 252 |
| Ex8R | 5′-ACTTGTCCATGATATAGTTAGACAC-3′ | |
| Ex9F | 5′-GGCAAGTATTTAGGAAAAATGTCAT-3′ | 401 |
| Ex9R | 5′-GCTATTTAGTGCCATTTTTCCTG-3′ | |
| Ex10F | 5′-GAGCAAGACATCTCTGTAACT-3′ | 276 |
| Ex10R | 5′-CTCTAAGGCCCTCCGAT-3′ | |
| Ex11F | 5′-AGGGCCTGCCAAATATAAT-3′ | 274 |
| Ex11R | 5′-CTGTACACTATGGGAAGGACC-3′ | |
| Ex12F | 5′-GTATGTGCTTTTACAGAATGTCTC-3′ | 260 |
| Ex12R | 5′-GCAGATGCAAGCCTCAA-3′ | |
| Ex13F | 5′-ATCCCAAAGCTTGAAATTGTC-3′ | 251 |
| Ex13R | 5′-CATTTGGCAGCACAACC-3′ |
5′-Tails were appended to each primer to simplify sequencing. Forward primers (F) started with 5′-ACGACGTTGTAAAACGAC-3′ and reverse primers (R) started with 5′-CAGGAAACAGCTATGACC-3′.
PCR
Hemizygous DNA of unknown male samples was mixed 1:1 with wild-type male DNA and amplified both mixed (for base substitutions and small insertions/deletions) and unmixed (for detection of deletions encompassing entire exons). Female DNA was tested without mixing. Because six DNA samples can be analyzed on one screening plate, the maximum capacity was three unknown male samples or six female samples. Typically, samples were analyzed in duplicate, so one unknown male DNA (mixed and unmixed) and one wild-type control were analyzed per plate. Once a mutation was detected, multiple family members were tested together for confirmation, using a PCR Master Mix containing only the primers for the responsible exon.
For initial screening using the primer plates, each well was resuspended to 10 μl with 50 ng of DNA and 1× LightScanner Master Mix (Idaho Technology) including the enzyme, antibody, dNTPs, LCGreen Plus dye, and necessary buffers for amplification. The filled plate was lightly sealed with adhesive film (Nunc 232702, Thermo Fisher Scientific, Rochester, NY) and vortexed (MixMate, Eppendorf, Westbury, NY) for 30 seconds at 1650 rpm. Each well was overlaid with 12 μl of mineral oil (Sigma-Aldrich), the film was sealed securely, and the plate was centrifuged at 2200 × g for 30 seconds (model 5430, Eppendorf).
PCR was performed on a 96-well plate thermocycler (PTC-200, Bio-Rad Laboratories) with an initial denaturation of 95°C for 15 seconds, followed by 40 cycles of 94°C for 10 seconds, 62°C for 10 seconds, and 75°C for 10 seconds, heating to 95°C for 5 seconds, and cooling to 15°C. The optimal annealing temperature across all exons was determined by annealing temperature gradient experiments of each exon, evaluated by both melting analysis and gel electrophoresis. After PCR, the plate was centrifuged again at 2200 × g for 30 seconds, and the adhesive film was removed. PCR required 60 minutes.
Melting Analysis
High-resolution melting was performed on a 96-well LightScanner (Idaho Technology), a melting instrument that resembles the LightTyper26 in physical form with extensive software and hardware changes that enable high resolution melting. The melting/cooling cycle requires only 10 minutes so that many thermal cyclers can feed one LightScanner. Heating is achieved by 16-bit pulse width control of a resistive heater board attached to a metal block that receives the microtiter tray. The 96- or 384-well block temperature is monitored with an embedded resistive thermal device and 16-bit digital conversion. Mounted above the plate are two banks of 61 superbright 470-nm LEDs that illuminate the tray through a bandpass filter. A charge-coupled device camera (1392 × 1040 pixels, 12-bit depth per pixel) monitors sample fluorescence through a longpass filter. During melting at 0.1°C/s, 5 to 10 consecutive camera images are continuously collected and averaged, storing about 12 data points every °C. Acquisition parameters were a hold temperature of 73°C, a start temperature of 78°C, an end temperature of 94°C, and the exposure set to “Auto.” Melting curves were displayed normalized with curve overlay (temperature shifting) and/or as difference plots of the normalized and overlaid curves, generated by subtraction of the abnormal melting curves from normal melting curves.27,28 Heterozygous samples were identified by automatic clustering of normalized, temperature overlaid melting curves using LightScanner software.
Predicted melting curves of the 13 CYBB PCR products were calculated using Poland's algorithm29 as modified by Fixman and Freire30 and implemented by Steger.31 The 75 mmol/L NaCl Blake and Delcourt parameters were used with a step size of 0.2°C. The “MeltPlot” data were inverted offline with a custom LabView program to convert the hypochromicity data to predicted, normalized fluorescent melting curves.
Cycle Sequencing
When sequence variants were detected, they were identified by cycle sequencing with dideoxy terminators. The PCR product of interest was recovered from the LightScanner and diluted 1:60 in water. One-sixth of the recommended amount of the dye terminator mixture (BigDye Terminator v1.1, Applied Biosystems, Foster City, CA) was combined with 1 μl of the diluted PCR product, 0.5 μmol/L of one of the common primer tails (Table 1, footnote), and 1× sequencing buffer (Applied Biosystems) in a total volume of 10 μl. After 25 cycles of extension (25 cycles of 96°C for 5 seconds, 50°C for 10 seconds, and 55°C for 20 seconds, PTC-200), unincorporated terminator dyes and salts were removed by gel filtration, and the eluate was loaded onto a capillary sequencer (3130 Genetic Analyzer, Applied Biosystems) and separated on a 36-cm array filled with POP-7 polymer using an UltraSeq36_POP7_1 protocol. The cycle sequencing required 40 minutes and the electrophoretic separation required 30 minutes.
CYBB mutations were compared against the CYBBbase browser (http://bioinf.uta.fi/CYBBbase/browser, last accessed June 27, 2009), the Human Gene Mutation Database at the Institute of Medical Genetics in Cardiff, UK (http://www.hgmd.cf.ac.uk/ac/all.php, last accessed October 21, 2009), and the Biobase Professional Release Database 2009.1 to determine whether mutations had been reported previously. Although benign polymorphisms within the coding region of CYBB are rare, some do occur and must be considered.10 Both coding and noncoding polymorphisms are cataloged on dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?locusId=1536, last accessed June 27, 2009).
Results
Predicted melting curves for the PCR products bracketing the 13 exons of CYBB are shown in Figure 1A. The number of melting domains was clearly defined in eight of the predicted curves with four single domain curves (exons 1, 2, 4, and 5), three double domain curves (exons 3, 8, and 11), and one triple domain curve (exon 9). The remaining five curves were difficult to classify because they show predominately a single domain with some indication of a second domain. This result compares well with the observed melting curves shown in Figure 1B, where nine of the curves were easy to classify (four single domain, four double domain, and one triple domain). The second domain in the PCR product bracketing exon 6 was more distinct by observation than prediction. Overall the shapes and relative positions of the observed curves were remarkably similar to the predicted curves. However, two differences are apparent. First, in multiple domain curves, lower melting domains seem to be exaggerated by prediction, whereas the higher domains dominate in experimental curves. Second, the absolute temperature position of the observed curves is 5 to 6°C higher than predicted. Supplemental Figure S1 (see http://jmd.amjpathol.org) shows the reproducibility of six replicate melting curves for each exon using the same DNA sample across the entire primer plate (Supplemental Figure S1, see http://jmd.amjpathol.org).
Figure 1.
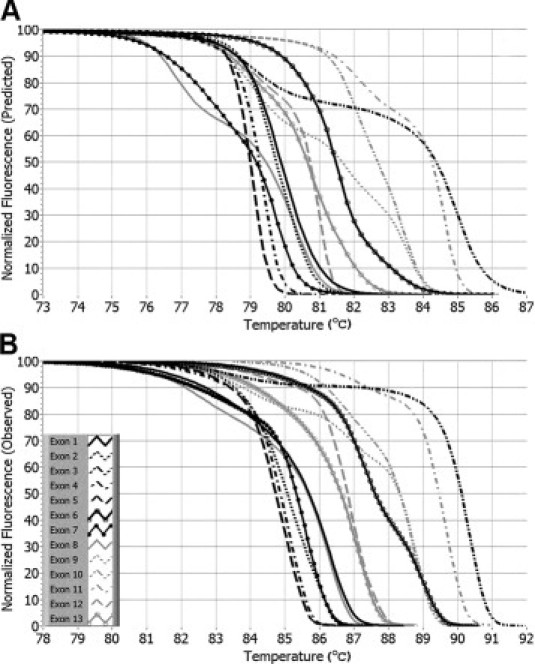
Predicted and observed normalized melting curves for PCR products bracketing the 13 exons of CYBB. A: Predicted melting curves were calculated using Poland's recursive algorithm, as described in Material and Methods. B: The observed melting curves were obtained by normalization using linear background extrapolation after PCR and high resolution melting of a normal DNA sample.
Because high-resolution melting detects any sequence variation, benign polymorphisms can complicate analysis. The population variation within each of the 13 PCR products covering CYBB was investigated using the DNA of 96 random blood donors. After PCR amplification and high-resolution melting analysis, no variants were detected (data not shown), indicating a low level of variation in the normal population.
Eighteen blinded validation samples were provided by one site (Sanquin Research Institute, Amsterdam, The Netherlands) to another (University of Utah) and were analyzed using the CYBB primer plates for PCR and high-resolution melting. In each case, aberrant melting curves were identified in 1 of the 13 PCR products by high-resolution melting, and that product was sequenced in both directions to identify the point mutation or small insertion/deletion. Mutations in all 18 samples were correctly identified, including 8 missense mutations, 4 substitutions leading to splicing defects, 2 single-base deletions, and 2 single-base insertions leading to frameshift termination and 2 large deletions of multiple exons (Table 2). Two of the mutations (c.134T>G and c.1462-2A>C) had not been previously reported; both of these had no neutrophil oxidase activity.
Table 2.
CYBB Mutations Detected by High-Resolution Melting
| Location | c.* | p. | No. M† | No. F† | Oxidase activity | Figure |
|---|---|---|---|---|---|---|
| Exon 1 | 1A>G | M1V | 1 | 0 | ||
| Intron 1 | 45 + 1G>T | 1 | 0 | None | ||
| Exon 2 | 90C>A‡ | Y30X | 1 | 0 | None | |
| 134T>G | L45R | 1 | 0 | None | ||
| Exon 3 | 177C>G | C59W | 1 | 0 | ||
| 190T>C | C64R | 1 | 0 | |||
| 252G>A | A84A | 1 | 3 | Decreased | S2§ | |
| 1 | 0 | None | S2§ | |||
| Intron 3 | 252 + 5G>A | 2 | 2 | Decreased | 3 | |
| Intron 4 | 338-2A>G | 1 | 1 | None | ||
| Exon 5 | 466G>A | A156T | 1 | 0 | ||
| 479dupT | N162EfsX2 | 1 | 0 | |||
| Intron 5 | 483 + 3A>T | 1 | 0 | |||
| Exon 6 | 636delT | F212LfsX1 | 1 | 2 | None | |
| Intron 6 | 674 + 4A>G | 1 | 0 | None | ||
| Exon 7 | 676C>T | R226X | 1 | 0 | None | |
| 1 | 1 | None | ||||
| 1 | 0 | None | ||||
| 703_704delAG | S235FfsX4 | 1 | 1 | None | ||
| 730T>A | C244S | 1 | 1 | Decreased | ||
| 731G>A | C244Y | 1 | 0 | |||
| 742dupA | I248NfsX35 | 1 | 0 | |||
| 755delG | G252EfsX2 | 0 | 1 | |||
| Exon 8 | 868C>T | R290X | 1 | 0 | None | |
| 897G>A | K299K | 1 | 0 | None | ||
| Intron 8 | 897 + 1G>T | 1 | 0 | |||
| 897 + 1G>A | 1 | 0 | None | |||
| Exon 9 | 1016C>A | P339H | 2 | 2 | None | 2 |
| 1032delC | A345PfsX40 | 1 | 0 | |||
| 1061A>G | H354R | 0 | 1¶ | |||
| Exon 10 | 1166G>C | G389A | 1 | 0 | ||
| Intron 11 | 1461 + 2delT | 1 | 0 | None | ||
| 1462-2A>C | 1 | 0 | None | |||
| Exon 13 | 1661_1662delCT | S554X | 1 | 0 | None | |
| Exons 6-8 | Deletion | 1 | 0 | None | 4 | |
| Exons 7-13 | Deletion | 1 | 0 | |||
| Exons 1-13 | Deletion | 1 | 0 | None | ||
| Exons 1-13 | Deletion | 1 | 0 | None | ||
| Exons 1-13 | Deletion | 1 | 0 | |||
| Exons 1-13 | Deletion | 1 | 0 | None |
Each family is shown on a separate line. Clinical samples are shown in bold and validation samples are not bolded. Novel mutations (previously unreported) are shown in italics.
Coding (c.) sequence numbering starting with the first base of the ATG initiation codon using NG_00965.1 as the reference sequence.
Number of affected males (M) and number of heterozygous females (F) studied in each family.
This sample also had an exon 12 variant, c.1551T>A (p.D517E) previously reported as a benign variant.10
Supplemental Figure S2 (see http://jmd.amjpathol.org).
Affected female.
Twenty-two clinical kindreds identified by decreased (3) or absent (19) oxidase activity were studied, comprising 37 individuals (24 males and 13 females). Nineteen different mutations were identified, of which 8 were novel (42%). Two mutations were present in multiple kindreds. Three families carried the c.676C>T mutation, and two families carried c.252G>A. One of the families with c.252G>A had no detectable oxidase activity, whereas the other with the same mutation had decreased, but not absent activity, indicating that both flow cytometry patterns and degrees of oxidase activity can arise from the same mutation (Supplemental Figure S2, see http://jmd.amjpathol.org). Two of the mutations were missense, four were nonsense, two were small deletions resulting in frameshift termination, seven involved splicing (six substitutions and one small deletion), and four were large deletions of multiple exons (Table 2). In one family, both a novel mutation (c.90C>A) and a known benign variant (c.1551T>A)10,11 were identified.
The pedigree and melting results of a variant X− kindred is shown in Figure 2. The affected cousins, aged 3 and 4 years, were evaluated for treatment and possible bone marrow transplant at Texas Children's Hospital, Baylor College of Medicine (Houston, TX). Their neutrophil oxidase activity was difficult to interpret, with broad peaks of mostly nonfluorescing cells after stimulation. The oxidase activity patterns of both mothers were similar to those of the unaffected fathers, complicating identification as an autosomal recessive or X-linked disorder. In contrast with this problematic phenotyping, high-resolution melting and targeted sequencing quickly established the molecular abnormality in this extended family. Melting curve analysis revealed no abnormalities in 12 of 13 exons. Melting curves of CYBB exon 9 segregated into two clusters, however. Both carrier mothers and the two affected males (mixed with normal male DNA) clustered together as heterozygotes, separate from the cluster of normal homozygous/hemizygous samples. Melting curves of the normal samples had three domains (Figure 2A). In contrast, only two domains were observed in the heterozygous samples resulting from heteroduplexes in the highest melting domain. The differences between the clusters are visually magnified by display as difference plots (Figure 2B). Targeted sequencing revealed a known point mutation in exon 9 (c.1016C>A), resulting in an amino acid change (p.P339H) in the FAD binding region of the CYBB gene product.32,33 The ability to use gene scanning by high-resolution melting analysis combined with targeted sequencing rapidly resulted in the appropriate molecular diagnosis in these candidates for bone marrow transplantation.
Figure 2.
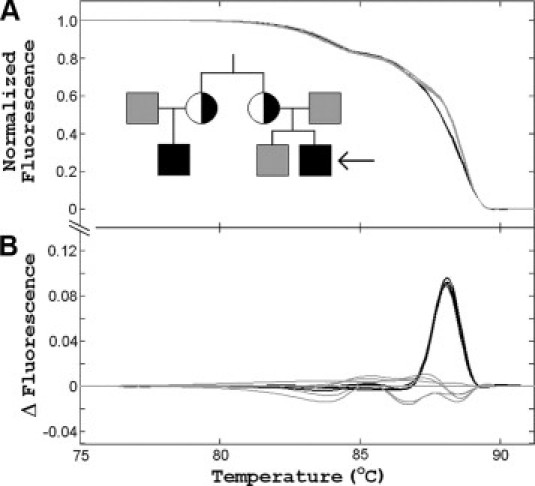
High-resolution melting analysis of a variant X− CGD family with a mutation in a three-domain melting curve. In the pedigree, black squares are affected males, gray squares are normal males, and half-black circles are heterozygous carrier females. Melting analysis of all family members and the wild-type control subjects clustered together except for exon 9. A: Normalized exon 9 melting curves after overlay reveals two clusters. Unaffected males, normal controls, and affected males without mixing are shown in gray. Heterozygous females and affected males mixed with wild-type DNA are shown in black. B: The differences between clusters are easier to see in “difference plots,” where all curves are subtracted from the wild-type average. Exon 9 sequencing of the affected males revealed a c.1016C>A mutation, also present in the carrier females in heterozygous form.
The pedigree of another variant X− kindred is shown in Figure 3. The 2-year-old male proband was hospitalized in the pediatric intensive care unit at Primary Children's Hospital (Salt Lake City, UT) with life-threatening Burkholderia cepacia pneumonia. The patient had an asymptomatic 2-month-old brother and an extended family available for study.34 The neutrophil oxidase activity of the two brothers was decreased but not absent, and the mother and an aunt had both normal and decreased populations by flow cytometry, suggesting X− CGD. Gene scanning of CYBB by high-resolution melting was normal except for the PCR product bracketing exon 3. For this PCR product, the melting curves for the patient and his younger brother (both mixed with wild-type male DNA), the mother, and the aunt clustered separately from the wild-type samples. Two domains were present with a minor domain 6 to 7°C lower than the predominate domain (Figure 3A). When both domains were analyzed together, the heterozygotes clustered above the wild-type samples, a counterintuitive result considering that heteroduplexes usually decrease the melting temperature. However, when the domains were analyzed separately, the two clusters segregated in the low-temperature domain with the heterozygotes below the normal samples as expected (Figure 3B). Only one cluster was present in the high-temperature domain when it was analyzed separately (Figure 3C). Targeted sequencing revealed an intron 3, c.252 + 5G>A variant (a known splicing mutation), in the patients and carriers. Genetic analysis allowed rapid institution of more appropriate antimicrobial therapy, interferon-γ administration, and detection of an unsuspecting carrier aunt.
Figure 3.
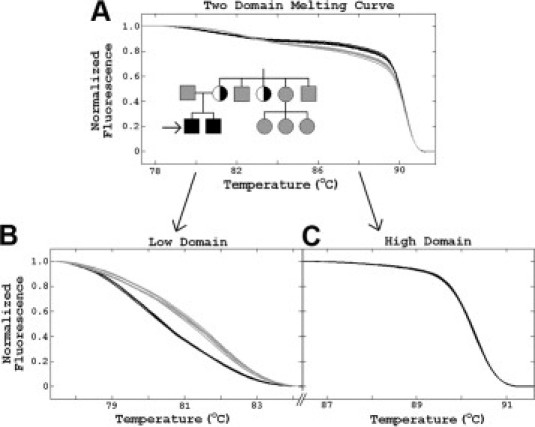
High-resolution melting analysis of a variant X− CGD family with a mutation isolated to a minor low temperature domain. In the pedigree, black squares are affected males, gray squares are unaffected males, gray circles are wild-type females, and half-black circles are heterozygous carrier females. Melting analysis revealed variation within exon 3. A: Two melting curve clusters are shown and a low-temperature domain is present that is easy to miss unless melting curve prediction is used (Figure 1). B and C: When the two domains are analyzed separately, the variation is isolated to the low-melting domain. Targeted sequencing revealed a c.252 + 5G splicing mutation in the patients and carriers.
Although high-resolution melting easily detects heterozygous variants that are between PCR primers, heterozygous deletions of entire exons are typically missed. However, in X-linked disorders, deleted exons are simple to detect because no specific product is amplified unless the sample is mixed with wild-type DNA. Figure 4 shows melting curves and the fluorescent plate image for an X0 CGD patient with exons 6 to 8 deleted. Using unmixed patient DNA, no specific products were detected on melting analysis of exons 6 to 8 (Figure 4A), and these exons did not fluoresce on scans of the plate (Figure 4B). In contrast, wild-type and mixed samples showed strong fluorescence and the expected melting curves. Deletion of all CYBB exons was observed in three kindreds. Two of these presented with the McLeod phenotype, classically resulting from a large deletion at Xp21 that includes CYBB.
Figure 4.
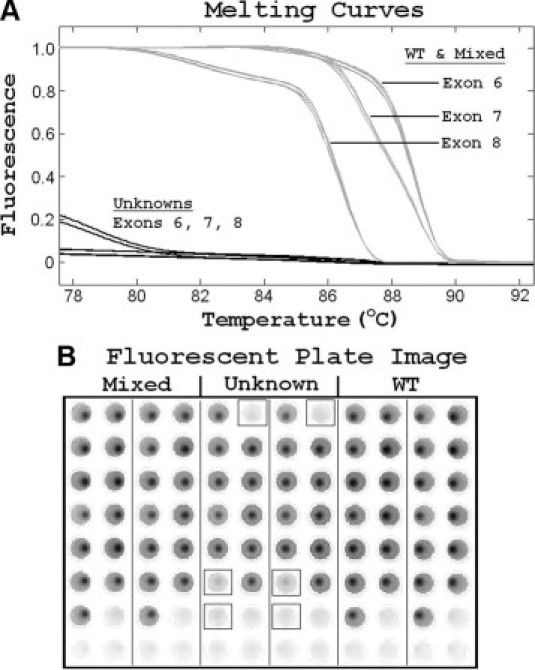
Deletion of exons 6 to 8 detected by PCR and high-resolution melting. A: Normalized melting curves for exons 6 to 8 are shown for both wild-type (WT) controls and a 1:1 mixture of unknown patient and wild-type DNA (gray traces). However, when the unknown sample is tested without mixing, no specific melting curves are observed (black traces). B: Fluorescent image of the plate. Primers for exons 1 to 7 are in columns 1, 3, 5, 7, 9, and 11 and primers for exons 8 to 13 are in columns 2, 4, 6, 8, 10, and 12. Mixed DNA is present in columns 1 to 4, the unknown sample without mixing is present in columns 5 to 8 (exons 6 to 8 are boxed), and wild-type male DNA is present in columns 9 to 12.
Discussion
For rare primary immunodeficiency disorders such as chronic granulomatous disease, high-throughput analysis of many samples is not as clinically useful as timely analysis of occasional single patients or a limited number of family members. Because many exons or multiple genes need to be analyzed, only a few samples can be amplified at once on a microtiter plate. High-resolution melting analysis of such “orphan” genetic disorders can be performed rapidly after PCR with minimal added cost or processing. With use of automated DNA extraction and previously prepared microtiter plates with dried primers, the complexity of performing such assays is reduced to the extent that reports can be generated within 3 hours after the start of DNA extraction for samples without sequence variations. Rapid cycle sequencing and short capillary electrophoresis protocols allow samples with mutations to be reported within 6 hours after the start of sample preparation.
High-resolution PCR product melting is a new scanning method that detects heterozygous point mutations and small insertions or deletions.25 After PCR, no separation or processing of the amplified sample is required. High-resolution melting is the only heterozygote scanning method that is performed in a closed tube and is generally recognized as superior to other options in terms of price, workload, sensitivity, and specificity.23,24,35
Genetic scanning by melting requires a high-resolution instrument, a DNA dye that detects heteroduplexes and dedicated software. Conventional real-time instruments perform poorly and do not have the software needed to detect the subtle changes in melting curve shape that identify sequence variations.36 Mutation detection also depends on a new class of “saturation” DNA dyes that detect heteroduplexes well, in contrast to the commonly used real-time dye, SYBR Green I.37 In genetic analysis, many exons are often scanned, so instruments with a large batch size are preferable. Although carousel-based instruments can be used for high-resolution melting,38 primers are easier to deposit, dry, and dissolve in a rectangular plate matrix. Finally, although PCR and high-resolution melting can be performed in one combined instrument, higher throughput and cost savings are obtained when the functions are separated by using two dedicated instruments.24 For these reasons, the LightScanner instrument and LCGreen Plus dye were used.
Genetic scanning by high-resolution melting has been applied to many genetic disorders, including BRCA1/2 and cystic fibrosis.39,40 In the case of X-linked genes, a known male wild-type sample is mixed 1:1 with the unknown sample so that any sequence difference (single base variants, small insertions, and small deletions) will create heterozygotes that alter the melting curve shape because of low-temperature heteroduplexes.41 To detect large deletions of one or more exons, the DNA is also amplified and melted without mixing. Deleted hemizygous exons lack the characteristic melting profiles of wild-type samples (Figure 4).
When 96 random individuals were analyzed by high-resolution melting, no polymorphisms were identified within CYBB. The low frequency of CYBB sequence variations in the random population has been observed previously10 and is in contrast with higher polymorphism frequencies in most other genes, including BRCA139 and CFTR.40 A single benign polymorphism (c.1551T>A, p.D517E) was detected in one clinical sample, together with a novel c.90C>A, p.Y30X mutation (Table 2). This low frequency of benign polymorphisms makes scanning analysis of CYBB attractive. Nevertheless, any unexpected sequence variation detected by scanning should be sequenced or otherwise genotyped for identification.
Eighteen previously genotyped, blinded samples were analyzed for method validation, including eight missense, four frameshift, four splicing variants, and two large deletions. All mutations were correctly identified, not surprising given the high sensitivity reported for high-resolution melting.24 Indeed, the rare false-negative results reported in the literature seem to have explanations such as sample tracking errors.42
CYBB mutations were found in 22 unrelated individuals identified by decreased or absent oxidase activity. Nineteen different variants were identified, with eight apparently novel mutations (Table 2). Four of these were nonsense, frameshift, or large deletion variants and are almost certainly disease-causing. The other four novel sequence variations are putative splicing or missense mutations. Sequence variations were detected in all PCR products except that bracketing exon 4, including a polymorphism in exon 12 (Table 2, footnote ‡). The longest PCR product was 401 bp long and had three melting domains as predicted (Figures 1 and 2). The optimal PCR product length and number of domains for high-resolution melting remain controversial.43 Sensitivity decreases somewhat above 400 bp,24 although longer products have been routinely analyzed.39,44
Melting simulation software has been previously applied to assay design and discrimination of different microbial species.45 Although the predicted number and shape of the melting transitions correlated well with observation, their relative magnitudes and absolute temperatures did not. In the CYBB scanning reported here, the relative magnitude of the lower temperature domains is always less than predicted. Furthermore, although the relative melting temperatures of the different PCR products generally follow predictions, there is an absolute temperature offset of 5 to 6°C. As previously suggested, this offset probably results from duplex stabilization by the dye, Mg2+, and other ionic components. Despite these limitations, melting simulation of CYBB predicted all of the low temperature melting domains. This is critical in genetic disease scanning, because there is a risk of missing variants in low-temperature domains that can be easily overlooked (Figure 3).
Molecular assessment of CGD is necessary for genetic counseling and prenatal diagnosis. Although carrier mothers are usually identified in X-linked disease, de novo mutations occur in 10 to 20% of cases.8,22 Rapid molecular confirmation of disease also helps to optimize treatment, including antimicrobial therapy and prophylaxis capable of penetrating phagocytes, interferon-γ administration, granulocyte transfusions, and even bone marrow transplantation or gene therapy.17
Some cases of variant X− CGD have decreased, but not absent, neutrophil oxidase activity. Because some reactive oxygen species are produced, infections may be milder and the disorder may not be detected until later in life. The probands of the three kindreds with decreased oxidase activity in Table 2 were 2.5, 7, and 17 years of age at diagnosis for mutations c.252G>A, c.252 + 5G>A, and 730T>A, respectively. Indeed, initial diagnosis of CGD in a 69-year-old man has been reported.46 Variable phenotypes can occur given identical CYBB mutations, suggesting that other genetic loci and/or environmental factors play a significant role in the course of disease.47 For example, two unrelated individuals with the same c.252G>A mutation had clearly different oxidase results (Table 2, Supplemental Figure S2, see http://jmd.amjpathol.org). This mutation induces abnormal splicing that can either be partial (X−) or total (X0).
High-resolution melting is a promising tool to reduce the time, cost, and complexity of genetic analysis. Evaluation of CGD is a good example of its application to rare disorders. Melting prediction and preparation of microtiter plates with dried primer pairs streamline the process. Definitive results can be returned within 1 working day. Rapid recognition, confirmation, and initiation of appropriate therapy can be critical in this life-threatening disease.
Acknowledgements
We thank Drs. Howard Rosenblatt (currently at Specially for Children, Dell Children's Hospital, Austin, TX and formerly of Texas Children's Hospital, Houston, TX), as well as Dr. Mary Ellen Conley (St. Jude Children Research Hospital, Memphis, TN), Dr. Susan Pacheco (University of Texas, Houston, TX), Dr. Maite de la Morena (University of Texas Southwestern School of Medicine, Dallas, TX), Dr. Thomas Rand (Boise, ID), and Dr. Lloyd Jensen (Pocatello, ID) for contributing one or more X-linked CGD cases and/or carriers to this study.
Footnotes
Supported by National Institutes of Health (grants GM072419 and GM073396), the ARUP Institute for Clinical and Experimental Pathology, and Idaho Technology.
Aspects of high resolution melting are licensed by the University of Utah to Idaho Technology. J.D.B. is an employee of Idaho Technology. C.T.W. has an equity interest in Idaho Technology. None of the other authors declare any relevant financial relationships.
Supplemental material for this article can be found on http://jmd.amjpathol.org.
Web Extra Material
References
- 1.Bridges RA, Berendes H, Good RA. A fatal granulomatous disease of childhood; the clinical, pathological, and laboratory features of a new syndrome. AMA J Dis Child. 1959;97:387–408. [PubMed] [Google Scholar]
- 2.Segal AW, Peters TJ. Characterization of the enzyme defect in chronic granulomatous disease. Lancet. 1976;1:1363–1365. doi: 10.1016/s0140-6736(76)93021-x. [DOI] [PubMed] [Google Scholar]
- 3.Roos D, Kuijpers TW, Curnutte JT. Chronic granulomatous disease. In: Ochs HD, Smith CIE, Puck JM, editors. Primary Immunodeficiency Diseases. Oxford University Press; New York: 2006. pp. 525–549. [Google Scholar]
- 4.Winkelstein JA, Marino MC, Johnston RB, Jr, Boyle J, Curnutte J, Gallin JI, Malech HL, Holland SM, Ochs H, Quie P, Buckley RH, Foster CB, Chanock SJ, Dickler H. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore) 2000;79:155–169. doi: 10.1097/00005792-200005000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Quie PG, White JG, Holmes B, Good RA. In vitro bactericidal capacity of human polymorphonuclear leukocytes: diminished activity in chronic granulomatous disease of childhood. J Clin Invest. 1967;46:668–679. doi: 10.1172/JCI105568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnston RB., Jr Clinical aspects of chronic granulomatous disease. Curr Opin Hematol. 2001;8:17–22. doi: 10.1097/00062752-200101000-00004. [DOI] [PubMed] [Google Scholar]
- 7.van den Berg JM, van Koppen E, Ahlin A, Belohradsky BH, Bernatowska E, Corbeel L, Español T, Fischer A, Kurenko-Deptuch M, Mouy R, Petropoulou T, Roesler J, Seger R, Stasia MJ, Valerius NH, Weening RS, Wolach B, Roos D, Kuijpers TW. Chronic granulomatous disease: the European experience. PLoS ONE. 2009;4:e5234. doi: 10.1371/journal.pone.0005234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine (Baltimore) 2000;79:170–200. doi: 10.1097/00005792-200005000-00004. [DOI] [PubMed] [Google Scholar]
- 9.Rothe G, Emmendorffer A, Oser A, Roesler J, Valet G. Flow cytometric measurement of the respiratory burst activity of phagocytes using dihydrorhodamine 123. J Immunol Methods. 1991;138:133–135. doi: 10.1016/0022-1759(91)90074-p. [DOI] [PubMed] [Google Scholar]
- 10.Heyworth PG, Curnutte JT, Rae J, Noack D, Roos D, van Koppen E, Cross AR. Hematologically important mutations: X-linked chronic granulomatous disease (second update) Blood Cells Mol Dis. 2001;27:16–26. doi: 10.1006/bcmd.2000.0347. [DOI] [PubMed] [Google Scholar]
- 11.Jirapongsananuruk O, Niemela JE, Malech HL, Fleisher TA. CYBB mutation analysis in X-linked chronic granulomatous disease. Clin Immunol. 2002;104:73–76. doi: 10.1006/clim.2002.5230. [DOI] [PubMed] [Google Scholar]
- 12.Roos D, de Boer M, Borregard N, Bjerrum OW, Valerius NH, Seger RA, Mühlebach T, Belohradsky BH, Weening RS. Chronic granulomatous disease with partial deficiency of cytochrome b558 and incomplete respiratory burst: variants of the X-linked, cytochrome b558-negative form of the disease. J Leukoc Biol. 1992;51:164–171. doi: 10.1002/jlb.51.2.164. [DOI] [PubMed] [Google Scholar]
- 13.Stasia MJ, Li XJ. Genetics and immunopathology of chronic granulomatous disease. Semin Immunopathol. 2008;30:209–235. doi: 10.1007/s00281-008-0121-8. [DOI] [PubMed] [Google Scholar]
- 14.Roos D, de Boer M, Köker MY, Dekker J, Singh-Gupta V, Ahlin A, Palmblad J, Sanal O, Kurenko-Deptuch M, Jolles S, Wolach B. Chronic granulomatous disease caused by mutations other than the common GT deletion in NCF1, the gene encoding the p47phox component of the phagocyte NADPH oxidase. Hum Mutat. 2006;27:1218–1229. doi: 10.1002/humu.20413. [DOI] [PubMed] [Google Scholar]
- 15.van Pelt LJ, van Zwieten R, Weening RS, Roos D, Verhoeven AJ, Bolscher BG. Limitations on the use of dihydrorhodamine 123 for flow cytometric analysis of the neutrophil respiratory burst. J Immunol Methods. 1996;191:187–196. doi: 10.1016/0022-1759(96)00024-5. [DOI] [PubMed] [Google Scholar]
- 16.Yu G, Hong DK, Dionis KY, Rae J, Heyworth PG, Curnutte JT, Lewis DB. Focus on FOCIS: the continuing diagnostic challenge of autosomal recessive chronic granulomatous disease. Clin Immunol. 2008;128:117–126. doi: 10.1016/j.clim.2008.05.008. [DOI] [PubMed] [Google Scholar]
- 17.Seger RA. Modern management of chronic granulomatous disease. Br J Haematol. 2008;140:255–266. doi: 10.1111/j.1365-2141.2007.06880.x. [DOI] [PubMed] [Google Scholar]
- 18.Kannengiesser C, Gérard B, El Benna J, Henri D, Kroviarski Y, Chollet-Martin S, Gougerot-Pocidalo MA, Elbim C, Grandchamp B. Molecular epidemiology of chronic granulomatous disease in a series of 80 kindreds: identification of 31 novel mutations. Hum Mutat. 2008;29:E132–E149. doi: 10.1002/humu.20820. [DOI] [PubMed] [Google Scholar]
- 19.Köker MY, Sanal O, De Boer M, Tezcan I, Metin A, Ersoy F, Roos D. Mutations of chronic granulomatous disease in Turkish families. Eur J Clin Invest. 2007;37:589–595. doi: 10.1111/j.1365-2362.2007.01828.x. [DOI] [PubMed] [Google Scholar]
- 20.Roos D, de Boer M, Kuribayashi F, Meischl C, Weening RS, Segal AW, Ahlin A, Nemet K, Hossle JP, Bernatowska-Matuszkiewicz E, Middleton-Price H. Mutations in the X-linked and autosomal recessive forms of chronic granulomatous disease. Blood. 1996;87:1663–1681. [PubMed] [Google Scholar]
- 21.Patiño PJ, Perez JE, Lopez JA, Condino-Neto A, Grumach AS, Botero JH, Curnutte JT, García de Olarte D. Molecular analysis of chronic granulomatous disease caused by defects in gp91-phox. Hum Mutat. 1999;13:29–37. doi: 10.1002/(SICI)1098-1004(1999)13:1<29::AID-HUMU3>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 22.Di Matteo G, Giordani L, Finocchi A, Ventura A, Chiriaco M, Blancato J, Sinibaldi C, Plebani A, Soresina A, Pignata C, Dellepiane RM, Trizzino A, Cossu F, Rondelli R, Rossi P, De Mattia D, Martire B, IPINET (Italian Network for Primary Immunodeficiencies) Molecular characterization of a large cohort of patients with chronic granulomatous disease and identification of novel CYBB mutations: an Italian multicenter study. Mol Immunol. 2009;46:1935–1941. doi: 10.1016/j.molimm.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 23.Erali M, Voelkerding KV, Wittwer CT. High resolution melting applications for clinical laboratory medicine. Exp Mol Pathol. 2008;85:50–58. doi: 10.1016/j.yexmp.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reed GH, Kent JO, Wittwer CT. High-resolution DNA melting analysis for simple and efficient molecular diagnostics. Pharmacogenomics. 2007;8:597–608. doi: 10.2217/14622416.8.6.597. [DOI] [PubMed] [Google Scholar]
- 25.Reed GH, Wittwer CT. Sensitivity and specificity of single-nucleotide polymorphism scanning by high-resolution melting analysis. Clin Chem. 2004;50:1748–1754. doi: 10.1373/clinchem.2003.029751. [DOI] [PubMed] [Google Scholar]
- 26.Bennett CD, Campbell MN, Cook CJ, Eyre DJ, Nay LM, Nielsen DR, Rasmussen RP, Bernard PS. The LightTyper: high-throughput genotyping using fluorescent melting curve analysis. Biotechniques. 2003;34:1288–1292. doi: 10.2144/03346pf01. 1294–1285. [DOI] [PubMed] [Google Scholar]
- 27.Wittwer CT, Reed GH, Gundry CN, Vandersteen JG, Pryor RJ. High-resolution genotyping by amplicon melting analysis using LCGreen. Clin Chem. 2003;49:853–860. doi: 10.1373/49.6.853. [DOI] [PubMed] [Google Scholar]
- 28.Palais R, Wittwer CT. Mathematical algorithms for high-resolution DNA melting analysis. Methods Enzymol. 2009;454:323–343. doi: 10.1016/S0076-6879(08)03813-5. [DOI] [PubMed] [Google Scholar]
- 29.Poland D. Recursion relation generation of probability profiles for specific-sequence macromolecules with long-range correlations. Biopolymers. 1974;13:1859–1871. doi: 10.1002/bip.1974.360130916. [DOI] [PubMed] [Google Scholar]
- 30.Fixman M, Freire JJ. Theory of DNA melting curves. Biopolymers. 1977;16:2693–2704. doi: 10.1002/bip.1977.360161209. [DOI] [PubMed] [Google Scholar]
- 31.Steger G. Thermal denaturation of double-stranded nucleic acids: prediction of temperatures critical for gradient gel electrophoresis and polymerase chain reaction. Nucleic Acids Res. 1994;22:2760–2768. doi: 10.1093/nar/22.14.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rae J, Newburger PE, Dinauer MC, Noack D, Hopkins PJ, Kuruto R, Curnutte JT. X-linked chronic granulomatous disease: mutations in the CYBB gene encoding the gp91-phox component of respiratory-burst oxidase. Am J Hum Genet. 1998;62:1320–1331. doi: 10.1086/301874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roesler J, Heyden S, Burdelski M, Schäfer H, Kreth HW, Lehmann R, Paul D, Marzahn J, Gahr M, Rösen-Wolff A. Uncommon missense and splice mutations and resulting biochemical phenotypes in German patients with X-linked chronic granulomatous disease. Exp Hematol. 1999;27:505–511. doi: 10.1016/s0301-472x(98)00024-1. [DOI] [PubMed] [Google Scholar]
- 34.Bender JM, Rand TH, Ampofo K, Pavia AT, Schober M, Tebo A, Pasi B, Augustine NH, Pryor RJ, Wittwer CT, Hill HR. Family clusters of variant X-linked chronic granulomatous disease. Pediatr Infect Dis J. 2009;28:529–533. doi: 10.1097/INF.0b013e318194cf09. [DOI] [PubMed] [Google Scholar]
- 35.Vossen RH, Aten E, Roos A, den Dunnen JT. High-resolution melting analysis (HRMA): more than just sequence variant screening. Hum Mutat. 2009;30:860–866. doi: 10.1002/humu.21019. [DOI] [PubMed] [Google Scholar]
- 36.Herrmann MG, Durtschi JD, Bromley LK, Wittwer CT, Voelkerding KV. Amplicon DNA melting analysis for mutation scanning and genotyping: cross-platform comparison of instruments and dyes. Clin Chem. 2006;52:494–503. doi: 10.1373/clinchem.2005.063438. [DOI] [PubMed] [Google Scholar]
- 37.Dujols VE, Kusukawa N, McKinney JT, Dobrowolski SF, Wittwer CT. High-resolution melting analysis for scanning and genotyping. In: Dorak MT, editor. Real-Time PCR. Garland Science; New York: 2006. pp. 157–171. [Google Scholar]
- 38.Seipp MT, Durtschi JD, Voelkerding KV, Wittwer CT. Multiplex amplicon genotyping by high-resolution melting. J Biomol Tech. 2009;20:160–164. [PMC free article] [PubMed] [Google Scholar]
- 39.van der Stoep N, van Paridon CD, Janssens T, Krenkova P, Stambergova A, Macek M, Matthijs G, Bakker E. Diagnostic guidelines for high-resolution melting curve (HRM) analysis: an interlaboratory validation of BRCA1 mutation scanning using the 96-well LightScanner. Hum Mutat. 2009;30:899–909. doi: 10.1002/humu.21004. [DOI] [PubMed] [Google Scholar]
- 40.Montgomery J, Wittwer CT, Kent JO, Zhou L. Scanning the cystic fibrosis transmembrane conductance regulator gene using high-resolution DNA melting analysis. Clin Chem. 2007;53:1891–1898. doi: 10.1373/clinchem.2007.092361. [DOI] [PubMed] [Google Scholar]
- 41.Dobrowolski SF, Ellingson CE, Caldovic L, Tuchman M. Streamlined assessment of gene variants by high resolution melt profiling utilizing the ornithine transcarbamylase gene as a model system. Hum Mutat. 2007;28:1133–1140. doi: 10.1002/humu.20558. [DOI] [PubMed] [Google Scholar]
- 42.Vandersteen JG, Bayrak-Toydemir P, Palais RA, Wittwer CT. Identifying common genetic variants by high-resolution melting. Clin Chem. 2007;53:1191–1198. doi: 10.1373/clinchem.2007.085407. [DOI] [PubMed] [Google Scholar]
- 43.Wittwer CT. High-resolution DNA melting analysis: advancements and limitations. Hum Mutat. 2009;30:857–859. doi: 10.1002/humu.20951. [DOI] [PubMed] [Google Scholar]
- 44.Dobrowolski SF, Gray J, Miller T, Sears M. Identifying sequence variants in the human mitochondrial genome using high-resolution melt (HRM) profiling. Hum Mutat. 2009;30:891–898. doi: 10.1002/humu.21003. [DOI] [PubMed] [Google Scholar]
- 45.Rasmussen JP, Saint CP, Monis PT. Use of DNA melting simulation software for in silico diagnostic assay design: targeting regions with complex melting curves and confirmation by real-time PCR using intercalating dyes. BMC Bioinformatics. 2007;8:107. doi: 10.1186/1471-2105-8-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schapiro BL, Newburger PE, Klempner MS, Dinauer MC. Chronic granulomatous disease presenting in a 69-year-old man. N Engl J Med. 1991;325:1786–1790. doi: 10.1056/NEJM199112193252506. [DOI] [PubMed] [Google Scholar]
- 47.Foster CB, Lehrnbecher T, Mol F, Steinberg SM, Venzon DJ, Walsh TJ, Noack D, Rae J, Winkelstein JA, Curnutte JT, Chanock SJ. Host defense molecule polymorphisms influence the risk for immune-mediated complications in chronic granulomatous disease. J Clin Invest. 1998;102:2146–2155. doi: 10.1172/JCI5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.