

Abstract
Spasticity, resulting in involuntary and sustained contractions of muscles, may evolve in patients with stroke, cerebral palsy, multiple sclerosis, brain injury, and spinal cord injury (SCI). The authors critically review the neural mechanisms that may contribute to spasticity after SCI and assess their likely degree of involvement and relative significance to its pathophysiology. Experimental data from patients and animal models of spasticity as well as computer simulations are evaluated. The current clinical methods used for the management of spasticity and the pharmacological actions of drugs are discussed in relation to their effects on spinal mechanisms. Critical assessment of experimental findings indicates that increased excitability of both motoneurons and interneurons plays a crucial role in pathophysiology of spasticity. New interventions, including forms of spinal electrical stimulation to suppress increased neuronal excitability, may reduce the severity of spasticity and its complications.
Keywords: spasticity, spasms, spinal cord injury, upper motor neuron syndrome, rehabilitation, neurological disorder
Introduction
Spasticity is one feature of the upper motor neuron syndrome and a major cause of disability in individuals with a variety of central nervous system diseases (including stroke, cerebral palsy, and multiple sclerosis) and trauma (such as spinal cord injury [SCI] and brain injury). Spasticity is usually defined as a velocity-dependent increase in the tonic stretch reflex (muscle tone) with exaggerated tendon jerks, clonus, and spasms, resulting from the hyperexcitability of the stretch reflex.1 Because spasticity results from lesions in the pyramidal and extrapyramidal pathways,2 its pathophysiology varies depending on the site of the lesion but commonly develops in the antigravity muscles. For instance, excessive muscle tone in the upper-extremity flexor muscles is prominent in spasticity following stroke, with relatively lesser involvement of muscle spasms. On the other hand, excessive muscle spasms in the lower-extremity extensor muscles are prominent following SCI. It is also believed that the mechanisms underlying spasticity in stroke and SCI are different. Neural mechanisms are thought to be the primary contributors to spasticity following SCI, whereas alterations in muscle contractile properties play a substantial role in spasticity following stroke. Accordingly, clinicians and researchers consider the classical definition of spasticity to be narrow and restrictive.3,4
The term spasticity has been increasingly used to refer to several features of the upper motor neuron syndrome.5 For example, in addition to muscle hypertonus, spasticity following SCI could also involve hyperreflexia, clonus, clasp-knife responses, long-lasting cutaneous reflexes, and muscle spasms evoked by brief nonnoxious cutaneous stimuli.4 In this article, we review the mechanisms contributing to these different features of the syndrome of spasticity after SCI and critically assess their possible involvement and significance to the pathophysiology of spasticity.
More than 80% of people with SCI have spasticity,6 and many have greater disability from it. Spasticity develops gradually over several months after injury. Immediately following SCI, the spinal cord becomes areflexic (spinal shock), a period characterized by loss of tendon reflexes below the level of the lesion, muscle paralysis, and flaccid muscle tone. Weeks after injury, various reflexes such as the tendon reflex, the flexor withdrawal reflex, and the Babinski sign appear.7 The threshold of the flexor reflexes, which are usually evoked by cutaneous stimulation, decreases over time to a point that brief stimulation of the foot plantar surface can evoke long-lasting flexor contractions.8 Extensor reflexes, which are usually evoked by proprioceptive stimuli, occur later and episodes of flexor/extensor muscle co-contraction become pronounced.8 Intense muscle spasms can also be triggered by various other stimuli such as heat/cold and bladder distention.9 Extensor spasms that cause episodic lower-extremity rigidity can at times provide assistance in dressing or walking but are usually painful and can be violent enough to expel a person out of a wheelchair. In individuals with incomplete SCI, spasticity may reduce the functional utility of residual voluntary motor control, compromising rehabilitation efforts.
Mechanisms Underlying Spasticity
The pathogenesis of spasticity following SCI in patients remains uncertain. The increased excitability of the stretch reflex in patients with spasticity directed research efforts toward investigating the spinal mechanisms modulating the excitability of this reflex (Figure 1) and the potential alteration in its excitability after SCI. We evaluated several proposed mechanisms and assessed their likelihood of involvement and relative significance in the pathophysiology of spasticity (Table 1). In this assessment, the likelihood of involvement of a particular mechanism in spasticity was based on 3 criteria: (1) detection of abnormality in the excitability of the mechanism in both humans and animal models of spasticity; (2) detection of abnormality in the same mechanism under different experimental conditions (ie, rest versus contraction); and (3) consistency in the mechanism’s alteration using multiple assessment measures. Pathologies present during movement (ie, during muscle contraction or functional tasks) were deemed to be especially relevant to spasticity because of the resulting movement impairment. A mechanism that is likely involved in spasticity was considered to be significant if an influential role of that mechanism in the manifestation of spasticity has been established experimentally and clinically.
Figure 1.
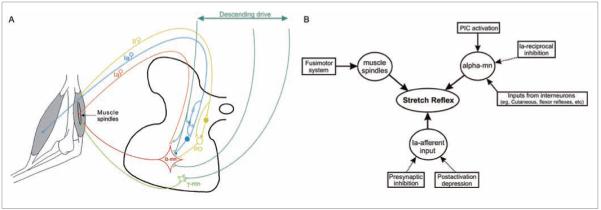
Schematic and block diagrams illustrating the main spinal circuits involved in the control of movement and the alteration in their excitability during spasticity. (A) Excitatory interneurons are represented by open circles and excitatory synapses by V-shaped bars. Inhibitory interneurons and inhibitory synapses are represented by filled circles. Pathways illustrated are the following: (1) the Ia-afferent fibers from 2 antagonistic muscles, their excitatory monosynaptic connection with the homonymous motoneuron (orange), and their inhibitory disynaptic reciprocal connection with the heteronymous motoneuron (blue); (2) the Ia-afferent inputs with their presynaptic inhibitory interneuron (PS, blue synapse); (3) group II afferents with their polysynaptic connections to the α-motoneuron through propriospinal neurons (PO, yellow); (4) γ-motoneurons innervating intrafusal muscle fibers (green); and (5) α-motoneurons innervating extrafusal muscle fibers (red). (B) Block diagram summarizing the alteration in excitability of spinal mechanisms contributing to the exaggerated reflexes in spasticity. Solid arrows indicate increased input and dotted arrows indicate reduced input after chronic spinal cord injury. PIC, persistent inward current; mn, motoneuron.
Table 1.
The Likelihood of Involvement of the Various Mechanisms Thought to Contribute to Spasticity After Spinal Cord Injury and Their Extent of Significance
| Mechanism | Involvement in Spasticity | Significance for Spasticity | References |
|---|---|---|---|
| Enhancement in the excitability of motoneurons | Most likely | High | 7, 10-12 |
| Enhancement in the excitability of interneurons | Most likely | High | 7, 13, 14 |
| Axonal sprouting | Likely | High | 15-17 |
| Reduction in presynaptic inhibition | Likely | Moderate | 18-20 |
| Reduction in postactivation depression | Likely | Uncertain | 21-24 |
| Reduction in Ia-reciprocal inhibition | Likely | Unclear | 25-27 |
| Fusimotor hyperexcitability | Unlikely | None | 28, 29 |
Fusimotor Hyperexcitability
The common characteristics between human spasticity and animal rigidity induced after intercollicular decerebration led to the assumption that the same spinal mechanisms contribute to the exaggeration of the stretch reflex in both conditions. In decerebrate animal rigidity, the exaggerated stretch reflex was attributed to the hyperactivity of γ-motoneurons that control the sensitivity of muscle spindles and stretch reflex gain.28 It was therefore posited that the hyperexcitability of the stretch reflex leading to spasticity after SCI is also because of an increase in the activity of γ-motoneurons (Figure 1). This hypothesis was refuted because direct recordings from muscle spindles in individuals with SCI showed no increases in activity during bouts of spasticity.29 Furthermore, despite the resemblance between spasticity and decerebrate animal rigidity, critical differences are found in the time course of development of each symptom. Spasticity develops weeks after SCI, whereas decerebrate animal rigidity appears within 1 hour after decerebration.30 The prolonged duration for the development of spasticity after SCI suggests the contribution of plastic changes in other spinal pathways to spasticity. Accordingly, fusimotor hyperexcitability is unlikely to be involved in the pathophysiology of spasticity after SCI.
Axonal Sprouting
Another mechanism for spasticity was proposed by McCouch et al.15 After SCI, terminals of the damaged axons on spinal neurons degenerate. By a few weeks postinjury, the remaining local afferents sprout new synaptic terminals in the vacated space and enhance the efficacy of the spinal connections. The prolongation in the time-to-peak of excitatory postsynaptic potentials (EPSPs) in spastic individuals supports the sprouting of Ia-afferents and the formation of new synapses on motoneurons.16 This proposed mechanism provides an explanation for the temporal changes in behavior after SCI and could contribute to spasticity by strengthening existing spinal circuits and/or increasing synaptic inputs to interneurons.17 It could also explain the disruption in the balance between excitatory and inhibitory inputs to motoneurons observed during spasticity as described in the following section. Thus, axonal sprouting is likely to contribute to the pathogenesis of spasticity after SCI. Given that axonal sprouting could contribute to several spinal mechanisms underlying spasticity, its significance to the pathophysiology of spasticity is high.
Alterations in Other Spinal Mechanisms
Evidence from experiments in humans and animal models of spasticity demonstrated that alterations in the excitability of numerous excitatory and inhibitory spinal pathways occur after SCI (Figure 1). These alterations are discussed below along with the pharmacological actions of antispasticity drugs used for management (Figure 2).
Figure 2.
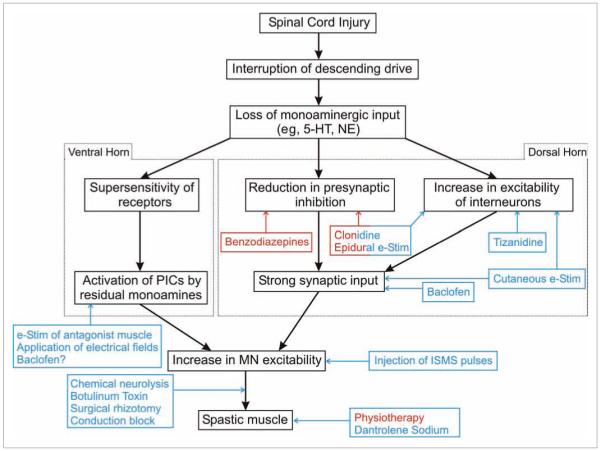
Block diagram illustrating the flow of events following spinal cord injury (SCI). The differential effect of monoamines on the dorsal and ventral horns is indicated. The site of action and nature of effect of the various drugs and interventions used for the management of spasticity after SCI are illustrated. Red color indicates a positive action, that is, increase, whereas a blue color indicates a negative action, that is, reduction. A description of the effect of each treatment method is provided in the text. 5-HT, serotonin; NE, norepinephrine; e-Stim, electrical stimulation; PICs, persistent inward currents; ISMS, intraspinal microstimulation
Reduction in postactivation depression
The size of the Hoffmann reflex (H-reflex) is usually reduced during repetitive stimulation of Ia muscle spindle fibers at stimulus rates above 0.3 pulses per second. This phenomenon is called postactivation dep ression (PAD) and results from the reduction in the amount of neurotransmitter release at the Ia–motoneuron synapse because of repeated activation.31 Reduction in PAD following SCI has been confirmed in humans as well as in animal models of SCI (Figure 1B).21,22 Therefore, it is likely that it is involved in the pathophysiology leading to spasticity. However, the significance of PAD to spasticity should be considered with caution as PAD was assessed in these studies only at rest, whereas the level of PAD is dependent on muscle activity (ie, rest vs contraction). In healthy individuals, the suppression of the H-reflex through PAD is diminished during volitional muscle contraction,23,24 and abolished during standing.24 Therefore, in spastic individuals, PAD might also be reduced during functional tasks; thus, the specific significance of its reduction at rest to spasticity following SCI is uncertain.
Reduction in presynaptic inhibition
Presynaptic inhibition (PSI) adjusts the strength of synaptic inputs to neurons by regulating the levels of neurotransmitter release (Figure 1). PSI is governed by descending drives in a state-dependent manner. Reduction in PSI has been reported in individuals with spasticity under both rest and muscle contraction conditions. In spastic individuals with SCI, PSI evoked by muscle or tendon vibration at rest was reduced,18 indicating an increase in the strength of the excitatory Ia–motoneuron synapse.19 The reduction in the magnitudes of the H-reflex and tendon jerks, however, could be a result of concurrent reduction in PAD, which is activated by vibration as well. The modulation of the H-reflex during muscle contraction was also altered in individuals with spasticity after incomplete SCI; the H-reflex disappeared during standing but was slightly depressed during walking relative to intact subjects.20 This lack of modulation of reflexes during walking probably contributes to the stiff gait observed after SCI.30 Thus, reduction in PSI is likely to be involved in the pathophysiology of spasticity following SCI. However, in contrast to findings in humans, no reduction in PSI was found in animal models of spasticity.32 Because of the inconsistency in results from humans and animal models, and the possibility of mixed actions with PAD, reduction in PSI appears to have moderate significance to spasticity. Nonetheless, PSI has been the target of many antispastic drugs. Baclofen (GABAB agonist), the most commonly used agent for the treatment of spasticity, acts in part by reducing neurotransmitter release from presynaptic terminals through the reduction of Ca2+ influx (Figure 2).33 Benzodiazepines (GABAA agonists) increase PSI at spinal and supraspinal sites (Figure 2)34 by opening chloride ion channels and inhibiting the presynaptic terminal.
Reduction in Ia-reciprocal inhibition
The reciprocal activation of antagonistic muscles during movement is mediated by a disynaptic inhibitory pathway called Ia-reciprocal inhibition (Figure 1). Alteration in the excitability of Ia-reciprocal inhibition in individuals with spasticity following SCI was reported, but contradictory results were obtained regarding the nature of this alteration. For instance, Ia-reciprocal inhibition was reduced in individuals with spasticity after incomplete SCI; they were unable to suppress the H-reflex when activating antagonistic muscles and showed abnormal coactivation of antagonistic muscles during isometric contractions.25 Further more, Crone et al26 found that Ia-reciprocal inhibition was replaced by reciprocal excitation of antagonistic muscles after SCI, which facilitated co-contraction of antagonistic muscles during movements. Conversely, Boorman et al27 found that Ia-reciprocal inhibition was increased in individuals with spasticity resulting from incomplete SCI. Therefore, alteration in Ia-reciprocal inhibition is likely to occur in spasticity following SCI, but the nature of alteration and its significance to the pathophysiology of spasticity is unclear.
Enhancement in the Excitability of Motoneurons
The enhancement in excitability of motoneurons is most likely involved in the pathophysiology of spasticity following SCI. First, it has been confirmed in spastic individuals and in animal models of spasticity.10,11,35,36 Second, it was observed during rest and muscle contraction. In people with spasticity caused by SCI, an increase in the ratio of potentials resulting from electrical stimulation of the sensory and motor axons (Hmax/Mmax) was observed at rest.12 An increase in the amplitude and persistence of the F-wave (the electrical potential resulting from antidromic activation of motoneurons) was also apparent,7 further supporting the enhancement of motoneuronal excitability. During muscle spasms, motor unit recordings indicated an increase in motoneuronal firing rate, causing vigorous muscle contraction.37 Third, the enhancement in motoneuronal excitability was consistently observed using multiple assessment measures.7,12,37 The enhancement in motoneuronal excitability was attributed to alterations in motoneuronal intrinsic properties, such as the activation of persistent inward currents (PICs) and depolarization of the membrane potential.10,11 These alterations have been extensively assessed in a validated chronic spinal rat model of spasticity,10,11,35 which established the ionic basis of the long-lasting exaggerated reflexes and muscle spasms associated with spasticity.
Activation of persistent inward currents
A PIC is a depolarizing current that does not inactivate with prolonged membrane depolarization. The activation of the motoneuronal PIC is regulated through the monoaminergic drive from the brainstem. After SCI, the PIC is no longer under the control of descending drive, leading to uncontrolled and high motoneuronal firing rates. The long-lasting exaggerated reflexes and muscle spasms seen during spasticity could be partly because of uncontrolled activation of PICs after injury.38 The activation of PICs transforms the response of motoneurons to brief volleys of Ia-afferent input from short to long-lasting firing activity.39 Therefore, activation of PICs causes self-sustained firing and maintained increases in motoneuronal excitability.
In spinal motoneurons, PICs are mediated by 2 types of ion channels with distinctive properties: the L-type calcium channels40 and persistent sodium channels.35,41 After SCI, the Ca2+ PIC, which has little time-dependent inactivation, underlies the long-lasting reflexes seen in spasticity,10,42 whereas the Na+ PIC, which becomes partially inactivated with time, is responsible for the initiation of the spontaneous firing commonly seen in association with a muscle spasm or voluntary contraction.10,42 Motor unit recordings from spastic individuals after SCI suggest that spasticity in humans involves similar Ca2+ and Na+ ionic mechanisms.42 Furthermore, recent computer models of motoneurons incorporating Ca2+ and/or Na+ PICs were able to exhibit self-sustained firing, plateau potentials, and sustained depolarization of the membrane potential that were similar to those seen during spasticity.43,44
Differential control of spinal neurons by descending drive
The monoaminergic drive from the brainstem to the spinal cord regulates the excitability of spinal neurons during different motor tasks. A number of neuromodulators have been shown to influence motoneurons, such as serotonin (5-HT), norepinephrine (NE), acetylcholine, thyroid releasing hormone, substance P, and adenosine. 5-HT and NE are the most extensively studied monoaminergic inputs. The monoaminergic drive has differential effects on the ventral and dorsal horns of the spinal cord (Figure 2). In the ventral horn, monoamines have excitatory effects on motoneurons and low-threshold muscle afferents (group I).45 Furthermore, 5-HT and NE enhance PIC activation,46 induce membrane depolarization,47 and decrease the amplitude of afterhyperpolarization.48 Collectively, these effects increase the excitability of the cell. Conversely, monoamines inhibit, through PSI, sensory inputs to motoneurons49 such as cutaneous50 and high-threshold muscle afferent inputs (groups III and IV).51 They also suppress inputs to deep dorsal horn interneurons from high-threshold afferents.52 This differential control by the monoaminergic drive in the spinal cord is attained through the effect of different monoaminergic receptors. For example, motoneuronal PIC facilitation is achieved through 5-HT253 and NE α1 receptors,46 whereas cutaneous and high-threshold muscle afferent inhibition is achieved through 5-HT1b/d and NE α2 receptors.54
This differential control is altered after SCI, a mechanism that contributes to the emergence of spasticity. Acutely, the loss of the monoaminergic drive reduces the excitability of motoneurons in the ventral horn but increases the size and duration of the polysynaptic EPSPs of sensory inputs mediated through the dorsal horn because of loss of inhibition.55 Despite the increase in the excitability of interneuronal pathways, long-lasting reflexes are not activated during this stage because of the reduced excitability of motoneurons. In the chronic stages of injury, motoneurons become highly sensitive to residual monoamines available below the level of the lesion. These facilitate the reactivation of the PICs and restore the motoneuronal excitability (Figure 2).56 Nonetheless, this reactivation of PICs is no longer regulated by the brainstem, which leads to uncontrolled motoneuronal firing evoked by synaptic inputs.10 In this stage, prolonged EPSPs generated by sensory stimuli can activate the PIC and trigger long-lasting reflexes and muscle spasms.55 Thus, spasticity does not emerge until motoneurons become hyperexcitable, regardless of the increases in interneuronal excitability early after SCI. This indicates that the enhancement in motoneuronal excitability plays a significant role in the pathophysiology of spasticity. Further support for this supposition is found in the action of the antispastic drug, baclofen, which partially acts on motoneurons and suppresses their excitability by reducing the Ca2+ PIC57 and shortens the duration of the monosynaptic EPSPs36 (Figure 2).
Enhanced Excitability of Interneurons
The enhancement in excitability of interneurons is also most likely involved in the pathophysiology of spasticity following SCI. This has been confirmed in humans and in animal models of spasticity. For example, the magnitude of flexor reflexes mediated by polysynaptic pathways was increased in individuals with spasticity after SCI.7 Mailis and Ashby13 reported an enhancement in the transmission through polysynaptic pathways from Ia-afferents to motoneurons. Schmit and Benz58 observed extensor spasms in individuals with SCI, similar to those seen clinically, when they shifted from a sitting to a supine position. The spasms were triggered by single-leg extensions about the hip. Thus, single-joint movements can also evoke multijoint responses after SCI.59 These observations support the contribution of increased excitability of interneuronal circuits to the hyperexcitability of reflexes in spasticity and the importance of hip afferents in triggering extensor spasms.60 Moreover, an increase in the ratio of excitatory-to-inhibitory inputs to motoneurons originating predominantly from spinal interneurons was observed during the development of spasticity.14 Collectively, evidence suggests that the enhancement in the excitability of interneurons plays a significant role in the pathophysiology of spasticity.
Several antispastic drugs have targeted the interneuronal polysynaptic pathways. Clonidine, an α2-adrenergic receptor agonist, suppresses polysynaptic reflexes11,61 and blocks the prolonged polysynaptic EPSPs that activate the PICs (Figure 2). It also enhances the PSI of sensory afferents.61 Tizanidine, another α2-adrenergic receptor agonist, acts presynaptically by reducing the release of excitatory neurotransmitters (glutamate and aspartate) from interneurons (Figure 2).62
In conclusion, multiple spinal mechanisms appear to be involved in the pathogenesis of spasticity after SCI. Therapeutic interventions targeting one or more of these mechanisms may be especially effective in reducing the severity of spasticity and related motor disabilities in people with SCI.
Current Treatments of Spasticity
Current clinical management of spasticity involves a wide variety of therapies ranging from noninvasive (eg, oral administration of antispastic drugs, physiotherapy) to invasive procedures (eg, surgical rhizotomy). The type and rate of treatment depend on the levels of spread (diffuse versus focal) and disability (mild vs severe) caused by spasticity.63 Common techniques for the management of spasticity and potential new interventions are summarized below.
Physical Techniques
Increased muscle tone causes spastic muscles to resist stretch and remain shortened for long durations. Prolonged muscle shortening leads to joint deformation and changes in the intrinsic properties of soft tissues and muscle fibers, which in turn restrict the range of motion63 and diminish the functional use of residual voluntary movements in individuals with incomplete SCI. These changes contribute a biomechanical component, in addition to the neural components discussed, to the disability resulting from spasticity. Whereas antispastic drugs act on the neural component of spasticity, physiotherapy minimizes the biomechanical side effects. Daily passive muscle stretching assists in reducing muscle tone and in maintaining joint mobility and range of motion (Figure 2). Orthoses are used to hold the limb in positions that resist contractures. Exercises are also performed to strengthen the spastic and synergistic muscles.63 Given the importance of hip afferents in triggering extensor spasms, careful positioning of the hip during patient manipulation (eg, transfers to/from wheelchairs) assists in minimizing the effects of those reflexes in individuals with SCI.64
Pharmacological and Surgical Approaches
Antispastic medications
Because oral medications produce systemic side effects, they are usually used in patients with diffuse spasticity. The most commonly used antispastic drugs are baclofen, benzodiazepine, clonidine, and tizanidine. These drugs could be used alone or in combination to obtain a desired effect and are administered orally or intrathecally. Because these medications produce nontargeted release of pharmacological agents, their major side effect is a general suppression of neuronal activity in individuals who already have a reduced voluntary drive. The majority of these drugs act by targeting the motoneuronal and/or interneuronal spinal mechanisms. Dantrolene sodium acts peripherally on intrafusal and extrafusal muscle fibers by decreasing the release of Ca2+ from the sarcoplasmic reticulum, which may reduce muscle force during contractions (Figure 2).65
Chemical neurolysis
Chemical neurolysis is usually used in individuals with focal spasticity or in those who want to assess the functional gain of future surgical treatments prior to the surgical procedure. A peripheral nerve innervating spastic muscles is injected with phenol or alcohol solutions (Figure 2), which destroy myelin. The effects of these solutions are proportional to their concentrations.63 Aqueous phenol is also used for motor point injection specifically in spastic muscles through an easy and safe procedure; however, the effectiveness of the injection diminishes over time and repeated injections are needed. The common complication is treatment failure resulting from poor localization of the nerve or an inadequate dose.63
Botulinum toxin
Botulinum toxin, injected intramuscularly, acts on the neuromuscular junction to inhibit the release of acetylcholine (Figure 2).66 The toxin causes chemical denervation of intrafusal and extrafusal muscle fibers, and its effect is reversible.67 The clinical effect of the toxin appears 24 to 72 hours following administration and lasts 2 to 6 months depending on the dosage. The treatment is effective in reducing pain and muscle spasm. The major side effect is excessive weakness of the treated muscle.
Surgical techniques
Surgical procedures are commonly used when spasticity cannot be managed by any of the aforementioned techniques. They involve the ablation of motor nerves and/or rhizotomy of sensory spinal roots to interrupt the sensory input (Figure 2).68 Ablation of motor nerves is effective when spasticity is focal in muscles innervated by the same nerve trunk and is usually selective in its suppression of spasticity without causing excessive muscle weakening. Preserving 25% of the motor fibers is necessary to maintain muscle tone.63 The main disadvantage of surgical procedures is that they are irreversible.
Electrical Stimulation
Various electrical stimulation modalities have been used to reduce the level of spasticity. These modalities range from surface electrical stimulation of muscles to electrical stimulation of the peripheral and central nerves.
Muscles
Surface electrical stimulation for the reduction of spasticity involves stimulation of the spastic muscles69 and/or their antagonists.70 In the former case, reduction of spasticity is attributed to stimulation of the cutaneous afferents, which could suppress motoneuronal excitability by depressing the propriospinal interneurons or induce long-term synaptic changes in primary afferents in the dorsal horn (Figure 2).71 In the latter case, reduction of spasticity may be attributed to the activation of the Ia-reciprocal inhibition pathway, which reduces the amplitude of the motoneuronal PIC and level of motoneuronal excitability (Figure 2).72
Peripheral nerves
Electrical stimulation of peripheral nerves to block exaggerated motor activity was examined in animal models (Figure 2). Application of direct current (DC)73 or high-frequency stimulation of various waveforms (sinusoidal and rectangular pulses) induced conduction block in peripheral nerves, which was complete and reversible.74 Despite their desired properties, electrical stimulation strategies of peripheral nerves lack specificity because of the fact that primary sensory fibers have larger diameters and lower extracellular activation thresholds than motor fibers and are more likely to be activated first. Therefore, this technique would block sensory and motor muscle activity and may prevent the transmission of residual voluntary activation of muscles that might remain after incomplete SCI.
Epidural
Electrical stimulation of the dorsal columns of the spinal cord through epidurally placed electrodes may reduce spasticity.75 Epidural stimulation can be effective in reducing mild spasticity,76 but its efficacy depends on electrode location and stimulation parameters.75 The clinical effect of epidural stimulation probably results from activation of inhibitory networks in the spinal cord,75 increasing the level of PSI,77 or activating inhibitory networks within the dorsal column–brainstem–spinal loop (Figure 2).78 However, epidural stimulation sometimes lacks long-term efficacy in reducing spasticity, pain, and muscle spasms after SCI.79
Intraspinal microstimulation
This technique aims to suppress the intrinsic excitability of spinal motoneurons through the application of extracellular current pulses using fine microwires implanted in the spinal cord (Figure 2).80 As demonstrated by computer simulations, the applied pulses regulate the membrane potential of the first node of Ranvier and modulate the increased motoneuronal firing activity after SCI. Various waveforms were tested in simulations for modulating the motoneuronal firing behavior, including subthreshold biphasic charge-imbalanced pulses and suprathreshold high-frequency alternating current (AC) pulses. The effect of these pulses ranged from a graded reduction to full blockade of the aberrantly high motoneuronal firing rate, respectively. The former pulses deactivate the Na+ channels at the first node of Ranvier and reduce axonal conduction, whereas the latter pulses inactivate the Na+ channels at the first node of Ranvier and block axonal conduction. These pulses are effective even in the presence of increased sprouting and changes in motoneuronal intrinsic properties encountered after long-term SCI.80 The proposed pulses could provide a means for reducing spasticity without prohibiting muscle activation through residual volitional drive. Figure 3 shows preliminary results from our laboratory demonstrating the effect of these pulses on the level of spasticity measured in vivo in the chronic spinal rat model. During the application of these pulses, the tail-resistive force to stretch (a measure of spasticity) was reduced by nearly 45% (Figure 3E).
Figure 3.

Effects of electrical stimulation techniques on the level of spasticity after spinal cord injury (SCI). Spasticity was assessed by measuring the tail-resistive forces through a force transducer connected to the rat tail, in response to sinusoidal side-to-side tail stretches. (A) Low tail-resistive forces measured from an adult intact rat, that is, no spasticity (control condition). (B) higher tail-resistive forces measured from an adult rat with chronic SCI at S2/S3 in response to the same level of sinusoidal tail stretches. (C) Intraspinal microstimulation (ISMS) current pulses (suprathreshold high-frequency sinusoidal pulses: 120 μA, 5 kHz) delivered through 4 microwires implanted in the sacral cord below the lesion of an adult rat with chronic (3 months) spinal injury; injection of ISMS pulses reduced the tail-resistive force in response to tail stretches. (D) AC electrical field (1 Hz, 100 mV/mm) applied along the mediolateral axis of the spinal cord through external metal plates during tail stretches of a chronic spinal rat (3 months) with spasticity; application of the electrical field slightly reduced the tail-resistive force. (E) Summary of mean tail-resistive forces in response to tail stretches under different conditions (before SCI, 3 weeks after SCI when spasticity was not fully developed, chronic SCI when spasticity was fully developed, chronic SCI during the injection of ISMS current pulses, and chronic SCI during the application of electrical fields). In these experiments, the fields were applied through external plates positioned noninvasively on the outside of the rat. The effectiveness of the fields is expected to increase substantially when applied through the metal plates/rods used to stabilize the spine. Data presented as mean ± SD. Asterisks represent the statistical significance level: **P < .01; ***P < .001.
Electrical fields
Electrical fields, in the form of DC or AC applied across the spinal cord through metal plates, have been shown through computer simulations to provide another means for modulating the firing rate of motoneurons after SCI.81 The electrical fields induce differential polarization in the various structures of the motoneuron and modulate the magnitude of the dendritic Ca2+ PIC, thereby suppressing their excitability (Figure 2). Application of electrical fields could alleviate spasticity by reducing the excessively high motoneuronal firing rates after chronic SCI. These proposed electrical fields could be practically implemented by passing electrical current between the metal plates used clinically to provide mechanical support to the spinal column after SCI. Figure 3D shows the effect of slow AC electrical fields, applied noninvasively in the chronic adult spinal rat model, on the level of spasticity. The tail-resistive force to stretch was reduced by nearly 14% during the application of the electrical field (Figure 3E).
In sum, available techniques for the clinical management of spasticity might mitigate one or few of the underlying pathological spinal mechanisms contributing to spasticity. Given the multifactorial origin of spasticity, a successful strategy would most likely require a combination of interventions to achieve the best clinical outcome.
Conclusion
Spasticity that leads to spasms, pain, and contractures is a debilitating secondary complication of SCI. Identification of potential mechanisms and their significance to the pathophysiology of spasticity is critical for the design of rehabilitation programs. The pathogenesis of spasticity resulting from SCI is multifactorial and extends beyond the stretch reflex. It also depends on the type, site, and duration of injury. The alteration in excitability of various inhibitory pathways has historically been studied the most, with less focus until recently on the alterations in excitability of motoneurons and interneurons themselves. This latter alteration appears to play critical roles in the manifestation of spasticity after SCI and could become the target of new pharmacological and clinical treatments. The new electrical stimulation interventions suggested by computer simulations for suppressing motoneuronal excitability seem promising, but more careful studies in animal models are required to establish the effectiveness of these techniques.
Acknowledgments
Funding
This work was funded by the National Institute for Neurological Disease and Stroke, National Institutes of Health (NINDS-NIH). S. M. Elbasiouny was supported by an Alberta Heritage Foundation for Medical Research (AHFMR) doctoral scholarship. He is currently supported by the Tim E. Noel fellowship from the Canadian Institutes of Health Research (CIHR) and the ALS Society of Canada. D. Moroz was supported by an AHFMR summer studentship. V. K. Mushahwar is an AHFMR Senior Scholar.
Footnotes
Declaration of Conflicting Interests
The authors declare that they do not have any conflict of interest.
References
- 1.Lance J. Pathophysiology of spasticity and clinical experience with baclofen. In: Lance J, Feldman R, Young R, Koella W, editors. Spasticity: Disordered Motor Control. Yearbook; Chicago, IL: 1980. pp. 185–204. [Google Scholar]
- 2.Burke D. Spasticity as an adaptation to pyramidal tract injury. Adv Neurol. 1988;47:401–423. [PubMed] [Google Scholar]
- 3.Pandyan A, Gregoric M, Barnes M, et al. Spasticity: clinical perceptions, neurological realities and meaningful measurement. Disabil Rehabil. 2005;27:2–6. doi: 10.1080/09638280400014576. [DOI] [PubMed] [Google Scholar]
- 4.Young RR. Spasticity: a review. Neurology. 1994;44:S12–S20. [PubMed] [Google Scholar]
- 5.Edwards S. Neurological Physiotherapy: A Problem-Solving Approach. 2nd ed. Churchill Livingstone; Edinburgh, NY: 2002. [Google Scholar]
- 6.Levi R, Hultling C, Seiger A. The Stockholm Spinal Cord Injury Study: 2. Associations between clinical patient characteristics and post-acute medical problems. Paraplegia. 1995;33:585–594. doi: 10.1038/sc.1995.125. [DOI] [PubMed] [Google Scholar]
- 7.Hiersemenzel L-P, Curt A, Dietz V. From spinal shock to spasticity: neuronal adaptations to a spinal cord injury. Neurology. 2000;54:1574–1582. doi: 10.1212/wnl.54.8.1574. [DOI] [PubMed] [Google Scholar]
- 8.Ashby P, McCrea D. Neurophysiology of spinal spasticity. In: Davidoff RA, editor. Handbook of the Spinal Cord. Marcel Dekker; New York, NY: 1987. pp. 120–143. [Google Scholar]
- 9.Little J, Micklesen P, Umlauf R, Britell C. Lower extremity manifestations of spasticity in chronic spinal cord injury. Am J Phys Med Rehabil. 1989;68:32–36. doi: 10.1097/00002060-198902000-00009. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, Gorassini MA, Bennett DJ. Role of persistent sodium and calcium currents in motoneuron firing and spasticity in chronic spinal rats. J Neurophysiol. 2004;91:767–783. doi: 10.1152/jn.00788.2003. [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Harvey PJ, Li X, Bennett DJ. Spastic long-lasting reflexes of the chronic spinal rat studied in vitro. J Neurophysiol. 2004;91:2236–2246. doi: 10.1152/jn.01010.2003. [DOI] [PubMed] [Google Scholar]
- 12.Delwaide PJ. Pathophysiological mechanisms of spasticity at the spinal cord level. In: Thilmann AF, Burke DJ, Rymer WZ, editors. Spasticity: Mechanisms and Management. Springer-Verlag; Heidelberg, Germany: 1993. pp. 296–308. [Google Scholar]
- 13.Mailis A, Ashby P. Alterations in group Ia projections to motoneurons following spinal lesions in humans. J Neurophysiol. 1990;64:637–647. doi: 10.1152/jn.1990.64.2.637. [DOI] [PubMed] [Google Scholar]
- 14.Kitzman P. Changes in vesicular glutamate transporter 2, vesicular GABA transporter and vesicular acetylcholine transporter labeling of sacrocaudal motoneurons in the spastic rat. Exp Neurol. 2006;197:407–419. doi: 10.1016/j.expneurol.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 15.McCouch GP, Austin GM, Liu CN, Liu CY. Sprouting as a cause of spasticity. J Neurophysiol. 1958;21:205–216. doi: 10.1152/jn.1958.21.3.205. [DOI] [PubMed] [Google Scholar]
- 16.Noguchi T, Homma S, Nakajima Y. Measurements of excitatory postsynaptic potentials in the stretch reflex of normal subjects and spastic patients. J Neurol Neurosurg Psychiatry. 1979;42:1100–1105. doi: 10.1136/jnnp.42.12.1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krenz NR, Weaver LC. Sprouting of primary afferent fibers after spinal cord transection in the rat. Neuroscience. 1998;85:443–458. doi: 10.1016/s0306-4522(97)00622-2. [DOI] [PubMed] [Google Scholar]
- 18.Faist M, Mazevet D, Dietz V, Pierrot-Deseilligny E. A quantitative assessment of presynaptic inhibition of la afferents in spastics: differences in hemiplegics and paraplegics. Brain. 1994;117:1449–1455. doi: 10.1093/brain/117.6.1449. [DOI] [PubMed] [Google Scholar]
- 19.Delwaide P. Human monosynaptic reflexes and presynaptic inhibition. In: Desmedt J, editor. New Developments in Electromyography and Clinical Neurophysiology. Karger; Basel, Switzerland: 1973. pp. 508–522. [Google Scholar]
- 20.Yang J, Fung J, Edamura M, Blunt R, Stein R, Barbeau H. H-reflex modulation during walking in spastic paretic subjects. Can J Neurol Sci. 1991;18:443–452. doi: 10.1017/s0317167100032133. [DOI] [PubMed] [Google Scholar]
- 21.Nielsen J, Petersen N, Ballegaard M, Biering-Sorensen F, Kiehn O. H-reflexes are less depressed following muscle stretch in spastic spinal cord injured patients than in healthy subjects. Exp Brain Res. 1993;97:173–176. doi: 10.1007/BF00228827. [DOI] [PubMed] [Google Scholar]
- 22.Thompson F, Parmer R, Reier P. Alteration in rate modulation of reflexes to lumbar motoneurons after midthoracic spinal cord injury in the rat: I. Contusion injury. J Neurotrauma. 1998;15:495–508. doi: 10.1089/neu.1998.15.495. [DOI] [PubMed] [Google Scholar]
- 23.Hultborn H, Nielsen J. Modulation of transmitter release from Ia afferents by their preceding activity: a “postactivation depression.”. In: Rudomin P, Romo R, Mendell LM, editors. Presynaptic Inhibition and Neural Control. Oxford University Press; New York, NY: 1998. pp. 178–191. [Google Scholar]
- 24.Stein R, Estabrooks K, McGie S, Roth M, Jones K. Quantifying the effects of voluntary contraction and inter-stimulus interval on the human soleus H-reflex. Exp Brain Res. 2007;182:309–319. doi: 10.1007/s00221-007-0989-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boorman GI, Lee RG, Becker WJ, Windhorst UR. Impaired “natural reciprocal inhibition” in patients with spasticity due to incomplete spinal cord injury. Electroencephalogr Clin Neurophysiol. 1996;101:84–92. doi: 10.1016/0924-980x(95)00262-j. [DOI] [PubMed] [Google Scholar]
- 26.Crone C, Johnsen LL, Biering-Sorensen F, Nielsen JB. Appearance of reciprocal facilitation of ankle extensors from ankle flexors in patients with stroke or spinal cord injury. Brain. 2003;126:495–507. doi: 10.1093/brain/awg036. [DOI] [PubMed] [Google Scholar]
- 27.Boorman G, Hulliger M, Lee RG, Tako K, Tanaka R. Reciprocal Ia inhibition in patients with spinal spasticity. Neurosci Lett. 1991;127:57–60. doi: 10.1016/0304-3940(91)90894-y. [DOI] [PubMed] [Google Scholar]
- 28.Rushworth G. Spasticity and rigidity: an experimental study and review. J Neurol Neurosurg Psychiatry. 1960;23:99–118. doi: 10.1136/jnnp.23.2.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hagbarth KE, Wallin G, Lofstedt L. Muscle spindle responses to stretch in normal and spastic subjects. Scand J Rehabil Med. 1973;5:156–159. [PubMed] [Google Scholar]
- 30.Pierrot-Deseilligny E, Burke D. The pathophysiology of spasticity and Parkinsonian rigidity. In: Pierrot-Deseilligny E, Burke D, editors. The Circuitry of the Human Spinal Cord: Its Role in Motor Control and Movement Disorders. Cambridge University Press; New York, NY: 2005. pp. 556–599. [Google Scholar]
- 31.Crone C, Nielsen J. Methodological implications of the post activation depression of the soleus H-reflex in man. Exp Brain Res. 1989;78:28–32. doi: 10.1007/BF00230683. [DOI] [PubMed] [Google Scholar]
- 32.Hultborn H, Malmsten J. Changes in segmental reflexes following chronic spinal cord hemisection in the cat: II. Conditioned monosynaptic test reflexes. Acta Physiol Scand. 1983;119:423–433. doi: 10.1111/j.1748-1716.1983.tb07358.x. [DOI] [PubMed] [Google Scholar]
- 33.Bussieres N, El Manira A. GABAB receptor activation inhibits N- and P/Q-type calcium channels in cultured lamprey sensory neurons. Brain Res. 1999;847:175–185. doi: 10.1016/s0006-8993(99)02002-8. [DOI] [PubMed] [Google Scholar]
- 34.Schlosser W. Action of diazepam on the spinal cord. Arch Int Pharmacodyn Ther. 1971;194:93–102. [PubMed] [Google Scholar]
- 35.Li Y, Bennett DJ. Persistent sodium and calcium currents cause plateau potentials in motoneurons of chronic spinal rats. J Neurophysiol. 2003;90:857–869. doi: 10.1152/jn.00236.2003. [DOI] [PubMed] [Google Scholar]
- 36.Li Y, Li X, Harvey PJ, Bennett DJ. Effects of baclofen on spinal reflexes and persistent inward currents in motoneurons of chronic spinal rats with spasticity. J Neurophysiol. 2004;92:2694–2703. doi: 10.1152/jn.00164.2004. [DOI] [PubMed] [Google Scholar]
- 37.Thomas CK, Ross BH. Distinct patterns of motor unit behavior during muscle spasms in spinal cord injured subjects. J Neurophysiol. 1997;77:2847–2850. doi: 10.1152/jn.1997.77.5.2847. [DOI] [PubMed] [Google Scholar]
- 38.Eken T, Hultborn H, Kiehn O. Possible functions of transmitter-controlled plateau potentials in alpha motoneurones. Prog Brain Res. 1989;80:257–267. doi: 10.1016/s0079-6123(08)62219-0. discussion 39-42. [DOI] [PubMed] [Google Scholar]
- 39.Hounsgaard J, Hultborn H, Jespersen B, Kiehn O. Intrinsic membrane properties causing a bistable behaviour of alpha-motoneurones. Exp Brain Res. 1984;55:391–394. doi: 10.1007/BF00237290. [DOI] [PubMed] [Google Scholar]
- 40.Hounsgaard J, Kiehn O. Ca++ dependent bistability induced by serotonin in spinal motoneurons. Exp Brain Res. 1985;57:422–425. doi: 10.1007/BF00236551. [DOI] [PubMed] [Google Scholar]
- 41.Lee RH, Heckman CJ. Essential role of a fast persistent inward current in action potential initiation and control of rhythmic firing. J Neurophysiol. 2001;85:472–475. doi: 10.1152/jn.2001.85.1.472. [DOI] [PubMed] [Google Scholar]
- 42.Gorassini MA, Knash ME, Harvey PJ, Bennett DJ, Yang JF. Role of motoneurons in the generation of muscle spasms after spinal cord injury. Brain. 2004;127:2247–2258. doi: 10.1093/brain/awh243. [DOI] [PubMed] [Google Scholar]
- 43.ElBasiouny SM, Bennett DJ, Mushahwar VK. Simulation of dendritic Cav1.3 channels in cat lumbar motoneurons: spatial distribution. J Neurophysiol. 2005;94:3961–3974. doi: 10.1152/jn.00391.2005. [DOI] [PubMed] [Google Scholar]
- 44.ElBasiouny SM, Bennett DJ, Mushahwar VK. Simulation of Ca+2 persistent inward currents in spinal motoneurons: mode of activation and integration of synaptic inputs. J Physiol (Lond) 2006;570:355–374. doi: 10.1113/jphysiol.2005.099119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hammar I, Jankowska E. Modulatory effects of alpha 1-, alpha 2-, and beta-receptor agonists on feline spinal interneurons with monosynaptic input from group I muscle afferents. J Neurosci. 2003;23:332–338. doi: 10.1523/JNEUROSCI.23-01-00332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee RH, Heckman CJ. Enhancement of bistability in spinal motoneurons in vivo by the noradrenergic alpha 1 agonist methoxamine. J Neurophysiol. 1999;81:2164–2174. doi: 10.1152/jn.1999.81.5.2164. [DOI] [PubMed] [Google Scholar]
- 47.Hsiao CF, Trueblood PR, Levine MS, Chandler SH. Multiple effects of serotonin on membrane properties of trigeminal motoneurons in vitro. J Neurophysiol. 1997;77:2910–2924. doi: 10.1152/jn.1997.77.6.2910. [DOI] [PubMed] [Google Scholar]
- 48.Berger A, Bayliss D, Viana F. Modulation of neonatal rat hypoglossal motoneuron excitability by serotonin. Neurosci Lett. 1992;143:164–168. doi: 10.1016/0304-3940(92)90257-8. [DOI] [PubMed] [Google Scholar]
- 49.Jacobs BL, Fornal CA. Serotonin and motor activity. Curr Opin Neurobiol. 1997;7:820–825. doi: 10.1016/s0959-4388(97)80141-9. [DOI] [PubMed] [Google Scholar]
- 50.Clarke RW, Eves S, Harris J, Peachey JE, Stuart E. Interactions between cutaneous afferent inputs to a withdrawal reflex in the decerebrated rabbit and their control by descending and segmental systems. Neuroscience. 2002;112:555–571. doi: 10.1016/s0306-4522(02)00093-3. [DOI] [PubMed] [Google Scholar]
- 51.Cleland CL, Rymer WZ. Neural mechanisms underlying the clasp-knife reflex in the cat: I. Characteristics of the reflex. J Neurophysiol. 1990;64:1303–1318. doi: 10.1152/jn.1990.64.4.1303. [DOI] [PubMed] [Google Scholar]
- 52.Garraway SM, Hochman S. Modulatory actions of serotonin, norepinephrine, dopamine, and acetylcholine in spinal cord deep dorsal horn neurons. J Neurophysiol. 2001;86:2183–2194. doi: 10.1152/jn.2001.86.5.2183. [DOI] [PubMed] [Google Scholar]
- 53.Perrier J-F, Hounsgaard J. 5-HT2 receptors promote plateau potentials in turtle spinal motoneurons by facilitating an l-type calcium current. J Neurophysiol. 2003;89:954–959. doi: 10.1152/jn.00753.2002. [DOI] [PubMed] [Google Scholar]
- 54.Bras H, Jankowska E, Noga B, Skoog B. Comparison of effects of various types of NA and 5-HT agonists on transmission from group II muscle afferents in the cat. Eur J Neurosci. 1990;2:1029–1039. doi: 10.1111/j.1460-9568.1990.tb00015.x. [DOI] [PubMed] [Google Scholar]
- 55.Bennett DJ, Sanelli L, Cooke CL, Harvey PJ, Gorassini MA. Spastic long-lasting reflexes in the awake rat after sacral spinal cord injury. J Neurophysiol. 2004;91:2247–2258. doi: 10.1152/jn.00946.2003. [DOI] [PubMed] [Google Scholar]
- 56.Harvey PJ, Li X, Li Y, Bennett DJ. 5-HT2 receptor activation facilitates a persistent sodium current and repetitive firing in spinal motoneurons of rats with and without chronic spinal cord injury. J Neurophysiol. 2006;96:1158–1170. doi: 10.1152/jn.01088.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Russo RE, Nagy F, Hounsgaard J. Inhibitory control of plateau properties in dorsal horn neurones in the turtle spinal cord in vitro. J Physiol (Lond) 1998;506:795–808. doi: 10.1111/j.1469-7793.1998.795bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schmit B, Benz E. Extensor reflexes in human spinal cord injury: activation by hip proprioceptors. Exp Brain Res. 2002;145:520–527. doi: 10.1007/s00221-002-1134-5. [DOI] [PubMed] [Google Scholar]
- 59.Schmit BD, McKenna-Cole A, Rymer WZ. Flexor reflexes in chronic spinal cord injury triggered by imposed ankle rotation. Muscle Nerve. 2000;23:793–803. doi: 10.1002/(sici)1097-4598(200005)23:5<793::aid-mus18>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 60.Wu M, Hornby TG, Hilb J, Schmit BD. Extensor spasms triggered by imposed knee extension in chronic human spinal cord injury. Exp Brain Res. 2005;162:239–249. doi: 10.1007/s00221-004-2173-x. [DOI] [PubMed] [Google Scholar]
- 61.Schomburg E, Steffens H. The effect of DOPA and clonidine on reflex pathways from group II muscle afferents to alpha-motoneurones in the cat. Exp Brain Res. 1988;71:442–446. doi: 10.1007/BF00247505. [DOI] [PubMed] [Google Scholar]
- 62.Davies J, Johnston S, Lovering R. Inhibition by DS 103-282 of D-(3H) aspartate release from spinal cord slices. Br J Pharmacol. 1983;78:2. [Google Scholar]
- 63.Barnes MP, Johnson GR. Upper Motor Neurone Syndrome and Spasticity: Clinical Management and Neurophysiology. Cambridge University Press; Cambridge, UK: 2001. [Google Scholar]
- 64.Onushko T, Schmit BD. Reflex response to imposed bilateral hip oscillations in human spinal cord injury. J Neurophysiol. 2007;98:1849–1861. doi: 10.1152/jn.00461.2007. [DOI] [PubMed] [Google Scholar]
- 65.Ellis K, Carpenter J. Mechanism of control of skeletal-muscle contraction by dantrolene sodium. Arch Phys Med Rehabil. 1974;55:362–369. [PubMed] [Google Scholar]
- 66.Burgen A, Dickens F, Zatman L. The action of botulinum toxin on the neuro-muscular junction. J Physiol. 1949;109:10–24. doi: 10.1113/jphysiol.1949.sp004364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rosales R, Arimura K, Takenaga S, Osame M. Extrafusal and intrafusal muscle effects in experimental botulinum toxin-A injection. Muscle Nerve. 1996;19:488–496. doi: 10.1002/(SICI)1097-4598(199604)19:4<488::AID-MUS9>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 68.McLaughlin JF, Bjornson KF, Astley SJ, et al. Selective dorsal rhizotomy: efficacy and safety in an investigator-masked randomized clinical trial. Dev Med Child Neurol. 1998;40:220–232. doi: 10.1111/j.1469-8749.1998.tb15454.x. [DOI] [PubMed] [Google Scholar]
- 69.Vodovnik L, Bowman B, Hufford P. Effects of electrical stimulation on spinal spasticity. Scand J Rehabil Med. 1984;16:29–34. [PubMed] [Google Scholar]
- 70.Alfieri V. Electrical treatment of spasticity: reflex tonic activity in hemiplegic patients and selected specific electrostimulation. Scand J Rehabil Med. 1982;14:177–182. [PubMed] [Google Scholar]
- 71.Dewald J, Given J, Rymer W. Long-lasting reductions of spasticity induced by skin electrical stimulation. IEEE Trans Rehabil Eng. 1996;4:231–242. doi: 10.1109/86.547923. [DOI] [PubMed] [Google Scholar]
- 72.Kuo JJ, Lee RH, Johnson MD, Heckman HM, Heckman C. Active dendritic integration of inhibitory synaptic inputs in vivo. J Neurophysiol. 2003;90:3617–3624. doi: 10.1152/jn.00521.2003. [DOI] [PubMed] [Google Scholar]
- 73.Bhadra N, Kilgore KL. Direct current electrical conduction block of peripheral nerve. IEEE Trans Neural Syst Rehabil Eng. 2004;12:313–324. doi: 10.1109/TNSRE.2004.834205. [DOI] [PubMed] [Google Scholar]
- 74.Bhadra N, Kilgore KL. High-frequency electrical conduction block of mammalian peripheral motor nerve. Muscle Nerve. 2005;32:782–790. doi: 10.1002/mus.20428. [DOI] [PubMed] [Google Scholar]
- 75.Pinter M, Gerstenbrand F, Dimitrijevic M. Epidural electrical stimulation of posterior structures of the human lumbo-sacral cord: 3. Control of spasticity. Spinal Cord. 2000;38:524–531. doi: 10.1038/sj.sc.3101040. [DOI] [PubMed] [Google Scholar]
- 76.Barolat G, Singh-Sahni K, Staas WJ, Shatin D, Ketcik B, Allen K. Epidural spinal cord stimulation in the management of spasms in spinal cord injury: a prospective study. Stereotact Funct Neurosurg. 1995;64:153–164. doi: 10.1159/000098744. [DOI] [PubMed] [Google Scholar]
- 77.Hunter JP, Ashby P. Segmental effects of epidural spinal cord stimulation in humans. J Physiol. 1994;474:407–419. doi: 10.1113/jphysiol.1994.sp020032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Saade NE, Tabet MS, Atweh SF, Jabbur SJ. Modulation of segmental mechanisms by activation of a dorsal column brainstem spinal loop. Brain Res. 1984;310:180–184. doi: 10.1016/0006-8993(84)90025-8. [DOI] [PubMed] [Google Scholar]
- 79.Midha M, Schmitt J. Epidural spinal cord stimulation for the control of spasticity in spinal cord injury patients lacks long-term efficacy and is not cost-effective. Spinal Cord. 1998;36:190–192. doi: 10.1038/sj.sc.3100532. [DOI] [PubMed] [Google Scholar]
- 80.ElBasiouny SM, Mushahwar VK. Modulation of motoneuronal firing behavior after spinal cord injury using intraspinal microstimulation current pulses: a modeling study. J Appl Physiol. 2007;103:276–286. doi: 10.1152/japplphysiol.01222.2006. [DOI] [PubMed] [Google Scholar]
- 81.ElBasiouny SM, Mushahwar VK. Suppressing the excitability of spinal motoneurons by extracellularly applied electrical fields: insights from computer simulations. J Appl Physiol. 2007;103:1824–1836. doi: 10.1152/japplphysiol.00362.2007. [DOI] [PubMed] [Google Scholar]