

Abstract
Objective
To report a novel mutation in TGFBI (Gen-Bank NM_000358), p.Met619Lys, associated with a variant of combined granular-lattice corneal dystrophy.
Methods
Slitlamp examination and DNA collection from the proband and affected and unaffected relatives. All 17 exons of TGFBI were amplified and sequenced in the proband. Exon 14 was amplified and sequenced in the proband’s family members and in 100 controls. Histopathologic examination of the excised corneal buttons from the proband and 3 family members was also performed.
Results
Affected individuals demonstrated an age-dependent phenotype, with the progression from central subepithelial needlelike deposits in younger individuals to polymorphic anterior stromal opacities in older family members. Screening of TGFBI in the proband demonstrated a novel mutation, p.Met619Lys, which was also present in all affected family members. Histopathologic examination revealed stromal deposits that stained with the Congo red and Masson trichrome stains as well as an antibody to the protein product of TGFBI.
Conclusions
We present a unique corneal dystrophy phenotype associated with the novel p.Met619Lys mutation in TGFBI.
It has been 10 years since Munier and colleagues1 published their seminal article that identified pathogenic mutations in the transforming growth factor β–induced gene (TGFBI) in families with a number of autosomal dominant corneal dystrophies, including lattice, granular, combined granular-lattice, and corneal dystrophy of the Bowman layer, type I. While a significant number of mutations have been identified in TGFBI during the last 10 years, many of which are associated with an atypical phenotype, most affected patients demonstrate a conserved mutation in either codon 124 or 555.2 Thus, a well-conserved genotype-phenotype correlation exists for the classic forms of the TGFBI dystrophies, with an atypical phenotype suggesting a less commonly encountered or novel underlying mutation.3-6
If one includes histopathologic with clinical findings in defining the affected phenotype, in no other TGFBI corneal dystrophy is the genotype-phenotype correlation as invariant as with combined granular-lattice corneal dystrophy. Dystrophic corneal stromal amyloid deposition is most commonly associated with the p.Arg124Cys mutation in the TGFBI gene, which defines the classic form of lattice corneal dystrophy. However, 26 other mutations in TGFBI have also been associated with variants of the lattice corneal dystrophy phenotype,3-5,7-39 18 of which have been confirmed histopathologically to be associated with stromal amyloid deposition.* Similarly, dystrophic stromal hyaline deposition in the absence of amyloid deposition is most commonly associated with the p.Arg555Trp mutation in the TGFBI gene, which defines the classic form of granular corneal dystrophy. However, 3 other mutations in TGFBI have been associated with variants of the granular corneal dystrophy phenotype,24-26,37,41,42 2 of which have been confirmed histopathologically to be associated with stromal hyaline deposition.24,25,37 In each of the 25 families reported to date in which histopathologic examination demonstrated corneal stromal amyloid and hyaline deposition and TGFBI screening has been performed, the p.Arg124His mutation has been identified.25,34,43-47 None of the other 34 reported mutations in TGFBI have been associated with corneal stromal amyloid and hyaline deposition, suggesting that the nature and location of the p.Arg124His mutation has a unique effect on the structure and function of the TGFBI protein (TGFBIp). Against the backdrop of this invariant phenotype-genotype relationship, we present a family with an atypical variant of lattice corneal dystrophy associated with a novel missense mutation in TGFBI, p.Met619Lys. The distinct clinical phenotype in several affected members, suggestive of an uncommon or novel underlying mutation, was confirmed to be associated with a novel TGFBI mutation through screening of the proband and affected and unaffected family members. However, the demonstration of both amyloid and hyaline deposits on histopathologic examination of several affected individuals violates the previously absolute phenotype-genotype correlation that had existed for combined granular-lattice corneal dystrophy. Thus, this article highlights that clinical and histopathologic features alone cannot be relied on to accurately diagnose and categorize the corneal dystrophies.
METHODS
REPORT OF CASES
Proband
The proband (II-4, Figure 1) is a 52-year-old Hispanic woman who was referred to one of the authors (A.J.A.) for corneal transplantation following an aborted corneal transplant in the left eye that had been associated with spontaneous expulsion of the crystalline lens. The patient stated that she initially noted reduced corrected visual acuity approximately 7 years before initial examination but denied a history of recurrent corneal erosions. Two of her siblings had previously undergone corneal transplantation as had her mother. Additionally, her maternal grandfather had “died blind,” and each of his 4 brothers had had poor eyesight that was not correctable with glasses.
Figure 1.

Pedigree of a 3-generation Hispanic family with a unique transforming growth factor β–induced (TGFBI)–associated corneal dystrophy. Presence of the wild-type allele (+) or the mutant allele is shown. * indicates members of the family who underwent examination, DNA collection, and TGFBI screening.
Uncorrected visual acuity measured 20/40 OD and hand motions OS. Slitlamp examination of the right eye revealed spine-like anterior stromal opacities characterized by a linear deposit with fine extensions along its length. In addition, polymorphic, semiconfluent opacities were axially distributed in the anterior corneal stroma and midcorneal stroma (Figure 2). A penetrating keratoplasty was successfully performed in the patient’s left eye, followed by a secondary intraocular lens placement 1 year later. Twenty months following corneal transplantation, no evidence of recurrent dystrophic deposition was noted in the corneal transplant, and the patient’s uncorrected visual acuity was 20/30 OS. The patient subsequently underwent an uneventful penetrating keratoplasty in the right eye as well.
Figure 2.
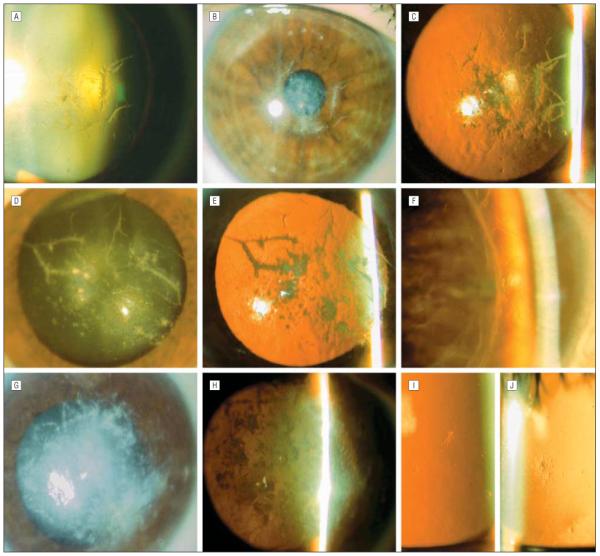
Slitlamp photomicrographs. The 52-year-old proband’s right cornea demonstrates spinelike and polymorphic central stromal opacities seen in retroillumination against the lens (A), with direct illumination (B), and against the red reflex (C). The proband’s 75-year-old mother has axially distributed branching linear opacities in the left cornea, seen with direct (D) and indirect (E) illumination. F, The proband’s 59-year-old brother, demonstrating thin branching anterior stromal lines in the host cornea adjacent to the edge of the corneal transplant in the right eye. G, The proband’s 55-year-old sister demonstrates confluent gray-white central stromal opacities seen with direct illumination. H, Retroillumination against the red reflex demonstrates that the deposits are in clumps, not linear aggregates, and cause significant diminution of the red reflex owing to the confluence of the deposits. The proband’s 46-year-old sister (I) and 26-year-old son (J) demonstrate focal stellate subepithelial opacities.
Additional Cases
Ten of the proband’s family members were examined (Figure 1). The proband’s 75-year-old mother (I-2) underwent a penetrating keratoplasty in her right eye at age 73 years. Slitlamp examination of her right eye demonstrated a few branching linear opacities in the peripheral host cornea but a clear graft, with no evidence of recurrent dystrophic deposits. Examination of the left cornea demonstrated axially distributed, linear branching anterior and midstromal deposits that appeared gray-white on direct illumination and translucent on retroillumination. In addition, discrete and semiconfluent anterior and midstromal polymorphic deposits, reminiscent of the stromal deposits associated with polymorphic amyloid degeneration, were noted (Figure 2).
The proband’s 59-year-old brother (II-1) developed decreased vision in each eye at approximately age 48 years without associated recurrent corneal erosions. He underwent corneal transplantation 5 (left eye) and 6 (right eye) years later, with no evidence of recurrence of dystrophic deposits in either corneal graft noted 5 years after the corneal transplant was performed in the right eye. The peripheral host corneal tissue demonstrated linear branching opacities that appeared similar to those characteristic of classic lattice corneal dystrophy (Figure 2).
The proband’s 55-year-old sister (II-3) underwent a penetrating keratoplasty in her right eye less than 1 year before examination. She denied a history of recurrent corneal erosions but did complain of significantly reduced vision in her left eye. Slitlamp examination demonstrated a clear graft in the right eye and dense, confluent, gray-while anterior and midstromal deposits involving the central 6 to 8 mm of the left cornea. Although a few linear extensions of the central opacity were noted superiorly, most of the deposits appeared in clumps and not in linear forms as noted in the proband. Superficial vascularization of the inferior cornea was also noted, giving an overall appearance reminiscent of gelatinous droplike corneal dystrophy (Figure 2). The patient subsequently underwent a penetrating keratoplasty in the left eye as well.
The proband’s 46-year-old sister (II-8) denied any history of ocular complaints, such as decreased vision, previous ocular trauma or surgery, and recurrent corneal erosions. Slitlamp examination of her right eye revealed a single fine stellate opacity in the cornea, located at the level of the Bowman layer, overlying the inferior pupillary border. Two similar-appearing opacities were noted in the subepithelial region of the left cornea, with an otherwise unremarkable anterior segment examination (Figure 2).
The proband’s eldest child (III-1), a 26-year-old man, denied a history of recurrent corneal erosions and decreased vision. On examination of the right cornea, a small, stellate-shaped subepithelial opacity was noted (Figure 2); no opacities were noted in the left cornea. The proband’s other 2 male off-spring, aged 25 (III-2) and 21 (III-4) years, had no ocular complaints and had normal corneal evaluations. The proband’s 25-year-old daughter (III-3) had no ocular complaints and no corneal abnormalities in her right eye. Careful evaluation of the left cornea revealed a single translucent, needle-shaped central subepithelial deposit shorter than 0.5 mm, which was sufficient to classify her as affected.
PATIENT IDENTIFICATION AND SPECIMEN COLLECTION
The researchers followed the tenets of the Declaration of Helsinki in the treatment of the patients described here. After institutional review board approval was granted, informed consent was obtained from the proband as well as affected and unaffected family members. A slitlamp examination was performed to determine each individual’s disease status (affected or unaffected).
DNA COLLECTION AND ANALYSIS
Buccal epithelial swab (CytoSoft Cytology Brush; Medical Packaging Corp, Camarillo, California) samples were collected from the proband and family members. Genomic DNA was prepared from the buccal epithelial cells using the QIAamp DNA Mini Kit spin protocol (Qiagen, Valencia, California). DNA previously collected from 100 healthy individuals served as control samples.
POLYMERASE CHAIN REACTION AMPLIFICATION AND DNA SEQUENCING
All 17 exons of TGFBI were amplified in the proband using previously reported primers and conditions.3 Exon 14 was amplified in the proband’s family members to screen for the p.Met619Lys mutation. Purification of the polymerase chain reaction products and automated DNA sequencing were then performed using the reagents and settings that have been reported previously.3 Nucleotide sequences, read manually and with Mutation Surveyor, version 2.2 (Softgenetics, State College, Pennsylvania), were compared with the published TGFBI complementary DNA sequence.
HISTOPATHOLOGIC EXAMINATION
The corneal buttons excised from the proband at the time of penetrating keratoplasty were fixed in neutral buffered formaldehyde, 10%. They were analyzed with light microscopy after staining with hematoxylin and eosin, Congo red, periodic acid–Schiff, and Masson trichrome.
IMMUNOHISTOCHEMICAL EXAMINATION
Immunohistochemistry was performed on slides prepared from paraffin-embedded sections of the corneal buttons from the proband using a goat polyclonal antibody directed against keratoepithelin (R&D Systems Inc, Minneapolis, Minnesota) at a concentration of 0.5 μg/mL. Antibody binding was detected using an RTU Vectastain Universal Quick Kit (Vector Laboratories, Burlingame, California) and a 3′3-diaminobenzidine hydrochloride peroxidase substrate kit (Vector Laboratories). Slides were then counterstained with Hematoxylin QS (Vector Laboratories).
RESULTS
TGFBI MUTATION ANALYSIS
Sequencing of all 17 exons of TGFBI in the proband revealed an unreported nucleotide substitution, c.1903T>A, in the heterozygous state, predicted to result in a novel missense mutation, p.Met619Lys, in exon 14 (Figure 3). In addition, 4 previously reported synonymous substitutions were identified, each in the heterozygous state: c.698C>G (p.Leu217Leu; rs1442); c.1028A>G (p.Val327Val; rs1054124); c.1463C>T (p.Leu472Leu; rs1133170); and c.1667T>C (p.Phe540Phe; rs4669).
Figure 3.
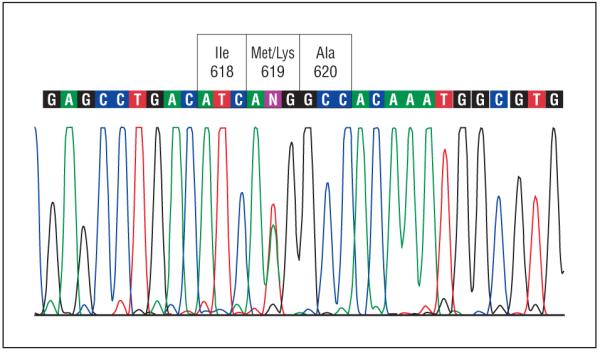
Portion of coding region of TGFBI exon 14 around codon 619 in the proband demonstrating heterozygous T to A transition at nucleotide 1903, resulting in a change in the encoded amino acid residue at codon 619 from methionine (Met) to lysine (Lys). Ala indicates alanine; Ile, isoleucine.
Screening of TGFBI exon 14 in the proband’s family members revealed the c.1903T>A nucleotide substitution in the heterozygous state in each of the 6 clinically affected individuals (Figure 1). The c.1903T>A substitution was also identified in 1 of the 4 clinically unaffected family members screened, the 25-year-old son (III-2) of the proband. None of the 100 samples from the controls demonstrated the c.1903T>A sequence variant.
LIGHT MICROSCOPY
Light microscopic examination of both excised corneal buttons from the proband revealed numerous large stromal deposits that appeared eosinophilic with the hematoxylineosin stain (Figure 4). Periodic acid–Schiff and Congo red stains also highlighted deposition of material at the level of the Bowman layer as well as the stromal deposits that stained with hematoxylin-eosin. The Congo red–stained deposits demonstrated birefringence and dichroism under polarized light with the use of polarizing filters characteristic of amyloid fibrils (Figure 4). The stromal deposits also demonstrated significant staining with Masson trichrome, characteristic of the granular or hyaline deposits in the TGFBI dystrophies.
Figure 4.
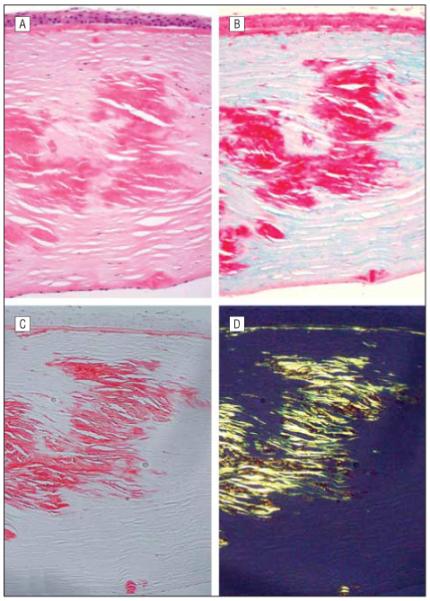
Histopathologic examination of the excised corneal button from the right eye of the proband. Large eosinophilic deposits are noted in the anterior, mid, and posterior corneal stroma (A, hematoxylin-eosin, original magnification ×200). The deposits noted stain brightly with Masson trichrome (B, original magnification ×200) and Congo red (C, original magnification ×250). The Congo red-stained section demonstrates dichroism with the use of polarizing filters (D), produced by the presence of amyloid fibrils in the stromal deposits.
Histopathologic examination of the excised corneal buttons from the other family members who underwent penetrating keratoplasty also had findings consistent with combined granular-lattice corneal dystrophy. In both of the corneal buttons from the proband’s 55-year-old sister (II-3) and the corneal button from the proband’s mother (I-2), eosinophilic stromal deposits were noted, which demonstrated staining with Congo red as well as birefringence and dichroism when viewed with a polarized light source. As observed in the proband’s cornea, these Congo red–stained deposits also demonstrated staining with Masson trichrome. However, in the corneal button from the proband’s 55-year-old sister (Figure 2), Masson trichrome revealed intensely bright staining deposits surrounding the amyloid deposits. Immunostaining of both corneal buttons from the proband with an antibody to TGFBIp showed intense reactivity of the Congo red– and Masson trichrome–staining stromal deposits, confirming that the stromal deposits consist of TGFBIp (Figure 5).
Figure 5.
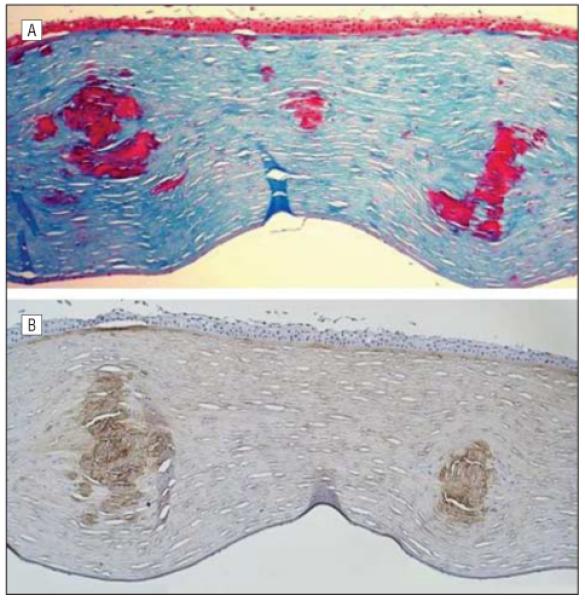
Histopathologic and immunohistochemical analysis of the excised corneal button from the left eye of the proband. A, Large stromal deposits stain bright red with Masson trichrome (original magnification ×200). B, The same deposits also stain with an antibody to transforming growth factor β–induced (TGFBI) protein, indicating that the deposits represent focal aggregations of the protein product of the TGFBI gene (original magnification ×200).
COMMENT
The corneal dystrophy associated with the p.Met619Lys mutation in TGFBI is best classified as a variant of combined granular-lattice corneal dystrophy. The clinical features in affected individuals are sufficiently similar to those of the previously reported classic and variant forms of lattice corneal dystrophy that the presumptive clinical diagnosis was a dystrophy associated with a TGBFI mutation. However, the clinical features were sufficiently atypical that a rare or novel TGFBI mutation was suspected and later confirmed. The presence of only a single focal corneal deposit in the 25- and 26-year-old affected children of the proband is not surprising, given that the TGFBI dystrophies are characterized by a progressive increase in the number and size of dystrophic deposits over time. Additionally, the identification of the p.Met619Lys mutation in the clinically unaffected 25-year-old son (III-2) of the proband is not surprising, given plausible explanations such as incomplete penetrance or a delayed onset of the affected phenotype. The development of a delayed-onset variant of lattice corneal dystrophy has been reported numerous times, most commonly in association with a mutation in exon 14 of TGFBI4,35,39,48 (as was identified in the family we report). Unexpected findings were that the proband’s 46-year-old sibling (II-8) demonstrated only very subtle corneal deposits and the proband’s 55-year-old sibling (II-3) demonstrated significantly greater corneal involvement than her 75-year-old mother (I-2). We previously reported4 the intrafamilial clinical variability observed in this pedigree in another family with a TGFBI dystrophy; this strengthens the contention that though the phenotypic expression is primarily determined by the effect of the identified mutation on the structure and function of the encoded protein, the genetic background of each individual as well as environmental factors likely influence the manner and degree of expression.
The p.Met619Lys mutation likely produces dystrophic corneal deposition through interference with an essential function of TGFBIp: cell adhesion. The TGFBIp contains 4 domains of high-sequence similarity that are also highly conserved across several species.49 Each of these domains, known as fas-1 domains owing to the original description of homologous domains in the insect cell-adhesion molecule fasciclin I,50 contains highly conserved sequences that are thought to be essential for cell adhesion.51 Kim and colleagues51 have identified 2 such conserved peptide sequences in TGFBIp, 1 of which is glutamic acid, proline, aspartic acid, isoleucine, and methionine (amino acids 615-619) in the fourth fas-1 domain, as the essential motifs for mediating cell adhesion. Mutation of either amino acid 617 (aspartic acid) or 618 (isoleucine) results in a loss of cell adhesion mediated by the fourth fas-1 domain. Thus, it is quite plausible that a mutation involving amino acid 619 (methionine), located in the curved β strand β6,52 would also result in loss of the cell adhesion mediated by the fourth fas-1 domain and thus in a partial or complete loss of function of TGFBIp.
What remains to be elucidated is the mechanism through which the p.Met619Lys mutation results in the clinical and histopathologic features observed in the family that we describe. It is inaccurate to assume that identification of the mutated amino acid residue in TGFBI is sufficient to predict the morphology and nature of the resultant corneal deposits, as is exemplified by the association of various missense substitutions involving the arginine residue at codon 124 with morphologically distinct dystrophies: p.Arg124Cys with classic lattice corneal dystrophy; p.Arg124His with combined granular-lattice corneal dystrophy; p.Arg124Leu with corneal dystrophy of the Bowman layer type I; and p.Arg124Ser with a variant of granular corneal dystrophy.26,37 Although the extracellular dystrophic deposits in each of these dystrophies contain mutant TGFBIp,44,53-55 each is characterized by clinically and histopathologically distinct amyloid or hyaline deposition, or both in the case of combined granular-lattice corneal dystrophy. Clout and Hohenester52 have proposed that mutations at codons 124 and 555 may produce dystrophic deposition through interference with protein-protein interactions, while the less commonly identified mutations in the fourth fas-1 domain of TGBFI may cause protein misfolding, resulting in abolition of secretion or significant protein destabilization. However, the novel p.Met619Lys mutation that we report, located in the fourth fas-1 domain of TGFBI, results in the deposition of both corneal stromal amyloid and hyaline, which has previously been associated only with the arginine to histidine mutation at codon 124. Thus, dystrophies that are produced by mutations in different domains of TGFBI, presumably through different mechanisms of inducing protein dysfunction, may have more clinical and histopathologic features in common than dystrophies that result from different substitutions of the same nucleotide. A complete understanding of the TGFBI dystrophies, therefore, involves not only an appreciation of the clinical and histopathologic features associated with the classic and variant forms of each but also knowledge of the underlying molecular genetic basis for the development of the dystrophic TGFBIp deposits.
Clinical Relevance.
The atypical and variable phenotype and the demonstration of both hyaline and amyloid stromal deposits indicate that neither clinical nor histopathologic features may be relied on to accurately diagnose and classify the corneal dystrophies.
Acknowledgments
Funding/Support: This study was supported by grant K08 EY016079 from the National Institutes of Health and by the Emily Plumb Estate and Trust (Dr Aldave).
Previous Presentation: This study was presented in part at the 2007 Annual Meeting of the Association for Research in Vision and Ophthalmology; May 10, 2007; Fort Lauderdale, Florida.
Footnotes
REFERENCES
- 1.Munier FL, Korvatska E, Djemai A, et al. Kerato-epithelin mutations in four 5q31-linked corneal dystrophies. Nat Genet. 1997;15(3):247–251. doi: 10.1038/ng0397-247. [DOI] [PubMed] [Google Scholar]
- 2.Aldave AJ, Sonmez B. Elucidating the molecular genetic basis of the corneal dystrophies: are we there yet? Arch Ophthalmol. 2007;125(2):177–186. doi: 10.1001/archopht.125.2.177. [DOI] [PubMed] [Google Scholar]
- 3.Aldave AJ, Rayner SA, Kim BT, Prechanond A, Yellore VS. Unilateral lattice corneal dystrophy associated with the novel His572del mutation in the TGFBI gene. Mol Vis. 2006;12:142–146. [PubMed] [Google Scholar]
- 4.Aldave AJ, Rayner SA, King JA, Affeldt JA, Yellore VS. A unique corneal dystrophy of Bowman’s layer and stroma associated with the Gly623Asp mutation in the transforming growth factor β-induced (TGFBI) gene. Ophthalmology. 2005;112(6):1017–1022. doi: 10.1016/j.ophtha.2004.12.044. [DOI] [PubMed] [Google Scholar]
- 5.Aldave AJ, Gutmark JG, Yellore VS, et al. Lattice corneal dystrophy associated with the Ala546Asp and Pro551Gln missense changes in the TGFBI gene. Am J Ophthalmol. 2004;138(5):772–781. doi: 10.1016/j.ajo.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 6.Kannabiran C, Klintworth GK. TGFBI gene mutations in corneal dystrophies. Hum Mutat. 2006;27(7):615–625. doi: 10.1002/humu.20334. [DOI] [PubMed] [Google Scholar]
- 7.Endo S, Nguyen TH, Fujiki K, et al. Leu518Pro mutation of the β ig-h3 gene causes lattice corneal dystrophy type I. Am J Ophthalmol. 1999;128(1):104–106. doi: 10.1016/s0002-9394(99)00053-7. [DOI] [PubMed] [Google Scholar]
- 8.Fujiki K, Hotta Y, Nakayasu K, et al. Six different mutations of TGFBI (βig-h3, keratoepithelin) gene found in Japanese corneal dystrophies. Cornea. 2000;19(6):842–845. doi: 10.1097/00003226-200011000-00015. [DOI] [PubMed] [Google Scholar]
- 9.Hirano K, Hotta Y, Fujiki K, Kanai A. Corneal amyloidosis caused by Leu518Pro mutation of βig-h3 gene. Br J Ophthalmol. 2000;84(6):583–585. doi: 10.1136/bjo.84.6.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirano K, Nakamura M, Yamamoto N, Hotta Y. Geographical feature of lattice corneal dystrophy patients in Aichi Prefecture: an analysis of the TGFBI gene [in Japanese] Nippon Ganka Gakkai Zasshi. 2002;106(6):352–359. [PubMed] [Google Scholar]
- 11.Fujiki K, Nakayasu K, Kanai A. Corneal dystrophies in Japan. J Hum Genet. 2001;46(8):431–435. doi: 10.1007/s100380170041. [DOI] [PubMed] [Google Scholar]
- 12.Ha NT, Fujiki K, Hotta Y, Nakayasu K, Kanai A. Q118X mutation of M1S1 gene caused gelatinous drop-like corneal dystrophy: the P501T of BIGH3 gene found in a family with gelatinous drop-like corneal dystrophy. Am J Ophthalmol. 2000;130(1):119–120. doi: 10.1016/s0002-9394(00)00596-1. [DOI] [PubMed] [Google Scholar]
- 13.Kawasaki S, Nishida K, Quantock AJ, Dota A, Bennett K, Kinoshita S. Amyloid and Pro501 Thr-mutated βig-h3 gene product colocalize in lattice corneal dystrophy type IIIA. Am J Ophthalmol. 1999;127(4):456–458. doi: 10.1016/s0002-9394(98)00360-2. [DOI] [PubMed] [Google Scholar]
- 14.Mashima Y, Yamamoto S, Inoue Y, et al. Association of autosomal dominantly inherited corneal dystrophies with BIGH3 gene mutations in Japan. Am J Ophthalmol. 2000;130(4):516–517. doi: 10.1016/s0002-9394(00)00571-7. [DOI] [PubMed] [Google Scholar]
- 15.Tian X, Fujiki K, Wang W, et al. Novel mutation (V505D) of the TGFBI gene found in a Chinese family with lattice corneal dystrophy, type I. Jpn J Ophthalmol. 2005;49(2):84–88. doi: 10.1007/s10384-004-0167-7. [DOI] [PubMed] [Google Scholar]
- 16.Tsujikawa K, Tsujikawa M, Yamamoto S, Fujikado T, Tano Y. Allelic homogeneity due to a founder mutation in Japanese patients with lattice corneal dystrophy type IIIA. Am J Med Genet. 2002;113(1):20–22. doi: 10.1002/ajmg.10709. [DOI] [PubMed] [Google Scholar]
- 17.Yamamoto S, Okada M, Tsujikawa M, et al. A kerato-epithelin (βig-h3) mutation in lattice corneal dystrophy type IIIA. Am J Hum Genet. 1998;62(3):719–722. doi: 10.1086/301765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujiki K, Hotta Y, Nakayasu K, et al. A new L527R mutation of the βig-h3 gene in patients with lattice corneal dystrophy with deep stromal opacities. Hum Genet. 1998;103(3):286–289. doi: 10.1007/s004390050818. [DOI] [PubMed] [Google Scholar]
- 19.Funayama T, Mashima Y, Kawashima M, Yamada M. Lattice corneal dystrophy type III in patients with a homozygous L527R mutation in the TGFBI gene. Jpn J Ophthalmol. 2006;50(1):62–64. doi: 10.1007/s10384-005-0260-6. [DOI] [PubMed] [Google Scholar]
- 20.Kawashima M, Yamada M, Funayama T, Mashima Y. Six cases of late-onset lattice corneal dystrophy associated with gene mutations induced by the transforming growth factor-β [in Japanese] Nippon Ganka Gakkai Zasshi. 2005;109(2):93–100. [PubMed] [Google Scholar]
- 21.Nakagawa E, Sakimoto T, Inada N, et al. Histopathological study of lattice corneal dystrophy with L 527 R mutation of transforming growth factor-β induced gene [in Japanese] Nippon Ganka Gakkai Zasshi. 2004;108(2):118–123. [PubMed] [Google Scholar]
- 22.Yamada N, Chikama TI, Morishige N, et al. Homozygous mutation (L527R) of TGFBI in an individual with lattice corneal dystrophy. Br J Ophthalmol. 2005;89(6):771–773. doi: 10.1136/bjo.2004.056168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chakravarthi SV, Kannabiran C, Sridhar MS, Vemuganti GK. TGFBI gene mutations causing lattice and granular corneal dystrophies in Indian patients. Invest Ophthalmol Vis Sci. 2005;46(1):121–125. doi: 10.1167/iovs.04-0440. [DOI] [PubMed] [Google Scholar]
- 24.Dighiero P, Drunat S, D’Hermies F, Renard G, Delpech M, Valleix S. A novel variant of granular corneal dystrophy caused by association of 2 mutations in the TGFBI gene: R124L and ΔT125-ΔE126. Arch Ophthalmol. 2000;118(6):814–818. doi: 10.1001/archopht.118.6.814. [DOI] [PubMed] [Google Scholar]
- 25.Dighiero P, Niel F, Ellies P, et al. Histologic phenotype-genotype correlation of corneal dystrophies associated with eight distinct mutations in the TGFBI gene. Ophthalmology. 2001;108(4):818–823. doi: 10.1016/s0161-6420(00)00662-x. [DOI] [PubMed] [Google Scholar]
- 26.Munier FL, Frueh BE, Othenin-Girard P, et al. BIGH3 mutation spectrum in corneal dystrophies. Invest Ophthalmol Vis Sci. 2002;43(4):949–954. [PubMed] [Google Scholar]
- 27.Nakagawa Asahina S, Fujiki K, Enomoto Y, Murakami A, Kanai A. Case of late onset and isolated lattice corneal dystrophy with Asn544Ser (N544S) mutation of transforming growth factor β-induced (TGFBI, BIGH3) gene [in Japanese] Nippon Ganka Gakkai Zasshi. 2004;108(10):618–620. [PubMed] [Google Scholar]
- 28.Rozzo C, Fossarello M, Galleri G, et al. A common β ig-h3 gene mutation (Δ f540) in a large cohort of Sardinian Reis Bucklers corneal dystrophy patients [Mutations in Brief no. 180] Hum Mutat. 1998;12(3):215–216. [PubMed] [Google Scholar]
- 29.Solari HP, Ventura MP, Perez AB, Sallum JM, Burnier MN, Belfort R. TGFBI gene mutations in Brazilian patients with corneal dystrophy. Eye. 2007;21(5):587–590. doi: 10.1038/sj.eye.6702264. published online ahead of print January 27, 2006. doi:10.1038/sj.eye.6702264. [DOI] [PubMed] [Google Scholar]
- 30.Stix B, Leber M, Bingemer P, et al. Hereditary lattice corneal dystrophy is associated with corneal amyloid deposits enclosing C-terminal fragments of keratoepithelin. Invest Ophthalmol Vis Sci. 2005;46(4):1133–1139. doi: 10.1167/iovs.04-1319. [DOI] [PubMed] [Google Scholar]
- 31.Eifrig DE, Jr, Afshari NA, Buchanan HW, III, Bowling BL, Klintworth GK. Polymorphic corneal amyloidosis: a disorder due to a novel mutation in the transforming growth factor β-induced (BIGH3) gene. Ophthalmology. 2004;111(6):1108–1114. doi: 10.1016/j.ophtha.2003.09.043. [DOI] [PubMed] [Google Scholar]
- 32.Klintworth GK, Bao W, Afshari NA. Two mutations in the TGFBI (BIGH3) gene associated with lattice corneal dystrophy in an extensively studied family. Invest Ophthalmol Vis Sci. 2004;45(5):1382–1388. doi: 10.1167/iovs.03-1228. [DOI] [PubMed] [Google Scholar]
- 33.Afshari N, Bahadur RP, Klintworth GK. Discovery of novel homozygous mutation in the TGFBI (BIGH3) gene (V624M) in a patient with unilateral lattice corneal dystrophy [ARVO abstract 1517] Invest Ophthalmol Vis Sci. 2004:45. doi: 10.1167/iovs.03-1228. [DOI] [PubMed] [Google Scholar]
- 34.Afshari NA, Mullally JE, Afshari MA, et al. Survey of patients with granular, lattice, avellino, and Reis-Bucklers corneal dystrophies for mutations in the BIGH3 and gelsolin genes. Arch Ophthalmol. 2001;119(1):16–22. [PubMed] [Google Scholar]
- 35.Stewart H, Black GC, Donnai D, et al. A mutation within exon 14 of the TGFBI (BIGH3) gene on chromosome 5q31 causes an asymmetric, late-onset form of lattice corneal dystrophy. Ophthalmology. 1999;106(5):964–970. doi: 10.1016/S0161-6420(99)00539-4. [DOI] [PubMed] [Google Scholar]
- 36.Warren JF, Abbott RL, Yoon MK, Crawford JB, Spencer WH, Margolis TP. A new mutation (Leu569Arg) within exon 13 of the TGFBI (BIGH3) gene causes lattice corneal dystrophy type I. Am J Ophthalmol. 2003;136(5):872–878. doi: 10.1016/s0002-9394(03)00541-5. [DOI] [PubMed] [Google Scholar]
- 37.Stewart HS, Ridgway AE, Dixon MJ, Bonshek R, Parveen R, Black G. Heterogeneity in granular corneal dystrophy: identification of three causative mutations in the TGFBI (BIGH3) gene-lessons for corneal amyloidogenesis. Hum Mutat. 1999;14(2):126–132. doi: 10.1002/(SICI)1098-1004(1999)14:2<126::AID-HUMU4>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 38.Chau HM, Ha NT, Cung LX, et al. H626R and R124C mutations of the TGFBI (BIGH3) gene caused lattice corneal dystrophy in Vietnamese people. Br J Ophthalmol. 2003;87(6):686–689. doi: 10.1136/bjo.87.6.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmitt-Bernard CF, Guittard C, Arnaud B, et al. BIGH3 exon 14 mutations lead to intermediate type I/IIIA of lattice corneal dystrophies. Invest Ophthalmol Vis Sci. 2000;41(6):1302–1308. [PubMed] [Google Scholar]
- 40.Dighiero P, Drunat S, Ellies P, et al. A new mutation (A546T) of the βig-h3 gene responsible for a French lattice corneal dystrophy type IIIA. Am J Ophthalmol. 2000;129(2):248–251. doi: 10.1016/s0002-9394(99)00324-4. [DOI] [PubMed] [Google Scholar]
- 41.Cung le X, Ha NT, Chau HM, et al. Mutation analysis of the TGFBI gene in Vietnamese with granular and Avellino corneal dystrophy. Jpn J Ophthalmol. 2004;48(1):12–16. doi: 10.1007/s10384-003-0009-z. [DOI] [PubMed] [Google Scholar]
- 42.Ha NT, Cung le X, Chau HM, et al. A novel mutation of the TGFBI gene found in a Vietnamese family with atypical granular corneal dystrophy. Jpn J Ophthalmol. 2003;47(3):246–248. doi: 10.1016/s0021-5155(03)00019-4. [DOI] [PubMed] [Google Scholar]
- 43.Konishi M, Yamada M, Nakamura Y, Mashima Y. Varied appearance of cornea of patients with corneal dystrophy associated with R124H mutation in the BIGH3 gene. Cornea. 1999;18(4):424–429. doi: 10.1097/00003226-199907000-00006. [DOI] [PubMed] [Google Scholar]
- 44.Konishi M, Yamada M, Nakamura Y, Mashima Y. Immunohistology of kerato-epithelin in corneal stromal dystrophies associated with R124 mutations of the BIGH3 gene. Curr Eye Res. 2000;21(5):891–896. doi: 10.1076/ceyr.21.5.891.5536. [DOI] [PubMed] [Google Scholar]
- 45.Korvatska E, Henry H, Mashima Y, et al. Amyloid and non-amyloid forms of 5q31-linked corneal dystrophy resulting from kerato-epithelin mutations at Arg-124 are associated with abnormal turnover of the protein. J Biol Chem. 2000;275(15):11465–11469. doi: 10.1074/jbc.275.15.11465. [DOI] [PubMed] [Google Scholar]
- 46.Korvatska E, Munier FL, Djemai A, et al. Mutation hot spots in 5q31-linked corneal dystrophies. Am J Hum Genet. 1998;62(2):320–324. doi: 10.1086/301720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mashima Y, Konishi M, Nakamura Y, et al. Severe form of juvenile corneal stromal dystrophy with homozygous R124H mutation in the keratoepithelin gene in five Japanese patients. Br J Ophthalmol. 1998;82(11):1280–1284. doi: 10.1136/bjo.82.11.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmitt-Bernard CF, Claustres M, Arnaud B, Demaille J, Argiles A. Lattice corneal dystrophy. Ophthalmology. 2000;107(9):1613–1614. doi: 10.1016/s0161-6420(00)00055-5. [DOI] [PubMed] [Google Scholar]
- 49.Kawamoto T, Noshiro M, Shen M, et al. Structural and phylogenetic analyses of RGD-CAP/βig-h3, a fasciclin-like adhesion protein expressed in chick chondrocytes. Biochim Biophys Acta. 1998;1395(3):288–292. doi: 10.1016/s0167-4781(97)00172-3. [DOI] [PubMed] [Google Scholar]
- 50.Zinn K, McAllister L, Goodman CS. Sequence analysis and neuronal expression of fasciclin I in grasshopper and Drosophila. Cell. 1988;53(4):577–587. doi: 10.1016/0092-8674(88)90574-0. [DOI] [PubMed] [Google Scholar]
- 51.Kim JE, Kim SJ, Lee BH, Park RW, Kim KS, Kim IS. Identification of motifs for cell adhesion within the repeated domains of transforming growth factor-β-induced gene, βig-h3. J Biol Chem. 2000;275(40):30907–30915. doi: 10.1074/jbc.M002752200. [DOI] [PubMed] [Google Scholar]
- 52.Clout NJ, Hohenester E. A model of FAS1 domain 4 of the corneal protein βig-h3 gives a clearer view on corneal dystrophies. Mol Vis. 2003;9:440–448. [PubMed] [Google Scholar]
- 53.Korvatska E, Munier FL, Chaubert P, et al. On the role of kerato-epithelin in the pathogenesis of 5q31-linked corneal dystrophies. Invest Ophthalmol Vis Sci. 1999;40(10):2213–2219. [PubMed] [Google Scholar]
- 54.Streeten BW, Qi Y, Klintworth GK, Eagle RC, Jr, Strauss JA, Bennett K. Immuno-localization of βig-h3 protein in 5q31-linked corneal dystrophies and normal corneas. Arch Ophthalmol. 1999;117(1):67–75. doi: 10.1001/archopht.117.1.67. [DOI] [PubMed] [Google Scholar]
- 55.Hedegaard CJ, Thogersen IB, Enghild JJ, Klintworth GK, Moller-Pedersen T. Transforming growth factor beta induced protein accumulation in granular corneal dystrophy type III (Reis-Bucklers dystrophy): identification by mass spectrometry in 15 year old two-dimensional protein gels. Mol Vis. 2003;9:355–359. [PubMed] [Google Scholar]