

Abstract
Mutations within exons are responsible for aberrant splicing of pre-mRNA in several human disease genes and in some viral systems. Nonsense, missense, and even synonymous mutations can induce aberrant skipping of the mutant exon, producing nonfunctional proteins. In this paper, we describe the effect on the splicing efficiency of the synonymous variant 2811 G>T [Gly893Gly] detected in a patient of Italian descent affected by a mild form of cystic fibrosis, until now mentioned as sequence variation with unknown functional consequences. The study, performed through DNA as well as RNA analyses, shows that this mutation creates a new 5′ splice site within exon 15, resulting in a transcript lacking 76 amino acid residues. Although this aberrant splicing causes a shorter exon 15, the downstream exonic sequence from exon 16 to the end of the open reading frame is in frame. This study indicates that apparently neutral polymorphism, which may be erroneously classified as nonpathogenic, may indeed led to aberrant splicing thereby resulting in defective protein.
Cystic fibrosis (CF; MIM# 219700) is the most frequent severe autosomic recessive disorder in the European population.1 Indeed, CF affects about 1 in 2500 births, and approximately 1 in 25 individuals are heterozygotes, with marked regional variations.2 CF is caused by mutations of the cystic fibrosis transmembrane conductance regulator (CFTR or ABCC7; MIM# 602421) gene, which is also involved in a broad spectrum of phenotypes including male infertility, due to congenital bilateral absence of the vas deferens,3,4 disseminated bronchiectasis,5,6 and chronic pancreatitis.7,8 The mutational spectrum of the disease is made up of more than 1600 different mutations, 98% of which consists of point mutations or microdeletions/insertions.9 A number of cases, however, still remain uncharacterized, even after extensive sequence of whole coding region.
Sequence analysis of the CFTR gene frequently identifies exon/intron sequence variations whose association with the disease phenotype is unclear. In fact, the pathological effects of apparently benign polymorphisms, such as codon third position variations, or nucleotide change/deletion in the intronic noncanonical splicing regulatory elements, are difficult to assess.
In this study, we describe the effect on the splicing efficiency of the 2811G>T [Gly-Gly] synonymous mutation found in an Italian patient affected by a mild form of CF.
Materials and Methods
Patient
The patient is a 31-year-old woman of Italian descent. The clinical features are typical of a mild phenotype including pancreatic sufficiency, hyponatremia, hypochloremia, metabolic alkalosis, positive sweat test, and mild pulmonary involvement.
DNA Analysis
The patient's genomic DNA was extracted from 5 ml of peripheral blood, according to standard procedure. Informed consent was obtained before collecting the patient's sample. Screening of the most frequent CF mutations was performed by INNO-LiPA CFTR 19, CFTR 17+Tn update, and CFTR Italian Regional kits (Innogenetics, Gent, Belgium), providing a multiparameter screening for 57 CFTR gene mutations.
Sequence analysis of all 27 exons and their flanking regions was performed according to the dideoxy-chain termination method of Sanger using the Big-Dye Termination cycle sequencing kit (Applied Biosystems, Foster City, CA). Sequencing reactions were run on ABI PRISM 3130XL (Applera).
Multiplex ligation-dependent probe amplification analysis of all 27 CFTR exons was performed to detect large rearrangements (MLPA CFTR kit, MRC-Holland, the Netherlands).
RNA Analysis
Using a cytobrush, nasal epithelial cells were collected from the patient and two non-CF control subjects. RNA was extracted using TRIzol LS (Life Technologies, Carlsbad, CA). cDNA synthesis was performed with the High Capacity cDNA Archive kit (Applied Biosystems) according to the manufacturer's instructions. The cDNA was amplified in six overlapping fragments as previously described.10 PCR products were electrophoresed on a 2% agarose gel.
As the patient's parents were not available, to define the phase of the W1282X and 2811 G>T mutations, extra-long PCR was performed in patient's cDNA using GeneAmp XL PCR kit (Applera), according to the manufacturer's instructions.
Extra-long PCR was performed using exon 14a forward 5′-GTGCTTAGTAATTTTTCTGGCAGAG-3′ and exon 21 reverse 5′-TTTCCATATTTCTTGATCA-3′ primers. PCR products were electrophoresed on a 0.8% agarose gel and sequenced as above described.
Results
After an extensive second level molecular screening (ie, direct sequencing for searching of point mutations and multiplex ligation-dependent probe amplification for detection of large rearrangements) was performed at the DNA level on 441 unrelated Italian CF patients, we selected one patient in which we detected the W1282X mutation and the synonymous mutation 2811 G>T. The first is a severe nonsense mutation localized on exon 20 (3978 G>T), the second is a synonymous mutation [Gly893Gly] localized on exon 15, reported by Bonizzato (unpublished data, 2006), as sequence variation with unknown functional consequences (http://www.genet.sickkids.on.ca/cftr) (Figure 1, A−C).
Figure 1.
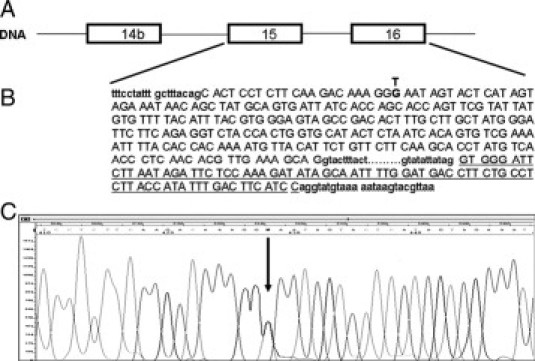
The patient's CFTR gene relevant fragment. A: CFTR DNA region spanning exons 14b to 16. B: DNA sequence of exons 15 and 16. The nucleotide 2811 which creates a new donor site is showed in bold. Sequence of exon 16 is underlined. C: Electropherogram of patient's DNA showing the 2811 G>T mutation in heterozygous state (arrow).
To define the effect at transcript level of the 2811G>T mutation, we performed RNA studies on the selected patient.
In the patient's cDNA, agarose gel electrophoresis of PCR product spanning exons 13 to 17 showed 714- and 484-base pair (bp) fragments (Figure 2A). Sequence analysis showed that the 714-bp fragment contained all five exons correctly spliced, while the 484-bp fragment lacked 230 nucleotides of exon 15 (Figure 2B) from nucleotide 2810 to nucleotide 3040. The same PCR procedure performed on cDNA of three non-CF control subjects produced only the normal 714-bp fragment, as expected. At DNA level, the 2811 G>T mutation creates a new 5′ splice site inside exon 15 causing a transcript lacking 76 amino acid residues. CFTR mRNA sequence analysis showed that, although this aberrant splicing caused a shorter exon 15, the downstream exonic sequence from exon 16 to the end of the open reading frame was in frame (Figure 2C).
Figure 2.
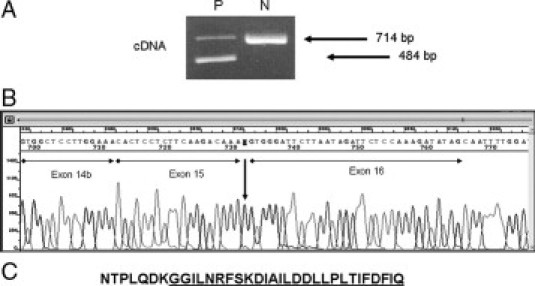
A: 2% agarose gel electrophoresis (ethidium bromide stained) of amplified cDNA fragment spanning exons 13 to 17, in the patients (P) and a normal control (N). The 484-bp fragment lacks of 230 nucleotides of exon 15. B: Sequence analysis of patient's cDNA showing the skipping of 230 bp of exon 15. C: Patient's amino acids residues of exons 15 to 16 showing that the downstream exonic sequence is in frame. Exon 16 amino acids residues are underlined.
As patient's parents were not available, we performed, in patient's cDNA, extra-long PCR spanning exons 14b-21 in order to define the phase of the 2811G>T and W1282X mutations.
Agarose gel electrophoresis of PCR products showed two fragments of 1124 and 839 base pairs. Sequence analysis showed the presence of the W1282X mutation in the 1124-bp fragments. On the other hand, sequence analysis of the 839-bp fragment showed that exon 15 lacked 230 nucleotides. These results indicate that the 2811G>T and W1282X mutations are in trans.
Molecular analysis of CFTR exon 15 performed in the remaining 440 CF patients did not show the 2811 G>T mutation. Furthermore, it was not found among the 266 control (non-CF) samples from the Italian population.
At the protein level, the 2811G>T mutation should remove part of the IV extracellular loop containing two glycosylated sites, transmembrane segment 8 in the second membrane spanning domain, and part of the third cytoplasmic loop.
There are some reports concerning the involvement of the CFTR membrane-spanning domains to the pore ion channel. In particular, the arginine 347 in transmembrane segment 6 of first membrane-spanning domain forms a salt bridge with asparagine 924 in transmembrane segment 8 of the second membrane-spanning domain. The salt bridge helps to stabilize the architecture of the pore to allow for ion conductance.11 Consequently, the protein produced by the 2811G>T mutation probably modify the structure and the regulation of the chloride (Cl−) channel.
Discussion
In this paper, we describe the effect on the splicing process of the synonymous variant 2811 G>T [Gly893Gly] detected in a patient of Italian descent who is affected by a mild form of CF. This mutation creates a new 5′ splice site within exon 15 causing a transcript lacking 76 amino acid residues.
Mutations within exons are responsible for aberrant splicing of pre-mRNA in several human disease genes, including ataxia telangiectasia,12 SMN2,13,14,15 BRCA1,16 neurofibromin 1,17 and CFTR18,19,20 genes, and in some viral systems such as HIV-1.21 Nonsense, missense, and even synonymous mutations can induce aberrant skipping of the mutant exon producing nonfunctional proteins.
In clinical diagnosis, synonymous variations have been routinely classified as innocuous polymorphisms and are assumed to be functionally neutral. A well-known exception regards synonymous changes that directly affect the splice sites, such as those that create a consensus splice site inside the exon22,23 or occur in the last nucleotide of the exon thereby resulting in exon skipping.24 For this reason CFTR mRNA analysis represents a good tool for the identification of unknown molecular defects of the CFTR gene and may allow to define the pathogenic role of sequence variations not yet characterized and splicing defects causing alternative products.10,25
However, since patients' RNA is not always available, evaluation of the role of sequence variations through dedicated bioinformatics tools may be useful.
This study indicates again that apparently neutral polymorphism, which may be erroneously classified as nonpathogenic, may indeed led to aberrant splicing thereby resulting in defective protein. These molecular mechanisms affecting the function of a protein are quite common considering that about one quarter of the synonymous changes studied in CFTR exon 918 and CFTR exon 1226 produce a splicing defect.
The growing interest on the potential effects of single-nucleotide changes in coding and noncoding regions on the extent and accuracy of pre-mRNA splicing is expected to have a significant impact on the diagnosis and treatment of genetic diseases. The correct classification of mutations is essential to understand structure−function relationship in the corresponding protein, to assess the prognosis in CF patients, and to devise new therapies.
Acknowledgements
We thank Prof. Carla Colombo (Centro Regionale di Riferimento-Fibrosicistica, Milano, Italy) for clinical evaluation.
Footnotes
Supported by grants from the Italian Cystic Fibrosis Research Foundation adopted by Silvana Ratti, Pesaro, Italy.
References
- 1.Welsh MJ, Tsui LC, Boat TF, Beaudet AI: Cystic fibrosis. The methabolic and molecular bases of inherited disease. Edited by CR Scriver, AI Beaudet, WS Sly, D Valle. New York, 1995, pp 3799–3876
- 2.Estivill X, Bancells C, Ramos C. The Biomed CF Mutation Analysis Consortium. Geographic distribution and regional origin of 272 cystic fibrosis mutations in European population. Hum Mutat. 1997;10:135–154. doi: 10.1002/(SICI)1098-1004(1997)10:2<135::AID-HUMU6>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 3.Chillon M, Casals T, Mercier B, Brassas L, Lissens W, Silber S, Romey M, Ruiz-Romero J, Verlingue C, Claustres M, Nunes V, Ferec C, Estvill X. Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. N Engl J Med. 1995;332:1475–1480. doi: 10.1056/NEJM199506013322204. [DOI] [PubMed] [Google Scholar]
- 4.Costes B, Girodon E, Ghanem N, Flori E, Jardin A, Soufir JC, Goossens M. Frequent occurrence of the CFTR intron 8 (TG)n5T allele in men with congenital absence of the vas deferens. Eur J Hum Genet. 1995;3:285–293. doi: 10.1159/000472312. [DOI] [PubMed] [Google Scholar]
- 5.Pignatti PF, Bombieri C, Marigo C, Benetazzo M, Luisetti M. Increased incidence of cystic fibrosis gene mutations in adults with disseminated bronchiectasis. Hum Mol Genet. 1995;4:635–639. doi: 10.1093/hmg/4.4.635. [DOI] [PubMed] [Google Scholar]
- 6.Girodon E, Cazeneuve C, Lebargy F, Chinet T, Costes B, Ghanem N, Martin J, Lemay S, Scheid P, Housset B, Bignon J, Goossens M. CFTR gene mutations in adults with disseminated bronchiectasis. Eur J Hum Genet. 1997;5:149–155. [PubMed] [Google Scholar]
- 7.Cohn JA, Friedman KJ, Noone PG, Knowels MR, Silverman LM, Jowel PS. Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N Engl J Med. 1998;339:653–658. doi: 10.1056/NEJM199809033391002. [DOI] [PubMed] [Google Scholar]
- 8.Sharer N, Schawrz M, Malone G, Howarth A, Painter J, Super M, Brazanga J. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N Engl J Med. 1998;339:645–652. doi: 10.1056/NEJM199809033391001. [DOI] [PubMed] [Google Scholar]
- 9.Bobadilla JL, Macek M, Jr, Fine JP, Farrell PM. Cystic fibrosis: A worldwide analysis of CFTR mutations-correlations with incidence data and application to screening. Hum Mut. 2002;19:575–606. doi: 10.1002/humu.10041. [DOI] [PubMed] [Google Scholar]
- 10.Faa' V, Pellegrini Bettoli P, Demurtas M, Zanda M, Ferri V, Cao A, Rosatelli MC. A new insertion/deletion of the cystic fibrosis transmembrane conductance regulator gene accounts for 3.4% of cystic fibrosis mutations in Sardinia: implications for population screening. J Mol Diagn. 2006;8:499–503. doi: 10.2353/jmoldx.2006.050146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cotten JF, Welsh MJ. Cystic fibrosis-associated mutations at arginine 347 alter the pore architecture of CFTR. J Biol Chem. 1999;274:5429–5435. doi: 10.1074/jbc.274.9.5429. [DOI] [PubMed] [Google Scholar]
- 12.Teraoka SN, Telatar M, Becker-Catania S, Liang T, Önengüt S, Tolun A, Chessa L, Sanal Ö, Bernatowska E, Gatti RA, Concannon P. Splicing defects in the ataxia-telangiectasia gene ATM: underlying mutations and consequences. Am J Hum Genet. 1999;64:1617–1631. doi: 10.1086/302418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lorson CL, Androphy EJ. An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum Mol Genet. 2000;9:259–265. doi: 10.1093/hmg/9.2.259. [DOI] [PubMed] [Google Scholar]
- 14.Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet. 2002;30:377–384. doi: 10.1038/ng854. [DOI] [PubMed] [Google Scholar]
- 15.Kashima T, Manley JL. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet. 2003;34:460–463. doi: 10.1038/ng1207. [DOI] [PubMed] [Google Scholar]
- 16.Liu HX, Cartegni L, Zhang MQ, Krainer AR. A mechanism for exon skipping caused by nonsense or missense mutations in BRCA1 and other genes. Nat Genet. 2001;27:55–58. doi: 10.1038/83762. [DOI] [PubMed] [Google Scholar]
- 17.Ars E, Serra E, Garcia J, Kruyer H, Gaona A, Lazaro C, Estivill X. Mutations affecting mRNA splicing are the most common molecular defects in patients with neurofibromatosis type 1. Hum Mol Genet. 2000;9:237–247. doi: 10.1093/hmg/9.2.237. [DOI] [PubMed] [Google Scholar]
- 18.Pagani F, Buratti E, Stuani C, Baralle FE. Missense, nonsense, and neutral mutations define juxtaposed regulatory elements of splicing in cystic fibrosis transmembrane regulator exon 9. J Biol Chem. 2003;278:26580–26588. doi: 10.1074/jbc.M212813200. [DOI] [PubMed] [Google Scholar]
- 19.Pagani F, Stuani C, Tzetis M, Kanavakis E, Efthymiadou A, Doudounakis S, Casals T, Baralle FE. New type of disease causing mutations: the example of the composite exonic regulatory elements of splicing in CFTR exon 12. Hum Mol Genet. 2003;12:1111–1120. doi: 10.1093/hmg/ddg131. [DOI] [PubMed] [Google Scholar]
- 20.Aznarez I, Chan EM, Zielenski J, Blencowe BJ, Tsui LC. Characterization of disease-associated mutations affecting an exonic splicing enhancer and two cryptic splice sites in exon 13 of the cystic fibrosis transmembrane conductance regulator gene. Hum Mol Genet. 2003;12:2031–2040. doi: 10.1093/hmg/ddg215. [DOI] [PubMed] [Google Scholar]
- 21.Caputi M, Zahler AM. SR proteins and hnRNP H regulate the splicing of the HIV-1 tev-specific exon 6D. EMBO J. 2002;21:845–855. doi: 10.1093/emboj/21.4.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richard I, Beckmann JS. How neutral are synonymous codon mutations? Nat Genet. 1995;10:259. doi: 10.1038/ng0795-259. [DOI] [PubMed] [Google Scholar]
- 23.Li X, Park W-J, Pyeritz RE, Jabs EW. Effect on splicing of a silent FGFR2 mutation in Crouzon syndrome. Nat Genet. 1995;10:232–233. doi: 10.1038/ng0395-232. [DOI] [PubMed] [Google Scholar]
- 24.Akli S, Chelly J, Mezard C, Gandy S, Kahn A, Poenaru L. A “G” to “A” mutation at position-1 of a 5′ splice site in a late infantile form of Tay-Sachs disease. J Biol Chem. 1990;265:7324–7330. [PubMed] [Google Scholar]
- 25.Faa' V, Incani F, Meloni A, Corda D, Masala M, Baffico AM, Seia M, Cao A, Rosatelli MC. Characterization of a disease-associated mutation affecting a putative splicing regulatory element in intron 6b of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene. J Biol Chem. 2009;284:30024–30031. doi: 10.1074/jbc.M109.032623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pagani F, Raponi M, Baralle FE. Synonymous mutations in CFTR exon 12 affect splicing and are not neutral in evolution. Proc Natl Acad Sci USA. 2005;102:6368–6372. doi: 10.1073/pnas.0502288102. [DOI] [PMC free article] [PubMed] [Google Scholar]