

Summary
Throughout most of history, epidemic and pandemic cholera was caused by Vibrio cholerae of the serogroup O1. In 1992, however, a V. cholerae strain of the serogroup O139 emerged as a new agent of epidemic cholera. Interestingly, V. cholerae O139 forms biofilms on abiotic surfaces more rapidly than V. cholerae O1 biotype El Tor, perhaps because regulation of exopolysaccharide synthesis in V. cholerae O139 differs from that in O1 El Tor. Here, we show that all flagellar mutants of V. cholerae O139 have a rugose colony morphology that is dependent on the vps genes. This suggests that the absence of the flagellar structure constitutes a signal to increase exopolysaccharide synthesis. Furthermore, although exopolysaccharide production is required for the development of a three-dimensional biofilm, inappropriate exopolysaccharide production leads to inefficient colonization of the infant mouse intestinal epithelium by flagellar mutants. Thus, precise regulation of exopolysaccharide synthesis is an important factor in the survival of V. cholerae O139 in both aquatic environments and the mammalian intestine.
Introduction
Vibrio cholerae is a natural inhabitant of estuarine environments (Colwell and Spira, 1992; Colwell and Huq, 1994). In the absence of human disease, these organisms most probably spend their entire life cycle in the estuarine environment. When a human host is encountered and the disease cholera ensues, the host develops massive diarrhoea that can be fatal. In the absence of adequate sanitation, this serves to disseminate V. cholerae quite efficiently to new freshwater and marine environments. Thus, survival in aquatic environments is essential to propagation of the bacterial species and the epidemiology of the disease, which is characterized by epidemics and pandemics. Because bacteria in aquatic environments are predominantly found in association with surfaces, the ability to form biofilms more rapidly and efficiently may provide a competitive advantage in these environments (Costerton et al., 1987).
In all parts of the world except the Indian subcontinent, the El Tor biotype of V. cholerae serogroup O1, which is responsible for the ongoing seventh pandemic, is the sole cause of cholera (Colwell, 1996). Interestingly, although V. cholerae O1 El Tor also predominates in the Indian subcontinent, there are periodic outbreaks of cholera caused by the classical biotype of V. cholerae O1, the bacterium that caused earlier pandemics, and V. cholerae O139, a bacterium from a new serogroup that emerged as an agent of epidemic cholera in 1992 (Samadi et al., 1983; Albert, 1994; Nair et al., 1994; Jalgaonkar et al., 1998; Kaur and Lal, 1999; Raut et al., 1999). This suggests that the environmental population of V. cholerae in this region, which serves as a reservoir for epidemic and pandemic strains, is dynamic. Thus, the acquisition of traits that improve survival in this aquatic reservoir may be a factor in determining the predominant cause of clinical disease.
Rugose colony morphology variants of pathogenic V. cholerae O1 were first characterized many years ago (White, 1938). Since then, rugose variants of V. cholerae O1 El Tor and V. cholerae O139 have also been isolated. Because rugose variants form thicker biofilms than smooth colony variants, they are believed to have a survival advantage in the environment outside the host (Wai et al., 1998; Mizunoe et al., 1999; Yildiz and Schoolnik, 1999). Recently, the exopolysaccharide and a number of the corresponding vps genes required for the rugose colony morphology have been identified (Yildiz and Schoolnik, 1999).
Much is known about the genetic basis of biofilm formation in Gram-negative bacteria. In Escherichia coli, Pseudomonas aeruginosa and V. cholerae O1 El Tor, initial attachment to a surface is accelerated by functional flagella, suggesting that motility may overcome some energetic barrier encountered by the bacterium en route to the surface (O’Toole and Kolter, 1998; Pratt and Kolter, 1998; Watnick and Kolter, 1999). Type IV pili are responsible for a surface-associated motility known as twitching motility (Whitchurch et al., 1991; Wu et al., 1997; Wolfgang et al., 1998; Semmler et al., 1999; Wall and Kaiser, 1999). In this type of motility, the pilus is thought to anchor to a surface and then propel the bacterium by retracting the pilus against the surface. In V. cholerae O1 El Tor, the mannose-sensitive haemagglutinin (MSHA), a type IV pilus, also accelerates attachment to a surface (Watnick and Kolter, 1999). This may result from the motility imparted to the bacterium by the pilus. Interestingly, in P. aeruginosa, a type IV pilus does not enhance attachment to the surface, but is required for surface-associated motility and microcolony formation once the cells are attached (O’Toole and Kolter, 1998).
Biofilm development by bacteria of many Gram-negative species requires exopolysaccharide synthesis. In both smooth and rugose colony variants of V. cholerae O1 El Tor, the construction of a three-dimensional biofilm structure, complete with water channels and pillars, follows attachment to a surface only if genes in the vps locus, which are also required for the colony morphology of rugose V. cholerae, are intact (Watnick and Kolter, 1999; Yildiz and Schoolnik, 1999). In P. aeruginosa, expression of the exopolysaccharide alginate is increased in biofilms (Davies et al., 1993). Furthermore, colanic acid mutants of E. coli form much lower profile biofilms than wild-type cells (Danese et al., 2000). Thus, exopolysaccharides appear to play an important role in creating the three-dimensional architecture of biofilms made by Gram-negative organisms.
In several microorganisms, there is evidence that flagellum and exopolysaccharide synthesis are inversely regulated. For instance, in E. coli, biofilm-associated cells repress the transcription of flagellar genes and increase the transcription of colanic acid biosynthetic genes (Prigent-Combaret et al., 1999). Furthermore, recent observations in P. aeruginosa suggest that alginate and flagellum synthesis are inversely regulated (Garrett et al., 1999). Taken together, these experiments suggest that some bacteria repress flagellar synthesis and increase exopolysaccharide synthesis in a biofilm and that these two functions may be co-ordinately regulated during biofilm development. Interestingly, for rugose V. cholerae O1 El Tor, wild-type levels of both motility and rugose exopolysaccharide depend on genes involved in the type II secretion of proteins (Ali et al., 2000). Thus, in V. cholerae O1 El Tor, rugose exopolysaccharide production and motility may have common steps in their regulatory and/or biosynthetic pathways.
As detailed above, there are several examples of bacteria that cease flagellum synthesis and initiate exopolysaccharide synthesis in a biofilm. The question remains, however, whether there is a regulatory relationship between the absence of flagellum synthesis and exopolysaccharide synthesis. The flagellum is involved in the regulation of other behavioural transitions such as the swimming to swarming transition of Vibrio parahaemolyticus and other organisms (Belas et al., 1986; Kawagishi et al., 1996). In this paper, we describe a role for the flagellar structure in the regulation of rugose polysaccharide biosynthesis by V. cholerae O139, and we show that increased production of rugose polysaccharide by flagellar mutants alters colony morphology, biofilm development and intestinal colonization.
Results
Steps in biofilm development by wild-type V. cholerae O139
When V. cholerae O139 is grown in a static system as described previously (Watnick and Kolter, 1999), a biofilm measurable by crystal violet staining is detected in ≈ 7 h. The biofilm becomes thicker over a period of days. Under these conditions, biofilm development by V. cholerae O139 proceeds much more rapidly than biofilm development by V. cholerae O1 El Tor and yields a biofilm that is 25–30 μM or 10–15 μM thicker than that of V. cholerae O1 El Tor. Crystal violet quantification of biofilm formation by V. cholerae O139 and O1 El Tor over a period of 2 days is compared in Fig. 1.
Fig. 1.

A comparison of biofilm formation by V. cholerae O1 El Tor (strain N16961) and O139 (strain MO10) over a period of days. The number of biofilm-associated cells was quantified by staining with crystal violet, solubilizing the crystal violet with DMSO and measuring the resultant OD570.
Because biofilm development by V. cholerae O139 takes place so rapidly, continuous monitoring of the early stages in the process of biofilm development is feasible. Using video microscopy, we monitored a polystyrene surface during the first 12 h of colonization and biofilm formation by V. cholerae O139. The micrographs shown in Fig. 2 represent the first frames in movie clips taken at different time points in biofilm development by V. cholerae O139. The corresponding videos are accessible at http://gasp.med.harvard.edu/biofilms/cholera/movies.html. As is clear from these videos, interaction with the surface occurred immediately, but immobilization of the bacteria and microcolony formation only began after 5 h of incubation with the surface.
Fig. 2.

Five frames taken at different time points in a 12 h video microscopic study of wild-type biofilm formation (bar = 5 μM). Associated movies may be accessed at the web address indicated in text. The micrographs shown were taken at the following time points: (A) 0 h; (B) 2 h; (C) 5.5 h; (D) 7 h; and (E) 12 h.
Three types of interactions with the surface were observed throughout the course of biofilm development. First, when bacteria approached the surface closely, a change in swimming pattern was noted. Planktonic V. cholerae usually swim forwards and then abruptly change direction and swim backwards, presumably because of a change in the direction of rotation of the flagellum. Near the surface, however, bacteria swam in circles. This is illustrated best in the movie corresponding to Fig. 2A. This type of behaviour may be the result of an attractive potential between the bacterium and the surface. We call this a planktonic bacterial interaction with the surface.
The second type of interaction observed was spatially restricted bacterial motion. This is best seen in the movie corresponding to Fig. 2B, although it occurs at all stages of biofilm development. One type of restricted bacterial motion was tethered spinning either on end or on a side, probably as a result of flagellar tethering to the surface. Bacteria were also observed to be fixed to the surface along their long axis for seconds to minutes. Stationary periods were interrupted by a brief jerky motion, as if the bacterium was struggling to break free of the surface. The motion was consistent with flagellar-driven motion hindered by an attraction to the surface. We call these cells surface associated. These cells remained associated with the surface during gentle rinsing but preserved the ability to escape the surface and swim off into the medium after seconds or minutes of association with the surface. This observation suggests that the flagellum remains intact and functional during this interaction.
As biofilm development continued, the number of surface-associated bacteria increased. Finally, completely immobilized bacteria began to appear after several hours of incubation with the surface (Fig. 2C movie). Microcolony formation proceeded rapidly after that time, and the three-dimensional biofilm structure developed (Fig. 2D and E movies).
In addition to surface-associated and immobilized cells, another population of cells was noted at the surface throughout the incubation. These cells underwent continuous random motion consistent with Brownian motion. The behaviour of these cells at the surface was indistinguishable from non-motile or heat-killed bacterial cells, although they often became motile after a period of time and disappeared from the microscopic field (data not shown). In addition, these cells were easily removed from the surface by gentle rinsing. Thus, we hypothesize that these cells do not represent surface-associated bacteria but, rather, bacteria with some type of flagellar insult. Consistent with our hypothesis that the flagellum is required for rapid attachment to a surface, these non-motile cells were not observed to become immobilized on the surface (Watnick and Kolter, 1999).
Genetic analysis of biofilm formation by V. cholerae O139
Because biofilm formation by V. cholerae O139 proceeds much more rapidly than biofilm formation by the O1 El Tor strain N16961, we questioned whether the genetic basis for biofilm formation in V. cholerae O139 differed from that in V. cholerae El Tor. In order to determine the genetic basis of biofilm formation in V. cholerae O139, we performed a limited screen of 1500 transposon insertion mutants for defects in biofilm formation, following the same protocol that we had used for the O1 El Tor strain, except that the incubation time was 18 h rather than 24 h (Watnick and Kolter, 1999). We defined biofilm-defective mutants as those that produced a biofilm exhibiting much less intense crystal violet staining when examined visually.
Forty-two biofilm-defective mutants were obtained. All 42 mutants were screened on motility agar, and eight of these 42 were non-motile. We performed sequence analysis on the transposon junctions of five of these non-motile mutants and confirmed transposon insertions in flagellar synthesis genes. Transposon junction sequences were obtained for 13 additional mutants. These mutants were found to have transposon insertions in the previously described vps locus (Yildiz and Schoolnik, 1999). We identified no mutants with insertions in genes required for the synthesis of the mannose-sensitive haemagglutinin type IV pilus.
Because MSHA accelerates biofilm formation in V. cholerae O1 El Tor, we constructed an MSHA deletion mutant (ΔmshA) of V. cholerae O139. Photographs as well as crystal violet quantifications of 18 h biofilms made by wild-type, fla, vps and ΔmshA mutants of V. cholerae O139 are shown in Fig. 3. As in the O1 El Tor background, the vps mutants are severely defective for biofilm formation in the O139 background. The behaviour of flagellar and ΔmshA mutants, however, differs between O1 El Tor and O139 V. cholerae. First of all, the biofilm made by the ΔmshA mutant is not significantly different from that made by the wild type, suggesting therefore that MSHA is not required for biofilm formation in V. cholerae O139. Secondly, in 18 h, the V. cholerae O139 flagellar mutants formed a biofilm detectable by crystal violet staining that rapidly increased in the next 6 h. This suggests that the V. cholerae O139 flagellar mutants are not as defective for attachment as V. cholerae O1 El Tor flagellar mutants.
Fig. 3.

A comparison of 18 h biofilms made by V. cholerae O139 wild-type (MO10) and mutants (Vps− = PW106, Fla− = KKV834, and MshA− = lC1). Biofilms stained with crystal violet are shown above, and quantification of biofilm-associated crystal violet is shown below.
Confocal analysis of biofilm-defective mutants
In order to determine the architecture of mature biofilms made by wild-type and mutant V. cholerae O139 strains, the respective strains were incubated with a glass coverslip for 3 days at room temperature. The biofilms formed under these conditions were examined using confocal microscopy. Confocal micrographs of 3-day-old biofilms made by wild-type and mutant V. cholerae O139 are shown in Fig. 4. The wild-type biofilm reached a thickness of 30 μm after 3 days. This is approximately twice as thick as an O1 El Tor biofilm formed under the same conditions (Watnick and Kolter, 1999). The vps mutants never progressed beyond the monolayer stage, suggesting that, as in V. cholerae O1 El Tor, exopolysaccharide is necessary for building the three-dimensional structure of the biofilm. The architecture of the biofilm made by the flagellar mutant is quite different from that of the wild-type biofilm. It is as thick as the wild-type biofilm, but the bacteria are much more noticeably organized into discrete microcolonies.
Fig. 4.

Confocal microscopy of biofilms made by V. cholerae O139 wild type (MO10) as well as representative biofilm-defective mutants (Vps− = PW106, Fla− = KKV834). Horizontal sections are shown on the left, and vertical sections are shown on the right (bar = 15 μM).
Video microscopy of biofilm-defective mutants
As a way of analysing further the developmental differences between these various biofilm-defective mutants, we used continuous video microscopy over a period of 8 h to record cells attaching to the bottom of polystyrene wells. The cells were grown on agar plates at room temperature for ≈ 18 h and then inoculated directly into fresh LB. Initial cell densities were ≈ 105 cells ml−1. Wild-type V. cholerae O139 and representative mutants were studied using both real-time and time-lapse video microscopy. Figure 5 shows the last frame in time-lapse movies tracing biofilm development by wild-type V. cholerae O139 as well as representative vps and flagellar mutants. Real-time movies that show early interactions with the surface as well as time-lapse movies that trace biofilm development for these three strains may be accessed at http://gasp.med.harvard.edu/biofilms/cholera/movies.html. In wild-type V. cholerae, the real-time movie depicts early surface association (see Fig. 5A, movie 1). The corresponding time-lapse movie shows that early surface association was followed by immobilization of single bacteria. These immobilized bacteria then nucleated micro-colony formation by recruitment of planktonic bacteria (see Fig. 5A, movie 2). The real-time movie of surface association by the vps mutant is very similar to that of the wild type (see Fig. 5B, movie 1). The time-lapse movie, however, demonstrates that the vps mutant cells never became immobilized, and microcolonies never formed (see Fig. 5B, movie 2). This suggests that exopolysaccharide is required and possibly responsible for immobilization of bacteria on the surface and subsequent microcolony formation. In contrast, an early, temporary association between the flagellar mutant cells and the surface was not observed (see Fig. 5C, movie 1). Instead, at early time points, these cells underwent a continuous and random shaking motion, which is consistent with Brownian motion. This observation is consistent with previous results suggesting that flagellar mutants of V. cholerae O1 El Tor are defective in initial attachment to abiotic surfaces (Watnick and Kolter, 1999).
Fig. 5.

The last frames in time-lapse movies demonstrating the difference in biofilm development between wild type and biofilm-defective mutants (Vps− = PW106, Fla− = KKV834; bar = 5 μM). Real-time movies were taken after 1 h of incubation with a surface. Time-lapse movies were acquired at one frame min−1 and are shown at 10 frames s−1.
Although the flagellar mutant cells eventually became immobilized on the surface, the sequence of events in microcolony formation was different from that in wild-type cells. In wild-type biofilm development, a surface is required for immobilization and subsequent microcolony formation. In other words, although aggregates are occasionally seen in the planktonic phase, they are formed primarily at the surface. Hence, we reason that the presence of a surface enhances exopolysaccharide biosynthesis. In contrast, the flagellar mutant aggregated in the planktonic phase. These small aggregates or microcolonies settled onto the surface, the clusters of bacteria were subsequently immobilized as a whole, and these became biofilm-associated microcolonies (see Fig. 5C, movie 2). Because the flagellar mutants are predisposed towards microcolony formation in liquid, we suggest that flagellar mutant cells produce increased amounts of exopolysaccharide in the planktonic phase and thus bypass the requirement for a surface in the earliest stages of biofilm development.
Characterization of flagellar mutants
In the process of conducting the screen for biofilm-defective mutants, it rapidly became clear that O139 strains with transposon insertions in flagellar synthesis genes also displayed a rugose colony morphology. The colony morphology of wild-type V. cholerae O139 is compared with that of a flagellar mutant in Fig. 6A and B. Yildiz and Schoolnik (1999) have demonstrated that the vps genes, which are responsible for exopolysaccharide synthesis, are also required for the production of the colony morphology of rugose V. cholerae O1 El Tor. Furthermore, it has been shown that this exopolysaccharide is present in colonies of rugose but not smooth V. cholerae O139 (Wai et al., 1998; Mizunoe et al., 1999; Yildiz and Schoolnik, 1999). Thus, we reasoned that exopolysaccharide synthesis might be increased in colonies of the O139 flagellar mutants.
Fig. 6.

Colony morphologies of V. cholerae O139 (MO10) as well as various motility mutants on LB-agar plates (Fla− = KKV834; Fla− Vps− = KKV923; motY− = KKV927; flrA− = KKV1004; flrC− = KKV850).
To test whether the vps genes are required for the rugose colony morphology of the flagellar mutants, we constructed a double mutant with insertions in both flagellar (flaA) and vps (vps59) genes. As shown in Fig. 6C, the non-rugose colony morphology of the double mutant is indistinguishable from that of wild-type O139, suggesting that the morphology of the flagellar mutant is, indeed, dependent on exopolysaccharide synthesis.
In order to compare the exopolysaccharide content of colonies formed by wild-type V. cholerae O139, flagellar mutant and flagellar/vps double mutant cells directly, we stained colonies with ruthenium red and then studied thin sections of these colonies using transmission electron microscopy. Representative micrographs are shown in Fig. 7. A small amount of electron-dense material is present between cells in the wild-type colony (Fig. 7A). In the flagellar mutant colony, however, this intercellular material forms a confluent mat from cell to cell (Fig. 7B). In the double mutant, no intercellular electron-dense material was observed (Fig. 7C). These results confirm that, in comparison with cells from the smooth colonies of wild-type V. cholerae O139, cells in the rugose colonies of flagellar mutants produce increased amounts of an exopolysaccharide that is dependent on the previously described vps genes.
Fig. 7.
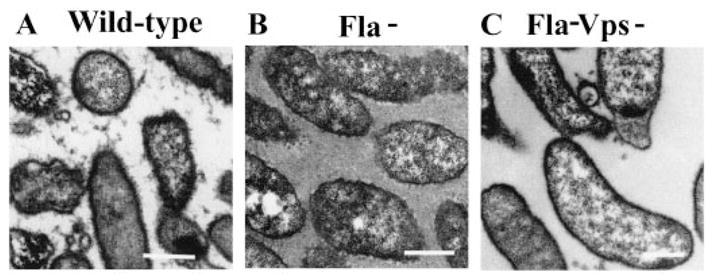
Electron micrographs of thin sections of colonies composed of V. cholerae O139 (MO10), flagellar mutant (Fla− = KKV834), and flagellar and vps double mutant (Fla− Vps− = KKV923) cells stained with ruthenium red (bar = 500 nm).
In E. coli and Salmonella typhimurium, synthesis of the flagellar structure requires hierarchical transcription of three classes of genes (Blair, 1995). Class I includes regulatory genes that promote the transcription of class II genes. Class II genes include all the structural genes necessary to assemble the basal body and hook. Class III genes include those encoding the flagellar filament and motor. Although homologues of known E. coli and S. typhimurium structural flagellar genes (levels II and III) are present in the V. cholerae chromosome and are required for synthesis of the single, polar flagellum, homologues of level I regulatory genes are not present. This suggests that, although the structure of the V. cholerae flagellum may be similar to that of E. coli and S. typhimurium, its regulation differs. Instead, FlrA, a σ54-dependent activator, appears to be the transcriptional activator of V. cholerae level II flagellar genes and therefore represents the likely level I flagellar regulatory factor (Klose and Mekalanos, 1998a; M. G. Prouty and K. E. Klose, unpublished results).
Flagellar mutants obtained from our screen for which transposon junctions were identified had transposon insertions in the following genes: fliK, flhB, fliF, fliE and fliR. By analogy with E. coli, these are all class II flagellar genes. In order to test whether mutations in genes from other levels of flagellar synthesis might have the same rugose phenotype, we constructed V. cholerae O139 mutants with insertions in flrA, flaA, motB and motY. flaA, the major flagellin subunit of the V. cholerae flagellum, as well as motB and motY, two of four genes encoding motor proteins that confer motility to the flagellum, are predicted to be class III genes by analogy with E. coli, S. typhimurium and other Vibrio species (McCarter, 1994; Okunishi et al., 1996; Klose and Mekalanos, 1998b).
As suspected by analogy with other Vibrio species, the V. cholerae motB and motY mutants we constructed possessed a flagellum but were non-motile, as confirmed by electron microscopy of negatively stained cells and motility assays respectively (data not shown). The colony morphologies of the motY and flrA mutants are shown in Fig. 6D and E. Interestingly, mutations in flagellar genes at any level that resulted in the absence of a complete flagellar filament caused a rugose colony morphology in the resultant strain. In contrast, the motB and motY mutants, which possess a completed flagellar filament, did not display a rugose colony morphology. We hypothesize that either an incomplete flagellum specifically and not the absence of motility is sensed in the signalling pathway leading to increased exopolysaccharide production or the motor itself is part of the signal transduction cascade.
FlrA is required for flagellar synthesis (Klose and Mekalanos, 1998a; Klose et al., 1998). FlrC is also a σ54-dependent activator and is the likely activator of level III flagellar genes (M. G. Prouty and K. E. Klose, unpublished results). We constructed a mutant of V. cholerae O139 in which flrC is locked into an inactive conformation, thus preventing transcription of FlrC-dependent genes such as flaA (Correa et al., 2000). As shown in Fig. 6F, the colony morphology of this mutant is also rugose. Thus, one would predict that, under environmental conditions that repress either FlrA- or FlrC-dependent transcription, wild-type V. cholerae O139 would cease flagellar synthesis and produce rugose polysaccharide.
Having defined the colony morphologies of several motility mutants, we then wanted to determine the correlation between the rugose colony morphology and the ability to form a biofilm. Biofilms formed by selected motility mutants after 18 h of incubation with a surface are shown in Fig. 8. The flrA mutant formed a biofilm similar to those of the flaA and flrC mutants, which are shown in Fig. 8. In contrast, the motY mutant, which did not exhibit a rugose colony morphology, also did not form a biofilm over a period of 18 h as assayed by crystal violet staining. The microscopic appearance of the biofilms formed by wild-type, flaA and motY mutants are shown in Fig. 9. The motY mutant clearly had a more severe defect in attachment to the surface than the flaA mutant, suggesting that there is a correlation between the rugose colony morphology and the ability to form a biofilm.
Fig. 8.
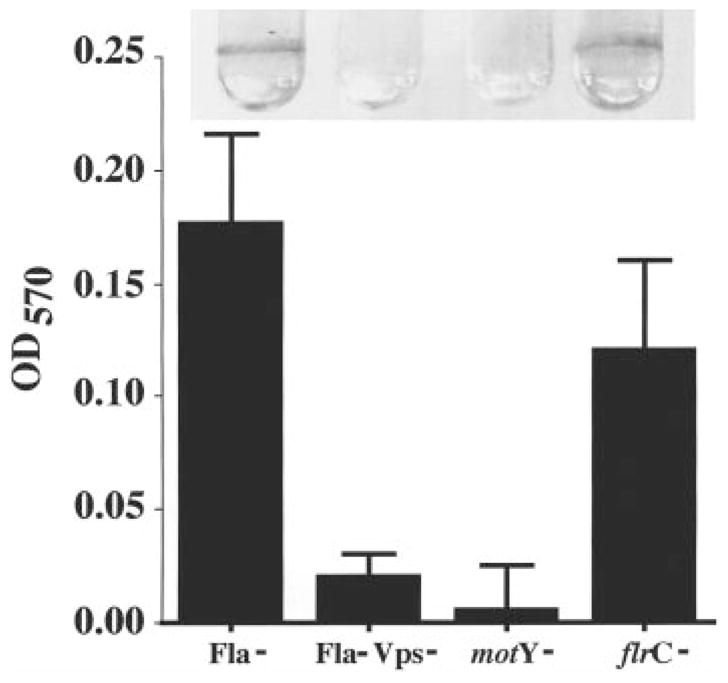
A comparison of 18 h biofilms made by V. cholerae O139 motility mutants (Fla− = KKV834; Fla− Vps− = KKV923; motY− = KKV927; flrC− = KKV850). Biofilms stained with crystal violet are shown above, and quantification of biofilm-associated crystal violet is shown below.
Fig. 9.
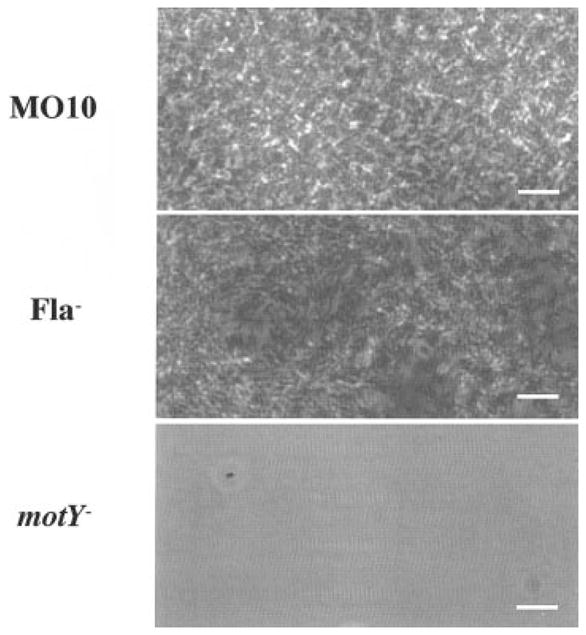
Phase-contrast micrographs of 24 h biofilms made by V. cholerae O139 wild type (MO10) and non-motile mutants (Fla− = KKV834; motY− = KKV927; bar = 5 μM).
Virulence studies of flagellar mutants
Although attachment to a surface plays a role in both colonization of the intestinal epithelium and biofilm formation on abiotic surfaces, very little overlap has been found between genes required for intestinal colonization and those required for biofilm formation on abiotic surfaces (Watnick et al., 1999). In accordance with this observation, motility itself, which is crucial for biofilm development, does not appear to be critical for colonization of the intestinal epithelium in the infant mouse model (although it may be important in human disease; Richardson, 1991; Gardel and Mekalanos, 1996; Klose and Mekalanos, 1998).
One might predict therefore that the genes necessary for persistence and survival within an environmental biofilm might be deleterious if expressed during an acute infection in the mammalian intestine. In order to test whether production of exopolysaccharide might confer a virulence defect on flagellar mutants, a competition assay of intestinal colonization was performed with wild-type V. cholerae O139 co-inoculated with wild-type, flaA, vps or flaA/vps mutants into infant mice as described below. The competitve indices, which indicate the level of intestinal colonization relative to the wild-type O139 strain, are shown in Table 1. The flaA mutant was ≈ 70 times less able to colonize than the wild-type strain. However, the competition defect of the flagellar mutant was eliminated in the non-rugose, non-motile flaA/vps double mutant. These results indicate that the rugose phenotype rather than the absence of motility is correlated with the observed intestinal colonization defect. In contrast, elimination of exopolysaccharide synthesis from wild-type V. cholerae O139 had no effect on intestinal colonization. This suggests that flagellar mutants of V. cholerae O139, but not wild-type cells, make exopolysaccharide in the intestine of the infant mouse. Furthermore, synthesis of exopolysaccharide, a crucial element in biofilm formation on abiotic structures, is not only unnecessary but, in fact, detrimental to colonization of the mammalian intestine.
Table 1.
Infant mouse colonization assay.
| Strain | Relevant genotypea | Competitive indexb | P-valuec |
|---|---|---|---|
| PW96 | Wild type | 2.93 ± 0.7 (n = 7) | |
| KKV1028 | vps59::pGP704 | 1.01 ± 0.3 (n = 7) | < 0.01 |
| KKV834 | ΔflaA::Cm | 0.04 ± 0.03 (n = 11) | < 0.01 |
| KKV1029 | ΔflaA::Cm; vps::pGP704 | 1.75 ± 1.3 (n = 5) | < 0.1 |
All strains additionally contain a lacZ::MCS mutation, which makes them phenotypically Lac−.
The competitive index is given as the ratio of output mutant to wild type (recovered from the intestine) divided by the ratio of input mutant to wild type (inoculated into a mouse). Thus, if a mutant has no colonization defect, we expect a competitive index close to 1. All strains were co-inoculated with MO10 (Lac+) as wild-type strain.
Using Student’s two-tailed t-test compared with the competitive index values of the wild-type strain.
Discussion
It is likely that, at least in the Indian subcontinent, V. cholerae O1 and V. cholerae O139 co-exist in the aquatic environment either through a dynamic competition for the same niche or by inhabiting distinct niches. Because surface-associated life predominates in the aquatic environment, the observation that V. cholerae O139 forms biofilms more quickly than V. cholerae O1 El Tor may have implications for either of these models of co-habitation in the aquatic environment (Watnick and Kolter, 2000). We were therefore encouraged to examine the basis of this difference in biofilm development.
In V. cholerae O139, we found that, as in V. cholerae O1 El Tor, both flagellar motility and exopolysaccharide production dependent on the vps genes are required for the development of a biofilm. All the flagellar mutants of V. cholerae O139, however, displayed a rugose colony morphology and were less defective for biofilm formation than V. cholerae O1 El Tor flagellar mutants. Because the rugose colony morphology has been correlated with exopolysaccharide production, this suggested to us that increased exopolysaccharide synthesis in V. cholerae O139 was somehow dependent on the flagellar structure (Wai et al., 1998; Mizunoe et al., 1999; Yildiz and Schoolnik, 1999).
Strains with mutations in flagellar genes at all levels of flagellar synthesis displayed a rugose colony phenotype. Interestingly, motB and motY mutants that possess a complete but paralyzed flagellum displayed a smooth colony morphology. This pattern is different from the swimming to swarming transition of Vibrio parahaemolyticus, in which the flagellum plays the role of a mechanosensor, signalling the production of lateral flagella when the viscosity of the medium is high (Belas et al., 1986; McCarter et al., 1988). In this case, both flaA and motY mutants produce lateral flagella, whereas only flaA V. cholerae O139 mutants produce exopolysaccharide (McCarter et al., 1988; McCarter and Silverman, 1990). In Caulobacter crescentus, the bacterium loses its flagellum by proteolytic cleavage in the swarmer to stalked cell transition, but there is no evidence that the loss of the flagellum plays a regulatory role in the generation of the stalk (Jenal and Shapiro, 1996; Aldridge and Jenal, 1999). Thus, V. cholerae O139 appears to possess a novel type of regulation by the flagellum, in which the bacterium somehow senses the presence or absence of the flagellar filament.
Non-motile mutants are rarely isolated from natural environments. Thus, it is important to show that environmental regulation in wild-type V. cholerae O139 can lead to repression of flagellar synthesis and stimulation of exopolysaccharide synthesis. Mutations in two regulatory genes involved in flagellar synthesis, flrA and flrC, were shown to induce exopolysaccharide synthesis in V. cholerae O139. Thus, under environmental conditions in which FlrA- or FlrC-regulated genes are not induced, one might expect surface attachment to be the favoured lifestyle. On the other hand, one might expect FlrA- or FlrC-regulated genes to be induced when environmental conditions favour the search for a new surface.
The developmental pathway followed by flagellar mutants in biofilm formation is different from that followed by wild-type V. cholerae O139. In these strains, micro-colony formation, which requires exopolysaccharide synthesis, occurs in solution. Because microcolony formation occurs primarily on the solid surface during wild-type O139 biofilm formation, we suggest that wild-type bacteria, but not flagellar mutants, require a surface for the synthesis of exopolysaccharide. Furthermore, because the biofilms made by flagellar mutants display much more empty space, we conclude that surface colonization by flagellar mutants is less efficient than that by wild type. Thus, the prolonged loose association phase displayed by wild-type V. cholerae before micro-colony formation may be important for uniform colonization. We suggest that, in the competition for a position on a surface, a balance must be struck between uniform colonization and rapid immobilization.
A flagellar (Fla−) mutant of V. cholerae O139, which displayed a rugose colony morphology, had a significant intestinal colonization defect in the infant mouse model of cholera. In contrast, non-motile mutants of V. cholerae O1 have previously been shown to have few or no colonization defects in infant mice (Richardson, 1991; Gardel and Mekalanos, 1996; Klose, 2000), even though motility appears to be important for intestinal colonization of rabbits and humans (Richardson, 1991; Kenner et al., 1995). Importantly, the non-rugose, non-motile Fla− Vps− O139 strain colonizes the infant mouse intestine at wild-type levels, demonstrating that the colonization defect of the Fla− O139 strain is the result of increased exopolysaccharide synthesis rather than the absence of motility. One possibility is that exopolysaccharide physically interferes with attachment to the intestinal epithelium. Alternatively, exopolysaccharide synthesis may interfere with colonization by overwhelming the type II protein secretion system of V. cholerae, which is required for the secretion of cholera toxin (Overbye et al., 1993; Sandkvist et al., 1993; 1997).
These results demonstrate that the appropriate activation of pathways that lead to environmental persistence is as critical to the pathogenesis of V. cholerae as the appropriate expression of factors that promote intestinal colonization. Critical repression of environmental colonization factors during host infection has been documented in other pathogenic bacteria. In Bordetella pertussis, for example, constitutive expression of the flagellar regulon, which is normally repressed during infection, disrupts colonization of the host (Akerley et al., 1995). Furthermore, the attenuation and immunogenicity of S. typhimurium lacking DNA adenine methylase is believed to result, at least in part, from expression of normally repressed factors within the host (Heithoff et al., 1999).
In this paper, we present evidence suggesting that the absence of a flagellum leads to increased exopolysaccharide synthesis by V. cholerae O139 in colonies, in planktonic bacteria and within the infant mouse intestine. This exopolysaccharide, although required for the development of a three-dimensional biofilm, interferes with virulence in an infant mouse model of cholera. Thus, precise regulation of exopolysaccharide is likely to be of critical importance to the survival of V. cholerae O139 in a variety of environments.
Experimental procedures
Bacterial strains and media
Bacterial strains used in these experiments are described in Table 2. All V. cholerae strains are isogenic with the wild-type O139 strain MO10. For experiments using green fluorescent protein (GFP)-expressing cells, biofilms were made in the presence of 150 μg ml−1 ampicillin. No difference in biofilm development was noted with and without ampicillin by phase microscopy. For biofilm experiments using KKV927, ampicillin was used at a concentration of 50 μg ml−1 because higher concentrations interfered with cell division. This concentration of ampicillin allowed for normal cell division but also maintained the plasmid insertion, as measured by the absence of cell motility. Colony morphology experiments were performed on 1.5% LB-agar plates with appropriate antibiotics added.
Table 2.
Bacterial strains and plasmids.
| Strains or plasmids | Genotype | Reference/source |
|---|---|---|
| E. coli | ||
| β2155 | ThrB1004 pro thi strA hsdS lacZΔM15 (F′ lacZΔM15 lacIq trajΔ36 proA+proB+) ΔdapA::erm (Ermr) pir::RP4 (::kan (Kmr) from SM10) | Dehio and Meyer (1997) |
| SM10λpir | Thi thr leu tonA lacY supE recA::RP4-2-Tc::Mu λpirR6K; Kmr | Miller and Mekalanos (1988) |
| V. cholerae | ||
| MO10 | 1992 clinical isolate of V. cholerae O139 from India, | Waldor and Mekalanos (1994) |
| KKV834 | ΔflaA::Cm; lacZ::MCS | This study |
| PW106 | MO10 Δvps::Tn10Kan | This study |
| LC1 | ΔmshA1 | This study |
| KKV923 | ΔflaA::Cm; vps::Tn10Kan | This study |
| KKV927 | motY::pGP704 | This study |
| KKV835 | Δ (flrBC)::Cm; lacZ::MCS | This study |
| KKV850 | flrCD54A | This study |
| KKV1004 | ΔflrA::Cm; lacZ::MCS | This study |
| KKV1028 | vps59::pGP704; lacZ::MCS | This study |
| KKV1029 | ΔflaA::Cm; vps59::pGP704; lacZ::MCS | This study |
| KKVMB | motB::pGP704 | This study |
| PW96 | lacZ::MCS | This study |
| Plasmids | ||
| pCVD442 | OriR6K mobRP4 sacB; Apr | Donnenberg and Kaper (1991) |
| pGP704 | OriR6K mobRP4; Apr | Miller and Mekalanos (1988) |
| pHT1 | pCVD442ΔmshA1; Apr | Thelin and Taylor (1996) |
| pBSL180 | OriR6K mobRP4 lacI pTAC tnp miniTn10Km; Kmr, Apr | Alexeyev and Shokolenko (1995) |
| p6891MCS | PBR327 with 8 kbp Sau3A fragment of V. cholerae lacZ gene interrupted by a multiple cloning site | Butterton et al. (1995) |
| pSMC2 | PKEN carrying bright mutant of gfp and 1.8 kb stabilizing fragment from pUC181.8; Apr | Bloemberg et al. (1997) |
| pKEK93 | ΔflaA::Cm in pCVD442 | Klose and Mekalanos (1998) |
| pKEK328 | ‘motY’ in pGP704 | This study |
| pCG1050 | ‘motB’ in pGP704 | Gardel and Mekalanos (1996) |
| pKEK85 | ΔflrA::Cm in pCVD442 | Klose and Mekalanos (1998) |
| pKEK151 | Δ (flrBC)::Cm in pCVD442 | Correa et al. (2000) |
| pKEK233 | flrBflrCD54A in pCVD442 | Correa et al. (2000) |
| pKEK349 | ‘vps59’ in pGP704 | This study |
The ΔmshA, ΔflrA::Cm, Δ (flrBC)::Cm, flrCD54A and ΔflaA::Cm chromosomal mutations were constructed in V. cholerae MO10 or PW96 using the suicide plasmids pHT1, pKEK85, pKEK151, pKEK233 and pKEK93, respectively, as described previously (Donnenberg and Kaper, 1991; Thelin and Taylor, 1996; Klose and Mekalanos, 1998a, b; Correa et al., 2000). MO10 lacZ::MCS (PW96) was constructed by electroporating plasmid p6891MCS into wild-type MO10, growing the resultant ampicillin-resistant colonies in LB broth overnight and then selecting colonies that were white on LB-Xgal plates and sensitive to ampicillin (Butterton et al., 1995).
Plasmid construction
Plasmids used in these experiments are described in Table 2. Internal fragments from the coding sequences of vps59 and motY were amplified by polymerase chain reaction (PCR) using MO10 chromosomal DNA and the paired primers vps59-1: 5′-GCGCGTCGACGCCCTGTTGATATTA TGGGTAG-3′ (SalI site underlined) and vps59-2: 5′-GCGGAATTCGTGTATAACGCATCAC-3′ (EcoRI site underlined); and motY1: 5′-CGCGGTCGACGCTGGCAAGAAAATCAAT CTCG-3′ (SalI site underlined) and motY2: 5′-GCGAATTCCGATAGATTTTGGCTATTGTTA-3′ (EcoRI site underlined); these sequences are based on the predicted homologous genes identified within The Institute for Genomic Research (TIGR) unfinished V. cholerae genome database. The resultant 417 bp ‘vps59’ and 525 bp ‘motY’ fragments were digested with EcoRI and SalI, then ligated into pGP704 that had been digested similarly to form pKEK349 and pKEK328 respectively (Miller and Mekalanos, 1988).
Transposon mutagenesis and screen for defects in biofilm formation
Transposon mutagenesis and the screen for biofilm formation by the resultant mutants was performed as described previously (Watnick and Kolter, 1999). Sequence analysis of transposon insertion mutations was also carried out as described previously.
Biofilm assays
For biofilm assays, bacteria were grown overnight on Luria–Bertani (LB) agar plates at room temperature. Cells were subsequently resuspended in LB broth at an OD600 of ≈0.6 and then added to 300 μl of LB in borosilicate glass tubes at a dilution of 1:100. Streptomycin (25 μg ml−1) and ampicillin (150 μg ml−1) were added as appropriate. Cells were allowed to incubate in the borosilicate tubes for ≈ 18 h. For longer incubations, spent medium was replaced with fresh medium daily. Tubes were then rinsed with distilled water and filled with crystal violet stain. After 5 min, the tubes were rinsed. For quantification, the biofilm-associated crystal violet was resuspended by several hours of incubation with dimethyl sulphoxide (DMSO), and the OD570 of the resultant suspension was measured.
Confocal microscopy
Samples for confocal microscopy were prepared by incubating GFP-expressing cells with a borosilicate coverslip in a 50 ml Falcon tube containing 6 ml of LB broth. Spent medium was exchanged for fresh medium every 24 h over a period of 3 days. An MRC-1024 confocal microscope (Bio-Rad) was used to collect x-, y- and z-sections through the biofilm of interest using a 488 nm excitation wavelength (magnification 600 ×).
Phase microscopy
For these experiments, biofilms were again formed on coverslips placed in Falcon tubes as described above. After 24 h, coverslips were rinsed with LB broth to remove loosely adherent bacteria, placed on a slide and observed using an Optiphot-2 microscope (Nikon) equipped with a CCD video camera system (Optronics Engineering) and computer interface (magnification 600 ×).
Video phase microscopy
For these experiments, biofilms were formed in 6-well microtitre dishes containing 2 ml of LB broth with appropriate antibiotics added. For long-term monitoring, wild-type cells were grown in several wells concurrently. The medium in the monitored well was exchanged with spent medium from another well every 2 hours. Bacteria were removed from the incoming medium before addition by gentle centrifugation. For shorter term concurrent observation of wild-type and mutants, the medium was not exchanged. Continuous phase microscopy was carried out using an inverted phase microscope equipped with a CCD video camera system (Optronics Engineering) interfaced with a computer and a VCR (magnification 400 ×). Selected images were digitized.
Ruthenium red staining of colonies
Ruthenium red staining of colonies was performed as described previously (method 5; Fassel and Edmiston, 1999). Briefly, the noted strains were plated on LB-agar with the appropriate antibiotics added. Colonies were allowed to form at 37°C for 48 h. At this point, the rugose colony morphology of the flagellar mutant strain was clearly visible. A single colony with underlying agar support was then excised from the plate using a spatula. This colony was fixed overnight at room temperature using a solution of 75 mM lysine, 0.075% (w/v) ruthenium red, 2% (w/v) paraformaldehyde, 2.5% (w/v) glutaraldehyde buffered in 0.1 M sodium cacodylate buffer, pH 7.2. The colony was rinsed with 0.1 M sodium cacodylate, post-fixed in 1% OsO4 for 2 h and subsequently dehydrated and sectioned according to standard procedures (Fassel and Edmiston, 1999). Sections were visualized using a transmission electron microscope.
In vivo colonization assay
The infant mouse colonization assay has been described previously (Gardel and Mekalanos, 1996). Briefly, V. cholerae strains PW96, KKV834, KKV1028 or KKV1029 (all Lac−) were mixed with wild-type strain MO10 (Lac+) and given in a peroral inoculum ratio of ≈ 105 mutant to 105 wild type to 5- to 6-day-old CD-1 suckling mice. After a 20–22-h period of colonization, homogenates from the isolated small intestines were collected, and the ratio of mutant to wild type was determined by plating on LB agar containing Xgal.
Acknowledgments
We thank George O’Toole, Stephen Finkel, Leslie Pratt, Paul Danese, Dianne Newman, Natalia Bomchil and Ned Ruby for helpful discussions. We also thank Shonna McBride for construction of KKV927, and John Mekalanos and Ronald Taylor for the generous gifts of strains as well as for helpful discussions. We also thank Maria Eriksson of the Harvard Medical School Electron Microscope Facility for her help and expertise. This work was supported by NIH K08 AI01588 to P.I.W., NIH AI43486 to K.E.K. and NIH GM58213 to R.K.
References
- Akerley BJ, Cotter PA, Miller JF. Ectopic expression of the flagellar regulon alters development of the Bordetella–host interaction. Cell. 1995;80:611–620. doi: 10.1016/0092-8674(95)90515-4. [DOI] [PubMed] [Google Scholar]
- Albert MJ. Vibrio cholerae O139 Bengal. J Clin Microbiol. 1994;32:2345–2349. doi: 10.1128/jcm.32.10.2345-2349.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldridge P, Jenal U. Cell cycle-dependent degradation of a flagellar motor component requires a novel-type response regulator. Mol Microbiol. 1999;32:379–391. doi: 10.1046/j.1365-2958.1999.01358.x. [DOI] [PubMed] [Google Scholar]
- Alexeyev MF, Shokolenko IN. Mini-Tn10 transposon derivatives for insertion mutagenesis and gene delivery into the chromosome of Gram-negative bacteria. Gene. 1995;160:59–62. doi: 10.1016/0378-1119(95)00141-r. [DOI] [PubMed] [Google Scholar]
- Ali A, Johnson JA, Franco AA, Metzger DJ, Connell TD, Morris JG, Sozhamannan S. Mutations in the extracellular protein secretion pathway genes (eps) interfere with rugose polysaccharide production in and motility of Vibrio cholerae. Infect Immun. 2000;68:1967–1974. doi: 10.1128/iai.68.4.1967-1974.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belas R, Simon M, Silverman M. Regulation of lateral flagella gene transcription in Vibrio parahaemolyticus. J Bacteriol. 1986;167:210–218. doi: 10.1128/jb.167.1.210-218.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair DF. How bacteria sense and swim. Annu Rev Microbiol. 1995;49:489–522. doi: 10.1146/annurev.mi.49.100195.002421. [DOI] [PubMed] [Google Scholar]
- Bloemberg GV, O’Toole GA, Lugtenberg BJ, Kolter R. Green fluorescent protein as a marker for Pseudomonas spp. Appl Environ Microbiol. 1997;63:4543–4551. doi: 10.1128/aem.63.11.4543-4551.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterton JR, Beattie DT, Gardel CL, Carroll PA, Hyman T, Killeen KP, et al. Heterologous antigen expression in Vibrio cholerae vector strains. Infect Immun. 1995;63:2689–2696. doi: 10.1128/iai.63.7.2689-2696.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwell RR. Global climate and infectious disease: the cholera paradigm. Science. 1996;274:2025–2031. doi: 10.1126/science.274.5295.2025. [DOI] [PubMed] [Google Scholar]
- Colwell RR, Huq A. Environmental reservoir of Vibrio cholerae. The causative agent of cholera. Ann NY Acad Sci. 1994;740:44–54. doi: 10.1111/j.1749-6632.1994.tb19852.x. [DOI] [PubMed] [Google Scholar]
- Colwell RR, Spira WM. The ecology of Vibrio cholerae. In: Barua D, Greenough WBI, editors. Cholera. New York: Plenum Press; 1992. pp. 107–127. [Google Scholar]
- Correa NE, Lauriano CM, McGee R, Klose KE. Phosphorylation of the flagellar regulatory protein FlrC is necessary for Vibrio cholerae motility and enhanced colonization. Mol Microbiol. 2000;35:743–755. doi: 10.1046/j.1365-2958.2000.01745.x. [DOI] [PubMed] [Google Scholar]
- Costerton JW, Cheng KJ, Geesey GG, Ladd TI, Nickel JC, Dasgupta M, Marrie TJ. Bacterial biofilms in nature and disease. Annu Rev Microbiol. 1987;41:435–464. doi: 10.1146/annurev.mi.41.100187.002251. [DOI] [PubMed] [Google Scholar]
- Danese PN, Pratt LA, Kolter R. Exopolysaccharide production is required for development of the Escherichia coli K-12 biofilm architecture. J Bacteriol. 2000;68:1967–1974. doi: 10.1128/jb.182.12.3593-3596.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies DG, Chakrabarty AM, Geesey GG. Exopolysaccharide production in biofilms: substratum activation of alginate gene expression by Pseudomonas aeruginosa. Appl Environ Microbiol. 1993;59:1181–1186. doi: 10.1128/aem.59.4.1181-1186.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehio C, Meyer M. Maintenance of broad-host-range incompatibility group P and group Q plasmids and transposition of Tn5 in Bartonella henselae following conjugal plasmid transfer from Escherichia coli. J Bacteriol. 1997;179:538–540. doi: 10.1128/jb.179.2.538-540.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnenberg MS, Kaper JB. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect Immun. 1991;58:4310–4317. doi: 10.1128/iai.59.12.4310-4317.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassel TA, Edmiston CE. Bacterial biofilms: strategies for preparing glycocalyx for electron microscopy. In: Doyle RJ, editor. Biofilms. San Diego: Academic Press; 1999. pp. 194–203. [DOI] [PubMed] [Google Scholar]
- Gardel CL, Mekalanos JJ. Alterations in Vibrio cholerae motility phenotypes correlate with changes in virulence factor expression. Infect Immun. 1996;64:2246–2255. doi: 10.1128/iai.64.6.2246-2255.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett ES, Perlegas D, Wozniak DJ. Negative control of flagellum synthesis in Pseudomonas aeruginosa is modulated by the alternative sigma factor AlgT (AlgU) J Bacteriol. 1999;181:7401–7404. doi: 10.1128/jb.181.23.7401-7404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heithoff DM, Sinsheimer RL, Low DA, Mahan MJ. An essential role for DNA ademine methylation in bacterial virulence. Science. 1999;284:976–970. doi: 10.1126/science.284.5416.967. [DOI] [PubMed] [Google Scholar]
- Jalgaonkar SV, Ingole KV, Ambhore NA, Fule RP. Re-emergence of Vibrio cholerae serogroup O139 during June–August, 1997 in Yavatmal (Maharashtra) Ind J Med Res. 1998;108:1–2. [PubMed] [Google Scholar]
- Jenal U, Shapiro L. Cell cycle-controlled proteolysis of a flagellar motor protein that is asymmetrically distributed in the Caulobacter predivisional cell. EMBO J. 1996;15:2393–2406. [PMC free article] [PubMed] [Google Scholar]
- Kaur H, Lal M. Reappearance of Vibrio cholerae O139 during March–August, 1998 in Ludhiana (Punjab), India. Ind J Med Res. 1999;109:3–4. [PubMed] [Google Scholar]
- Kawagishi I, Imagawa M, Imae Y, McCarter L, Homma M. The sodium-driven polar flagellar motor of marine Vibrio as the mechanosensor that regulates lateral flagellar expression. Mol Microbiol. 1996;20:693–699. doi: 10.1111/j.1365-2958.1996.tb02509.x. [DOI] [PubMed] [Google Scholar]
- Kenner JR, Coster TS, Taylor DN, Trofa AF, Barrera-Oro M, Hyman T, et al. Peru-15, an improved live-attenuated vaccine candidate for Vibrio cholerae O1. J Infect Dis. 1995;172:1126–1129. doi: 10.1093/infdis/172.4.1126. [DOI] [PubMed] [Google Scholar]
- Klose KE. The suckling mouse model of cholera. Trends Microbiol. 2000;8:189–191. doi: 10.1016/s0966-842x(00)01721-2. [DOI] [PubMed] [Google Scholar]
- Klose KE, Mekalanos JJ. Distinct roles of an alternative sigma factor during both free-swimming and colonizing phases of the Vibrio cholerae pathogenic cycle. Mol Microbiol. 1998a;28:501–520. doi: 10.1046/j.1365-2958.1998.00809.x. [DOI] [PubMed] [Google Scholar]
- Klose KE, Mekalanos JJ. Differential regulation of multiple flagellins in Vibrio cholerae. J Bacteriol. 1998b;180:303–316. doi: 10.1128/jb.180.2.303-316.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose KE, Novik V, Mekalanos JJ. Identification of multiple sigma54-dependent transcriptional activators in Vibrio cholerae. J Bacteriol. 1998;180:5256–5259. doi: 10.1128/jb.180.19.5256-5259.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarter LL. MotY, a component of the sodium-type flagellar motor. J Bacteriol. 1994;176:4219–4225. doi: 10.1128/jb.176.14.4219-4225.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarter L, Silverman M. Surface-induced swarmer cell differentiation of Vibrio parahaemolyticus. Mol Microbiol. 1990;4:1057–1062. doi: 10.1111/j.1365-2958.1990.tb00678.x. [DOI] [PubMed] [Google Scholar]
- McCarter L, Hilmen M, Silverman M. Flagellar dynamometer controls swarmer cell differentiation of Vibrio parahaemolyticus. Cell. 1988;54:345–351. doi: 10.1016/0092-8674(88)90197-3. [DOI] [PubMed] [Google Scholar]
- Miller VL, Mekalanos JJ. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J Bacteriol. 1988;170:2575–2583. doi: 10.1128/jb.170.6.2575-2583.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizunoe Y, Wai SN, Takade A, Yoshida SI. Isolation and characterization of rugose form of Vibrio cholerae O139 strain MO10. Infect Immun. 1999;67:958–963. doi: 10.1128/iai.67.2.958-963.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair GB, Ramamurthy T, Bhattacharya SK, Mukhopadhyay AK, Garg S, Bhattacharya MK, et al. Spread of Vibrio cholerae O139 Bengal in India. J Infect Dis. 1994;169:1029–1034. doi: 10.1093/infdis/169.5.1029. [DOI] [PubMed] [Google Scholar]
- Okunishi I, Kawagishi I, Homma M. Cloning and characterization of motY, a gene coding for a component of the sodium-driven flagellar motor in Vibrio alginolyticus. J Bacteriol. 1996;178:2409–2415. doi: 10.1128/jb.178.8.2409-2415.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Toole GA, Kolter R. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol Microbiol. 1998;30:295–304. doi: 10.1046/j.1365-2958.1998.01062.x. [DOI] [PubMed] [Google Scholar]
- Overbye LM, Sandkvist M, Bagdasarian M. Genes requires for extracellular secretion of enterotoxin are clustered in Vibrio cholerae. Gene. 1993;132:101–106. doi: 10.1016/0378-1119(93)90520-d. [DOI] [PubMed] [Google Scholar]
- Pratt LA, Kolter R. Genetic analysis of Escherichia coli biofilm formation: roles of flagella, motility, chemotaxis and type I pili. Mol Microbiol. 1998;30:285–293. doi: 10.1046/j.1365-2958.1998.01061.x. [DOI] [PubMed] [Google Scholar]
- Prigent-Combaret C, Vidal O, Dorel C, Lejeune P. Abiotic surface sensing and biofilm-dependent regulation of gene expression in Escherichia coli. J Bacteriol. 1999;181:5993–6002. doi: 10.1128/jb.181.19.5993-6002.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raut S, Jalgaonkar SV, Tankhiwale NS, Agarwal VA. Re-emergence of Vibrio cholerae O139 serogroup during 1998 in Nagur (Maharashtra), India. Ind J Med Res. 1999;109:1–2. [PubMed] [Google Scholar]
- Richardson K. Roles of motility and flagellar structure in pathogenicity of Vibrio cholerae: analysis of motility mutants in three animal models. Infect Immun. 1991;59:2727–2736. doi: 10.1128/iai.59.8.2727-2736.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samadi AR, Huq MI, Shahid N, Khan MU, Eusof A, Rahman AS, et al. Classical Vibrio cholerae biotype displaces El Tor in Bangladesh. Lancet. 1983;1:805–807. doi: 10.1016/s0140-6736(83)91860-3. [DOI] [PubMed] [Google Scholar]
- Sandkvist M, Morales V, Bagdasarian M. A protein required for secretion of cholera toxin through the outer membrane of Vibrio cholerae. Gene. 1993;123:81–86. doi: 10.1016/0378-1119(93)90543-c. [DOI] [PubMed] [Google Scholar]
- Sandkvist M, Overbye LM, Hough LP, Morales VM, Bagdasarian M, Koomey M, et al. General secretion pathway (eps) genes required for toxin secretion and outer membrane biogenesis in Vibrio cholerae. J Bacteriol. 1997;179:6994–7003. doi: 10.1128/jb.179.22.6994-7003.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semmler AB, Whitchurch CB, Mattick JS. A re-examination of twitching motility in Pseudomonas aeruginosa. Microbiology. 1999;145:2863–2873. doi: 10.1099/00221287-145-10-2863. [DOI] [PubMed] [Google Scholar]
- Thelin KH, Taylor RK. Toxin-coregulated pilus, but not mannose-sensitive hemagglutinin, is required for colonization by Vibrio cholerae O1 El Tor biotype and O139 strains. Infect Immun. 1996;64:2853–2856. doi: 10.1128/iai.64.7.2853-2856.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai SN, Mizunoe Y, Takade A, Kawabata SI, Yoshida SI. Vibrio cholerae O1 strain TSI-4 produces the exopolysaccharide materials that determine colony morphology, stress resistance, and biofilm formation. Appl Environ Microbiol. 1998;64:3648–3655. doi: 10.1128/aem.64.10.3648-3655.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldor MK, Mekalanos JJ. ToxR regulates virulence gene expression in non-O1 strains of Vibrio cholerae that cause epidemic cholera. Infect Immun. 1994;62:72–78. doi: 10.1128/iai.62.1.72-78.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall D, Kaiser D. Type IV pili and cell motility. Mol Microbiol. 1999;32:1–10. doi: 10.1046/j.1365-2958.1999.01339.x. [DOI] [PubMed] [Google Scholar]
- Watnick PI, Kolter R. Steps in the development of a Vibrio cholerae biofilm. Mol Microbiol. 1999;34:586–595. doi: 10.1046/j.1365-2958.1999.01624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watnick PI, Kolter R. Biofilm, city of microbes. J Bacteriol. 2000;182:2675–2679. doi: 10.1128/jb.182.10.2675-2679.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watnick PI, Fullner KJ, Kolter R. A role for the mannose-sensitive hemagglutinin in biofilm formation by Vibrio cholerae El Tor. J Bacteriol. 1999;181:3606–3609. doi: 10.1128/jb.181.11.3606-3609.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitchurch CB, Hobbs M, Livingston SP, Krishapillai V, Mattick JS. Characterisation of a Pseudomonas aeruginosa twitching motility gene and evidence for a specialised protein export system widespread in eubacteria. Gene. 1991;101:33–44. doi: 10.1016/0378-1119(91)90221-v. [DOI] [PubMed] [Google Scholar]
- White PB. The rugose variant of vibrios. J Pathol Bacteriol. 1938;46:1–6. [Google Scholar]
- Wolfgang M, Lauer P, Park H, Brossay L, Hebert J, Koomey M. PilT mutations lead to simultaneous defects in competence for natural transformation and twitching motility in piliated Neisseria gonorrhoeae. Mol Microbiol. 1998;29:321–330. doi: 10.1046/j.1365-2958.1998.00935.x. [DOI] [PubMed] [Google Scholar]
- Wu SS, Wu J, Kaiser D. The Myxococcus xanthus pilT locus is required for social gliding motility although pili are still produced. Mol Microbiol. 1997;23:109–121. doi: 10.1046/j.1365-2958.1997.1791550.x. [DOI] [PubMed] [Google Scholar]
- Yildiz FH, Schoolnik GK. Vibrio cholerae O1 El Tor: identification of a gene cluster required for the rugose colony type, exopolysaccharide production, chlorine resistance, and biofilm formation. Proc Natl Acad Sci USA. 1999;96:4028–4033. doi: 10.1073/pnas.96.7.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]