

Abstract
Steroid hormones participate in organ development, reproduction, body homeostasis, and stress responses. The steroid machinery is expressed in a development- and tissue-specific manner, with the expression of these factors being tightly regulated by an array of transcription factors (TFs). Epigenetics provides an additional layer of gene regulation through DNA methylation and histone tail modifications. Evidence of epigenetic regulation of key steroidogenic enzymes is increasing, though this does not seem to be a predominant regulatory pathway. Steroid hormones exert their action in target tissues through steroid nuclear receptors belonging to the NR3A and NR3C families. Nuclear receptor expression levels and post-translational modifications regulate their function and dictate their sensitivity to steroid ligands. Nuclear receptors and TFs are more likely to be epigenetically regulated than proteins involved in steroidogenesis and have secondary impact on the expression of these steroidogenic enzymes. Here we review evidence for epigenetic regulation of enzymes, transcription factors, and nuclear receptors related to steroid biogenesis and action.
Keywords: Epigenetics, nuclear receptors, steroid hormones, steroidogenesis, DNA methylation, transcription factors
Introduction
Despite its complexity, the human body comprises only 200 different cell types or so that originate from a unique DNA sequence. Each cell type arises from a carefully orchestrated gene expression pattern that produces an exclusive specialized cell phenotype. Patterns of expression, which originate from the same genomic template, are epigenetically controlled, dynamically changing, and influenced by the environment (1–3). Epigenetics, literally meaning “above the genome,” refers to a change in gene expression that is independent of the DNA sequence. DNA methylation, histone tail modifications, and microRNAs are ways in which cell type identity can be conferred despite a common DNA makeup.
Sources of the major steroid production supplying the body are the ovary, testis, adrenal cortex, and during fetal development, the placenta. In the ovary, granulosa and theca cells make estrogen and progesterone. In the testis, Leydig cells localized in the interstitial space produce testosterone. The adrenal cortex is subdivided into three morphological distinct zones: the glomerulosa synthesizing aldosterone, the fasciculata producing cortisol, and the reticularis primarily making dehydroepiandrosterone (DHEA) and sulfated DHEA (DHEAS). The central nervous system (4–6) and heart (7–9) have been found to locally produce steroids, and the mechanisms governing steroid biosynthesis in these tissues are likely similar to those operating in the main steroidogenic cells.
Cholesterol is the precursor of all steroids, and its transport from intracellular sources into the mitochondria is the rate-limiting step in steroid biosynthesis [For a review refer to Jefcoate et at. (10) and Rone et al. (11)] (Fig. 2). Steroid production can be divided in two broad stages, 1) cholesterol transport into mitochondria and formation of pregnenolone 2) pregnenolone metabolism by tissue-specific steroidogenic enzymes. In the first stage, the steroidogenic acute regulatory protein (STAR) facilitates transport of cholesterol to the outer mitochondrial membrane where the translocator protein (18 kDa, TSPO), formerly known as the peripheral-type benzodiazepine receptor, acts in concert with other proteins to bind and transport cholesterol into the mitochondrial matrix (11). CYP11A then metabolizes cholesterol into pregnenolone, which freely diffuses out of the mitochondria. In the second stage, the expression of tissue-specific enzymes downstream of pregnenolone production ends in steroid biosynthesis. Steroid specificity is conferred by tissue-specific expression of CYP17, leading to the biosynthesis of DHEA, DHEAS, testosterone, estradiol, and cortisol. The expression of 3βHSD is also tissue-specific, leading to progesterone and ultimately aldosterone formation. In rodents, corticosterone is the major glucocorticoid due to lack of CYP17 expression in the adrenal gland (12–14). These two stages (i.e., the control of cholesterol transport into mitochondria and the expression of tissue-specific steroidogenic enzymes) are the key determinants of steroid biosynthesis and are likely to be the main points of regulation of the expression of the steroidogenic machinery (15).
Fig. 2.

Steroid biosynthesis. Cholesterol is the precursor of all steroids and is delivered into the inner mitochondrial membrane by STAR and TSPO where is cleaved into pregnenolone. Pregnenolone freely diffuses out of the mitochondria and is used for the biosysnthesis of testosterone and estradiol in gonads or cortisol and aldosterone in the adrenal glands.
The effects of steroid hormones are mainly mediated through steroid nuclear receptors. Although steroid binding sites and receptors have been identified in the plasma membrane and other organelles, the focus of this review is on the main mediators of steroid hormone action, the nuclear receptors. Tissue-specific expression and modulation of nuclear receptor function by co-regulators determine the action of steroid hormones. The critical influence of steroid hormones is illustrated by the inactivating mutations of the androgen receptor (AR) resulting in a phenotypic female with XY chromosomes (16). Stress responses and blood pressure are also influenced by the nuclear receptors, and mutations like M604L in the human glucocorticoid receptor (GR) can increase sensitivity to glucocorticoids, resulting in increased blood pressure in response to dexamethasone (17).
The purpose of this review is to gather evidence for epigenetic regulation of the steroid hormone synthesis and action pathways. For information on tissue-specific expression, reactions, and nomenclature of steroidogenic enzymes, refer to Payne and Hales (18, 19). For a discussion of the transcriptional regulation of the steroidogenic machinery, refer to LaVoie and King (15).
Epigenetic control of gene expression
The first layer of gene regulation is mediated by transcription factors (TFs) that are selectively expressed in the cell. These TFs and the genes themselves are subject to a second layer of gene expression regulation that occurs independently of the DNA sequence. As noted earlier, DNA methylation, histone tail modifications, and microRNA expression are ways in which cell type identity can be conferred in the presence of a shared DNA makeup.
DNA methylation
DNA methylation refers to the transfer of a methyl group to the carbon-5 position of cytosine and occurs in cytosine-phosphodiester-guanine (CpG) dinucleotides (20) (Fig. 1B). It is estimated that humans have 37,729 CpG islands (i.e., CpG-rich regions) that are ~1 kb in size, with 35% existing in promoter regions (21). DNA methyl-transferases (DNMTs) transfer a methyl group to CpGs using S-adenosyl-methionine as a methyl group donor. CpG islands in promoter regions are mostly unmethylated, and their methylation is correlated with gene transcription suppression and chromatin structure condensation (20).
Fig. 1.
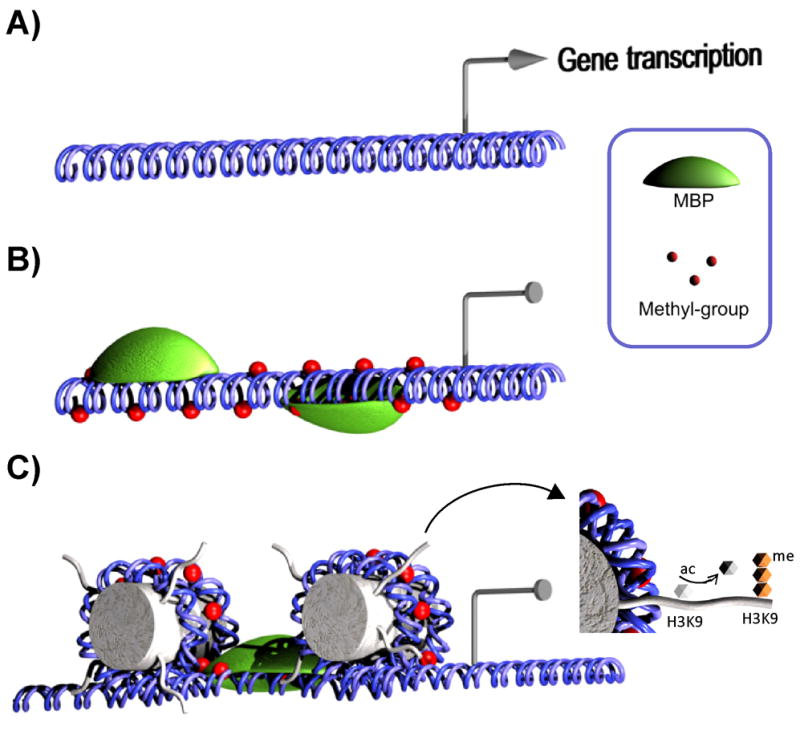
Role of DNA methylation in gene silencing. (A) Methylation-free DNA allows binding of transcription factors and is transcriptionally active. (B) Methyl groups are added to CpGs in promoter regions by DNMTs and are recognized by methyl-CpG-binding proteins (MBPs). (C) MBPs are coupled to chromatin remodeling complexes that lead to a closed promoter configuration through histone modifications such as deacetylation and trimethylation of H3K9.
The proteins MeCP2, MBD1, MBD2, MBD3, and MBD4 form part of a family of CpG-binding proteins that share a methyl-CpG-binding domain (MDB) and recognize genomic areas marked by DNA methylation (Fig. 1B). Kaiso, another methylated CpG (mCpG)-binding protein, does not contain a MDB but instead has zinc fingers (22). The binding affinity for mCpG depends on the properties of the mCpG-binding proteins, which form part of larger transcriptional repression complexes. DNA methylation and chromatin remodeling are linked through the recruitment of chromatin remodeling proteins like histone deacetylases (HDACs) that are present in the repressor complexes (23, 24) (Fig. 1C). For instance, MeCP2 CpG binding leads to decreased gene transcription though chromatin remodeling mediated by a repressor complex containing the corepressor mSin3A known to recruit HDACs (23, 24). Transcriptional activity suppression has also been proposed to occur through decreased binding of TFs to mCpG (25, 26).
Four mammalian DNA methyltransferases sharing a conserved catalytic domain have been characterized. DNMT1, commonly known as the maintenance methyltransferase, exhibits preference for hemimethylated DNA and is essential to maintain double strand methylation after genomic replication (27). Inactivating mutations of DNMT1 cause genomic instability and are embryonic lethal at E9.5 (28). DNMT2 has been shown to efficiently methylate tRNAAsp rather than DNA (29). However, its role remains unclear since mutants have no apparent phenotypic abnormalities (30). DNMT3A and DNMT3B participate in de novo DNA methylation, and inactivating mutations of both genes are embryonic lethal at E9.5 (31). DNMT3L lacks methyltransferase activity due to the absence of catalytic residues but has been shown to play a role in DNA imprinting (32, 33).
Nucleoside analogs that inhibit CpG methylation have formed the basis for DNA methylation studies and anti-tumor therapies. The commonly used analogs, 5-aza-2′deoxycytidine (5-aza-dC) and 5-azacytidine (5-aza), are incorporated into DNA during replication and inhibit methyltransferase activity. This results in global hypomethylation and consequently, transcriptional activation of many genes. However, because the use of demethylating agents results in global hypomethylation, establishing a direct correlation with hypomethylation of the gene of interest or an indirect correlation through expression of TFs regulating its expression is difficult.
Histone modifications
Histones aid in the packing of the genome and are an integral component of the expression architecture of a cell by virtue of their ability to undergo covalent modification at the tail region (34). Electrostatic interactions between DNA and positively charged histone complexes comprised of H1, H2A, H3, and H4 form the nucleosome. Each histone, in turn, has a number of histone variants that are associated with transcriptional repression or activity, increasing chromatin complexity (35). Histone tails, mainly H3 and H4, can be modified by acetylation (36), methylation (37), phosphorylation, sumoylation (38), or ubiquitination (39). The most studied histone modification is acetylation of H3K9, which is associated with transcriptional activation through inactivation of the positive lysine charge (Fig. 1C). Histone acetylation aids in chromatin remodeling and is maintained by the activity of the histone acetyl transferases (HATs) and HDACs that are associated with TFs. Histone acetylation has been linked to DNA methylation, as seen by the finding that DNMT3L mediates trimethylation of H3K9, a mark associated with transcriptional repression. This relationship was shown to be unidirectional and underscores the interdependence of DNA and histone modifications (40).
Similar to DNA methylation inhibitors, the use of HDACs inhibitors such as trichostatin A (TSA) increases gene expression though hyperacetylation of histones (41, 42). Interestingly, evidence for epigenetic regulation of the enzymes involved in steroid biosynthesis is indirect and comes from the finding that clinical use of HDAC inhibitors is associated with the side effect of reduced steroid production (43, 44).
Role of steroidogenic factor-1 (SF-1; NR5A1) in steroidogenesis
The orphan nuclear receptor, SF-1, is expressed in all steroidogenic tissues (45, 46). SF-1 has been shown to increase expression of the steroidogenic machinery by binding to its response element site found in the promoter regions of the genes encoding for STAR, CYPs, and 3βHSD (47, 48). The action of SF-1 is needed but not essential for steroid production, and SF-1, together with other TFs, accounts for the control of tissue-specific steroid protein levels. For instance, in the human adrenal corticocarcinoma cell line, NCI-H295R, a reduction in SF-1 decreases STAR protein expression but does not affect the ability of cAMP to increase STAR expression through the action of the cAMP-response element modulator (49). Aside from its role in steroidogenesis, SF-1 is also involved in the development of the adrenals and testis (50).
SF-1 expression is temporally regulated and tissue specific, suggesting that it is tightly regulated, perhaps in part epigenetically (48). The human NR5A1 gene is found on chromosome 9q33 and contains a CpG island that spans the promoter region and the first exon. DNA methylation has been shown to control NR5A1 gene expression, as seen by the finding that methylation of the SF-1 promoter is associated with SF-1 non expression in normal endometrial tissue, while promoter hypomethylation is associated with SF-1 expression in endometrial stromal cells (51). The presence of SF-1 is accompanied by the expression of steroid machinery proteins capable of producing estrogen, including CYP19.
Further evidence of epigenetic regulation of SF-1 expression comes from a study of the HDAC inhibitor valproate, which is commonly used for the treatment of epilepsy. This study found that valproate decreases SF-1 and CYP11A1 levels in mouse Y1 and human H295 adrenocortical tumor cell lines (52). Unexpectedly, this decrease was shown to result from an indirect epigenetic mechanism triggered by the treatment. The authors concluded that the decreased protein levels were due to ubiquitin-mediated degradation of SF-1 (52). This observation is relevant, as HDAC inhibitors are under clinical trials and their impact on steroidogenesis is a concern for their use.
Star
The rate-limiting step in cholesterol synthesis is the transport of cholesterol into the inner mitochondrial membrane. STAR is one of the proteins involved in bringing endogenous cholesterol to the mitochondrial outer membrane. The Star gene does not contain a CpG island but proximal promoter modifications have been reported (53, 54). Star mRNA is rapidly induced in MA-10 cells after stimulation with 8-Br-cAMP reaching maximum nascent RNA transcription 60 min post-stimulation (53, 54). Quantitative chromatin immunoprecipitation assays performed in MA-10 cells stimulated with 8-Br-cAMP showed increased binding of GATA4, SF-1, and CBP to the proximal Star promoter within 30 minutes of stimulation and coincided with the increase in nascent Star RNA. Chromatin remodeling was made evident by the decrease in tri-methyl histone H3 (K4), di-methyl histone H3 (K9), and an increase in acetyl histone H3 within the fist 30 mins (54). Taken all together, the evidence suggests that TF binding to the Star proximal promoter induced a change in chromatin structure that allowed rapid transcription.
CYP17A1
The expression of the CYP17A1 gene is both species and tissue specific (55). In humans, CYP17 expression in the adrenal cortex leads to cortisol production (56). This is in contrast to rodents, which do not express CYP17 in the adrenals and instead express 3βHSD to produce corticosterone from pregnenolone (57, 58). CpG islands have been identified in the rat (-905/-692) and mouse (-2791/-2644) but not in humans (55). However, as pointed out by the authors, 1.5 kb of the human promoter areas has ~45% homology to the rodent promoter and could share common regulatory mechanisms (55).
The first reports of CpG methylation came from studies of bovine adrenals showing that two CpG sites become demethylated upon placement of adrenocortical cells in culture (Fig. 3A) (59, 60). In vivo evidence for the epigenetic regulation of Cyp17a1 comes from an ingenious experiment in which tetrahydrocortisol, a cortisol metabolite found in low levels in rodents, was monitored in rat urine after treatment of these animals with 5-aza-dC (55). In rodent adrenals, methylation of the Cyp17a1 promoter region is correlated with the silencing of gene expression. The absence of CYP17 leads to the production of corticosterone as the main glucocorticoid. If adrenal Cyp17a1 expression is controlled by methylation then treatment of rodents with 5-aza should demethylate the Cyp17a1 promoter resulting in the expression of CYP17 and thus cortisol formation that can be indirectly detected by the presence of tetrahydrocortisol in the urine. Indeed, Cyp17a1 mRNA was detected in the adrenals and treatment with 5-aza-dC induced changes in the levels of tetrahydrocortisol present in the urine of the treated animals (Fig. 3B) (55).
Fig. 3.

Demethylation of Cyp17a1 in rat adrenals leads to the production of cortisol. (A) In rodent adrenals, methylation of the Cyp17a1 promoter region is correlated with the silencing of gene expression. The absence of CYP17 leads to the production of corticosterone as the main glucocorticoid. (B) Treatment of rodents with 5-aza demethylates the Cyp17a1 promoter and is associated with re-expression of CYP17, resulting in cortisol production that can be indirectly detected by the presence of the metabolite tetrahydrocortisol in the urine.
The absence of a CpG island in the human CYP17A1 gene suggests that direct epigenetic regulation of the Cyp17a1 promoter is more essential in rodents. In humans, expression of CYP17A1 is driven by a complex interaction of TFs (61). Indirect evidence of epigenetic regulation of human CYP17A1 is tied to the induction of the GATA-4 and GATA-6 TFs by 5-aza-dC in human NCI-H295A cells (62). These TFs were shown to be required for CYP17A1 expression, adding to the mounting evidence that expression of the steroidogenic machinery is governed by tissue-specific TF expression. Of note, the epigenetic studies of Cyp17a1 have relied on the use of methylation-sensitive enzymes that have a limited ability to identify mCpGs. Bisulfite- or pyro-sequencing will be helpful in further characterizing the methylation pattern of the Cyp17a1 promoter across species and tissues.
CYP19A1
In humans, the CYP19A1 gene encodes aromatase, which is expressed in a tissue-, developmental stage-, and time-specific manner. This enzyme is controlled by multiple promoters distributed along a region that is 136 kb upstream of the translation initiation site in exon II (63). Aromatase metabolizes testosterone into estradiol and androstenedione to estrone, both products that are capable of activating estrogen receptor. Atypic or deregulated expression of aromatase is relevant to estrogen-dependent pathologies such as endometriosis and breast cancer, and such abnormal expression could be mediated, in part, by epigenetic modifications.
The use of the multiple CYP19A1 promoters and the distribution of these elements vary across species, but data relating to the modulation and level of promoter activity comes from multi-species studies. In one study, CpG methylation of promoter I.1 (PI.1) and PII was compared between bovine granulose cells and corpora lutea (64). PII showed high transcriptional activity in granulose cells and was largely unmethylated. In contrast, this promoter was methylated and inactive in corpora lutea (64). The responsiveness of the CYP19A1 promoter to cAMP also seems be regulated by CpG methylation. In human skin fibroblasts, the cAMP sensitivity of the cAMP response element (CRE) element located -211/-199 upstream of exon II was found to be related to DNA methylation. That is, cAMP responsiveness was associated with DNA hypermethylation and irresponsiveness with DNA methylation (65). This CRE element is in PI.3 and near PII, which are thought to have common regulatory elements (66, 67). Further evidence of epigenetic regulation of the CYP19A1 promoter is provided by a study of sheep placentomes. In this tissue, P1.5/2 methylation correlated with expression of aromatase at different developmental stages and between placental structures (68). Nevertheless, the authors concluded that CpG methylation was not the main factor driving tissue-specific CYP19A1 expression (68, 69).
Until now, evidence of epigenetic modification is limited to Cyp17a1 and CYP19A1. Although relevant, epigenetic modifications, such as DNA methylation, have not emerged as a predominant widespread regulatory mechanism in the expression of key steroidogenic enzymes.
Epigenetic changes in the steroid nuclear receptors affect physiological responses
Steroidogenic hormones act on their gene targets through steroid nuclear receptors (70). The nuclear receptor groups NR3A and NR3C contain the steroid nuclear receptors (71). Most nuclear receptors are found in the cytoplasm and migrate to the nucleus upon binding hormone (Fig. 4). In the nucleus, consensus sequences found in the promoters of target genes, otherwise known as hormone response elements, serve as docking sites for the hormone-receptor complex. The steroid nuclear receptors are distributed throughout the body, and disruption of their function leads to an insensitivity or malfunction of steroid hormone endocrine action (17, 72, 73). It is worth mentioning that some compounds, such as doiseynolic acids, can trigger estrogenic-like activities while exhibiting low binding activity for ER alpha and beta receptors (74, 75). The role that DNA methylation, and other epigenetic modifications, may play on the levels of the nuclear receptors and action of compounds like doiseynelic remains to be examined.
Fig. 4.

Epigenetic modifications of nuclear receptors impact steroid hormone signaling. The nuclear receptor subfamily NR3 and NR4 is known to be epigenetically regulated (shown by the arrow). Demethylation of promoter region of the nuclear receptor (NR) leads to normal production of protein that binds to steroid hormones and translocates to the nucleus to activate target promoter areas. Methylation of promoter regions of NRs leads to decrease expression, hormone-binding, and activation of target promoters.
Glucocorticoid receptor
The dynamic nature of epigenetic regulation and its link with the environment has been observed in hypothalamic-pituitary-adrenal programming of the stress response (76, 77). Maternal licking and grooming (LG) as well as arched-back nursing (ABN) occurring within the first days postpartum correlate with the expression of the GR (NR3C1) in the rat forebrain. During stress, corticotrophin-releasing factor is secreted from the paraventricular nucleus. This stimulates ACTH secretion, which is followed by increased glucocorticoid production. The magnitude of the stress response is related to a negative feedback loop that is dependent on the level of GR expression in the hypothalamus (78). Higher levels of GR, like those found in offspring exposed to high LG-ABN, impart increased sensitivity to glucocorticoids and as consequence, are associated with a moderate stress response. The opposite is true for offspring exposed to low LG-ABN (76, 77, 79, 80). These observations led to the discovery of epigenetic modifications in exon 17 of brain-expressed GR. This promoter area contains the NGFI-A binding site thought to be involved in brain GR expression. This region undergoes a dynamic change in DNA methylation during embryonic development, with the area being unmethylated at gestational day 20 and fully methylated by postnatal day (PND) 1 (81). Interestingly, changes in DNA methylation persist into adulthood depending upon exposure to low/high LG-ABN. In the case of high LG-ABN, the region is demethylated during the first week of life, and this methylation state, along with GR expression level changes, persists into adulthood. In contrast, in animals exposed to low LG-ABN, the region remains methylated, and this is associated with decreased GR expression and an increased stress response (81). The authors postulate that a mechanism exists in which NGFI-A binding to the GR promoter is dependent on DNA methylation programmed in the early postnatal period. NGFI-A binding to the GR promoter will bring HATs to the promoter area and make the chromatin state more transcriptional-favorable, leading to high expression of GR in the hypothalamus (82).
One of the first studies to correlate these findings in rats to humans was performed in victims of child abuse (83). Rat GR exon 17 is homologous to human GR1F (84), which is also expressed in the hippocampus (85). Victims with history of child abuse were found to have decreased hippocampal expression of GR and increased GR1F methylation (83). Taken together, all available data suggest that maternal fostering during the perinatal period bears its epigenetic mark well into adulthood and affects the stress response of offspring.
Mineralocorticoid receptor
Environmental exposure during fetal development can reprogram the epigenome. We have previously shown that in utero exposure to the plasticizer di-(2-ethylhexyl) phthalate (DEHP) decreases testosterone production in PND60 offspring (86). In the rat, Leydig cells develop in two waves. The fetal-Leydig cells, responsible for primary sex organ development, exhibit peak production of testosterone at gestational day (GD) 19 and disappear by PND10. The adult-Leydig cells are responsible for the onset of puberty and the levels of testosterone thereafter (Fig. 5A) (87, 88). Interestingly, the effects of DEHP observed in the adult are not directly caused by DEHP exposure since DEHP metabolites are cleared from the body within 3 days after contact with DEHP (89). Furthermore, the adult-Leydig cell population was not present during treatment, suggesting that the effects seen in the adult result from disruption of adult-Leydig cell progenitors. In search of mechanisms that would explain these observations, we analyzed expression of steroid nuclear receptors in PND60 animals and found the mineralocorticoid receptor (NR3C2; MR) to be the only steroid nuclear receptor undergoing decreased expression in adult offspring after in utero DEHP exposure (Fig. 5A) (90). The MR is widely distributed in the body and is best known for its role in the renin-angiotensin-aldosterone system (91). However, the MR has been shown to be expressed in Leydig cells, where its activation by aldosterone increases testosterone production (92). In the rat, the MR gene is found in the long arm of chromosome 19 (19q11). Similar to the human MR gene, the rat gene contains two 5′ exons that form part of the 5′ UTR and are spliced into the MR coding sequence to form MRα and MRβ (93). It also contains two large CpG islands that enclose both UTRs. Pyrosequencing revealed that the two areas upstream of the transcription start sites are methylated in the adult (90). MRβ sequences contain moderate methylation immediately upstream of the transcription start site in ~20% of the testis population. MRα sequences contain four CpGs immediately upstream of the transcription start site that are differentially methylated in animals subjected to in utero DEHP exposure (Fig. 5B) (90). These differentially methylated CpGs contained putative AP1 and SP1 binding sites, leading us to hypothesize that their deregulation could alter the activity of the whole promoter resulting in the observed decrease in MR expression.
Fig. 5.

In utero exposure to the plasticizer DEHP decreases MR expression in adult Leydig cells. (A) In utero DEHP treatment from GD14 until birth targets fetal-Leydig cells (yellow) and progenitor cells of adult Leydig cells (blue), leading to decreased MR expression and testosterone production in adult-Leydig cells. (B) Pyrosequencing of the MR promoter region revealed a differentially methylated region (DMR) that correlates with the decrease in MR expression. (C) Methylation-sensitive restriction fingerprinting (MSRF) identified two regions that were hypomethylated in the adult (PND60) after in utero exposure to DEHP. (D) Schematic diagram depicting the possible MSRF patterns.
Estrogen receptors
Brain expression of the estrogen receptor α (NR3A1; ERα) has also been associated with maternal licking and grooming as well as sexual behavior. The medial preoptic area (MPOA), medial preoptic nucleus subdivisions, parastrial nucleus, posterodorsal preoptic nucleus, and ventral medial hypothalamus (VMH) exhibit high expression of ERα (94, 95). ERα knock-out in females is associated with increased infanticide and a reduced motivation to retrieve pups, suggesting that nurturing impairments exist. This has been attributed to a deregulation of oxytocin (96) and alterations in dopaminergic signaling (97) acting on the MPOA and VMH, which are known to regulate nurturing (98) and reproductive (99) behavior.
Similar to the findings related to differential methylation in the GR rat cross-fostering study, immediate postnatal care was found to correlate with ERα expression in female offspring exposed to high or low licking and grooming behavior (100). ERα1b promoter methylation was also found to be correlated with ERα expression in the adult brain (100). The differentially methylated DNA contains consensus sequences for Stat5b, which would not be expected to bind methylated sequences (100).
In humans, the ERα gene promoter is found on chromosome 6q25 and contains a dense CpG island that encompasses the promoter and first exon regions (101, 102). ERα has been widely studied in breast cancer. The disease can be broadly classified into two groups [i.e., ERα expressing (ERα+) and non expressing tumor cells (ERα-)] that are related to the epigenomic state of ERα. Promoter regions are mostly unmethylated in ERα+ cells, which are associated with a better outcome. On the other hand, ERα- cells tend to have epigenetically silenced promoters and are considered to be more aggressive and resistant to hormonal treatments. Interestingly, ERα+ cells eventually give rise to the more aggressive ERα- cells, suggesting that cancer aggressiveness is correlated with the epigenomic state of the cell. The methylation activity on this ERα promoter area has been correlated with the expression of ERα in various cell lines, tumors, and metastases, suggesting that DNA methylation plays an important role in the regulation of ERα expression (103, 104). Further support for this role comes from the finding that treatment of a non ERα-expressing cancer tumor cell line (MD-MB-231 cell line) with 5-aza or 5-aza-dC results in ERα re-expression (105), a result also observed following TSA treatment (106).
The human estrogen receptor beta (NR3A2; ERβ) is found on chromosome 14q23. Exposure to the endocrine disruptor, methoxychlor during fetal and perinatal development decreases ERβ, but not ERα, expression in the ovary, and this effect is accompanied by early pubertal onset and first estrus, reduced litter size, and increased irregular cyclicity (107). Bisulfate sequencing and methylation-specific PCR have revealed that decreased ERβ expression correlates with DNA hypermethylation at PND50-60, while the ERα promoter remains unaffected at this time (108). This study suggests that the epigenetic regulation of the two receptors is controlled by different pathways. ERβ participates in granulosa cell differentiation induced by FSH and responsiveness to LH (109, 110). The decreased expression of this receptor could explain the early puberty, increased irregular cyclicity, reduced litter sizes, and premature reproductive aging when exposed to a high dose of methoxychlor (108). ERβ methylation has also been implicated in prostate and ovarian cancer (111). TSA or 5-aza has been shown to elicit re-expression of ERβ, and this effect is accompanied by induction of apoptosis in the prostate cancer cell lines LNCaP, DU-145, and PC-3 (112).
Androgen receptor
The human AR (NR3C4) is found on chromosome Xq12 and is widely expressed throughout the body. Evidence of epigenetic regulation of AR comes from the finding that advance-stage and androgen-insensitive prostate cancers are associated with AR gene hypermethylation and loss of AR expression. The first evidence of AR epigenetic regulation came from the observation that the AR-deficient (AR-) cell lines Du145, DuPro, TSU-PR1, and PPC1 have hypermethylated AR promoters, while the AR-expressing cell lines LNCaP and PC3 cells used in these studies have hypomethylated promoters (113). Treatment of AR- cells with 5-aza restores AR expression, an effect that is associated with AR promoter hypomethylation. Similarly, loss of AR expression and AR promoter hypermethylation has been observed in invasive prostate adenocarcinomas chemically induced by treatment of rats with 3,2′-dimethyl-4-aminobiphenyl (114). AR hypermethylation also seems to play a significant role in human prostate cancer. AR hypermethylation has been detected in 20% of primary cell lines from prostate cancer patients and 28% of hormone-refractory prostate cancers (115). Prostate cancer is associated with many genome-wide effects, and AR methylation is likely one of multiple epigenetic reprogramming events (116).
Like methylation, histone modifications also participate in the regulation of AR-driven genes. Studies have linked the activation of AR-dependent genes with lysine-specific demethylase 1 (LSD1), a demethylase that removes transcriptionally repressive mono-, di-, and tri-methylation marks from H3K4 and H3K9 to favor gene expression. LSD1 knockdown decreases transcriptional activation of AR-driven genes as well as cell proliferation in LNCaP cells (117). Similar results have been obtained with the LSD1 inhibitor, parygyline (117).
Infection and inflammation are associated with early prostate cancer progression (118). However, it is not known whether infection and inflammation are leading to AR methylation. As pointed out by Nelson (118), iNOS-producing macrophages are found in earlier stages of prostate cancer progression. Moreover, interleukin 1β silences FMR1 and HPRT, an event associated with production of nitric oxide (119). Taken together these findings suggest that epigenomic changes may occur early in the development of cancer and defining the role that inflammation plays in the 40 or so genes modified in prostate cancer could be of great relevance for future treatments (120).
Challenges and methods of studying epigenetic changes in vivo
The epigenome is tissue-, developmental stage-, and species-specific. In addition, it dynamically changes in the adult in response to the environment (3, 121, 122). Its dynamic nature is best exemplified, in one hand, by the reset of all the epigenomic marks during gametogenesis, effectively erasing cell memory before passing it on to the next generation (123), and in the other, by the generation of unique configurations of epigenomic marks that are laid down to give rise to the different cell types and their expression patterns. Even within the same cell type, subtle differences in the epigenome exist. For example, bisulfate sequencing of DNA from tissues or cell lines often shows that not all clones have the same methylated pattern. This implies that the epigenome is adaptable, with differences in the epigenome arising not only from cell type, but also from the cellular microenvironment, such as closeness to vascular nutrition and neighboring cells. The challenge is how to study the epigenome in an efficient, flexible, and affordable way.
For the study of small genomic regions, bisulfate sequencing is the gold standard in determining DNA methylation. Improvements in bisulfate treatment of the genomic DNA have enhanced conversion efficiency and ease of use (124). However, genome-wide methylation studies with single CpG resolution are expensive, time consuming, and challenging to perform. Multiple genome-wide methods have been developed and are constantly being perfected for the study of the epigenome (125–127). Shotgun bisulfate sequencing is among the promising technologies that uses high-throughput sequencing combined with bisulfate-treated DNA to offer high-resolution mCpG reading (128). Other approaches like microarrays (129) have had relative success, and development of algorithms to analyze the data will improve their use (130). Nevertheless, the costs and availability of these technologies limit their wide spread use.
Restriction landmark genomic scanning (RGLS) (131) and methylation-sensitive restriction fingerprinting (MSRF) (132) are popular techniques for genome-wide methylation analysis. The materials for these techniques are readily available and can be setup in any laboratory. RGLS is a restriction endonuclease-based epigenomic approach that has been successfully used in the discovery of methylated genes in cancer (133) and investigation of mouse germ cell development (134). RGLS relies on methyl-sensitive enzymes to generate genomic fragments, which can be separated by 2D gel electrophoresis to reveal a pattern of spots representing the global methylation status. The data output obtained from RGLS can be combined with bioinformatics algorithms to identify changing targets (135, 136). The power of this technique is limited by the ability to separate fragments and the amount of restriction sites present in the genome for a given enzyme. In the case of NotI, this is ~2,000 fragments (137).
MSRF relies upon a methylation-sensitive enzyme that is frequently found in the genome to fragment non-methylated consensus sites in the genome. The DNA fragments not digested by the enzyme are then PCR-amplified under low stringent conditions using two short primers. The fragments are separated in a sequencing gel, and the differences between digested and undigested DNA samples orient to areas undergoing methylation changes (132). We have used MSRF to identify epigenetic changes in adult rats exposed to DEHP in utero. In this experiment, pregnant dams were administered the endocrine disruptor DEHP by gavage from GD14 until birth, and testes were collected at PND60. DNA was extracted, digested with the methylation-sensitive enzyme BstUI, and PCR-amplified using a combination of MRSF primers. The products were then separated in a sequencing gel. The resulting band pattern reflects the methylation status of a particular area in the genome. We identified several regions that were differentially methylated and are in the processes of cloning and characterizing the expression of the targets (Fig. 5C). We hope that a similar analysis in purified cell populations will uncover new epigenetically modified targets and shed more light into the long-term effects of DEHP.
Conclusions
Interplay exists among steroid hormone action, biosynthesis, and steroid receptor function. Epigenetic modifications to either branch are likely to alter their associated signaling and depending on age, impact development. Promoter regulation similar to that observed in GR and ERα genes likely occurs across the genome. The identification of genes and characterization of their response to the environment is of high clinical relevance. The development of new sequencing technologies will likely aid in obtaining a high-resolution CpG map.
Table 1.
Epigenetic changes in transcription factors and enzymes involved in steroid biosynthesis.
| Gene | Evidence | Reference |
|---|---|---|
|
NR5A1 SF-1 |
Restoration of SF-1 expression in endometrial tissue is associated with SF-1 promoter hypomethylation | (51) |
| The HDAC inhibitor valproate decreases SF-1 and Cyp11a1 levels by an indirect mechanism in Y1 and H295 cell lines | (52) | |
| Star | Decrease in tri-methyl histone H3 (K4) and di-methyl histone H3 (K9) and an increase in acetyl histone H3 within the fist 30 mins post 8-Br-cAMP stimulation of MA-10 cells. | (53, 54) |
| Cyp17 | CpG demethylation in primary bovine adrenal cell lines | (59, 60) |
| Rodents treated with 5-aza-dC express Cyp17 in adrenals and produce cortisol | (55) | |
| Cyp19 | Hypermethylation in bovine granulose cells and hypomethylation in corpora lutea at PI.1/PII | (64) |
| Demethylation of the CRE element at -211/-199 confers camp responsiveness to human skin fibroblast at PI.3/II | (65) | |
| Methylation of CpGs in P1.5/2 of sheep placentomes correlates with aromatase expression but is not the only mechanism of regulation | (68, 69) | |
|
Nr3c1 GR |
Offspring exposed to high LG-ABN exhibit demethylation at exon 17, while those exposed to low LG-ABN exhibit methylation in this region | (81) |
| Victims of child abuse exhibit hypermethylation of GR1F and low GR expression in the hippocampus | (83) | |
|
Nr3c2 MR |
A differentially methylated area in the MR promoter is associated with decreased MR expression in the testis after in utero DEHP exposure | (90) |
|
Nr3a1 ERα |
Differential methylation in the ERα1b promoter is correlated with ERα expression in the adult brain | (100) |
| ERα promoter methylation is correlated with the loss of Erα expression in human breast cancer | (103, 104) | |
| 5-aza and 5-aza-dC treatment of the ERα-negative cell line, MD-MB-231, results in ERα re-expression | (105) | |
| TSA treatment of the MD-MB-231 cell line induces ERα re-expression | (106) | |
|
Nr3a2 ERβ |
Fetal and perinatal exposure to methoxychlor hypermethylates the ERβ promoter and decreases ERβ expression in the ovary at PND50-60 | (108) |
| TSA treatment of the ERβ-negative cell lines LNCaP, DU-145, and PC-3 induces ERβ expression | (112) | |
|
NR3C4 AR |
5-aza treatment induces re-expression of AR in the AR-negative cell lines Du145, DuPro, TSU-PR1, and PPC1 | (113) |
| Rat invasive adenocarcinomas contain hypermethylation and lose AR expression | (114) | |
| 20% of human primary cancer cell lines and 28% of hormone-refractory prostate cancers contain hypermethylation | (115) | |
Acknowledgments
We are grateful to Dr. M. Culty (McGill University) for her critical review of the manuscript. This work was supported by grant R01 ES013495 from the National Institutes of Health, a Canada Research Chair in Biochemical Pharmacology (V.P.), and a Georgetown University-Consejo Nacional de Ciencia y Tecnologia fellowship (D.M-A.). The Research Institute of McGill University Health Centre is supported in part by a center grant from Fonds de la Recherche en Santé Quebec.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Barros SP, Offenbacher S. Epigenetics: connecting environment and genotype to phenotype and disease. J Dent Res. 2009;88:400–408. doi: 10.1177/0022034509335868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meaney MJ, Szyf M. Environmental programming of stress responses through DNA methylation: life at the interface between a dynamic environment and a fixed genome. Dialogues Clin Neurosci. 2005;7:103–123. doi: 10.31887/DCNS.2005.7.2/mmeaney. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simmons RA. Developmental origins of adult disease. Pediatr Clin North Am. 2009;56:449–466. doi: 10.1016/j.pcl.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baulieu EE. Neurosteroids: a novel function of the brain. Psychoneuroendocrinology. 1998;23:963–987. doi: 10.1016/s0306-4530(98)00071-7. [DOI] [PubMed] [Google Scholar]
- 5.Mellon SH, Griffin LD. Neurosteroids: biochemistry and clinical significance. Trends Endocrinol Metab. 2002;13:35–43. doi: 10.1016/s1043-2760(01)00503-3. [DOI] [PubMed] [Google Scholar]
- 6.Brown RC, Cascio C, Papadopoulos V. Pathways of neurosteroid biosynthesis in cell lines from human brain: regulation of dehydroepiandrosterone formation by oxidative stress and beta-amyloid peptide. J Neurochem. 2000;74:847–859. doi: 10.1046/j.1471-4159.2000.740847.x. [DOI] [PubMed] [Google Scholar]
- 7.Kayes-Wandover KM, White PC. Steroidogenic enzyme gene expression in the human heart. J Clin Endocrinol Metab. 2000;85:2519–2525. doi: 10.1210/jcem.85.7.6663. [DOI] [PubMed] [Google Scholar]
- 8.Young MJ, Clyne CD, Cole TJ, Funder JW. Cardiac steroidogenesis in the normal and failing heart. J Clin Endocrinol Metab. 2001;86:5121–5126. doi: 10.1210/jcem.86.11.7925. [DOI] [PubMed] [Google Scholar]
- 9.White PC. Aldosterone: direct effects on and production by the heart. J Clin Endocrinol Metab. 2003;88:2376–2383. doi: 10.1210/jc.2003-030373. [DOI] [PubMed] [Google Scholar]
- 10.Jefcoate CR. Liver X receptor opens a new gateway to StAR and to steroid hormones. J Clin Invest. 2006;116:1832–1835. doi: 10.1172/JCI29160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rone MB, Fan J, Papadopoulos V. Cholesterol transport in steroid biosynthesis: Role of protein-protein interactions and implications in disease states. Biochim Biophys Acta. 2009;1791:646–658. doi: 10.1016/j.bbalip.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brock BJ, Waterman MR. Biochemical differences between rat and human cytochrome P450c17 support the different steroidogenic needs of these two species. Biochemistry. 1999;38:1598–1606. doi: 10.1021/bi9821059. [DOI] [PubMed] [Google Scholar]
- 13.Pelletier G, Li S, Luu-The V, Tremblay Y, Belanger A, Labrie F. Immunoelectron microscopic localization of three key steroidogenic enzymes (cytochrome P450(scc), 3 beta-hydroxysteroid dehydrogenase and cytochrome P450(c17)) in rat adrenal cortex and gonads. J Endocrinol. 2001;171:373–383. doi: 10.1677/joe.0.1710373. [DOI] [PubMed] [Google Scholar]
- 14.Perkins LM, Payne AH. Quantification of P450scc, P450(17) alpha, and iron sulfur protein reductase in Leydig cells and adrenals of inbred strains of mice. Endocrinology. 1988;123:2675–2682. doi: 10.1210/endo-123-6-2675. [DOI] [PubMed] [Google Scholar]
- 15.Lavoie HA, King SR. Transcriptional regulation of steroidogenic genes: STARD1, CYP11A1 and HSD3B. Exp Biol Med (Maywood ) 2009;234:880–907. doi: 10.3181/0903-MR-97. [DOI] [PubMed] [Google Scholar]
- 16.Brinkmann AO. Molecular basis of androgen insensitivity. Mol Cell Endocrinol. 2001;179:105–109. doi: 10.1016/s0303-7207(01)00466-x. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J, Ge R, Matte-Martone C, Goodwin J, Shlomchik WD, Mamula MJ, Kooshkabadi A, Hardy MP, Geller D. Characterization of a novel gain of function glucocorticoid receptor knock-in mouse. J Biol Chem. 2009;284:6249–6259. doi: 10.1074/jbc.M807997200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller WL. Steroidogenic enzymes. Endocr Dev. 2008;13:1–18. doi: 10.1159/000134751. [DOI] [PubMed] [Google Scholar]
- 19.Payne AH, Hales DB. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr Rev. 2004;25:947–970. doi: 10.1210/er.2003-0030. [DOI] [PubMed] [Google Scholar]
- 20.Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 21.Han L, Zhao Z. CpG islands or CpG clusters: how to identify functional GC-rich regions in a genome? BMC Bioinformatics. 2009;10:65. doi: 10.1186/1471-2105-10-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prokhorchuk AV, Aitkhozhina DS, Sablina AA, Ruzov AS, Prokhorchuk EB. [KAISO--a new member of the BTB/POZ family specifically bindsto methylated DNA sequences] Genetika. 2001;37:737–744. [PubMed] [Google Scholar]
- 23.Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 24.Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 25.Watt F, Molloy PL. Cytosine methylation prevents binding to DNA of a HeLa cell transcription factor required for optimal expression of the adenovirus major late promoter. Genes Dev. 1988;2:1136–1143. doi: 10.1101/gad.2.9.1136. [DOI] [PubMed] [Google Scholar]
- 26.Tate PH, Bird AP. Effects of DNA methylation on DNA-binding proteins and gene expression. Curr Opin Genet Dev. 1993;3:226–231. doi: 10.1016/0959-437x(93)90027-m. [DOI] [PubMed] [Google Scholar]
- 27.Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet. 1998;19:219–220. doi: 10.1038/890. [DOI] [PubMed] [Google Scholar]
- 28.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 29.Goll MG, Kirpekar F, Maggert KA, Yoder JA, Hsieh CL, Zhang X, Golic KG, Jacobsen SE, Bestor TH. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science. 2006;311:395–398. doi: 10.1126/science.1120976. [DOI] [PubMed] [Google Scholar]
- 30.Jeltsch A, Nellen W, Lyko F. Two substrates are better than one: dual specificities for Dnmt2 methyltransferases. Trends Biochem Sci. 2006;31:306–308. doi: 10.1016/j.tibs.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 31.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 32.Hata K, Okano M, Lei H, Li E. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development. 2002;129:1983–1993. doi: 10.1242/dev.129.8.1983. [DOI] [PubMed] [Google Scholar]
- 33.Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, Sasaki H. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature. 2004;429:900–903. doi: 10.1038/nature02633. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y, Reinberg D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 2001;15:2343–2360. doi: 10.1101/gad.927301. [DOI] [PubMed] [Google Scholar]
- 35.Henikoff S, Ahmad K. Assembly of variant histones into chromatin. Annu Rev Cell Dev Biol. 2005;21:133–153. doi: 10.1146/annurev.cellbio.21.012704.133518. [DOI] [PubMed] [Google Scholar]
- 36.Wade PA, Pruss D, Wolffe AP. Histone acetylation: chromatin in action. Trends Biochem Sci. 1997;22:128–132. doi: 10.1016/s0968-0004(97)01016-5. [DOI] [PubMed] [Google Scholar]
- 37.Jenuwein T. Re-SET-ting heterochromatin by histone methyltransferases. Trends Cell Biol. 2001;11:266–273. doi: 10.1016/s0962-8924(01)02001-3. [DOI] [PubMed] [Google Scholar]
- 38.Shiio Y, Eisenman RN. Histone sumoylation is associated with transcriptional repression. Proc Natl Acad Sci U S A. 2003;100:13225–13230. doi: 10.1073/pnas.1735528100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shilatifard A. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem. 2006;75:243–269. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- 40.Tamaru H, Zhang X, McMillen D, Singh PB, Nakayama J, Grewal SI, Allis CD, Cheng X, Selker EU. Trimethylated lysine 9 of histone H3 is a mark for DNA methylation in Neurospora crassa. Nat Genet. 2003;34:75–79. doi: 10.1038/ng1143. [DOI] [PubMed] [Google Scholar]
- 41.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 42.Chen WY, Bailey EC, McCune SL, Dong JY, Townes TM. Reactivation of silenced, virally transduced genes by inhibitors of histone deacetylase. Proc Natl Acad Sci U S A. 1997;94:5798–5803. doi: 10.1073/pnas.94.11.5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gregoraszczuk E, Wojtowicz AK, Tauboll E, Ropstad E. Valproate-induced alterations in testosterone, estradiol and progesterone secretion from porcine follicular cells isolated from small- and medium-sized ovarian follicles. Seizure. 2000;9:480–485. doi: 10.1053/seiz.2000.0443. [DOI] [PubMed] [Google Scholar]
- 44.Mikkonen K, Vainionpaa LK, Pakarinen AJ, Knip M, Jarvela IY, Tapanainen JS, Isojarvi JI. Long-term reproductive endocrine health in young women with epilepsy during puberty. Neurology. 2004;62:445–450. doi: 10.1212/01.wnl.0000106942.35533.62. [DOI] [PubMed] [Google Scholar]
- 45.Hatano O, Takakusu A, Nomura M, Morohashi K. Identical origin of adrenal cortex and gonad revealed by expression profiles of Ad4BP/SF-1. Genes Cells. 1996;1:663–671. doi: 10.1046/j.1365-2443.1996.00254.x. [DOI] [PubMed] [Google Scholar]
- 46.Bamberger AM, Ezzat S, Cao B, Wong M, Parker KL, Schulte HM, Asa SL. Expression of steroidogenic factor-1 (SF-1) mRNA and protein in the human placenta. Mol Hum Reprod. 1996;2:457–461. doi: 10.1093/molehr/2.6.457. [DOI] [PubMed] [Google Scholar]
- 47.Morohashi K, Honda S, Inomata Y, Handa H, Omura T. A common trans-acting factor, Ad4-binding protein, to the promoters of steroidogenic P-450s. J Biol Chem. 1992;267:17913–17919. [PubMed] [Google Scholar]
- 48.Parker KL, Rice DA, Lala DS, Ikeda Y, Luo X, Wong M, Bakke M, Zhao L, Frigeri C, Hanley NA, Stallings N, Schimmer BP. Steroidogenic factor 1: an essential mediator of endocrine development. Recent Prog Horm Res. 2002;57:19–36. doi: 10.1210/rp.57.1.19. [DOI] [PubMed] [Google Scholar]
- 49.Sugawara T, Sakuragi N, Minakami H. CREM confers cAMP responsiveness in human steroidogenic acute regulatory protein expression in NCI-H295R cells rather than SF-1/Ad4BP. J Endocrinol. 2006;191:327–337. doi: 10.1677/joe.1.06601. [DOI] [PubMed] [Google Scholar]
- 50.Lin L, Achermann JC. Steroidogenic factor-1 (SF-1, Ad4BP, NR5A1) and disorders of testis development. Sex Dev. 2008;2:200–209. doi: 10.1159/000152036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xue Q, Lin Z, Yin P, Milad MP, Cheng YH, Confino E, Reierstad S, Bulun SE. Transcriptional activation of steroidogenic factor-1 by hypomethylation of the 5′ CpG island in endometriosis. J Clin Endocrinol Metab. 2007;92:3261–3267. doi: 10.1210/jc.2007-0494. [DOI] [PubMed] [Google Scholar]
- 52.Chen WY, Weng JH, Huang CC, Chung BC. Histone deacetylase inhibitors reduce steroidogenesis through SCF-mediated ubiquitination and degradation of steroidogenic factor 1 (NR5A1) Mol Cell Biol. 2007;27:7284–7290. doi: 10.1128/MCB.00476-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hiroi H, Christenson LK, Strauss JF., III Regulation of transcription of the steroidogenic acute regulatory protein (StAR) gene: temporal and spatial changes in transcription factor binding and histone modification. Mol Cell Endocrinol. 2004;215:119–126. doi: 10.1016/j.mce.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 54.Hiroi H, Christenson LK, Chang L, Sammel MD, Berger SL, Strauss JF., III Temporal and spatial changes in transcription factor binding and histone modifications at the steroidogenic acute regulatory protein (stAR) locus associated with stAR transcription. Mol Endocrinol. 2004;18:791–806. doi: 10.1210/me.2003-0305. [DOI] [PubMed] [Google Scholar]
- 55.Missaghian E, Kempna P, Dick B, Hirsch A, ikhani-Koupaei R, Jegou B, Mullis PE, Frey BM, Fluck CE. Role of DNA methylation in the tissue-specific expression of the CYP17A1 gene for steroidogenesis in rodents. J Endocrinol. 2009;202:99–109. doi: 10.1677/JOE-08-0353. [DOI] [PubMed] [Google Scholar]
- 56.Chung BC, Picado-Leonard J, Haniu M, Bienkowski M, Hall PF, Shively JE, Miller WL. Cytochrome P450c17 (steroid 17 alpha-hydroxylase/17,20 lyase): cloning of human adrenal and testis cDNAs indicates the same gene is expressed in both tissues. Proc Natl Acad Sci U S A. 1987;84:407–411. doi: 10.1073/pnas.84.2.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Johnson DC, Sen M. The cytochrome P450(17) alpha (17 alpha-hydroxylase/C17,20-lyase) activity of the junctional zone of the rat placenta. J Endocrinol. 1990;125:217–224. doi: 10.1677/joe.0.1250217. [DOI] [PubMed] [Google Scholar]
- 58.Namiki M, Kitamura M, Buczko E, Dufau ML. Rat testis P-450(17)alpha cDNA: the deduced amino acid sequence, expression and secondary structural configuration. Biochem Biophys Res Commun. 1988;157:705–712. doi: 10.1016/s0006-291x(88)80307-3. [DOI] [PubMed] [Google Scholar]
- 59.Hornsby PJ, Yang LQ, Raju SG, Cheng CY. Changes in gene expression and DNA methylation in adrenocortical cells senescing in culture. Mutat Res. 1991;256:105–113. doi: 10.1016/0921-8734(91)90004-u. [DOI] [PubMed] [Google Scholar]
- 60.Hornsby PJ, Yang L, Raju SG, Maghsoudlou SS, Lala DS, Nallaseth FS. Demethylation of specific sites in the 5′-flanking region of the CYP17 genes when bovine adrenocortical cells are placed in culture. DNA Cell Biol. 1992;11:385–395. doi: 10.1089/dna.1992.11.385. [DOI] [PubMed] [Google Scholar]
- 61.Sewer MB, Jagarlapudi S. Complex assembly on the human CYP17 promoter. Mol Cell Endocrinol. 2009;300:109–114. doi: 10.1016/j.mce.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fluck CE, Miller WL. GATA-4 and GATA-6 modulate tissue-specific transcription of the human gene for P450c17 by direct interaction with Sp1. Mol Endocrinol. 2004;18:1144–1157. doi: 10.1210/me.2003-0342. [DOI] [PubMed] [Google Scholar]
- 63.Chen D, Reierstad S, Lu M, Lin Z, Ishikawa H, Bulun SE. Regulation of breast cancer-associated aromatase promoters. Cancer Lett. 2009;273:15–27. doi: 10.1016/j.canlet.2008.05.038. [DOI] [PubMed] [Google Scholar]
- 64.Vanselow J, Pohland R, Furbass R. Promoter-2-derived Cyp19 expression in bovine granulosa cells coincides with gene-specific DNA hypo-methylation. Mol Cell Endocrinol. 2005;233:57–64. doi: 10.1016/j.mce.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 65.Demura M, Bulun SE. CpG dinucleotide methylation of the CYP19 I.3/II promoter modulates cAMP-stimulated aromatase activity. Mol Cell Endocrinol. 2008;283:127–132. doi: 10.1016/j.mce.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 66.Zhou J, Gurates B, Yang S, Sebastian S, Bulun SE. Malignant breast epithelial cells stimulate aromatase expression via promoter II in human adipose fibroblasts: an epithelial-stromal interaction in breast tumors mediated by CCAAT/enhancer binding protein beta. Cancer Res. 2001;61:2328–2334. [PubMed] [Google Scholar]
- 67.Zhou D, Chen S. Identification and characterization of a cAMP-responsive element in the region upstream from promoter 1.3 of the human aromatase gene. Arch Biochem Biophys. 1999;371:179–190. doi: 10.1006/abbi.1999.1454. [DOI] [PubMed] [Google Scholar]
- 68.Furbass R, Selimyan R, Vanselow J. DNA methylation and chromatin accessibility of the proximal Cyp 19 promoter region 1.5/2 correlate with expression levels in sheep placentomes. Mol Reprod Dev. 2008;75:1–7. doi: 10.1002/mrd.20756. [DOI] [PubMed] [Google Scholar]
- 69.Vanselow J, Selimyan R, Furbass R. DNA methylation of placenta-specific Cyp19 promoters of cattle and sheep. Exp Clin Endocrinol Diabetes. 2008;116:437–442. doi: 10.1055/s-2008-1058083. [DOI] [PubMed] [Google Scholar]
- 70.Tsai MJ, O’Malley BW. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu Rev Biochem. 1994;63:451–486. doi: 10.1146/annurev.bi.63.070194.002315. [DOI] [PubMed] [Google Scholar]
- 71.Robinson-Rechavi M, Escriva GH, Laudet V. The nuclear receptor superfamily. J Cell Sci. 2003;116:585–586. doi: 10.1242/jcs.00247. [DOI] [PubMed] [Google Scholar]
- 72.Gottlieb B, Beitel LK, Wu J, Elhaji YA, Trifiro M. Nuclear receptors and disease: androgen receptor. Essays Biochem. 2004;40:121–136. doi: 10.1042/bse0400121. [DOI] [PubMed] [Google Scholar]
- 73.Komesaroff PA, Funder JW, Fuller PJ. Hormone-nuclear receptor interactions in health and disease. Mineralocorticoid resistance. Baillieres Clin Endocrinol Metab. 1994;8:333–355. doi: 10.1016/s0950-351x(05)80256-3. [DOI] [PubMed] [Google Scholar]
- 74.Meyers CY, Hou Y, Winters TA, Banz WJ, Adler S. Activities of a non-classical estrogen, Z-bis-dehydrodoisynolic acid, with ERalpha and ERbeta. J Steroid Biochem Mol Biol. 2002;82:33–44. doi: 10.1016/s0960-0760(02)00150-4. [DOI] [PubMed] [Google Scholar]
- 75.Scribner AW, Jonson SD, Welch MJ, Katzenellenbogen JA. Synthesis, estrogen receptor binding, and tissue distribution of [18F]fluorodoisynolic acids. Nucl Med Biol. 1997;24:209–224. doi: 10.1016/s0969-8051(97)00058-9. [DOI] [PubMed] [Google Scholar]
- 76.Liu D, Diorio J, Tannenbaum B, Caldji C, Francis D, Freedman A, Sharma S, Pearson D, Plotsky PM, Meaney MJ. Maternal care, hippocampal glucocorticoid receptors, and hypothalamic-pituitary-adrenal responses to stress. Science. 1997;277:1659–1662. doi: 10.1126/science.277.5332.1659. [DOI] [PubMed] [Google Scholar]
- 77.Francis D, Diorio J, Liu D, Meaney MJ. Nongenomic transmission across generations of maternal behavior and stress responses in the rat. Science. 1999;286:1155–1158. doi: 10.1126/science.286.5442.1155. [DOI] [PubMed] [Google Scholar]
- 78.Kolber BJ, Wieczorek L, Muglia LJ. Hypothalamic-pituitary-adrenal axis dysregulation and behavioral analysis of mouse mutants with altered glucocorticoid or mineralocorticoid receptor function. Stress. 2008;11:321–338. doi: 10.1080/10253890701821081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Meaney MJ, Aitken DH, Viau V, Sharma S, Sarrieau A. Neonatal handling alters adrenocortical negative feedback sensitivity and hippocampal type II glucocorticoid receptor binding in the rat. Neuroendocrinology. 1989;50:597–604. doi: 10.1159/000125287. [DOI] [PubMed] [Google Scholar]
- 80.Meaney MJ. Maternal care, gene expression, and the transmission of individual differences in stress reactivity across generations. Annu Rev Neurosci. 2001;24:1161–1192. doi: 10.1146/annurev.neuro.24.1.1161. [DOI] [PubMed] [Google Scholar]
- 81.Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- 82.Weaver IC, D’Alessio AC, Brown SE, Hellstrom IC, Dymov S, Sharma S, Szyf M, Meaney MJ. The transcription factor nerve growth factor-inducible protein a mediates epigenetic programming: altering epigenetic marks by immediate-early genes. J Neurosci. 2007;27:1756–1768. doi: 10.1523/JNEUROSCI.4164-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McGowan PO, Sasaki A, D’Alessio AC, Dymov S, Labonte B, Szyf M, Turecki G, Meaney MJ. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 2009;12:342–348. doi: 10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McCormick JA, Lyons V, Jacobson MD, Noble J, Diorio J, Nyirenda M, Weaver S, Ester W, Yau JL, Meaney MJ, Seckl JR, Chapman KE. 5′-heterogeneity of glucocorticoid receptor messenger RNA is tissue specific: differential regulation of variant transcripts by early-life events. Mol Endocrinol. 2000;14:506–517. doi: 10.1210/mend.14.4.0438. [DOI] [PubMed] [Google Scholar]
- 85.Turner JD, Muller CP. Structure of the glucocorticoid receptor (NR3C1) gene 5′ untranslated region: identification, and tissue distribution of multiple new human exon 1. J Mol Endocrinol. 2005;35:283–292. doi: 10.1677/jme.1.01822. [DOI] [PubMed] [Google Scholar]
- 86.Culty M, Thuillier R, Li W, Wang Y, Martinez-Arguelles DB, Benjamin CG, Triantafilou KM, Zirkin BR, Papadopoulos V. In utero exposure to di-(2-ethylhexyl) phthalate exerts both short-term and long-lasting suppressive effects on testosterone production in the rat. Biol Reprod. 2008;78:1018–1028. doi: 10.1095/biolreprod.107.065649. [DOI] [PubMed] [Google Scholar]
- 87.Davidoff MS, Middendorff R, Enikolopov G, Riethmacher D, Holstein AF, Muller D. Progenitor cells of the testosterone-producing Leydig cells revealed. J Cell Biol. 2004;167:935–944. doi: 10.1083/jcb.200409107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Benton L, Shan LX, Hardy MP. Differentiation of adult Leydig cells. J Steroid Biochem Mol Biol. 1995;53:61–68. doi: 10.1016/0960-0760(95)00022-r. [DOI] [PubMed] [Google Scholar]
- 89.Koo HJ, Lee BM. Toxicokinetic relationship between di(2-ethylhexyl) phthalate (DEHP) and mono(2-ethylhexyl) phthalate in rats. J Toxicol Environ Health A. 2007;70:383–387. doi: 10.1080/15287390600882150. [DOI] [PubMed] [Google Scholar]
- 90.Martinez-Arguelles DB, Culty M, Zirkin BR, Papadopoulos V. In utero exposure to di-(2-ethylhexyl) phthalate decreases mineralocorticoid receptor expression in the adult testis. Endocrinology. 2009 doi: 10.1210/en.2009-0847. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stier CT, Jr, Rocha R, Chander PN. Effect of aldosterone and MR blockade on the brain and the kidney. Heart Fail Rev. 2005;10:53–62. doi: 10.1007/s10741-005-2349-x. [DOI] [PubMed] [Google Scholar]
- 92.Ge RS, Dong Q, Sottas CM, Latif SA, Morris DJ, Hardy MP. Stimulation of testosterone production in rat Leydig cells by aldosterone is mineralocorticoid receptor mediated. Mol Cell Endocrinol. 2005;243:35–42. doi: 10.1016/j.mce.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 93.Zennaro MC, Keightley MC, Kotelevtsev Y, Conway GS, Soubrier F, Fuller PJ. Human mineralocorticoid receptor genomic structure and identification of expressed isoforms. J Biol Chem. 1995;270:21016–21020. doi: 10.1074/jbc.270.36.21016. [DOI] [PubMed] [Google Scholar]
- 94.Laflamme N, Nappi RE, Drolet G, Labrie C, Rivest S. Expression and neuropeptidergic characterization of estrogen receptors (ERalpha and ERbeta) throughout the rat brain: anatomical evidence of distinct roles of each subtype. J Neurobiol. 1998;36:357–378. doi: 10.1002/(sici)1097-4695(19980905)36:3<357::aid-neu5>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 95.Simerly RB, Chang C, Muramatsu M, Swanson LW. Distribution of androgen and estrogen receptor mRNA-containing cells in the rat brain: an in situ hybridization study. J Comp Neurol. 1990;294:76–95. doi: 10.1002/cne.902940107. [DOI] [PubMed] [Google Scholar]
- 96.Young LJ, Wang Z, Donaldson R, Rissman EF. Estrogen receptor alpha is essential for induction of oxytocin receptor by estrogen. Neuroreport. 1998;9:933–936. doi: 10.1097/00001756-199803300-00031. [DOI] [PubMed] [Google Scholar]
- 97.Kuppers E, Krust A, Chambon P, Beyer C. Functional alterations of the nigrostriatal dopamine system in estrogen receptor-alpha knockout (ERKO) mice. Psychoneuroendocrinology. 2008;33:832–838. doi: 10.1016/j.psyneuen.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 98.Numan M. Neural basis of maternal behavior in the rat. Psychoneuroendocrinology. 1988;13:47–62. doi: 10.1016/0306-4530(88)90006-6. [DOI] [PubMed] [Google Scholar]
- 99.Flanagan-Cato LM, Calizo LH, Daniels D. The synaptic organization of VMH neurons that mediate the effects of estrogen on sexual behavior. Horm Behav. 2001;40:178–182. doi: 10.1006/hbeh.2001.1679. [DOI] [PubMed] [Google Scholar]
- 100.Champagne FA, Weaver IC, Diorio J, Dymov S, Szyf M, Meaney MJ. Maternal care associated with methylation of the estrogen receptor-alpha1b promoter and estrogen receptor-alpha expression in the medial preoptic area of female offspring. Endocrinology. 2006;147:2909–2915. doi: 10.1210/en.2005-1119. [DOI] [PubMed] [Google Scholar]
- 101.Green S, Kumar V, Krust A, Walter P, Chambon P. Structural and functional domains of the estrogen receptor. Cold Spring Harb Symp Quant Biol. 1986;51(Pt 2):751–758. doi: 10.1101/sqb.1986.051.01.088. [DOI] [PubMed] [Google Scholar]
- 102.Piva R, Gambari R, Zorzato F, Kumar L, del SL. Analysis of upstream sequences of the human estrogen receptor gene. Biochem Biophys Res Commun. 1992;183:996–1002. doi: 10.1016/s0006-291x(05)80289-x. [DOI] [PubMed] [Google Scholar]
- 103.Ottaviano YL, Issa JP, Parl FF, Smith HS, Baylin SB, Davidson NE. Methylation of the estrogen receptor gene CpG island marks loss of estrogen receptor expression in human breast cancer cells. Cancer Res. 1994;54:2552–2555. [PubMed] [Google Scholar]
- 104.Lapidus RG, Nass SJ, Butash KA, Parl FF, Weitzman SA, Graff JG, Herman JG, Davidson NE. Mapping of ER gene CpG island methylation-specific polymerase chain reaction. Cancer Res. 1998;58:2515–2519. [PubMed] [Google Scholar]
- 105.Ferguson AT, Lapidus RG, Baylin SB, Davidson NE. Demethylation of the estrogen receptor gene in estrogen receptor-negative breast cancer cells can reactivate estrogen receptor gene expression. Cancer Res. 1995;55:2279–2283. [PubMed] [Google Scholar]
- 106.Yang X, Ferguson AT, Nass SJ, Phillips DL, Butash KA, Wang SM, Herman JG, Davidson NE. Transcriptional activation of estrogen receptor alpha in human breast cancer cells by histone deacetylase inhibition. Cancer Res. 2000;60:6890–6894. [PubMed] [Google Scholar]
- 107.Armenti AE, Zama AM, Passantino L, Uzumcu M. Developmental methoxychlor exposure affects multiple reproductive parameters and ovarian folliculogenesis and gene expression in adult rats. Toxicol Appl Pharmacol. 2008;233:286–296. doi: 10.1016/j.taap.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zama AM, Uzumcu M. Fetal and neonatal exposure to the endocrine disruptor methoxychlor causes epigenetic alterations in adult ovarian genes. Endocrinology. 2009;150:4681–4691. doi: 10.1210/en.2009-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Couse JF, Korach KS. Estrogen receptor null mice: what have we learned and where will they lead us? Endocr Rev. 1999;20:358–417. doi: 10.1210/edrv.20.3.0370. [DOI] [PubMed] [Google Scholar]
- 110.Couse JF, Yates MM, Deroo BJ, Korach KS. Estrogen receptor-beta is critical to granulosa cell differentiation and the ovulatory response to gonadotropins. Endocrinology. 2005;146:3247–3262. doi: 10.1210/en.2005-0213. [DOI] [PubMed] [Google Scholar]
- 111.Yap OW, Bhat G, Liu L, Tollefsbol TO. Epigenetic modifications of the Estrogen receptor beta gene in epithelial ovarian cancer cells. Anticancer Res. 2009;29:139–144. [PMC free article] [PubMed] [Google Scholar]
- 112.Walton TJ, Li G, Seth R, McArdle SE, Bishop MC, Rees RC. DNA demethylation and histone deacetylation inhibition co-operate to re-express estrogen receptor beta and induce apoptosis in prostate cancer cell-lines. Prostate. 2008;68:210–222. doi: 10.1002/pros.20673. [DOI] [PubMed] [Google Scholar]
- 113.Jarrard DF, Kinoshita H, Shi Y, Sandefur C, Hoff D, Meisner LF, Chang C, Herman JG, Isaacs WB, Nassif N. Methylation of the androgen receptor promoter CpG island is associated with loss of androgen receptor expression in prostate cancer cells. Cancer Res. 1998;58:5310–5314. [PubMed] [Google Scholar]
- 114.Takahashi S, Inaguma S, Sakakibara M, Cho YM, Suzuki S, Ikeda Y, Cui L, Shirai T. DNA methylation in the androgen receptor gene promoter region in rat prostate cancers. Prostate. 2002;52:82–88. doi: 10.1002/pros.10099. [DOI] [PubMed] [Google Scholar]
- 115.Nakayama T, Watanabe M, Suzuki H, Toyota M, Sekita N, Hirokawa Y, Mizokami A, Ito H, Yatani R, Shiraishi T. Epigenetic regulation of androgen receptor gene expression in human prostate cancers. Lab Invest. 2000;80:1789–1796. doi: 10.1038/labinvest.3780190. [DOI] [PubMed] [Google Scholar]
- 116.Schulz WA, Hoffmann MJ. Epigenetic mechanisms in the biology of prostate cancer. Semin Cancer Biol. 2009;19:172–180. doi: 10.1016/j.semcancer.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 117.Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, Gunther T, Buettner R, Schule R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- 118.Nelson WG. Prostate cancer prevention. Curr Opin Urol. 2007;17:157–167. doi: 10.1097/MOU.0b013e3280eb110f. [DOI] [PubMed] [Google Scholar]
- 119.Hmadcha A, Bedoya FJ, Sobrino F, Pintado E. Methylation-dependent gene silencing induced by interleukin 1beta via nitric oxide production. J Exp Med. 1999;190:1595–1604. doi: 10.1084/jem.190.11.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bastian PJ, Yegnasubramanian S, Palapattu GS, Rogers CG, Lin X, De Marzo AM, Nelson WG. Molecular biomarker in prostate cancer: the role of CpG island hypermethylation. Eur Urol. 2004;46:698–708. doi: 10.1016/j.eururo.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 121.Martin DI, Cropley JE, Suter CM. Environmental influence on epigenetic inheritance at the Avy allele. Nutr Rev. 2008;66(Suppl 1):S12–S14. doi: 10.1111/j.1753-4887.2008.00057.x. [DOI] [PubMed] [Google Scholar]
- 122.Liu L, Li Y, Tollefsbol TO. Gene-environment interactions and epigenetic basis of human diseases. Curr Issues Mol Biol. 2008;10:25–36. [PMC free article] [PubMed] [Google Scholar]
- 123.Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, Reik W, Walter J, Surani MA. Epigenetic reprogramming in mouse primordial germ cells. Mech Dev. 2002;117:15–23. doi: 10.1016/s0925-4773(02)00181-8. [DOI] [PubMed] [Google Scholar]
- 124.Warnecke PM, Stirzaker C, Song J, Grunau C, Melki JR, Clark SJ. Identification and resolution of artifacts in bisulfite sequencing. Methods. 2002;27:101–107. doi: 10.1016/s1046-2023(02)00060-9. [DOI] [PubMed] [Google Scholar]
- 125.Ho SM, Tang WY. Techniques used in studies of epigenome dysregulation due to aberrant DNA methylation: an emphasis on fetal-based adult diseases. Reprod Toxicol. 2007;23:267–282. doi: 10.1016/j.reprotox.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Beck S, Rakyan VK. The methylome: approaches for global DNA methylation profiling. Trends Genet. 2008;24:231–237. doi: 10.1016/j.tig.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 127.Brena RM, Huang TH, Plass C. Quantitative assessment of DNA methylation: Potential applications for disease diagnosis, classification, and prognosis in clinical settings. J Mol Med. 2006;84:365–377. doi: 10.1007/s00109-005-0034-0. [DOI] [PubMed] [Google Scholar]
- 128.Taylor KH, Kramer RS, Davis JW, Guo J, Duff DJ, Xu D, Caldwell CW, Shi H. Ultradeep bisulfite sequencing analysis of DNA methylation patterns in multiple gene promoters by 454 sequencing. Cancer Res. 2007;67:8511–8518. doi: 10.1158/0008-5472.CAN-07-1016. [DOI] [PubMed] [Google Scholar]
- 129.Gitan RS, Shi H, Chen CM, Yan PS, Huang TH. Methylation-specific oligonucleotide microarray: a new potential for high-throughput methylation analysis. Genome Res. 2002;12:158–164. doi: 10.1101/gr.202801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Graf S, Nielsen FG, Kurtz S, Huynen MA, Birney E, Stunnenberg H, Flicek P. Optimized design and assessment of whole genome tiling arrays. Bioinformatics. 2007;23:i195–i204. doi: 10.1093/bioinformatics/btm200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hatada I, Hayashizaki Y, Hirotsune S, Komatsubara H, Mukai T. A genomic scanning method for higher organisms using restriction sites as landmarks. Proc Natl Acad Sci U S A. 1991;88:9523–9527. doi: 10.1073/pnas.88.21.9523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Huang TH, Laux DE, Hamlin BC, Tran P, Tran H, Lubahn DB. Identification of DNA methylation markers for human breast carcinomas using the methylation-sensitive restriction fingerprinting technique. Cancer Res. 1997;57:1030–1034. [PubMed] [Google Scholar]
- 133.Wu M, Ho SM. PMP24, a gene identified by MSRF, undergoes DNA hypermethylation-associated gene silencing during cancer progression in an LNCaP model. Oncogene. 2004;23:250–259. doi: 10.1038/sj.onc.1207076. [DOI] [PubMed] [Google Scholar]
- 134.Oakes CC, La SS, Smiraglia DJ, Robaire B, Trasler JM. Developmental acquisition of genome-wide DNA methylation occurs prior to meiosis in male germ cells. Dev Biol. 2007;307:368–379. doi: 10.1016/j.ydbio.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 135.Rouillard JM, Erson AE, Kuick R, Asakawa J, Wimmer K, Muleris M, Petty EM, Hanash S. Virtual genome scan: a tool for restriction landmark-based scanning of the human genome. Genome Res. 2001;11:1453–1459. doi: 10.1101/gr.181601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Smiraglia DJ, Kazhiyur-Mannar R, Oakes CC, Wu YZ, Liang P, Ansari T, Su J, Rush LJ, Smith LT, Yu L, Liu C, Dai Z, Chen SS, Wang SH, Costello J, Ioshikhes I, Dawson DW, Hong JS, Teitell MA, Szafranek A, Camoriano M, Song F, Elliott R, Held W, Trasler JM, Plass C, Wenger R. Restriction landmark genomic scanning (RLGS) spot identification by second generation virtual RLGS in multiple genomes with multiple enzyme combinations. BMC Genomics. 2007;8:446. doi: 10.1186/1471-2164-8-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Costello JF, Hong C, Plass C, Smiraglia DJ. Restriction landmark genomic scanning: analysis of CpG islands in genomes by 2D gel electrophoresis. Methods Mol Biol. 2009;507:131–148. doi: 10.1007/978-1-59745-522-0_11. [DOI] [PubMed] [Google Scholar]