

Abstract
Dystrophic epidermolysis bullosa is a heritable skin disorder with dominant and recessive genetic patterns. Numerous studies underline that both forms are caused by mutations of the COL7A1 gene, which encodes collagen type VII. It has been reported that most mutations detected in the recessive disease form are nonsense mutations or small insertions or deletions leading to frameshift and premature translational termination, which tend to produce severe phenotypes. In contrast, missense mutations causing amino acid substitutions, which result in variable phenotypes, predominate in the dominant form of dystrophic epidermolysis bullosa. Genomic DNA from the patient and parents was subjected to PCR amplification of the coding region of the COL7A1 gene. Direct sequencing of the PCR products revealed a homozygous single-base deletion in the patient (c.6269-6270delC). The parents were heterozygous for the same mutation. This deletion is a novel mutation in the human COL7A1 gene based on comparisons with the Human Genome Mutation Database. To our knowledge, this is the first report of dystrophic epidermolysis bullosa in an Iranian patient confirmed by molecular diagnosis.
Epidermolysis bullosa (EB) is a group of heritable and acquired skin disorders with highly variable clinical severity. The most severe forms can cause mortality during the early postnatal period, whereas milder variants are characterized by protracted skin involvement that does not influence the overall life span of the affected individuals.1 The unifying diagnostic feature of EB is skin fragility, which manifests itself as blistering and skin erosion after mechanical trauma.2 In addition to skin symptoms, a variety of extracutaneous manifestations can be encountered in different forms of EB, including corneal erosions, enamel hypoplasia, nail dystrophy, scarring alopecia, tracheal epithelial erosion, development of esophageal strictures, and muscular dystrophy.3
Hereditary forms of EB include epidermolysis bullosa simplex (autosomal dominant), junctional epidermolysis bullosa (autosomal recessive), and dystrophic epidermolysis bullosa (DEB, autosomal dominant or recessive).4 DEB is characterized by sublamina densa tissue separation and abnormalities in the anchoring fibrils, which result from mutations in the COL7A1 gene and subsequent defects in type VII collagen.7 Genetic analyses of the COL7A1 gene in affected individuals revealed that most mutations detected in recessive DEB (RDEB) are nonsense mutations or small insertions or deletions leading to frameshift and a premature termination codon, which often results in severe phenotypes.8 Dominant DEB is characterized by missense COL7A1 mutations resulting in amino acid substitutions that often result in a milder disease form.9 Molecular genetic analysis of the COL7A1 gene is important for accurate diagnosis of the EB type.
We present here a molecular genetic study of a young girl referred to our laboratory suspected to have EB. Despite the lack of a skin biopsy for histological examination, and based purely on symptoms and family history, we searched for putative mutations in the COL7A1 gene to confirm the diagnosis of DEB.
Materials and Methods
Patient History
The proband, an 11-year-old girl, was referred to our diagnostic laboratory to confirm clinical diagnosis and determine the mode of inheritance. The primary diagnosis was made on the basis of typical skin symptoms including scarring and blisters on trauma-exposed body sites, including the hands and feet, and resulting in pseudosyndactyly. No skin biopsy was performed for the patient. At birth, reddened areas were present on the hands and toe tips of the patient, which blistered by 2 to 3 days of age. Shortly thereafter, scarring began on the entire body leading to disfigurement that continued to progress throughout adolescence (Figure 1). The parents are relatives of the third degree without similar clinical symptoms, and they have another symptom-free 15-year-old daughter.
Figure 1.
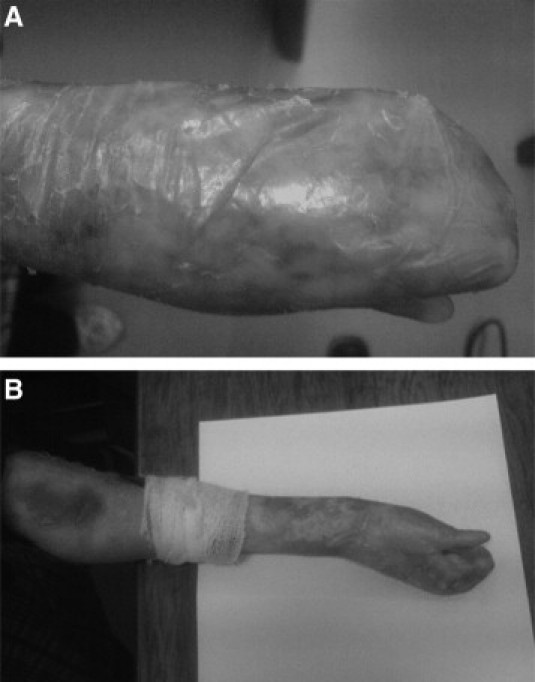
Patient exhibits blistering with scarring leading to pseudosyndactyly of the hands (A and B), which is a hallmark of the recessive form of DEB and fuses the digits into “mitten” hands and feet with severe loss of function.
Molecular Analysis
Genomic DNA extracted from peripheral blood from the patient, parents, and the unaffected sister was used as a template for amplifying individual exons of the COL7A1 gene. Primer pairs for entire exons and the flanking intron sequences were designed using the Primer3out software based on the NT02217 gene sequence (NCBI). PCR was performed (Eppendorf: master Gradient) with 100 ng of genomic DNA in 25 μl of reaction mixture containing 10 mmol/L Tris-HCl pH 8.3, 1 mmol/L MgCl2, 35 pmol of each primer, 200 μmol/L dNTP, and 2.5 units of superTaq (Fermentase). Purified PCR products were subjected to direct sequencing on an ABI automated sequencer 3770.
Results and Discussion
A young girl was diagnosed with DEB on the basis of symptoms and genetic counseling. Screening of the entire coding as well as flanking intron regions of the COL7A1 gene in this individual revealed the out-of-frame deletion of a single cytosine at codon 2090 within exon 74. The mutation was designated according to the Human Genome Variation Sequence (http://www.hgvs.org) as c.6269-70delC. The asymptomatic parents possess the same deletion in a heterozygous state (Figure 2). To verify the pathogenetic consequence of the detected mutation, we sequenced exon 74 of COL7A1 from genomic DNA from 24 randomly selected healthy persons as well as the oldest daughter of the family, none of whom showed any remarkable symptoms that are characteristics for EB. None of the individuals possessed mutations in exon 74, including the deletion in the affected girl described here.
Figure 2.
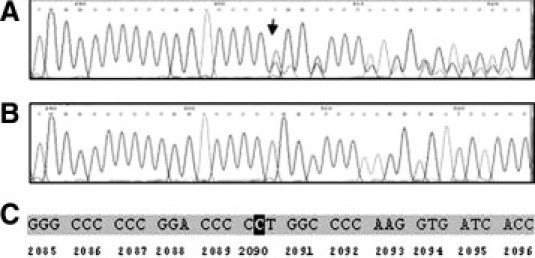
Partial sequence of exon 74 of the COL7A1 gene revealed a single cytosine deletion in a heterozygous mode in the parents (A) and homozygous mode in their affected daughter (B). This frameshift mutation at codon 2090 (C) creates a stop codon at codon 2116, which may cause a truncated gene product.
The human COL7A1 gene encodes type VII collagen and is expressed in keratinocytes, including the basal keratinocytes of the epidermis, where the protein products are assembled into homotrimeric molecules with a triple helical collagen structure.10 Mutations in the COL7A1 gene cause both recessive and dominant forms of DEB,3 which are different in terms of clinical manifestations and severity.10
Currently, more than 400 mutations have been described for the mild and severe forms of DEB.11 The severest forms of RDEB are caused by mutations on both alleles that result in either null alleles or out-of-frame mutations from insertions/deletions, single-base substitutions, and splice junction alterations.6,10,12,13,14,15 The severity may be related to the position of the stop codon; however, the presence of some functional protein appears to be the most important factor in ameliorating the disease severity.16
Here we report a novel homozygous single-base deletion in the COL7A1 gene of a young girl from southwest Iran with a severe form of DEB. The parents were heterozygous for the same mutation and, therefore, obligate carriers. This out-of-frame mutation at codon 2090 within exon 74 creates a premature termination codon at codon 2116 within the collagenous subdomains.
Homozygous mutations creating premature termination in this area occur in the majority of cases with severe mutilating RDEB.3 Premature termination leads consecutively to nonsense-mediated mRNA decay or truncated collagen VII polypeptides that are degraded within the cell.12,17
However, it has been described that approximately 75% of the dominant DEB mutations occur in exons 73, 74, and 75.6
According to the classification of recessive forms of DEB, there are Hallopeau-Siemens RDEB (HS-RDEB) and non-Hallopeau-Siemens RDEB (Non-HS RDEB).
Because of the failure of skin biopsy in the present case, which is necessary for a precise clinical diagnosis, and without analysis of the affected specific dermal structures, it is difficult to distinguish between the DEB types definitively.
HS-RDEB is one of the most severe subtypes of EB.18 Mutilations on the hands and feet are characteristic for HS-RDEB and can develop early in life.18 Generalized blistering, as is the case in the patient described here, is already present at birth in HS-RDEB patients and increases progressively with age. The blistering is the result of reduced resistance to minor trauma, whereby the type and location of the mutation determine the severity of blistering phenotype.19 Nucleotide deletions occurring within the central collagenous domain allow preservation of the noncollagenous extensions serving adhesive functions. Milder disease forms result because the anchoring fibrils may be morphologically altered but still partially functional.13 In contrast, the non–Hallopeau-Siemens RDEB (Non-HS RDEB) is less severe than HS-RDEB.18 The phenotype may be mild, with mild blistering localized to hands, feet, knees, and elbows and dystrophic nails, or relatively more widespread including flexural areas and trunk, but without the severe, mutilating scarring seen in HS-RDEB.5
One product of basic research on DEB is the identification of specific mutations in the COL7A1 gene. This expanding mutational database has the power to improve accurate diagnosis and refine DEB classification, which has implications for patient prognosis.20 DNA-based prenatal testing of families at risk for recurrence is another benefit of this genetic information base. To our knowledge, the present study is the first molecular diagnostic report of DEB from Iran. Studies of a larger number of patients are needed to show the types and frequencies of COL7A1 gene mutations in Iranian patients suffering from DEB.
This case report demonstrates that molecular diagnostic studies can be beneficial in some situations, such as the present case, if the clinical diagnosis is indeterminate or suspicious.
References
- 1.Tosti A, Piraccini BM, Scher RK. Isolated nail dystrophy suggestive of dominant dystrophic epidermolysis bullosa. Pediatr Dermatol. 2003;20:456–457. doi: 10.1046/j.1525-1470.2003.20422.x. [DOI] [PubMed] [Google Scholar]
- 2.Serrano-Martinez MC, Bagan JV, Silvestre FJ, Viguer MT. Oral lesions in recessive dystrophic epidermolysis bullosa. Oral Dis. 2003;9:264–268. doi: 10.1034/j.1601-0825.2003.03971.x. [DOI] [PubMed] [Google Scholar]
- 3.Smith FJ, Eady RA, Leigh IM, McMillan JR, Rugg EL, Kelsell DP, Bryant SP, Spurr NK, Geddes JF, Kirtschig G, Milana G, de Bono AG, Owaribe K, Wiche G, Pulkkinen L, Uitto J, McLean WH, Lane EB. Plectin deficiency results in muscular dystrophy with epidermolysis bullosa. Nat Genet. 1996;13:450–457. doi: 10.1038/ng0896-450. [DOI] [PubMed] [Google Scholar]
- 4.Uitto J, Richard G. Progress in epidermolysis bullosa: from eponyms to molecular genetics classification. Clin Dermatol. 2005;23:33–40. doi: 10.1016/j.clindermatol.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 5.De Benedittis M, Petruzzi M, Favia G, Serpico R. Oro-dental manifestations in Hallopeau-Siemens-type recessive dystrophic epidermolysis bullosa. Clin Exp Dermatol. 2004;29:128–132. doi: 10.1111/j.1365-2230.2004.01485.x. [DOI] [PubMed] [Google Scholar]
- 6.Mallipeddi R, Bleck O, Mellerio JE, Ashton GH, Eady RA, McGrath JA. Dilemmas in distinguishing between dominant and recessive forms of dystrophic epidermolysis bullosa. Br J Dermatol. 2003;149:810–818. doi: 10.1046/j.1365-2133.2003.05315.x. [DOI] [PubMed] [Google Scholar]
- 7.Murata T, Masunaga T, Ishiko A, Shimizu H, Nishikawa T. Differences in recurrent COL7A1 mutations in dystrophic epidermolysis bullosa: ethnic-specific and worldwide recurrent mutations. Arch Dermatol Res. 2004;295:442–447. doi: 10.1007/s00403-003-0444-1. [DOI] [PubMed] [Google Scholar]
- 8.Pfendner E, Uitto J, Fine JD. Epidermolysis bullosa carrier frequencies in the US population. J Invest Dermatol. 2001;116:483–484. doi: 10.1046/j.1523-1747.2001.01279-11.x. [DOI] [PubMed] [Google Scholar]
- 9.Nakamura H, Sawamura D, Goto M, Sato-Matsumura KC, LaDuca J, Lee JY, Masunaga T, Shimizu H. The G2028R glycine substitution mutation in COL7A1 leads to marked inter-familiar clinical heterogeneity in dominant dystrophic epidermolysis bullosa. J Dermatol Sci. 2004;34:195–200. doi: 10.1016/j.jdermsci.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 10.Gardella R, Zoppi N, Zambruno G, Barlati S, Colombi M. Different phenotypes in recessive dystrophic epidermolysis bullosa patients sharing the same mutation in compound heterozygosity with two novel mutations in the type VII collagen gene. Br J Dermatol. 2002;147:450–457. doi: 10.1046/j.1365-2133.2002.04914.x. [DOI] [PubMed] [Google Scholar]
- 11.Sawamura D, Goto M, Yasukawa K, Sato-Matsumura K, Nakamura H, Ito K, Nakamura H, Tomita Y, Shimizu H. Genetic studies of 20 Japanese families of dystrophic epidermolysis bullosa. J Hum Genet. 2005;50:543–546. doi: 10.1007/s10038-005-0290-4. [DOI] [PubMed] [Google Scholar]
- 12.Christiano AM, Fine JD, Uitto J. Genetic basis of dominantly inherited transient bullous dermolysis of the newborn: a splice site mutation in the type VII collagen gene. J Invest Dermatol. 1997;109:811–814. doi: 10.1111/1523-1747.ep12341013. [DOI] [PubMed] [Google Scholar]
- 13.Cserhalmi-Friedman PB, McGrath JA, Mellerio JE, Romero R, Salas-Alanis JC, Paller AS, Dietz HC, Christiano AM. Restoration of open reading frame resulting from skipping of an exon with an internal deletion in the COL7A1 gene. Lab Invest. 1998;78:1483–1492. [PubMed] [Google Scholar]
- 14.Mellerio JE, Salas-Alanis JC, Amaya-Guerra M, Tamez E, Ashton GH, Mohammedi R, Eady RA, McGrath JA. A recurrent frameshift mutation in exon 19 of the type VII collagen gene (COL7A1) in Mexican patients with recessive dystrophic epidermolysis bullosa. Exp Dermatol. 1999;8:22–29. doi: 10.1111/j.1600-0625.1999.tb00344.x. [DOI] [PubMed] [Google Scholar]
- 15.Gardella R, Castiglia D, Posteraro P, Bernardini S, Zoppi N, Paradisi M, Tadini G, Barlati S, McGrath JA, Zambruno G, Colombi M. Genotype-phenotype correlation in italian patients with dystrophic epidermolysis bullosa. J Invest Dermatol. 2002;119:1456–1462. doi: 10.1046/j.1523-1747.2002.19606.x. [DOI] [PubMed] [Google Scholar]
- 16.Tamai K, Murai T, Mayama M, Kon A, Nomura K, Sawamura D, Hanada K, Hashimoto I, Shimizu H, Masunaga T, Nishikawa T, Mitsuhashi Y, Ishida-Yamamoto A, Ikeda S, Ogawa H, McGrath JA, Pulkkinen L, Uitto J. Recurrent COL7A1 mutations in Japanese patients with dystrophic epidermolysis bullosa: positional effects of premature termination codon mutations on clinical severity. Japanese Collaborative Study Group on Epidermolysis Bullosa. J Invest Dermatol. 1999;112:991–993. doi: 10.1046/j.1523-1747.1999.00601.x. [DOI] [PubMed] [Google Scholar]
- 17.Järvikallio A, Pulkkinen JL, Uitto J. Molecular basis of dystrophic epidermolysis bullosa: mutations in the type VII collagen gene (COL7A1) Hum Mutat. 1997;10:338–347. doi: 10.1002/(SICI)1098-1004(1997)10:5<338::AID-HUMU2>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 18.Gayraud B, Hopfner B, Jassim A, Aumailley M, Bruckner-Tuderman L. Characterization of a 50-kDa component of epithelial basement membranes using GDA-J/F3 monoclonal antibody. J Biol Chem. 1997;272:9531–9538. doi: 10.1074/jbc.272.14.9531. [DOI] [PubMed] [Google Scholar]
- 19.Rimoin DL, Connor MJ, Pyeritz RE, Korf BR, Emery AEH, editors. Principles and Practice of Medical Genetics. 4 ed. Churchill Livingstone Publishers; NY: 2002. [Google Scholar]
- 20.Pulkkinen L, Uitto J. Mutation analysis and molecular genetics of epidermolysis bullosa. Matrix Biol. 1999;18:29–42. doi: 10.1016/s0945-053x(98)00005-5. [DOI] [PubMed] [Google Scholar]