

Abstract
A robust innate immune response is essential to the protection of all vertebrates from infection, but it often comes with the price tag of acute inflammation. If unchecked, a runaway inflammatory response can cause significant tissue damage, resulting in myriad disorders, such as dermatitis, toxicshock, cardiovascular disease, acute pelvic and arthritic inflammatory diseases, and various infections. To prevent such pathologies, cells have evolved mechanisms to rapidly and specifically shut off these beneficial inflammatory activities before they become detrimental. Our review of recent literature, including our own work, reveals that the most dominant and common mechanism is translational silencing, in which specific regulatory proteins or complexes are recruited to cis-acting RNA structures in the untranslated regions of single or multiple mRNAs that code for the inflammatory protein(s). Enhancement of the silencing function may constitute a novel pharmacological approach to prevent immunity-related inflammation.
Regulating gene expression at the level of protein translation offers several benefits, including rapidity of response and reversibility. Translational control can be subdivided into two major classes: global and transcript specific. Although the first mechanism regulates most transcripts, the second permits translation regulation of a limited ensemble of proteins. Transcript-specific regulation is generally mediated by the interaction of RNA-binding proteins with cognate cis-elements in the untranslated region (UTR) of the target transcripts, causing translational repression (1, 2). Although not significantly emphasized, an important role of translational control in regulating acute inflammation is evident from emerging studies. It is obligatory for the cells of the immune system to rapidly activate the inflammatory protein synthesis in response to infection or its blockage when they are no longer needed. Translational control offers a strategic advantage to these cells, because the use of the pre-existing mRNAs bypass the lengthy nuclear control mechanisms (e.g., transcription, splicing, and transport). At the same time, it provides the reversibility through modifications of the regulatory intermediates, mainly via reversible phosphorylation. Together, these two features allow rapid activation or termination of synthesis of a specific protein or group of proteins required for inflammation.
Acute inflammation is a highly orchestrated, tissue-based response to trauma incited by tissue injury or microbial invasion of the host (3, 4). Initiated by responsive leukocytes and lymphocytes, a key component of the process is the trafficking of inflammatory cells to the sites of injury or infection. The cytokine/receptor interactions on the surface of these cells culminate in the expression of new gene products that efficiently kill or injure the invading organisms. However, uncontrolled production of inflammatory products is injurious to host cells and even leads to neoplastic transformation (5). Therefore, endogenous mechanisms have evolved to limit the production of inflammatory molecules and permit the resolution of the inflammatory response (3, 6). In-depth studies of these mechanisms are important because defects in the pathway may contribute to the progression of chronic inflammatory disorders, and the pathway itself may present targets for novel anti-inflammatory therapeutic strategies.
Outstanding past reviews summarized the resolution of inflammation (3, 6), as well as stress-mediated, global translational control mechanisms (2, 7–9). A recent article reviewed how a number of cytokine mRNAs can be regulated at the level of mRNA stability (10). In addition, the role of one particular RNA-binding complex in the control of inflammation was the subject of a recent review (11). In this article, our goal is to present and defend the hypothesis that transcript-specific translational control is the prime determinant of the onset, progression, and resolution of acute inflammation.
Translational regulations offer stringent control of highly inflammatory cytokines
TNF-α, which is synthesized and secreted by lymphocytes, mast cells, and activated macrophages, has multiple proinflammatory functions geared toward protection of the host from infection. TNF-α induces fever via induction of PGE2 synthesis by the hypothalamic vascular endothelium (12), activates synthesis of complement factors (3), and recruits leukocytes to the sites of infection via induction of adhesion molecules (13, 14). The high potency and diversity of proinflammatory functions require stringent control of TNF-α expression. Indeed, expression of TNF-α in unstimulated mouse peritoneal macrophages is extremely low, in part because of low mRNA expression and as a result of a block in translation (15). Treatment of macrophages with LPS, a cell wall component of Gram-negative bacteria, induces a 10,000-fold increase in TNF-α protein secretion resulting from simultaneous activation of transcription and derepression of the translational blockade (16).
Our knowledge of the translational control mechanisms has been advanced by the discovery of several proteins that bind AU-rich elements (AREs). Found in the 3′ UTRs of various mRNAs, AREs are of enormous physiological significance because they recruit protein factors that regulate the stability of the target mRNA. Interestingly, a number of ARE-containing mRNAs, which include TNF-α and cyclooxygenase-2 (COX-2), are associated with the inflammatory response. The embedded ARE sequence clearly offers protection against the overexpression of TNF-α, because transgenic mice that express TNF-α mRNA lacking ARE exhibit elevated levels of circulating TNF-α and developed joint- and gut-associated immunopathology (17). Although TNF-α is an arm of innate immunity, its highly inflammatory property must have necessitated multiple checkpoints for stringent control.
The ARE-based mechanisms are orchestrated by a complex ensemble of RNA-binding proteins that are recruited to ARE, such as heterogeneous nuclear ribonucleoprotein (hnRNP)-A1, HuR, T cell intracellular Ag (TIA)-1, TIA-1–related protein (TIAR), and TTP. Basal translation of TNF-α mRNA can be silenced by hnRNP-A1 binding to the ARE element. Phosphorylation of hnRNP-A1 by Mnk, a kinase downstream of p38 MAPK, decreases its affinity for the ARE and, thus, reactivates TNF-α translation (18). Two other ARE-binding proteins, TIA-1 and TIAR, constitutively bind to TNF-α mRNA in unstimulated and stimulated macrophages (19, 20). TIA-1 has multiple roles in the regulation of TNF-α expression that can be negative or positive, depending on context. More TNF-α mRNA associates with polysomes in peritoneal macrophages isolated from TIA-1–null mice compared with control mice, demonstrating its function in transcript-specific translational silencing (19). The inhibitory mechanism has yet to be determined, but it may involve sequestration in cytoplasmic mRNA storage depots, known as stress granules, which contain TIA-1 and TIAR (21).
Translational silencing by TIA-1/TIAR is not limited to TNF-α mRNA. COX-2, which catalyzes the rate-limiting step of PG synthesis during inflammation, is also silenced by TIA-1 binding to an ARE in the 3′ UTR (22). Like TNF-α, the powerful, multi-factorial inflammatory protein COX-2 is under translational control by multiple pathways. In epithelial cells, COX-2 translation is silenced by yet another protein CUG-binding protein 2 (23) that is predominantly nuclear but rapidly translocates to the cytoplasm following stress. Interestingly, CUG-binding protein 2 binds to two sets of AREs within the COX-2 3′ UTR, and unlike the previous mechanism, it stabilizes COX-2 mRNA, although it inhibits its translation nonetheless. Another critical target of TIAR is human matrix metalloproteinase (MMP)-13, which is expressed in chronic inflammation and cancer and may remodel the extracellular matrix (24). MMP-13 mRNA is translationally silenced by an alternately spliced form of TIAR (25). Genome-wide analysis of TIA-1–bound transcripts identified a bipartite stem-loop as the target element and predicted that up to 3000 transcripts may contain this element (26). An important physiological role of the TIA-1/TIAR translational silencing pathway is suggested by the uncontrolled cartilage inflammation and arthritis in TIA-1–null mice (19) and in transgenic mice that overexpress ARE-less TNF-α (17). Studies with TIA-1–deficient macrophages also showed that HuR-mediated translational silencing requires TIA-1. Furthermore, myeloid-restricted overexpression of HuR inhibits the macrophage inflammatory response in vivo (27). In summary, translational control by TIA-1/TIAR and others is critical for the maintenance of the basal, noninflammatory state in several cell types. Reciprocally, its derepression is necessary for maximal induction of target transcripts during the early initiation phase of acute inflammation.
The first line of innate antiviral defense in host cells is a robust production of type I IFNs (-α and -β) (28) induced by the transcription factor IFN regulatory factor (IRF)7, which, in turn, is regulated by the translational repressors 4E-binding protein (4E-BP)1 and 4E-BP2 (29). Mouse embryonic fibroblasts lacking these repressors are resistant to multiple viruses, such as influenza, encephalomyocarditis, vesicular stomatitis, and Sindbis. This enhanced antiviral response is caused exclusively by the significant upregulation of IRF7 translation (29). Contrary to its positive role in innate and adaptive immunity, the activation of mammalian target of rapamycin (mTOR) in phagocytic cells can also limit the inflammatory response. mTOR downregulates the expression of proinflammatory cytokines IL-12 and -23; reciprocally, the inhibition of mTOR by rapamycin promotes the expression of IL-6, -12, and -23 and TNF-α and downregulates the expression of the anti-inflammatory cytokine IL-10 (30). Despite the multifaceted role of mTOR in immunity (31), it remains unknown whether its activation directly influences the de novo translation of the mRNAs coding for these cytokines, perhaps via a common RNA element. The lack of knowledge is probably due to the preferential attention bestowed on the upstream and downstream signaling cascade of mTOR, rather than identifying the target mRNAs. It is also possible that mTOR targets a critical transcription factor common to this array of cytokine genes. The recent report of mTOR-mediated direct translational repression of IRF7 mRNA is consistent with this notion (29).
Studies of IL-10 have provided strong evidence for the role of translational control in anti-and proinflammatory pathways (Fig. 1). mRNAs of inflammatory proteins (e.g., TNF-α, COX-2, and MMP-2) are translationally repressed by ARE-binding proteins in the 3′ UTRs. Inflammatory stimuli activate p38 MAPK, which targets ARE-binding proteins and derepresses the translation. As an anti-inflammatory protein, IL-10 blocks this process by inhibiting p38 MAPK (32) (Fig. 1). Interestingly, occupancy of the adenosine receptor by its ligands activates translation of IL-10 mRNA, thus playing an important role in the anti-inflammatory activity of these ligands (33). However, in a proinflammatory role, IL-10 stimulates ferritin translation by reducing the affinity of the translational repressor, the iron response element-binding protein, to the 5′ UTR of ferritin mRNA (34). Considering the role of ferritin in iron sequestration, this may lead to hyperferritinemia and limited iron availability for the progenitor cells in bone marrow, a condition typically seen in the anemia of chronic inflammation (Fig. 1).
FIGURE 1.

Pro- and anti-inflammatory axis of IL-10 depends on translation regulation. Left, Inflammatory stimuli activate p38 MAPK that disengages ARE binding proteins and derepresses the translation of inflammatory proteins. In a global anti-inflammatory role, IL-10 negatively regulates this process by abrogating the activation by p38 MAPK. Right, Occupancy of the adenosine receptors by its ligands relieves the imposed translational repression of IL-10 mRNA and activates the synthesis of IL-10 protein (blue circles). As a proinflammatory molecule, IL-10 stimulates ferritin translation by removing the translational repressor (the iron response element-binding protein) from 5′ UTR. Overproduction of ferritin causes sequestration of available iron (rust-colored circles) for bone marrow, leading to the characteristic anemia of inflammation.
Activation of T lymphocytes
The proliferation and migration of T lymphocytes promote inflammation and the eukaryotic translation initiation factor (eIF)4E-BP kinase mTOR plays a pivotal role in this process. mTOR targets Kruppel-like factor 2, an essential transcription factor of CCR7 and L-selectin that is crucial for the migration of CD8+ T lymphocytes into lymph nodes (35). Indeed, G1 to S phase progression in IL-2–stimulated T lymphocytes can be blocked by rapamycin, an inhibitor of mTOR (36). In apparent contrast, a recent report revealed that inhibition of mTOR has immunostimulatory effects. This report showed that rapamycin significantly enhances the quantity and quality of virus-specific CD8+ T cells and identified eIF4E as a downstream effector molecule of mTOR (37). In addition, a recent study showed that activation of mTOR is required for regulatory and effector T cell lineage commitment (38). A subsequent study also showed that single IgG IL-1–related receptor, a negative regulator of IL-1R and TLR signaling, controls Th17 cell development and effector function by blocking the IL-1–induced activation of mTOR (39). Together, these results show that translational control by mTOR is critical for the regulation of adaptive immunity and raise new hope for the management of inflammation by its pharmacological manipulations.
Chemoattraction of T lymphocytes
The chemoattraction of T lymphocytes to the site of injury is an obligatory component of inflammation, and RANTES is a major player in this process (40). A rheostat function of regulating RANTES expression at the site of inflammation is crucial for the rapid response to stress, cytokines, and other proinflammatory factors in the local microenvironment of T lymphocytes (41). This precise control is achieved by regulating the translation of RANTES factor of late-activated T lymphocytes (RFLAT)-1 (Fig. 2). RFLAT-1 is a transcription factor that activates the RANTES gene; although its mRNA remains constant in resting and activated T cells, the protein is expressed only 3–5 d after T cell activation (42). This occurs through an interesting mechanism, in which two upstream open-reading frames and a highly structured 5′ UTR of RFLAT-1 mRNA promote translational silencing. Conversely, phosphorylation of 4E-BP and eIF4E itself by mTOR and p38 MAPK/ERK rescues RFLAT-1 mRNA from silencing (Fig. 2) (41). The extracellular signals that activate these kinases are unknown. Nevertheless, the translational control of RFLAT clearly helps to precisely control RANTES, which, in turn, may be required to constantly monitor the environment as T lymphocytes move through extracellular spaces to the sites of injury (41).
FIGURE 2.
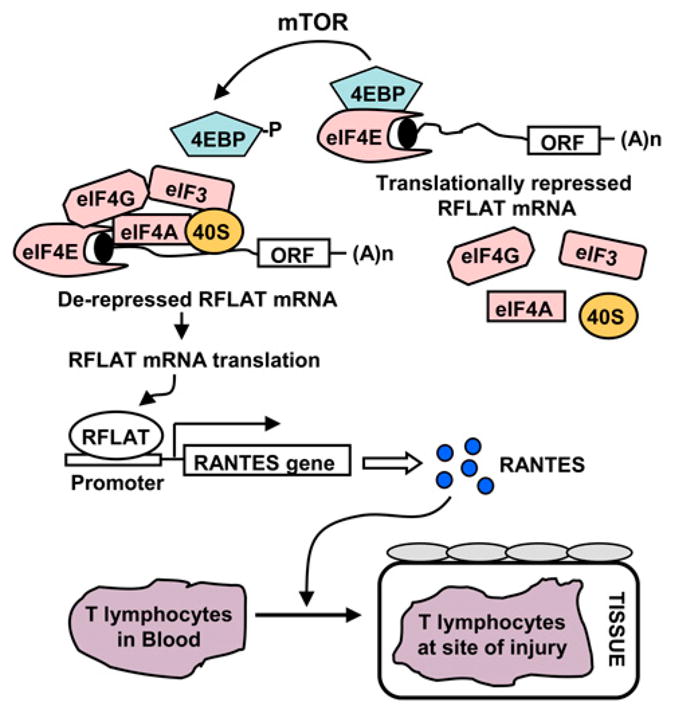
Chemoattraction of T lymphocytes by RANTES is under translation control. RFLAT, a transcription factor of RANTES, is silenced at the level of translation in unstimulated cells by 4E-BP. Phosphorylation of 4E-BP by mTOR promotes dissociation of 4E-BP, allowing assembly of the translation initiation factors (eIFs) on RFLAT mRNAs. This activates the translation of RFLAT protein and promotes the transcription of RANTES gene. Expression of RANTES (blue circles) is required for the chemoattraction of T lymphocytes to the sites of injury.
Neutrophil influx
Neutrophils are major players in innate immunity and provide early links between the innate and adaptive inflammatory responses. Upon activation by cognate signals, the neutrophils rapidly translate their constitutive mRNAs. For example, translation of IL-6 soluble receptor is induced when neutrophils are activated by platelet-activating factor (43), a signaling phosphopeptide implicated in homeostatic and injurious inflammation (44). The mechanisms that limit neutrophil influx demonstrate nicely how the signal to resolve inflammation has been embedded in the signal for initiation. Neutrophil-derived arachidonate serves as substrate for neutrophil 5-lipoxygenase to generate the inflammatory leukotriene B4. However, the infiltrating neutrophils also deliver arachidonate to tissue cells that express 15-lipoxygenase; this catalyzes the conversion of arachidonate to lipoxins, which inhibit neutrophil influx (3, 45) (Fig. 3). Consistent with these findings, a defect in lipoxin-mediated anti-inflammatory activity was reported in the cystic fibrosis airway (46). 15-Lipoxygenase that produces lipoxins, a signal for inflammation resolution, can be regulated at the level of translation by the binding of hnRNP-K and E1 to the differentiation induced control element in its 3′ UTR (47), which blocks the 60S ribosomal subunit from joining (Fig. 3) (48). Unexpectedly, the activation of c-Src kinase causes phosphorylation of hnRNP-K, abrogating its RNA-binding activity and derepressing 15-lipoxygenase mRNA translation (Fig. 3) (49). These results add to the mounting evidence for the importance of translational control in the interface between pro- and anti-inflammatory responses, in this case from neutrophils using arachidonate in a context-specific fashion.
FIGURE 3.

A translationally regulated enzyme 15-lipoxygenase controls neutrophil influx. A neutrophil (magenta) is recruited to the tissue from the lumen. This process is mediated by leukotriene B4 synthesized from arachidonate and catalyzed by 5-lipoxygenase within the neutrophil. The recruitment of neutrophil in the tissue is depicted to bring in arachidonate that produces lipoxin using translationally regulated enzyme, 15-lipoxygenase. Note that this enzyme is different from the 5-lipoxygenase within the neutrophil. The regulation requires recruitment of the hRNPK complex (green) to DICE, a novel RNA element in the 3′ UTR of 15-lipoxygenase mRNA that can block 60S ribosomal subunit joining and inhibit translation. Tissue lipoxin (blue circles), generated by 15-lipoxygenase, exits and blocks further neutrophil influx. DICE, differentiation induced control element.
NK cell-mediated cytotoxicity
Contact-dependent cytotoxicity is critical for innate immunity and depends upon the rapid synthesis of perforin and granzymes (50). There is compelling evidence that blockage of perforins and granzyme mRNA translation underlies the minimal cytotoxicity of resting NK cells and that removal of this blockage is crucial for NK cell activation (51). The finding that the activation signal(s) cause an immediate release of the translation brake of the killer proteases could offer a novel opportunity for therapeutic intervention.
Monocyte adhesion and platelet activation
Adhesion of monocytes and activation of platelets are vital steps in hemostasis and inflammation (52). A series of observations made by Zimmerman and colleagues showed that both processes pass through checkpoints executed by translational control of some key molecules. Engagement of P-selectin gp1 to the plasma membrane of monocytes delivers outside-in signals that activate mTOR and, hence, phosphorylate 4E-BP. The latter then activates translation of urokinase plasminogen activator receptor, a critical cell surface protease receptor required for cell adhesion and cell surface phenotype (53). Thrombin stimulation induces platelet adhesion and aggregation, and the activated platelets cause translational activation of Bcl-3 (54), the oncogene product of B cell lymphoma-3. The secretion of Bcl-3 mediates adhesion and outside-in signaling of these cells by engagement of αIIβ/β3 integrin. The lack of transcriptional activity in the platelets, a longstanding observation, argues that Bcl-3 may be regulated at the level of translation. Metabolic labeling of thrombin-activated platelet showed that Bcl-3 is indeed activated directly at the level of translation and requires activation of mTOR and 4E-BP phosphorylation (54).
Macrophage survival in injured tissue
Low levels of oxygen and glucose, as well as high concentrations of lactate and reductive metabolites, characterize the surroundings of the injured tissues (55). To maintain viability and activity in such challenging conditions, the cells of the innate immune system must adopt a strategy to maintain the cellular ATP pool. The longstanding question of how the cells achieve this feat has been answered by studies from Johnson and colleagues (56). Using gene knockout mice, these studies showed the essential role of hypoxia-inducible factor (HIF)-1α in the process of myeloid cell survival and inflammation. The expression of the mRNAs of the glycolytic enzymes phosphoglycerate kinase (57), lactate dehydrogenase (57), pyruvate kinase M (58), and glucose transporter-1 (59) is HIF-1α –dependent and required for survival of the macrophages. In agreement with this hypothesis, genetic abrogation of HIF-1α causes profound defects in myeloid cell aggregation, motility, invasiveness, and bacterial killing (56). The 5′ UTR of HIF-1α mRNA is highly structured and contains an internal ribosome entry site (IRES) responsible for translational activation (60, 61). The IRES-mediated translation of HIF-1α under hypoxic conditions is particularly important, because hypoxia inhibits cap-dependent translation by reducing the availability of eIF4E and by phosphorylation of translation initiation factor eIF2α (62, 63), thus creating a highly compromised condition. Under these conditions, IRES-mediated translation might be an effective means to sustain the synthesis of the survival proteins for the macrophages. It is tempting to speculate that resolution of inflammation results from inhibition of IRES activity, downregulation of HIF-1α, and the subsequent loss of monocytes/macrophages from the site of injury.
Controlling the immune and inflammatory responses by microRNAs
A major breakthrough in translational regulation is the recent recognition of the RNA interference pathway, variations of which are found in all metazoan cells. In the RNA interference pathway, small noncoding RNA molecules, broadly called microRNAs (miRNAs), bind to partially complementary sequences in the 3′ UTR of target mRNAs, called miRNA response element (MRE), and repress translation (64). In the dendritic cells, bacterial LPS or viral dsRNA binds to TLRs, triggering a signaling cascade that ultimately leads to T cell activation and elaboration of a plethora of inflammatory cytokines, including the type I IFNs. Recent studies showed how miRNAs could impose restraints on this process to protect the cells from uncontrolled immune response. Microarray analysis of macrophages after exposure to dsRNA (or IFN-β) revealed that miR-155 is the only miRNA that was substantially upregulated by LPS and dsRNA, suggesting that miR-155 is a common target of a broad range of inflammatory mediators (65). In-depth studies revealed that miR-155 silenced transcripts that code for several proinflammatory proteins, such as the Fas-associated death domain protein, I-κ B kinase-ε, TNFR superfamily-interacting serine-threonine kinase 1, and TAK-1–associated binding protein-2. An unexpected observation was that miR-155 also targeted the TNF-α mRNA 3′ UTR and actually enhanced TNF-α translation, the mechanism of which remained unexplored (66). In fact, transgenic mice expressing recombinant miR-155 produced higher levels of TNF-α when exposed to LPS and were hypersensitive to LPS-induced septic shock, suggesting that the TNF-α–stimulating role of miR-155 may have a dominant effect in pathology (66). Altogether, these results show a balancing act of miR-155 in acute inflammation and sepsis, perhaps in a context-dependent manner.
In another study (67), miR-9 was found to be upregulated in polymorphonuclear neutrophils and monocytes by multiple pro-inflammatory signals, such as TLR4, TLR2, and TLR7/8 agonists, and by the proinflammatory molecules LPS, TNF-α, and IL-1β but not by IFN-γ. A number of these agonists activate the proinflammatory transcription factor NF-κB that, in turn, acts as a transcription factor to induce miR-9 expression. Interestingly, NF-κB translation was reciprocally silenced by miR-9 via an MRE in the 3′ UTR of the NF-κB mRNA. Overall, these data suggest that NF-κB rapidly increases the expression of miR-9 that operates a feedback control of the NF-κB–dependent acute inflammatory response triggered by multiple signals.
Evidence of miRNA-mediated silencing of inflammatory genes in nonimmune cells has only begun to appear, and a recent example comes from studies of pulmonary inflammation. The epithelial lining of the mammalian lungs is constantly exposed to inflammatory signals in the inhaled air. A recent study revealed that stimulation with IL-1β, a proinflammatory cytokine, results in a rapid increase in miR-146a expression in pulmonary cells (68). The increased miRNA-146a promoted translational silencing of the proinflammatory chemokines IL-8 and RANTES. In an interesting twist, IL-1β also activates NF-κB (69), and NF-κB activates the transcription of miR-146a (70) and IL-8 genes (71). Clearly, this is an intricate feedback inhibition operating at multiple levels, such that the initial trigger, IL-1β, rapidly activates the two chemokines via NF-κB and, at the same time, induces the synthesis of a silencer miRNA of the chemokines. Such controls limit the severe inflammation that would otherwise accompany the innate immune response of the pulmonary epithelia.
Translational control of the negative regulators of inflammation
Response to ILs and IFNs are negatively regulated by the members of a family of Src homology 2 domain-containing adaptor molecules, suppressor of cytokine signaling (SOCS). Expression of SOCS-1 is repressed at the level of translation initiation by two upstream open reading frames in the 5′ UTR (72). Induction of RNA helicase activity relieves the repression, permitting an increase in SOCS-1 protein expression and subsequent inhibition of the cytokine-induced response. Regulation of SOCS-1 exemplifies how translational control of a single gene can be crucial for simultaneous control of an array of inflammatory genes.
The proinflammatory cytokine IL-6 is negatively regulated by an unsuspected player, the tumor suppressor p53,a defect in which leads to the pathogenesis of rheumatoid arthritis (73, 74) and autoimmune inflammation (75). In both diseases, lack of p53 function leads to overexpression of IL-6 (74) and a heightened response to LPS and IFN (75). Interestingly, p53 mRNA itself is a target of the translational silencing mechanism that requires a region of its 3′ UTR (76). This region is also targeted by the RNA-binding protein HuR that relieves the translational repression (77, 78). Together, these findings show that translation upregulation of p53 can be important in resolving inflammation.
Translational control of IFN-γ expression
The entire IFN-γ axis, including the synthesis of IFN-γ and its downstream signaling, is under translational control, as we describe in this section and the next. Production of IFN-γ by T lymphocytes is subject to at least two distinct translational control mechanisms. IL-18 (originally designated as IFN-γ–inducing factor) is an epithelial and stromal cell product and is a potent and specific inducer of IFN-γ transcription in T lymphocytes. In a rat glomerular nephritis model, IL-18 mRNA is translationally silenced by its 5′ UTR, resulting in minimal induction of IFN-γ (Fig. 4) (79). Additionally, T cell expression of IFN-γ is autoregulated at the level of translation, again by a 5′ UTR element, but using an unusual and indirect mechanism. A pseudoknot in the IFN-γ 5′ UTR recruits and activates dsRNA-activated protein kinase R (PKR) (Fig. 4) (80), which, in turn, phosphorylates eIF2α and inhibits IFN-γ translation. Unexpectedly, global protein synthesis remains unaltered, suggesting that the cellular region of PKR activation remains highly restricted. Therefore, in this feedback mechanism, the pseudoknot essentially acts as a PKR sensor, preventing the overproduction of IFN-γ.
FIGURE 4.
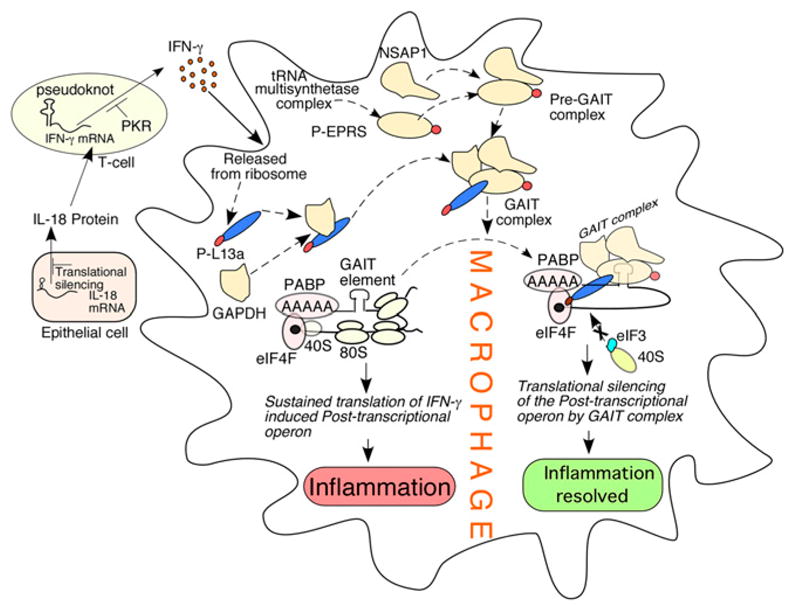
Control of the IFN-γ axis and the GAIT paradigm of translational silencing. IL-18, an epithelial cell-derived factor, induces IFN-γ in T cells. The synthesis of both IL-18 and IFN-γ is under translational control by sequence elements at the 5′ UTR of their cognate mRNAs (left). In myeloid cells (such as macrophages), IFN-γ induces the initial inflammatory signal that is eventually silenced by an ordered assembly of the GAIT complex (subunits in yellow). Formation of GAIT complex requires the release of its subunit proteins from their parental complex triggered by phosphorylation events (phosphate groups in red). The binding of silencing-competent GAIT complex at the GAIT elements of the 3′ UTRs of the mRNAs (the members of the posttranscriptional operon) encoding inflammatory proteins directly blocks translation by intercepting the recruitment of 40S ribosomal subunit. Translational silencing of a group of inflammatory proteins by the GAIT pathway resolves inflammation.
IFN-γ–activated inhibitor of translation pathway: the translational master regulator of IFN-γ–induced inflammation
IFN-γ is a major cytokine activator of macrophages that contributes to innate immunity toward pathogens (81). A series of studies from our group and other investigators discovered a unique translational silencing mechanism that is triggered by IFN-γ exposure of monocytic cells (Fig. 4) (82–89). We found robust transcriptional activation of ceruloplasmin (Cp) within 2 h of IFN-γ treatment, but this was followed by translation silencing 16–24 h later. Cp is an inflammatory, acute-phase protein made by liver, monocytes, and macrophages, and it has a documented role in monocyte-mediated lipoprotein oxidation, bactericidal activity, and iron homeostasis (90). Translational silencing of Cp requires the binding of a protein complex (IFN-γ–activated inhibitor of translation [GAIT] complex) to a specific element (GAIT element) in the 3′ UTR of Cp mRNA (82, 85). The GAIT element is a degenerate 29-nucleotide bipartite hairpin with features that distinguish it from other translational control elements. Using genetic and biochemical approaches, four protein constituents of the GAIT complex have been identified: ribosomal protein L13a, glutamyl-prolyl-tRNA synthetase (EPRS), NS1-associated protein-1, and GAPDH (Fig. 4) (84, 86). Interestingly, two of the proteins are released from pre-existing cytosolic macromolecular complexes that are critical components of the protein synthesis pathway: EPRS is released from the aminoacyl-tRNA multisynthetase complex (86), and L13a is released from the large ribosomal subunit (84). In both cases, release is initiated by IFN-γ and requires phosphorylation of the released protein (84, 91). Assembly of the GAIT complex occurs in two stages (Fig. 4). During the first stage, which is complete within ~4 h, phosphorylated EPRS binds to NS1-associated protein-1 to form a nonfunctional, pre-GAIT complex. Approximately 12 h later, ribosome-free, phosphorylated L13a joins GAPDH and the pre-GAIT complex to form the functional quaternary GAIT complex that binds the GAIT element and blocks translation. Surprisingly, despite the phosphorylation and release of L13a and EPRS from their parent complexes, global protein synthesis remains unaltered. As part of the GAIT complex, phospho-L13a binds the translation initiation factor eIF4G of the cap-binding eIF4F complex, thereby preventing the formation of the 48S preinitiation complex (88). Recent studies identified Ser77 of L13a as the single site for IFN-γ–inducible phosphorylation (92) and death-associated protein kinase (DAPK) and zipper-interacting protein kinase (ZIPK) as the responsible kinases (92). Interestingly, DAPK-ZIPK itself is also a target of GAIT-mediated translational silencing. Thus, the GAIT mechanism provides a self-limiting pathway that relies on the DAPK-ZIPK-L13a negative feedback module.
A clinically significant GAIT-containing gene, the translation of which is also silenced by the GAIT complex described above, is vascular endothelial growth factor (VEGF) (93). The sustained expression of VEGF is obligatory for the hypoxic response. In a recent report, hypoxia was shown to override GAIT-mediated repression of VEGF translation observed in normoxia (93, 94) through the formation of alternate RNA secondary structure in the GAIT region. The ability of the hypoxia to override the translational repression could help the decision-making process of macrophages simultaneously exposed to opposing inflammatory and hypoxic signals.
GAIT pathway regulates an inflammation-responsive posttranscriptional operon
Posttranscriptional operons in eukaryotes represent a powerful mechanism for regulating functionally related genes simultaneously on the same cue. This mechanism uses specific RNA-binding proteins that recognize structural elements common to the mRNAs of that family (95, 96). With this concept in mind, and knowing the inflammatory property of Cp, we recently hypothesized that the GAIT pathway may regulate a whole cohort of mRNAs belonging to an inflammation-related posttranscriptional operon. This hypothesis is also based on the high abundance of GAIT complex components and stoichiometric release of L13a and EPRS from their parental complexes (84, 86). To test this, we conducted a genome-wide screen for target mRNAs of GAIT-mediated translational silencing in IFN-γ–activated monocytes (89) through an analysis of polysome-bound and -unbound mRNA pools. This led us to identify a cohort of mRNAs that follow the same pattern of regulation as Cp. Functional mapping of the GAIT-like elements in the 3′ UTRs of these putative targets validated the translational silencing mechanism. Importantly, many of these mRNAs coded for inflammatory proteins, such as chemokines and their receptors and proteins important in cytokine response and signaling. The chemokines and their receptors, for example, are responsible for activation-directed migration of leukocytes to the sites of inflammation during immune surveillance (97), which is now also recognized as a major contributor to the pathogenesis of inflammatory disease, such as atherosclerosis (98, 99), and neoplastic, infectious, allergic, and autoimmune diseases (100). A potential therapeutic approach would be to enhance the formation of the GAIT complex, perhaps by triggering the phosphorylation and release of L13a from its 60S ribosomal depot. This approach could be used to identify a powerful anti-inflammatory drug because of its ability to silence all inflammatory mRNAs of this posttranscriptional operon using a natural mechanism.
Conclusion and Future Directions
Translational regulation, primarily through the recruitment of specific proteins to cognate sequences in the UTRs of the target mRNAs, is gaining new recognition. By virtue of its mechanism, this form of regulation is fast and capable of being mRNA and pathway specific (7, 9, 10). The silencing is rapid because the molecular components involved in the process are constitutively present in the cytoplasm or form rapidly once the commitment to silencing is made. In general, the feedback silencing force slowly builds up while the immune response is functioning and then is rapidly deployed when the time is right. It seems that the onset of the silencing is programmed into the initiation of immune response itself. In the prototype GAIT-mediated silencing, the DAPK-ZIPK kinase cascade that phosphorylates L13a exhibits a rapid induction of kinase activities between 12 and 16 h of IFN-γ treatment, despite essentially unchanged kinase levels (92). This delay imposes a deliberate restriction to form the silencing-competent GAIT complex and allows the expression of the IFN-γ–induced inflammatory proteins at early hours. Clearly, these mechanisms offer new targets of pharmacological interventions against the overproduction of many inflammatory proteins by promoting the modifications of the regulatory intermediates.
An interesting new area of study will be to understand whether and how two silencing elements on the same proinflammatory mRNA interact. This may include two homologous elements (i.e., belonging to the same family), such as two GAIT sequences, or heterologous ones (of different families), such as a GAIT and an MRE. The cis-acting location of both in the same 3′ UTR may allow novel forms of interaction between their cognate complexes. We envision that such interactions may also affect specific genes of the inflammatory pathways, allowing control by multiple physiological signals. Insights gained from the translational-silencing mechanisms targeting single or multiple mRNAs should ultimately lead to novel anti-inflammatory approaches exploiting the endogenous cellular pathways of inflammation resolution.
Acknowledgments
This work was supported by National Institutes of Health Grant RO1HL79164 and American Heart Association Grant-in-Aid 0855555D (to B.M.), RO1AI060632 (to X. L.), and RO1AI059267 (to S.B.).
We offer our sincere apology to many scientists whose work could not be included in the references because of space limitations. We are extremely thankful to Dr. Joseph D. Fontes, University of Kansas School of Medicine, for critical reading and proofreading of the late-stage manuscript.
Abbreviations used in this paper
- 4E-BP
4E-binding protein
- ARE
AU-rich element
- COX-2
cyclooxygenase-2
- Cp
ceruloplasmin
- DAPK
death-associated protein kinase
- eIF
eukaryotic translation initiation factor
- DICE
differentiation induced control element
- EPRS
glutamyl-prolyl-tRNA synthetase
- GAIT
IFN-γ–activated inhibitor of translation
- HIF
hypoxia-inducible factor
- hnRNP
heterogeneous nuclear ribonucleoprotein
- IRES
internal ribosome entry site
- IRF
IFN regulatory factor
- miRNA
micro-RNA
- MMP
matrix metalloproteinase
- MRE
microRNA response element
- mTOR
mammalian target of rapamycin
- PKR
protein kinase R
- RFLAT
RANTES factor of late-activated T lymphocytes
- SOCS
suppressor of cytokine signaling
- TIA
T cell intracellular Ag
- TIAR
T cell intracellular Ag-1–related protein
- UTR
untranslated region
- VEGF
vascular endothelial growth factor
- ZIPK
zipper-interacting protein kinase
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Mazumder B, Seshadri V, Fox PL. Translational control by the 3′-UTR: the ends specify the means. Trends Biochem Sci. 2003;28:91–98. doi: 10.1016/S0968-0004(03)00002-1. [DOI] [PubMed] [Google Scholar]
- 2.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 4.Kumar V, Abbas AK, Fausto N. Robbins and Cotran’s Pathologic Basis of Disease. 7. Saunders; Philadelphia: 2004. [Google Scholar]
- 5.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 6.Gilroy DW, Lawrence T, Perretti M, Rossi AG. Inflammatory resolution: new opportunities for drug discovery. Nat Rev Drug Discov. 2004;3:401–416. doi: 10.1038/nrd1383. [DOI] [PubMed] [Google Scholar]
- 7.Dever TE. Gene-specific regulation by general translation factors. Cell. 2002;108:545–556. doi: 10.1016/s0092-8674(02)00642-6. [DOI] [PubMed] [Google Scholar]
- 8.Holcik M, Sonenberg N. Translational control in stress and apoptosis. Nat Rev Mol Cell Biol. 2005;6:318–327. doi: 10.1038/nrm1618. [DOI] [PubMed] [Google Scholar]
- 9.Sonenberg N, Hinnebusch AG. New modes of translational control in development, behavior, and disease. Mol Cell. 2007;28:721–729. doi: 10.1016/j.molcel.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 10.Anderson P. Post-transcriptional control of cytokine production. Nat Immunol. 2008;9:353–359. doi: 10.1038/ni1584. [DOI] [PubMed] [Google Scholar]
- 11.Mukhopadhyay R, Jia J, Arif A, Ray PS, Fox PL. The GAIT system: a gatekeeper of inflammatory gene expression. Trends Biochem Sci. 2009;34:324–331. doi: 10.1016/j.tibs.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Warren JS. Interleukins and tumor necrosis factor in inflammation. Crit Rev Clin Lab Sci. 1990;28:37–59. doi: 10.3109/10408369009105897. [DOI] [PubMed] [Google Scholar]
- 13.Tsang CM, Wong CK, Ip WK, Lam CW. Synergistic effect of SCF and TNF-α on the up-regulation of cell-surface expression of ICAM-1 on human leukemic mast cell line (HMC)-1 cells. J Leukoc Biol. 2005;78:239–247. doi: 10.1189/jlb.0704400. [DOI] [PubMed] [Google Scholar]
- 14.Edelstein LC, Pan A, Collins T. Chromatin modification and the endothelial-specific activation of the E-selectin gene. J Biol Chem. 2005;280:11192–11202. doi: 10.1074/jbc.M412997200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beutler B, Krochin N, Milsark IW, Luedke C, Cerami A. Control of cachectin (tumor necrosis factor) synthesis: mechanisms of endotoxin resistance. Science. 1986;232:977–980. doi: 10.1126/science.3754653. [DOI] [PubMed] [Google Scholar]
- 16.Han J, Brown T, Beutler B. Endotoxin-responsive sequences control cachectin/tumor necrosis factor biosynthesis at the translational level. J Exp Med. 1990;171:465–475. doi: 10.1084/jem.171.2.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10:387–398. doi: 10.1016/s1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]
- 18.Buxadé M, Parra JL, Rousseau S, Shpiro N, Marquez R, Morrice N, Bain J, Espel E, Proud CG. The Mnks are novel components in the control of TNF α biosynthesis and phosphorylate and regulate hnRNP A1. Immunity. 2005;23:177–189. doi: 10.1016/j.immuni.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 19.Piecyk M, Wax S, Beck AR, Kedersha N, Gupta M, Maritim B, Chen S, Gueydan C, Kruys V, Streuli M, Anderson P. TIA-1 is a translational silencer that selectively regulates the expression of TNF-α. EMBO J. 2000;19:4154–4163. doi: 10.1093/emboj/19.15.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gueydan C, Droogmans L, Chalon P, Huez G, Caput D, Kruys V. Identification of TIAR as a protein binding to the translational regulatory AU-rich element of tumor necrosis factor α mRNA. J Biol Chem. 1999;274:2322–2326. doi: 10.1074/jbc.274.4.2322. [DOI] [PubMed] [Google Scholar]
- 21.Kedersha NL, Gupta M, Li W, Miller I, Anderson P. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J Cell Biol. 1999;147:1431–1442. doi: 10.1083/jcb.147.7.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dixon DA, Balch GC, Kedersha N, Anderson P, Zimmerman GA, Beauchamp RD, Prescott SM. Regulation of cyclooxygenase-2 expression by the translational silencer TIA-1. J Exp Med. 2003;198:475–481. doi: 10.1084/jem.20030616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mukhopadhyay D, Houchen CW, Kennedy S, Dieckgraefe BK, Anant S. Coupled mRNA stabilization and translational silencing of cyclooxygenase-2 by a novel RNA binding protein, CUGBP2. Mol Cell. 2003;11:113–126. doi: 10.1016/s1097-2765(03)00012-1. [DOI] [PubMed] [Google Scholar]
- 24.Oh J, Takahashi R, Kondo S, Mizoguchi A, Adachi E, Sasahara RM, Nishimura S, Imamura Y, Kitayama H, Alexander DB, et al. The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis. Cell. 2001;107:789–800. doi: 10.1016/s0092-8674(01)00597-9. [DOI] [PubMed] [Google Scholar]
- 25.Yu Q, Cok SJ, Zeng C, Morrison AR. Translational repression of human matrix metalloproteinases-13 by an alternatively spliced form of T-cell-restricted intracellular antigen-related protein (TIAR) J Biol Chem. 2003;278:1579–1584. doi: 10.1074/jbc.M203526200. [DOI] [PubMed] [Google Scholar]
- 26.López de Silanes I, Galbán S, Martindale JL, Yang X, Mazan-Mamczarz K, Indig FE, Falco G, Zhan M, Gorospe M. Identification and functional outcome of mRNAs associated with RNA-binding protein TIA-1. Mol Cell Biol. 2005;25:9520–9531. doi: 10.1128/MCB.25.21.9520-9531.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Katsanou V, Papadaki O, Milatos S, Blackshear PJ, Anderson P, Kollias G, Kontoyiannis DL. HuR as a negative posttranscriptional modulator in inflammation. Mol Cell. 2005;19:777–789. doi: 10.1016/j.molcel.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 28.García-Sastre A, Biron CA. Type 1 interferons and the virus-host relationship: a lesson in détente. Science. 2006;312:879–882. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- 29.Colina R, Costa-Mattioli M, Dowling RJ, Jaramillo M, Tai LH, Breitbach CJ, Martineau Y, Larsson O, Rong L, Svitkin YV, et al. Translational control of the innate immune response through IRF-7. Nature. 2008;452:323–328. doi: 10.1038/nature06730. [DOI] [PubMed] [Google Scholar]
- 30.Weichhart T, Costantino G, Poglitsch M, Rosner M, Zeyda M, Stuhlmeier KM, Kolbe T, Stulnig TM, Hörl WH, Hengstschläger M, et al. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity. 2008;29:565–577. doi: 10.1016/j.immuni.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 31.Weichhart T, Säemann MD. The multiple facets of mTOR in immunity. Trends Immunol. 2009;30:218–226. doi: 10.1016/j.it.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 32.Kontoyiannis D, Kotlyarov A, Carballo E, Alexopoulou L, Blackshear PJ, Gaestel M, Davis R, Flavell R, Kollias G. Interleukin-10 targets p38 MAPK to modulate ARE-dependent TNF mRNA translation and limit intestinal pathology. EMBO J. 2001;20:3760–3770. doi: 10.1093/emboj/20.14.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Németh ZH, Lutz CS, Csóka B, Deitch EA, Leibovich SJ, Gause WC, Tone M, Pacher P, Vizi ES, Haskó G. Adenosine augments IL-10 production by macrophages through an A2B receptor-mediated posttranscriptional mechanism. J Immunol. 2005;175:8260–8270. doi: 10.4049/jimmunol.175.12.8260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tilg H, Ulmer H, Kaser A, Weiss G. Role of IL-10 for induction of anemia during inflammation. J Immunol. 2002;169:2204–2209. doi: 10.4049/jimmunol.169.4.2204. [DOI] [PubMed] [Google Scholar]
- 35.Sinclair LV, Finlay D, Feijoo C, Cornish GH, Gray A, Ager A, Okkenhaug K, Hagenbeek TJ, Spits H, Cantrell DA. Phosphatidylinositol-3-OH kinase and nutrient-sensing mTOR pathways control T lymphocyte trafficking. Nat Immunol. 2008;9:513–521. doi: 10.1038/ni.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morice WG, Brunn GJ, Wiederrecht G, Siekierka JJ, Abraham RT. Rapamycin-induced inhibition of p34cdc2 kinase activation is associated with G1/S-phase growth arrest in T lymphocytes. J Biol Chem. 1993;268:3734–3738. [PubMed] [Google Scholar]
- 37.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, Worley PF, Kozma SC, Powell JD. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gulen MF, Kang Z, Bulek K, Youzhong W, Kim TW, Chen Y, Altuntas CZ, Bak-Jenson K, McGeachy MJ, Do JS, et al. The Receptor SIGIRR Suppresses Th17 Cell Proliferation via Inhibition of the Interleukin-1 Receptor Pathway and mTOR Kinase Activation. Immunity. 2010 doi: 10.1016/j.immuni.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schall TJ, Bacon K, Toy KJ, Goeddel DV. Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature. 1990;347:669–671. doi: 10.1038/347669a0. [DOI] [PubMed] [Google Scholar]
- 41.Nikolcheva T, Pyronnet S, Chou SY, Sonenberg N, Song A, Clayberger C, Krensky AM. A translational rheostat for RFLAT-1 regulates RANTES expression in T lymphocytes. J Clin Invest. 2002;110:119–126. doi: 10.1172/JCI15336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song A, Chen YF, Thamatrakoln K, Storm TA, Krensky AM. RFLAT-1: a new zinc finger transcription factor that activates RANTES gene expression in T lymphocytes. Immunity. 1999;10:93–103. doi: 10.1016/s1074-7613(00)80010-2. [DOI] [PubMed] [Google Scholar]
- 43.Lindemann SW, Yost CC, Denis MM, McIntyre TM, Weyrich AS, Zimmerman GA. Neutrophils alter the inflammatory milieu by signal-dependent translation of constitutive messenger RNAs. Proc Natl Acad Sci USA. 2004;101:7076–7081. doi: 10.1073/pnas.0401901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prescott SM, Zimmerman GA, Stafforini DM, McIntyre TM. Platelet-activating factor and related lipidmediators. Annu Rev Biochem. 2000;69:419–445. doi: 10.1146/annurev.biochem.69.1.419. [DOI] [PubMed] [Google Scholar]
- 45.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol. 2001;2:612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 46.Karp CL, Flick LM, Park KW, Softic S, Greer TM, Keledjian R, Yang R, Uddin J, Guggino WB, Atabani SF, et al. Defective lipoxin-mediated anti-inflammatory activity in the cystic fibrosis airway. Nat Immunol. 2004;5:388–392. doi: 10.1038/ni1056. [DOI] [PubMed] [Google Scholar]
- 47.Ostareck DH, Ostareck-Lederer A, Wilm M, Thiele BJ, Mann M, Hentze MW. mRNA silencing in erythroid differentiation: hnRNP K and hnRNP E1 regulate 15-lipoxygenase translation from the 3′ end. Cell. 1997;89:597–606. doi: 10.1016/s0092-8674(00)80241-x. [DOI] [PubMed] [Google Scholar]
- 48.Ostareck DH, Ostareck-Lederer A, Shatsky IN, Hentze MW. Lipoxygenase mRNA silencing in erythroid differentiation: The 3′UTR regulatory complex controls 60S ribosomal subunit joining. Cell. 2001;104:281–290. doi: 10.1016/s0092-8674(01)00212-4. [DOI] [PubMed] [Google Scholar]
- 49.Ostareck-Lederer A, Ostareck DH, Cans C, Neubauer G, Bomsztyk K, Superti-Furga G, Hentze MW. c-Src-mediated phosphorylation of hnRNP K drives translational activation of specifically silenced mRNAs. Mol Cell Biol. 2002;22:4535–4543. doi: 10.1128/MCB.22.13.4535-4543.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yokoyama WM, Kim S, French AR. The dynamic life of natural killer cells. Annu Rev Immunol. 2004;22:405–429. doi: 10.1146/annurev.immunol.22.012703.104711. [DOI] [PubMed] [Google Scholar]
- 51.Fehniger TA, Cai SF, Cao X, Bredemeyer AJ, Presti RM, French AR, Ley TJ. Acquisition of murine NK cell cytotoxicity requires the translation of a pre-existing pool of granzyme B and perforin mRNAs. Immunity. 2007;26:798–811. doi: 10.1016/j.immuni.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 52.Weyrich AS, Lindemann S, Zimmerman GA. The evolving role of platelets in inflammation. J Thromb Haemost. 2003;1:1897–1905. doi: 10.1046/j.1538-7836.2003.00304.x. [DOI] [PubMed] [Google Scholar]
- 53.Mahoney TS, Weyrich AS, Dixon DA, McIntyre T, Prescott SM, Zimmerman GA. Cell adhesion regulates gene expression at translational checkpoints in humanmyeloid leukocytes. Proc Natl Acad Sci USA. 2001;98:10284–10289. doi: 10.1073/pnas.181201398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weyrich AS, Dixon DA, Pabla R, Elstad MR, McIntyre TM, Prescott SM, Zimmerman GA. Signal-dependent translation of a regulatory protein, Bcl-3, in activated human platelets. Proc Natl Acad Sci USA. 1998;95:5556–5561. doi: 10.1073/pnas.95.10.5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saadi S, Wrenshall LE, Platt JL. Regional manifestations and control of the immune system. FASEB J. 2002;16:849–856. doi: 10.1096/fj.01-0690hyp. [DOI] [PubMed] [Google Scholar]
- 56.Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, et al. HIF-1α is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Firth JD, Ebert BL, Pugh CW, Ratcliffe PJ. Oxygen-regulated control elements in the phosphoglycerate kinase 1 and lactate dehydrogenase A genes: similarities with the erythropoietin 3′ enhancer. Proc Natl Acad Sci USA. 1994;91:6496–6500. doi: 10.1073/pnas.91.14.6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem. 1994;269:23757–23763. [PubMed] [Google Scholar]
- 59.Ebert BL, Firth JD, Ratcliffe PJ. Hypoxia and mitochondrial inhibitors regulate expression of glucose transporter-1 via distinct Cis-acting sequences. J Biol Chem. 1995;270:29083–29089. doi: 10.1074/jbc.270.49.29083. [DOI] [PubMed] [Google Scholar]
- 60.Lang KJ, Kappel A, Goodall GJ. Hypoxia-inducible factor-1α mRNA contains an internal ribosome entry site that allows efficient translation during normoxia and hypoxia. Mol Biol Cell. 2002;13:1792–1801. doi: 10.1091/mbc.02-02-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou J, Callapina M, Goodall GJ, Brüne B. Functional integrity of nuclear factor kappaB, phosphatidylinositol 3′-kinase, and mitogen-activated protein kinase signaling allows tumor necrosis factor alpha-evoked Bcl-2 expression to provoke internal ribosome entry site-dependent translation of hypoxia-inducible factor 1alpha. Cancer Res. 2004;64:9041–9048. doi: 10.1158/0008-5472.CAN-04-1437. [DOI] [PubMed] [Google Scholar]
- 62.Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, Koromilas A, Wouters BG. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol Cell Biol. 2002;22:7405–7416. doi: 10.1128/MCB.22.21.7405-7416.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tinton SA, Buc-Calderon PM. Hypoxia increases the association of 4E-binding protein 1 with the initiation factor 4E in isolated rat hepatocytes. FEBS Lett. 1999;446:55–59. doi: 10.1016/s0014-5793(99)00185-4. [DOI] [PubMed] [Google Scholar]
- 64.Hannon GJ, Rossi JJ. Unlocking the potential of the human genome with RNA interference. Nature. 2004;431:371–378. doi: 10.1038/nature02870. [DOI] [PubMed] [Google Scholar]
- 65.O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci USA. 2007;104:1604–1609. doi: 10.1073/pnas.0610731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tili E, Michaille JJ, Cimino A, Costinean S, Dumitru CD, Adair B, Fabbri M, Alder H, Liu CG, Calin GA, Croce CM. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol. 2007;179:5082–5089. doi: 10.4049/jimmunol.179.8.5082. [DOI] [PubMed] [Google Scholar]
- 67.Bazzoni F, Rossato M, Fabbri M, Gaudiosi D, Mirolo M, Mori L, Tamassia N, Mantovani A, Cassatella MA, Locati M. Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc Natl Acad Sci USA. 2009;106:5282–5287. doi: 10.1073/pnas.0810909106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Perry MM, Moschos SA, Williams AE, Shepherd NJ, Larner-Svensson HM, Lindsay MA. Rapid changes in microRNA-146a expression negatively regulate the IL-1beta-induced inflammatory response in human lung alveolar epithelial cells. J Immunol. 2008;180:5689–5698. doi: 10.4049/jimmunol.180.8.5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yoshida Y, Kumar A, Koyama Y, Peng H, Arman A, Boch JA, Auron PE. Interleukin 1 activates STAT3/nuclear factor-kappaB cross-talk via a unique TRAF6- and p65-dependent mechanism. J Biol Chem. 2004;279:1768–1776. doi: 10.1074/jbc.M311498200. [DOI] [PubMed] [Google Scholar]
- 70.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ahn KS, Aggarwal BB. Transcription factor NF-kappaB: a sensor for smoke and stress signals. Ann N Y Acad Sci. 2005;1056:218–233. doi: 10.1196/annals.1352.026. [DOI] [PubMed] [Google Scholar]
- 72.Gregorieff A, Pyronnet S, Sonenberg N, Veillette A. Regulation of SOCS-1 expression by translational repression. J Biol Chem. 2000;275:21596–21604. doi: 10.1074/jbc.M910087199. [DOI] [PubMed] [Google Scholar]
- 73.Firestein GS, Echeverri F, Yeo M, Zvaifler NJ, Green DR. Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. Proc Natl Acad Sci USA. 1997;94:10895–10900. doi: 10.1073/pnas.94.20.10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yamanishi Y, Boyle DL, Rosengren S, Green DR, Zvaifler NJ, Firestein GS. Regional analysis of p53 mutations in rheumatoid arthritis synovium. Proc Natl Acad Sci USA. 2002;99:10025–10030. doi: 10.1073/pnas.152333199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zheng SJ, Lamhamedi-Cherradi SE, Wang P, Xu L, Chen YH. Tumor suppressor p53 inhibits autoimmune inflammation and macrophage function. Diabetes. 2005;54:1423–1428. doi: 10.2337/diabetes.54.5.1423. [DOI] [PubMed] [Google Scholar]
- 76.Fu L, Benchimol S. Participation of the human p53 3′UTR in translational repression and activation following gamma-irradiation. EMBO J. 1997;16:4117–4125. doi: 10.1093/emboj/16.13.4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mazan-Mamczarz K, Galbán S, López de Silanes I, Martindale JL, Atasoy U, Keene JD, Gorospe M. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc Natl Acad Sci USA. 2003;100:8354–8359. doi: 10.1073/pnas.1432104100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Galbán S, Martindale JL, Mazan-Mamczarz K, López de Silanes I, Fan J, Wang W, Decker J, Gorospe M. Influence of the RNA-binding protein HuR in pVHL-regulated p53 expression in renal carcinoma cells. Mol Cell Biol. 2003;23:7083–7095. doi: 10.1128/MCB.23.20.7083-7095.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Garcia GE, Xia Y, Ku G, Johnson RJ, Wilson CB, Feng L. IL-18 translational inhibition restricts IFN-γ expression in crescentic glomerulonephritis. Kidney Int. 2003;64:160–169. doi: 10.1046/j.1523-1755.2003.00077.x. [DOI] [PubMed] [Google Scholar]
- 80.Ben-Asouli Y, Banai Y, Pel-Or Y, Shir A, Kaempfer R. Human interferon-γ mRNA autoregulates its translation through a pseudoknot that activates the interferon-inducible protein kinase PKR. Cell. 2002;108:221–232. doi: 10.1016/s0092-8674(02)00616-5. [DOI] [PubMed] [Google Scholar]
- 81.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-γ: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 82.Mazumder B, Fox PL. Delayed translational silencing of ceruloplasmin transcript in gamma interferon-activated U937 monocytic cells: role of the 3′ untranslated region. Mol Cell Biol. 1999;19:6898–6905. doi: 10.1128/mcb.19.10.6898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mazumder B, Seshadri V, Imataka H, Sonenberg N, Fox PL. Translational silencing of ceruloplasmin requires the essential elements of mRNA circularization: poly(A) tail, poly(A)-binding protein, and eukaryotic translation initiation factor 4G. Mol Cell Biol. 2001;21:6440–6449. doi: 10.1128/MCB.21.19.6440-6449.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mazumder B, Sampath P, Seshadri V, Maitra RK, DiCorleto PE, Fox PL. Regulated release of L13a from the 60S ribosomal subunit as a mechanism of transcript-specific translational control. Cell. 2003;115:187–198. doi: 10.1016/s0092-8674(03)00773-6. [DOI] [PubMed] [Google Scholar]
- 85.Sampath P, Mazumder B, Seshadri V, Fox PL. Transcript-selective translational silencing by gamma interferon is directed by a novel structural element in the ceruloplasmin mRNA 3′ untranslated region. Mol Cell Biol. 2003;23:1509–1519. doi: 10.1128/MCB.23.5.1509-1519.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sampath P, Mazumder B, Seshadri V, Gerber CA, Chavatte L, Kinter M, Ting SM, Dignam JD, Kim S, Driscoll DM, Fox PL. Non-canonical function of glutamyl-prolyl-tRNA synthetase: gene-specific silencing of translation. Cell. 2004;119:195–208. doi: 10.1016/j.cell.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 87.Chaudhuri S, Vyas K, Kapasi P, Komar AA, Dinman JD, Barik S, Mazumder B. Human ribosomal protein L13a is dispensable for canonical ribosome function but indispensable for efficient rRNA methylation. RNA. 2007;13:2224–2237. doi: 10.1261/rna.694007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kapasi P, Chaudhuri S, Vyas K, Baus D, Komar AA, Fox PL, Merrick WC, Mazumder B. L13a blocks 48S assembly: role of a general initiation factor in mRNA-specific translational control. Mol Cell. 2007;25:113–126. doi: 10.1016/j.molcel.2006.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vyas K, Chaudhuri S, Leaman DW, Komar AA, Musiyenko A, Barik S, Mazumder B. Genome-wide polysome profiling reveals an inflammation-responsive posttranscriptional operon in gamma interferon-activated monocytes. Mol Cell Biol. 2009;29:458–470. doi: 10.1128/MCB.00824-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Musci G. Ceruloplasmin, the unique multi-copper oxidase of vertebrates. Protein Pept Lett. 2001;8:159–169. [Google Scholar]
- 91.Arif A, Jia J, Mukhopadhyay R, Willard B, Kinter M, Fox PL. Two-site phosphorylation of EPRS coordinates multimodal regulation of non-canonical translational control activity. Mol Cell. 2009;35:164–180. doi: 10.1016/j.molcel.2009.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mukhopadhyay R, Ray PS, Arif A, Brady AK, Kinter M, Fox PL. DAPK-ZIPK-L13a axis constitutes a negative-feedback module regulating inflammatory gene expression. Mol Cell. 2008;32:371–382. doi: 10.1016/j.molcel.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ray PS, Fox PL. A post-transcriptional pathway represses monocyte VEGF-A expression and angiogenic activity. EMBO J. 2007;26:3360–3372. doi: 10.1038/sj.emboj.7601774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ray PS, Jia J, Yao P, Majumder M, Hatzoglou M, Fox PL. A stress-responsive RNA switch regulates VEGFA expression. Nature. 2009;457:915–919. doi: 10.1038/nature07598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Keene JD. RNA regulons: coordination of post-transcriptional events. Nat Rev Genet. 2007;8:533–543. doi: 10.1038/nrg2111. [DOI] [PubMed] [Google Scholar]
- 96.Keene JD, Tenenbaum SA. Eukaryotic mRNPs may represent posttranscriptional operons. Mol Cell. 2002;9:1161–1167. doi: 10.1016/s1097-2765(02)00559-2. [DOI] [PubMed] [Google Scholar]
- 97.Weber C, Koenen RR. Fine-tuning leukocyte responses: towards a chemokine ‘interactome’. Trends Immunol. 2006;27:268–273. doi: 10.1016/j.it.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 98.Lambert MP, Sachais BS, Kowalska MA. Chemokines and thrombogenicity. Thromb Haemost. 2007;97:722–729. doi: 10.1160/th07-01-0046. [DOI] [PubMed] [Google Scholar]
- 99.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 100.Gerard C, Rollins BJ. Chemokines and disease. Nat Immunol. 2001;2:108–115. doi: 10.1038/84209. [DOI] [PubMed] [Google Scholar]