

Abstract
Narcolepsy is a chronic sleep disorder, characterized by excessive daytime sleepiness (EDS), cataplexy, hypnagogic hallucinations, and sleep paralysis. Both sporadic (95%) and familial (5%) forms of narcolepsy exist in humans. The major pathophysiology of human narcolepsy has been recently discovered based on the discovery of narcolepsy genes in animals; the genes involved in the pathology of the hypocretin/orexin ligand and its receptor. Mutations in hypocretin-related genes are rare in humans, but hypocretin-ligand deficiency is found in a large majority of narcolepsy with cataplexy.
Hypocretin ligand deficiency in human narcolepsy is likely due to the postnatal cell death of hypocretin neurons. Although tight association between human leukocyte antigen (HLA) association and human narcolepsy with cataplexy suggests an involvement of autoimmune mechanisms, this has not yet been proven. Hypocretin deficiency is also found in symptomatic cases of narcolepsy and EDS with various neurological conditions, including immune-mediated neurological disorders, such as Guillain-Barre syndrome, MA2-positive paraneoplastic syndrome and neuromyelitis optica (NMO) related disorder. These findings likely have significant clinical relevance and for understanding the mechanisms of hypocretin cell death and choice of treatment option.
These series of discoveries in humans lead to the establishment of the new diagnostic test of narcolepsy (i.e. low cerebrospinal fluid [CSF] hypocretin-1 levels for narcolepsy with cataplexy and narcolepsy due to medical condition). Since a large majority of human narcolepsy patients are ligand deficient, hypocretin replacement therapy may be a promising new therapeutic option, and animal experiments using gene therapy and cell transplantations are in progress.
Keywords: narcolepsy, orexin, hypocretin, REM sleep, cataplexy, CSF, histamine
Introduction
Narcolepsy is a chronic sleep disorder, characterized by excessive daytime sleepiness (EDS), cataplexy, hypnagogic hallucinations, and sleep paralysis. Although the disease was first described towards the end of the 19th century, the underlying pathophysiological mechanisms have only been described within the last decade (Nishino and Kanbayashi, 2005). Central to the pathology of narcolepsy is an impairment of hypocretin neurotransmission (Nishino, 2007, Mignot et al., 2002). Our understanding of the role of hypocretin in narcolepsy is based on the discovery of narcolepsy genes (i.e., hypocretin and hypocretin receptor genes) in animals, followed by the discovery of the major pathophysiology of human narcolepsy (i.e. postnatal hypocretin ligand deficiency). Since review articles covering a series of these discoveries are available elsewhere (Nishino, 2007, Nishino, 2003), this review mostly focuses on new basic and clinical insights that help to understand the current elucidation of etiology/pathophysiology of human narcolepsy and its related disorders.
Basic sleep physiology and sleep abnormalities in narcolepsy
In order to understand primary symptoms of narcolepsy, basic sleep physiology will be briefly discussed. Normal sleep is a structured process that is divided into two distinct states, rapid eye movement (REM), and non–REM (NREM) sleep (Nishino et al., 2004, Espana and Scammell, 2004). NREM sleep is characterized by the slow oscillation of thalamocortical neurons, partly detected as cortical slow waves. Based on characteristic EEG signals, NREM sleep is divided into 4 stages (S1, S2, S3, and S4) in humans. Human sleep alternates sequentially between NREM stages S1 to S4, followed by REM sleep; this sleep cycle takes place approximately every 90 minutes and is repeated 4 to 5 times a night (Nishino et al., 2004). During the course of a normal night of sleep, there is a change from a predominance of slow wave NREM sleep during the first part of the night to a predominance of REM sleep during the second part of the night (Nishino et al., 2004). During NREM sleep, reductions of muscle tonus occur, and complete muscle atonia is seen during REM sleep.
It is hypothesized that sleep involves the interactions of facilitating sleep centers and inhibiting arousal centers in the brain (Nishino et al., 2004, Saper et al., 2005, Espana and Scammell, 2004). Wakefulness is promoted by an ascending arousal pathway that begins in the rostral pons and runs through the midbrain reticular formation (Nishino et al., 2004, Espana and Scammell, 2004). Brainstem and hypothalamic neurons that produce acetylcholine, norepinephrine, dopamine, serotonin, histamine, excitatory amino acids, and hypocretin/orexin may compose this ascending arousal pathway (Espana and Scammell, 2004, Nishino et al., 2004). These neurotransmitters are also likely to be involved in the control of muscle tonus during sleep, directly or indirectly. Each of these arousal networks can increase wakefulness, but coordinated activity is required for complete alertness and cortical activation (Nishino et al., 2004). A switch in the hypothalamus shuts off this arousal system during sleep (Saper et al., 2005, Espana and Scammell, 2004). Sleep and wake state must be stable and be sustained a certain length to act and to rest effectively. Narcolepsy represents a major neurologic malfunction of this control system.
Knowledge of the pathological mechanisms of narcolepsy has evolved over the last 40 to 50 years (see (Nishino and Mignot, 1997)). In the 1960s, many of its features were shown to be related to dysregulation of REM sleep; specifically, people with narcolepsy entered REM sleep more rapidly when falling asleep than those without narcolepsy (Nishino and Mignot, 1997). This phenomenon is called a reduced REM sleep latency or a sleep onset REM period (SOREMP). Sometimes this transition to REM sleep occurred immediately upon falling asleep, without an entry into NREM sleep (Nishino and Mignot, 1997). The cataplexy, sleep paralysis and hypnagogic hallucinations associated with narcolepsy may all result from REM sleep intruding into wakefulness. However, it should be also noted that cataplexy is pathognomic for narcolepsy, while sleep paralysis and hypnagogic hallucinations often occur in other sleep disorders, such as sleep apnea syndromes or even in normal populations when their sleep patterns are disturbed (Aldrich et al., 1997, Takeuchi et al., 1992), and thus cataplexy should have a unique pathophysiological mechanism.
Discovering the pathophysiology of narcolepsy
The next major discovery came in the 1980s, when it was found that many people with narcolepsy have the tissue type human leukocyte antigen (HLA) DR2 (Nishino and Mignot, 1997, Juji et al., 1984). The high resolution typing revealed that narcolepsy has the closest association with HLA DQB1*0602, which is found in 95% of narcoleptic patients with cataplexy, 41% of patients with narcolepsy without cataplexy, but only 18–35% of the general population (Nishino and Mignot, 1997, Mignot et al., 1997). While the significance of this association with immune system antigens is not well understood, it suggests that autoimmune processes may play a role in narcolepsy since many autoimmune diseases exhibit tight associations with class II HLA haplotypes. There is, however, no strong evidence of inflammatory processes or immune abnormalities associated with narcolepsy, and studies have found neither classical auto-antibodies nor an increase in oligoclonal cerebrospinal fluid (CSF) bands in narcoleptics (see also below, mechanisms involved in hypocretin neuron death) (Nishino, 2007, Scammell, 2006).
In 1998, 2 groups of researchers independently discovered a novel hypothalamic peptide neurotransmitter named by one group hypocretin and orexin by the other (Sakurai et al., 1998, De Lecea et al., 1998). Hypocretins 1 and 2 are produced exclusively by a group of several thousand neurons localized in the lateral hypothalamus. These neurons project widely to the olfactory bulb, cerebral cortex, thalamus, hypothalamus, and brainstem, and more densely to the locus coeruleus, tuberomamillary nucleus, raphe nucleus, and bulbar reticular formation (Peyron et al., 1998).
The following year, using positional cloning of a naturally-occurring familial canine narcolepsy model, the Stanford researchers identified an autosomal recessive mutation responsible for narcolepsy in dogs (Lin et al., 1999). Narcoleptic dogs have cataplexy (primarily elicited by the presentation of food), sleepiness (i.e. reduced sleep latency), and SOREMPs (Nishino and Mignot, 1997, Nishino et al., 2000a). The authors determined that canine narcolepsy is caused by null mutation of the hypocretin (orexin) receptor 2 gene (Hcrtr2) (Lin et al., 1999). Almost at the same time, Chemelli et al reported that hypocretin ligand deficient mice (prepro-orexin gene knockout mice) exhibit narcolepsy phenotype, characterized with sleep fragmentation and cataplexy-like behavior (Chemelli et al., 1999). These results identified hypocretins as major sleep-modulating neurotransmitters.
Following the discovery of the hypocretin gene-narcolepsy association in dogs and mice, the role of the hypocretin system in human narcolepsy was examined. Systematic screening of mutations in the hypocretin system in patients with cataplexy with high risk (including familial, early onset and HLA negative case) has so far identified only one patient with a mutation in hypocretin related genes, and this patient was atypical with very early disease onset (6 months old) (Peyron et al., 2000b). Hence, most human cases of narcolepsy are not caused by gene mutations.
Whereas gene mutations were not identified as a major cause of narcolepsy in almost all humans, narcoleptic patients were found to have low levels of hypocretin-1 in their CSF (Fig. 1). Experiments using in situ hybridization, immunochemistry, and radioimmunological assays of peptides in the post-mortem brain tissue of narcoleptic patients found undetectable levels of pre-hypocretin RNA, and loss of hypocretin peptides, all representing significant disparities from people without narcolepsy (Fig. 1) (Peyron et al., 2000b, Thannickal et al., 2000b). The loss of hypocretin function was not the result of a generalized neuronal degeneration in these brain regions, because melanin-concentrating hormone neurons that are normally located within the same region as the hypocretin neurons were intact (Peyron et al., 2000b). Together, these results indicate that hypocretin production or hypocretin neurons are selectively damaged in narcoleptic patients.
Fig. 1. CSF hypocretin-1 levels in narcoleptic and control subjects.

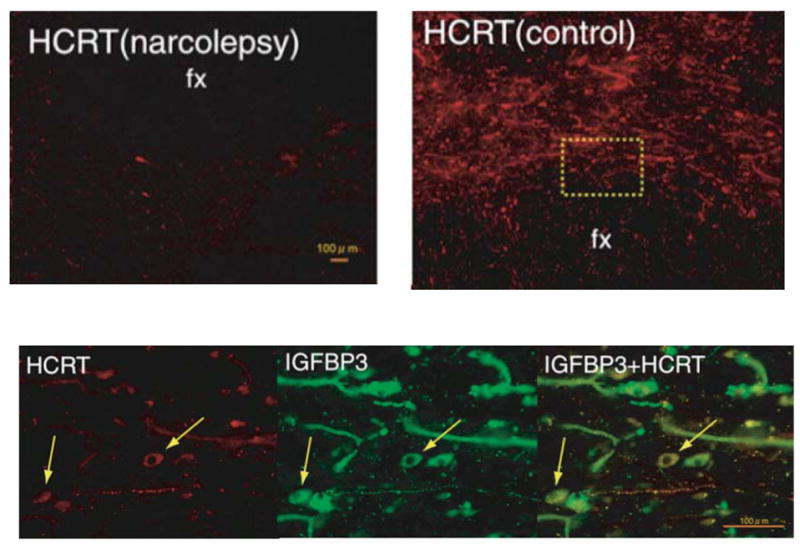
(A) CSF hypocretin-1 levels are undetectably low in most narcoleptic subjects (84.2%). Note that two HLA DqB1*0602-negative and one familial case have normal or high CSF hypocretin levels. (B) Preprohypocretin transcripts are detected in the hypothalamus of control (b) but not in narcoleptic subjects (a). Melanin-concentrating hormone (MCH) transcripts are detected in the same region in both control (d) and narcoleptic (c) sections. (C) Colocalization of IGFBP3 in HCRT cells in control and narcolepsy human brain. Upper panel: Distribution of hypocretin cells and fibers in the perifornical area of human hypothalamus. (e) In control brains, HCRT cells and fibers were densely stained by an anti-HCRT monoclonal antibody (red fluorescence: VectorRed), while in narcolepsy brains, staining was markedly reduced (f) Lower panel: HCRT immunoreactivity (g: red fluorescence) and IGFBP3 immunoreactivity (h: green fluorescene; Q-dot525) and a composite picture (i) arrows indicate HCRT cells colocalized with IGFBP3). Note non-neuronal autofluorescent elements. f and fx, fornix. Scale bar represents 10 mm (a–d), 500 mm in e and f, 100 mm in g, h and i (from (Peyron et al., 2000b) and (Honda et al., 2009)).
Hypocretin ligand deficiency is now used to help diagnose narcolepsy, and this can be clinically detected by CSF hypocretin-1 measures (Fig. 1, 2) (Nishino et al., 2000c, Nishino et al., 2001). CSF hypocretin-1 concentrations lower than 110 pg/ml have a high positive predictive value (94%) for narcolepsy with cataplexy (and thus HLA positivity) (Mignot et al., 2002). In controls or in individuals with other sleep and neurological disorders, hypocretin-1 concentrations in the CSF were almost always above 200 pg/ml (Fig. 2) (Mignot et al., 2002). When used to assess patients for narcolepsy, low CSF hypocretin-1 appears to be a more specific test than the multiple sleep latency test (MSLT, a polygraphic measure of daytime sleepiness). Therefore, low CSF hypocretin-1 levels (less than 110 pg/ml) were included for the diagnosis of narcolepsy with cataplexy in the second edition of ICSD (ICSD-2) (ICSD-2, 2005). Previously, no specific and sensitive diagnostic test for narcolepsy based on the pathophysiology of the disease was available, and the final diagnosis was often delayed for several years after the disease onset, typically until adolescence (Alaila, 1992). Of note, sensitivity of HLA positivity is equal to that of hypocretin deficiency, but specificity of HLA positively is very low, since up to 30% of the general population share the HLA haplotype narcoleptics are susceptible to. Many patients with narcolepsy and related EDS disorders, therefore, are likely to obtain immediate benefits from this new specific diagnostic test. Furthermore, hypocretin receptor agonists (or cell transplantation) may be promising in the treatment of narcolepsy and CSF hypocretin-1 measures can also be used for the selection of the patient for this therapeutic option (see the section for the hypocretin replacement therapy).
Fig. 2. Lumbar CSF hypocretin-1 concentrations in controls, narcoleptics, and other pathologies.

Each data point is the concentration of hypocretin-1 in the crude (unfiltered) lumbar CSF of a single individual. Represented are controls (samples taken both during night and day) and narcoleptics, including those with typical cataplexy, with atypical cataplexy, with cataplexy but who are HLA-negative, and without cataplexy, as well as narcolepsy family probands. Individuals with hypersomnia due to idiopathic hypersomnia, periodic hypersomnia, or hypersomnia due to secondary etiology are also shown, as are those with other diagnostically described sleep disorders (obstructive sleep apnea (n=17), restless legs syndrome (n=12), insomnia (n=12)) and those with a variety of neurological disorders. Specific pathologies are described for individuals with low (<110 pg/mL) or intermediate (110 – 200 pg/mL) concentrations of hypocretin-1. Data is derived from (Mignot et al., 2002).
The availability of this test is also challenging our view of the nosology of narcolepsy. As emphasized by Honda et al. (Honda, 1988), narcolepsy with cataplexy may be more a homogeneous etiological entity, as reflected by low CSF hypocretin-1, and to be differentiated from narcolepsy without cataplexy or the related syndrome of idiopathic hypersomnia, with generally normal hypocretin levels (Mignot et al., 2002, Kanbayashi et al., 2002) (see also FIG. 2). In the ICSD-2, “narcolepsy with cataplexy” and “narcolepsy without cataplexy” are coded separately, and low CSF hypocretin-1 was considered as a positive diagnostic result for “narcolepsy with cataplexy” (ICSD-2, 2005).
It was not known at the time of the discovery of hypocretin deficiency in human if this is due to the problem of transcription/biosynthesis and/or processing of hypocretin peptide or selective loss of hypocretin neurons. Shortly after, several investigators identified several substances/genes that colocalize in hypocretin-containing neurons, such as dynorphin, neuronal activity-regulated pentraxin (NARP) (Blouin et al., 2005, Crocker et al., 2005) and most recently, insulin-like growth factor binding protein 3 (IGF BP3) (Honda et al., 2009). Since these substances are also deficient in the LHA in the postmortem brains of hypocretin deficient narcoleptic subjects (Crocker et al., 2005, Blouin et al., 2005, Honda et al., 2009) (see Fig. 1 for IGF BP3), selective cell death of hypocretin neurons are suspected in human narcolepsy. These results taken together with the fact that onsets of the sporadic narcoleptic cases are generally later than those of familial cases (in both human and dogs) (see (Nishino, 2007), suggests that the postnatal cell death of the hypocretin neurons is the major pathophysiological process in human narcolepsy with cataplexy.
Possible mechanisms involved in hypocretin neuron death
The cause of hypocretin cell death is not known yet, but an autoimmune mechanism has been a primary hypothesis since the disease is tightly associated with HLA haplotypes. Some earlier studies testing a variety of serological tests in narcoleptics yielded higher levels of antistreptolysine 0 and anti-DNase antibodies in narcoleptics than in controls (Billiard et al., 1989, Montplaisir et al., 1989). There is, however, no strong evidence of the inflammatory processes or immune abnormalities associated with narcolepsy (Mignot et al., 1992), and studies have found no classical auto-antibodies and no increase in oligoclonal CSF bands in narcoleptics (Frederickson et al., 1990). Typical autoimmune pathologies (e.g., erythrocyte sedimentation rates, serum immunoglobulin levels, C-reactive protein levels, complement levels and lymphocyte subset ratios) are apparently normal in narcoleptic patients (Matsuki et al., 1988). Recent studies by Black et al. examined the presence of many neuron-specific and organ-specific auto-antibodies, but any auto-antibody was not associated with narcolepsy patients (HLA DQB1*0602-positive and -negative) (Black et al., 2002). Furthermore, the same authors tested for immunoglobulin (Ig) G reactive to preprohypocretin and its major cleavage products (including hypocretin 1 and 2) in serum or CSF in DQB1*0602-positive narcoleptic subjects with cataplexy, but no evidence for IgG reactive to preprohypocretin or its cleavage products was found in narcoleptic subjects (Black et al., 2005). However, these negative results do not exclude the possibility that a transient autoimmune reaction restricted to the CNS could have occurred around the disease onset, but disappeared later (Dauvilliers et al., 2007).
Among the substances colocalizing in hypocretin neurons (see above), functional analysis of the IGFBP3 was carried out using several different methods and the result indicated that IGFBP3 is a new regulator of hypocretin cell physiology that may be involved not only in the pathophysiology of narcolepsy, but also in the regulation of sleep in normal individuals (Honda et al., 2009). Studies of transgenic mice overexpressing human IGFBP3 indicated decreased hypocretin messenger RNA and peptide content, and increased sleep, an effect possibly mediated through decreased hypocretin promoter activity in the presence of excessive IGFBP3. Although no IGFBP3 auto-antibodies nor a genetic association with IGFBP3 polymorphisms was found in human narcolepsy, an IGFBP3 polymorphism known to increase serum IGFBP3 levels was associated with lower CSF hypocretin-1 in normal individuals (Honda et al., 2009). However, it is not known if excessive IGFBP3 expression may initiate hypocretin cell death and cause narcolepsy.
Gene polymorphisms in narcolepsy
The results of familial and twins studies had suggested that genes other than HLA are also likely involved in human narcolepsy (Mignot, 1998), Therefore, genetic linkage and association studies have been conducted by several authors, and several susceptible genes have been identified (Hohjoh et al., 1999, Hohjoh et al., 2000, Dauvilliers et al., 2001, Miyagawa et al., 2008). The predictive genes of narcolepsy may exacerbate the narcolepsy sleep phenotype or involve mechanisms of hypocretin cell death, but the multifactorial trait of the disease occurrence suggests that influence of these genes alone may not be large.
A series of association studies suggested that tumor necrosis factor (TNF) and TNF receptor 2 have been implicated in sporadic cases of narcolepsy (Hohjoh et al., 1999, Hohjoh et al., 2000, Park et al., 2004, Wieczorek et al., 2003).
TNF is produced by macrophages and circulating monocytes and is involved in host defense and pathogenesis of various diseases (Aggarwal, 1992, Beutler and Grau, 1993). In the central nervous system (CNS), TNF is produced in astrocytes and microglial macrophages (Lieberman et al., 1989) and influences the peripheral immune system through the lymphatic pathway (Dickstein et al., 1999). The TNF-α gene sits within the HLA class III region on chromosome 6, and polymorphisms in (TNF-α [−857T]) are associated with narcolepsy in Japan (Hohjoh et al., 1999). A polymorphism in the TNF receptor 2 gene on chromosome 1 also occurs more frequently among Japanese narcoleptics with cataplexy (Hohjoh et al., 2000). This susceptibility appears especially strong among narcoleptics who have specific polymorphisms in both the TNF-α gene and the TNF receptor 2 gene. The positive association between TNF-α [−857T] polymorphisms was reproduced in 60 Korean patients with narcolepsy-cataplexy and 200 healthy controls (Park et al., 2004). A positive association was also reported in HLA negative narcolepsy in European countries (Wieczorek et al., 2003).
Since TNF-α polymorphisms increase the risk of other HLA-linked diseases such as multiple sclerosis or systemic lupus erythematosis, altered TNF signaling in susceptible subjects may contribute to an autoimmune inflammatory process and develop narcolepsy, but this has not been proven.
Beside the TNF-related genes, a different distribution of the catechol-O-methyl transferase genotype, a key enzyme in dopaminergic and noradrenergic degradation, was found as well as a correlation between this genotype and the severity of narcolepsy in female narcoleptic subjects (Dauvilliers et al., 2001). Recent genome wide association analysis of Japanese narcoleptic subjects also revealed the narcolepsy candidate regions on chromosomes 21 (three predictive genes on 21q 22.3) (Kawashima et al., 2006) and two new narcolepsy susceptible genes CPT1B and CHKB on chromosomes 22 (Miyagawa et al., 2008). CPT1B regulates β-oxidation, a pathway involved in regulating theta frequency during REM sleep (Tafti et al., 2003). CHKB is an enzyme involved in the metabolism of choline (Rao and Qureshi, 1997), a precursor of the REM- and wake-regulating neurotransmitter acetylcholine. Either of the two genes may modify the narcolepsy sleep phenotype. However, their involvements in mechanisms of hypocretin cell death are uncertain at this moment.
This study was followed by a remarkable study of another genome wide association for the HLA matched (DQB1*0602 positive) subjects and for across different ethnicities (1,830 cases, 2,164 controls). Hallmayer et al (Hallmayer et al., 2009) found that association between narcolepsy and polymorphisms in the TRA@ (T-cell receptor alpha) locus, with highest signi cance at rs1154155 (average allelic odds ratio 1.69, genotypic odds ratios 1.94 and 2.55, P < 10–21). Therefore, it is now more credible that postnatal cell death of hypocretin neurons of human narcolepsy occurs by organ-specific autoimmune targeting with HLA-TCR interactions, but exact mechanisms involved need to be determined.
Hypocretin deficiency and symptomatic narcolepsy and EDS
The discovery of hypocretin ligand deficiency in idiopathic narcolepsy also led to new insights into the pathophysiology of symptomatic (or secondary) narcolepsy and EDS. The symptoms of narcolepsy can also occur during the course of other neurological conditions (i.e., symptomatic narcolepsy) (Nishino and Kanbayashi, 2005). In a recent meta analysis, 116 symptomatic cases of narcolepsy reported in the literature were analyzed (cases that met with the ICSD criteria for narcolepsy and were also associated with a significant underlying neurological disorder that accounts for EDS and temporal associations) (Nishino and Kanbayashi, 2005). As several authors have previously reported, inherited disorders (n=38), tumors (n=33), and head trauma (n=19) are the three most frequent causes for symptomatic narcolepsy. Of the 116 cases, 10 are associated with multiple sclerosis (MS), one case with acute disseminated encephalomyelitis, and relatively rare cases were reported with vascular disorders (n=6), encephalitis (n=4), degeneration (n=1), and heterodegenerative disorder (autosomal dominant cerebrospinal ataxia with deafness, three cases in a family). Although it is difficult to rule out the comorbidity of idiopathic narcolepsy in some cases, a review of the literature reveals numerous unquestionable cases of symptomatic narcolepsy (Nishino and Kanbayashi, 2005). These include cases with HLA negative and/or late onset, and cases in which the occurrences of the narcoleptic symptoms are parallel with the rise and fall of the causative disease. Interestingly, a review of these cases (especially those with brain tumors), illustrates clearly that the hypothalamus is most often involved (see (Nishino and Kanbayashi, 2005)).
A number of EDS cases without cataplexy or any REM sleep abnormalities is also often associated with these neurological conditions, defined as symptomatic cases of EDS (Nishino and Kanbayashi, 2005). It is often difficult to evaluate whether most of the old cases meet with ICSD II (hypersomnia due to medical condition), since objective measures for sleepiness are lacking in most of these cases. In the same review article (Nishino and Kanbayashi, 2005), we listed a total of 71 symptomatic EDS cases. However, prevalence of symptomatic EDS is likely to be much higher. For example, about several million subjects in the USA suffered from chronic brain injury, and 75% of those people have sleep problems, and about a half claim sleepiness (Verma et al., 2007). Symptomatic EDS may thus have a significant clinical relevance.
CSF hypocretin-1 measurement were also carried out in these symptomatic narcolepsy and EDS (Nishino and Kanbayashi, 2005), and reduced CSF hypocretin-1 levels were seen in most symptomatic narcolepsy and EDS cases with various etiologies. EDS in these cases is sometimes reversible with an improvement of the causative neurological disorder and an improvement of the hypocretin status, suggesting a functional link between hypocretin deficiency and sleep symptoms in these patients.
Von Economo (1931) (von Economo, 1931.) was probably the first person to suggest that narcolepsy may have its origins in the posterior hypothalamus and in some cases, is a secondary etiology. Neuropathological studies on encephalitis lethargica pandemic (1916–1923) revealed involvements of the midbrain periaqueductal grey matter and posterior hypothalamus in the hypersomnolent variant, with frequent extensions to the oculomotor nuclei. This led von Economo to speculate that the anterior hypothalamus contained a sleep-promoting area while an area spanning from the posterior wall of the third ventricle to the third nerve was involved in actively promoting wakefulness. Along with von Economo’s cases, three case reports for narcolepsy-cataplexy after encephalitis lethargica were also available by Stiefler (1924) (Stiefler, 1924), Adie (1926) (Adie, 1926) and Lhermitte and Rouques (1928) (Lhermitte and Roques, 1928).
The cause of idiopathic narcolepsy was also speculated to be involved in this general area by von Economo (von Economo, 1930). A postulated hypothalamic cause of narcolepsy was widespread until the 1940s (Gill, 1941), but was then ignored during the psychoanalytic boom (Barker, 1948, Smith and Hamilton, 1959), thereafter replaced by a brainstem hypothesis (Mamelak, 1992) along with establishments of the brainstem roles of generating REM sleep and REM sleep atonia (Siegel, 2000). The involvement of the hypothalamus in the occurrence of narcoleptic symptoms was nicely refined by Aldrich et al. (1989) (Aldrich and Naylor, 1989), who noted that tumors or other lesions located close to the third ventricle were associated with symptomatic narcolepsy and hypothesized that the posterior hypothalamic region may be the culprit. Von Economo’s hypothesis is finally confirmed by the discovery of hypocretin deficiency in idiopathic cases of human narcolepsy (Peyron et al., 2000a, Nishino et al., 2000b, Thannickal et al., 2000a)
From von Economo’s point of view, it is appealing to introduce a case report by Scammell et al. of a 23 year-old male who developed narcolepsy-cataplexy due to a large hypothalamic stroke after a resection of a craniopharyngioma (Scammell et al., 2001). This lesion included 2/3 of the caudal hypothalamus except for the most lateral component on the right, and extended into the mediodorsal thalamus bilaterally, the left amygdala, and parts of the basal forebrain and rostral midbrain, mirroring the structures responsible for hypersomnia pointed out by von Economo (Fig. 3). His HLA was negative for DQB1*0602 and CSF hypocretin-1 level was 167 pg/ml. The hypocretin-1 level was intermediate, but hypocretin projections to the brainstem structures are also likely damaged in this case.
Fig. 3. A hypersomnia case with hypothalamic tumor and low hypocretin level (167 pg/ml).
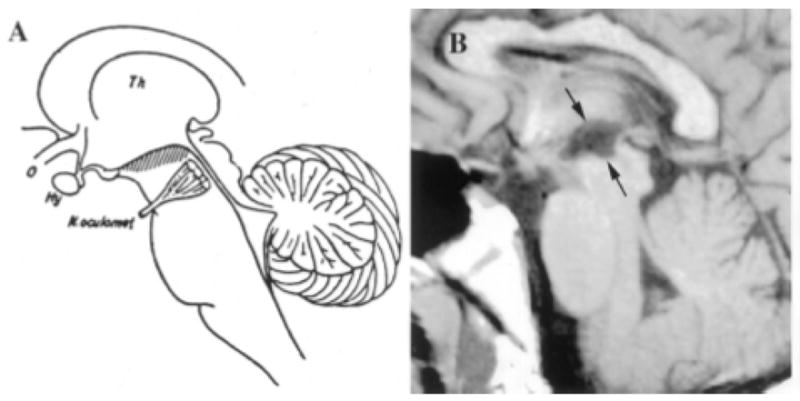
A 23 year-old man who developed narcolepsy-cataplexy after a large hypothalamic stroke. His lesion included much of the hypothalamic region in which orexin is produced. (A) Diagram of a sagittal section of the brain adapted from von Economo 1; the hatching highlights those regions in which inflammatory lesions produced hypersomnia. (B) T1-weighted sagittal and horizontal MR images demonstrating ex vacuo changes (between arrows) in the posterior hypothalamus and rostral midbrain of the patient with secondary narcolepsy. Th = thalamus; o = optic nerve; Hy = hypothalamus; N. oculomot = occulomotor nerve. A permission from ((Scammell et al., 2001)).
Another important symptomatic case we need to be aware is a case of 11-year-old boy in a vegetative state (Glasgow coma scale of 8) following astrocytoma resection and CNS hemorrhage reported by Marcus et al. (Marcus et al., 2002). An MRI revealed a large suprasellar mass that extended into the sella inferiorly and was displaced posteriorly. The boy developed hypothyroidism and syndrome of inappropriate secretion of antidiuretic hormone (SIADH). The boy appeared to be asleep most of the day, but would open his eyes when stimulated. He did not speak or obey commands, and was fed by nasogastric tube. The EEG was abnormal, with generalized slowing and increased amplitude. Although it was difficult to distinguish the different NREM sleep stages, nighttime sleep was fragmented, with 16 short REM cycles. The daytime EEG also showed frequent REM periods, suggesting altered NREM-REM sleep regulation. HLA-DR2 and DQB1*0602 was negative. Hypocretin-1 was undetectably low. His vegetative state improved with 200 mg modafinil and 5 mg methylphenidate. He was able to ambulate with support and be fed by mouth. He had returned to school. This result suggests that, sleepiness, possibly due to the hypocretin deficiency, at least partially contribute to the major symptom of the patient. This case may be an extreme case. However, it is possible that sleepiness may worsen the neurological symptoms of the organic brain impairment and the overall condition can be improved by treatments of the sleepiness.
Neuromyelitis Optica (NMO), autoimmune involvements for bilateral hypothalamic lesions and hypocretin deficiency
Low CSF hypocretin-1 concentrations were also found in some immune mediated neurological conditions, namely subsets of Guillain-Barre syndrome (Nishino et al., 2003), Ma2-positive paraneoplastic syndrome (Overeem et al., 2001) and MS (Nishino and Kanbayashi, 2005), and EDS are often associated with the patients with low CSF hypocretin-1 levels.
Basic knowledge of symptomatic narcolepsy/EDS in immune mediated conditions should be useful for understanding the etiological mechanism of narcolepsy. Kanbayashi et al. recently experienced seven cases of EDS occurring in the course of MS patients initially diagnosed with symmetrical hypothalamic inflammatory lesions together with hypocretin ligand deficiency (Kanbayashi et al., 2009) that contrasts with the characteristics of classic MS cases (Fig. 4).
Fig. 4. MRI findings (FLAIR or T2) of MS/NMO patients with hypocretin deficiency and EDS.

A typical horizontal slice including the hypothalamic periventricular area from each case is presented. All cases were female (f) and age (y) listed in the parenthesis. * met with ICSDII criteria for narcolepsy due to medical condition, and ** met with ICSDII criteria for hypersomnia due to medical condition. All cases were initially diagnosed as MS. Case 3, 4 and 5 exhibited optic neuritis and/or spinal cord lesions and are seropositive for anti-AQP4 antibody and thus being diagnosed as NMO. CSF hypocretin levels are listed below the MRI image. Cases 1–5 (Kato et al., 2003, Oka et al., 2004, Nozaki et al., 2008, Baba et al., 2008) and (Nakamura et al., 2005), respectively.
Symptomatic narcolepsy in MS patients has been reported from several decades ago. Since both MS and narcolepsy are associated with the HLA-DR2 positivity, an autoimmune target on the same brain structures has been proposed to be a common etiology for both diseases (Poirier et al., 1987). However, the discovery of the selective loss of hypothalamic hypocretin neurons in narcolepsy rather indicates that narcolepsy coincidently occurs in MS patients when MS plaques appear in the hypothalamic area and secondarily damage the hypocretin/orexin neurons. In favor of this interpretation, the hypocertin system are not impaired in MS subjects who do not exhibit narcolepsy (Ripley et al., 2001b), although MS patients frequently show other sleep problems such as insomnia, parasomnia, and sleep-related movement disorders (Tachibana et al., 1994). Nevertheless, it is also the case that a subset of MS patients predominantly shows EDS and REM sleep abnormalities, and it is likely that specific immune-mediated mechanisms may be involved in these cases.
CSF hypocretin measures revealed that marked (≤110 pg/ml, n=3) or moderate (110 to 200 pg/ml, n=4) hypocretin deficiency was observed in all 7 cases (Kanbayashi et al., 2009). Therefore, four cases met with ICSDII criteria (ICSD-2, 2005) for narcolepsy due to medical condition, and three cases met with the hypersomnia due to medical condition. Interestingly, four of them had either or both optic neuritis and spinal cord lesions, sharing the clinical characteristics of Neuromyelitis optica (NMO). HLA was evaluated in only two cases (case 2 and case 4) and was negative for DQB1*0602. Repeated evaluations of the hypocretin status were carried out in six cases, and CSF hypocretin-1 levels returned to the normal levels or significantly increased with marked improvements of EDS and hypothalamic lesions in all six cases. Since four of them exhibited clinical characterization of NMO, anti-AQP4 antibody was evaluated and it was found that 3 out of 7 cases were anti-AQP4 antibody positive, thus being diagnosed as NMO-related disorder (Kanbayashi et al., 2009).
AQP4, a member of the AQP super-family, is an integral membrane protein that forms pores in the membrane of biological cells (Amiry-Moghaddam and Ottersen, 2003). Aquaporins selectively conduct water molecules in and out of the cell, while preventing the passage of ions and other solutes and is known as water channels. AQP4 is expressed throughout the central nervous system, especially in periaqueductal and periventricular regions (Amiry-Moghaddam and Ottersen, 2003, Pittock et al., 2006) and is found in non-neuronal structures such as astrocytes and ependymocytes, but is absent from neurons. Recently, the NMO-IgG, which can be detected in the serum of patients with NMO, has been shown to selectively bind to AQP4 (Lennon et al., 2005).
Since AQP4 are enriched in periventricular regions in the hypothalamus where hypocretin-containing neurons are primarily located, symmetrical hypothalamic lesions associated with reduced CSF hypocretin-1 levels in our 3 NMO cases with anti-AQP4 antibody might be caused by the immuno-attack to the AQP4, and this may secondarily affect the hypocretin neurons.
However, our other four MS cases with EDS and hypocretin deficiency were anti-AQP4 antibody negative at the time of blood testing. This leaves a possibility that other antibody-mediated mechanisms are additionally responsible for the bilateral symmetric hypothalamic damage causing EDS in the MS/NMO subjects. There is a possibility that the four MS cases whose anti-AQP4 antibody was negative could be NMO, since anti-AQP4 antibody was tested only once for each subject during the course of the disease and the assay was not standardized among the institutes (Kanbayashi et al., 2009). It is thus essential to further determine the immunological mechanisms that cause the bilateral hypothalamic lesions with hypocretin deficiency and EDS, and their association with NMO and AQP4. This effort may lead to establishment of a new clinical entity, and the knowledge is essential to prevent and treat EDS associated with MS and its related disorders. It should also be noted that none of our cases exhibited cataplexy, contrary to the 9 out of 10 symptomatic narcoleptic MS cases reported in the past (Nishino and Kanbayashi, 2005). Early therapeutic intervention with steroids and other immunosuppressants may thus prevent irreversible damage of hypocretin neurons and prevent chronic sleep-related symptoms.
A new therapeutic option, ligand replacement therapy
EDS associated with narcolepsy has been treated for over several decades with amphetamines, amphetamine-like compounds, and more recently with modafinil (Nishino, 2007). These treatments are merely symptomatic, and do not act on the pathophysiology (impairments of hypocretin neurotransmission). In addition these compounds do not treat cataplexy, and additional anticataplectic antidepressants (monoaminergic uptake inhibitors) are prescribed (Nishino, 2007).
Since a large majority of human narcolepsy patients are ligand deficient, hypocretin replacement therapy may be a new therapeutic option. This may be effective for both sleepiness (i.e. fragmented sleep/wake pattern) and cataplexy.
Animal experiments using ligand deficient narcoleptic dogs suggest that stable and centrally active hypocretin analogs (possibly non-peptide synthetic hypocretin ligands) need to be developed in order to be peripherally effective (Fujiki et al., 2003, Schatzberg et al., 2004) (Table 1). This is also substantiated by a mice study that found normalization of sleep/wake patterns and behavioral arrest episodes (equivalent to cataplexy and REM sleep onset) in the hypocretin cell ablated narcoleptic mice model (i.e. orexin/ataxin-3 mice) supplemented by central administration of hypocretin-1 (Mieda et al., 2004). These authors also demonstrated that exogenous expression of prepro-hypocretin gene in orexin/ataxin-3 mice rescue sleep fragmentation and cataplexy in orexin/ataxin-3 mouse model (Mieda et al., 2004) (Table 1).
Table 1.
Evaluations of hypocretin ligand replacement therapy in the animal model of narcolepsy
| Approaches | Animal models | Methods | Effect on Cataplexy | Effects on Sleep | References |
|---|---|---|---|---|---|
| Peptide replacement | |||||
| Ligand deficient narcoleptic dog | IV | very short lasting anticataplectic effect | na | (Fujiki et al., 2003) | |
| Ligand deficient narcoleptic dog | Intrathecal | no effect | na | (Schatzberg et al., 2004) | |
| Ligand deficient mice | ICV | improve | wake-promoting | (Mieda et al., 2004) | |
| Gene therapy | |||||
| Ligand deficient mice | Diffuse expression of ligand (TG with beta-actin promoter) | improve | more consolidated | (Mieda et al., 2004) | |
| Ligand deficient mice | Transient expression of ligand (with AAV vector) in the LH | improve | no effect on sleep fragmentation | APSS presentation | |
| Cell Transplantation | |||||
| Cells transplanted (LH from 10-day old rats) survive for a short period (up to36 days). | na | na | (Arias-Carrion et al., 2004) | ||
IV: intravenous, ICV: intracerebroventriclular, AAV: adeno-associated virus, TG: transgenic, LH: lateral hypothalamus
If exogenously administered hypocretin receptor agonists rescue the symptoms of narcolepsy, cell transplantations and gene therapy may be developed in the future, and animal experiments aiming for these options are in progress. One of the concerns for these is the receptor function in ligand deficient narcolepsy. A significant degree of ligand deficiency is already evident at the disease onset. Life-long treatment of narcolepsy is required, and thus preserved receptor functions, many years after the loss of ligand, are essential for the replacement therapy. In order to evaluate changes in hypocretin receptors in hypocretin deficient narcolepsy, Mishima et al. (Mishima et al., 2008) recently studied hypocretin receptor gene expressions of ligand deficient narcolepsy in mice, dogs, and humans. Substantial decline (by 50–71%) in the expression of hypocretin receptor genes were observed in both ligand deficient humans and dogs (Fig. 5). The result in the mice study (8-week-old and 27-week-old orexin/ataxin-3 transgenic hypocretin cell ablated mice and wild-type mice) suggested that the decline is progressive over age (Mishima et al., 2008). However, about 50% of the original expression was still observed in old human subjects (average age 77.0 year old). It is unknown if this is beneficial for the patients. However, since narcoleptic Dobermans heterozygous for hypocretin receptor 2 mutation (they are supposed to express 50% of hypocretin receptor 2 genes and normal levels of hypocretin (Ripley et al., 2001a)) are asymptomatic, it is likely that an adequate ligand supplement prevents narcoleptic symptoms of hypocretin deficient patients.
Fig. 5. hcrtR1 and hcrtR2 expression in Hypocretin Ligand deficient Human and Canine (sporadic) Narcoleptic Subjects.

Data are expressed as relative values to average expression level (= 1) in the corresponding control (CNT) group. Blue and red closed circles represent the CNT and narcoleptics (NAC), respectively. The number of the control subjects for the pontine analysis is four, due to the lack of the availability of the tissue. Columns with solid horizontal bars cover mean ± SEM. Data are from (Mishima et al., 2008).
Summary
The major pathophysiology of human narcolepsy has been recently discovered based on the discovery of narcolepsy genes in animals; these genes are involved in the pathology of the hypocretin/orexin ligand and its receptor. Mutations in hypocretin-related genes are rare in humans, but hypocretin-ligand deficiency, possibly due to postnatal cell death of hypocretin neurons, is found in many cases of narcolepsy with cataplexy.
Hypocretin ligand deficiency can be clinically diagnosed with CSF hypocretin-1 measures, and this is now included in the positive diagnosis of narcolepsy with cataplexy in the ICSD II.
Since HLA DQB1*0602 positivity is associated with narcolepsy with cataplexy and thus with hypocretin deficiency, an involvement of autoimmune mechanisms in cell death of hypocretin neurons has been suggested. There is, however, no strong evidence of inflammatory processes or immune abnormalities associated with narcolepsy, and studies have found no classical auto-antibodies and no increase in oligoclonal CSF bands in narcoleptics However, these results do not exclude the possibility that a transient autoimmune reaction restricted to the CNS could have occurred around the disease onset, but disappeared later.
Hypocretin deficiency is also found in symptomatic cases of narcolepsy and EDS in various neurological conditions, including immune-mediated neurological disorders, such as with Guillain-Barre syndrome, MA2-positive paraneoplastic syndrome and NMO related disorder. Symptomatic narcolepsy in MS patients has been reported from several decades ago. Although only small number of MS patients develop narcolepsy (and a large majority of MS subjects exhibit neither EDS/cataplexy nor hypocretin deficiency), a subset of MS patients predominantly shows EDS and REM sleep abnormalities. It is likely that specific immune-mediated mechanisms may be involved in these cases. Recent case reports of MS and its related disorders associated with bilateral symmetrical hypothalamic lesions, hypocretin deficiency, and EDS often share the clinical symptoms of NMO. Some of these patients are seropositive for anti-AQP4 antibody, suggesting a functional link between anti-AQP4 antibody; bilateral symmetrical hypothalamic lesions and anti-AQP4 antibody might be caused by the immuno-attack to the AQP4, and this may secondarily affect the hypocretin neurons.
Since a large majority of human narcolepsy patients are ligand deficient, hypocretin replacement therapy may be a new therapeutic option, and animal experiments using gene therapy and cell transplantations are in progress.
Substantial decline (by 50–71%) in the expression of hypocretin receptor genes were observed in both deficient humans. It is unknown if this is beneficial for the patients. However, since narcoleptic Dobermans heterozygous for hypocretin receptor 2 mutation (supposed to express 50% of hypocretin receptor 2 genes and normal levels of hypocretin) are asymptomatic, it is likely that an adequate ligand supplement prevents narcoleptic symptoms of hypocretin deficient patients.
Acknowledgments
This study was supported by a NIH Grant R01MH072525.
Footnotes
Conflict of Interest
There are no conflicts of interests for any of the authors for the contents described in the paper.
References
- Adie J. Idiopathic narcolepsy: a disease sui generis, with remarks on the mechanism of sleep. Brain. 1926;49:257–306. [Google Scholar]
- Aggarwal BB. Comparative analysis of the structure and function of TNF-alpha and TNF-beta. Immunol Ser. 1992;56:61–78. [PubMed] [Google Scholar]
- Alaila SL. Life effects of narcolepsy: measures of negative impact, social support and psychological well-being. In: GOSWANMI M, POLLAK CP, COHEN FL, THORPY MJ, KAVEY NB, editors. Loss, Grief and Care: Psychosocial Aspects of Narcolepsy. New York: Haworth Press; 1992. [Google Scholar]
- Aldrich M, Naylor M. Narcolepsy associated with lesions of the diencephalon. Neurology. 1989;39:1505–8. doi: 10.1212/wnl.39.11.1505. [DOI] [PubMed] [Google Scholar]
- Aldrich MS, Chervin RD, Malow BA. Value of the multiple sleep latency test (MSLT) for the diagnosis of narcolepsy. Sleep. 1997;20:620–629. [PubMed] [Google Scholar]
- Amiry-Moghaddam M, Ottersen OP. The molecular basis of water transport in the brain. Nat Rev Neurosci. 2003;4:991–1001. doi: 10.1038/nrn1252. [DOI] [PubMed] [Google Scholar]
- Baba T, Nakashima I, Kanbayashi T, Konno M, Takahashi T, Fujihara K, MIsu T, Takeda A, Shiba Y, Ogawa H, Itoyama Y. Narcolepsy as an initial manifestation of neuromyelitis optica with anti-aquaporin 4 antibody Journal of Neurology. 2008 doi: 10.1007/s00415-009-0139-4. In press. [DOI] [PubMed] [Google Scholar]
- Barker W. Studies in epilepsy: personality patterns, situational stress and the symptoms of narcolepsy. Psychosom Med. 1948;10:193–202. doi: 10.1097/00006842-194807000-00001. [DOI] [PubMed] [Google Scholar]
- Beutler B, Grau GE. Tumor necrosis factor in the pathogenesis of infectious diseases. Crit Care Med. 1993;21:S423–35. [PubMed] [Google Scholar]
- Billiard M, Laaberki MF, Reygrobellet C, Seignalet J, Brissaud L, Besset A. Elevated antibodies to streptococcal antigens in narcoleptic subjects. Sleep Res. 1989;18:201. [Google Scholar]
- Black JL, 3rd, Krahn LE, Pankratz VS, Silber M. Search for neuron-specific and nonneuron-specific antibodies in narcoleptic patients with and without HLA DQB1*0602. Sleep. 2002;25:719–23. doi: 10.1093/sleep/25.7.719. [DOI] [PubMed] [Google Scholar]
- Black JL, 3rd, Silber MH, Krahn LE, Avula RK, Walker DL, Pankratz VS, Fredrickson PA, Slocumb NL. Studies of Humoral Immunity to Preprohypocretin in Human Leukocyte Antigen DQB1*0602-Positive Narcoleptic Subjects with Cataplexy. Biol Psychiatry. 2005 doi: 10.1016/j.biopsych.2005.04.026. [DOI] [PubMed] [Google Scholar]
- Blouin AM, Thannickal TC, Worley PF, Baraban JM, Reti IM, Siegel JM. Narp immunostaining of human hypocretin (orexin) neurons: loss in narcolepsy. Neurology. 2005;65:1189–92. doi: 10.1212/01.wnl.0000175219.01544.c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, Fitch TE, Nakazato M, Hammer RE, Saper CB, Yanagisawa M. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- Crocker A, Espana RA, Papadopoulou M, Saper CB, Faraco J, Sakurai T, Honda M, Mignot E, Scammell TE. Concomitant loss of dynorphin, NARP, and orexin in narcolepsy. Neurology. 2005;65:1184–8. doi: 10.1212/01.wnl.0000168173.71940.ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauvilliers Y, Arnulf I, Mignot E. Narcolepsy with cataplexy. Lancet. 2007;369:499–511. doi: 10.1016/S0140-6736(07)60237-2. [DOI] [PubMed] [Google Scholar]
- Dauvilliers Y, Neidhart E, Lecendreux M, Billiard M, Tafti M. MAO-A and COMT polymorphisms and gene effects in narcolepsy. Mol Psychiatry. 2001;6:367–72. doi: 10.1038/sj.mp.4000911. [DOI] [PubMed] [Google Scholar]
- De Lecea L, Kilduff TS, Peyron C, Gao XB, Foye PE, Danielson PE, Fukuhara C, Battenberg ELF, Gautvik VT, Barlett FS, Frankel WN, Van Den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci USA. 1998;95:322–327. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickstein JB, Moldofsky H, Lue FA, Hay JB. Intracerebroventricular injection of TNF-alpha promotes sleep and is recovered in cervical lymph. Am J Physiol. 1999;276:R1018–22. doi: 10.1152/ajpregu.1999.276.4.R1018. [DOI] [PubMed] [Google Scholar]
- Espana RA, Scammell TE. Sleep neurobiology for the clinician. Sleep. 2004;27:811–20. [PubMed] [Google Scholar]
- Frederickson S, Carlander B, Billiard M, Link H. CSF immune variable in patients with narcolepsy. Acta Neurol Scand. 1990;81:253–254. doi: 10.1111/j.1600-0404.1990.tb00978.x. [DOI] [PubMed] [Google Scholar]
- Fujiki N, Ripley B, Yoshida Y, Mignot E, Nishino S. Effects of IV and ICV hypocretin-1 (orexin A) in hypocretin receptor-2 gene mutated narcoleptic dogs and IV hypocretin-1 replacement therapy in a hypocretin ligand deficient narcoleptic dog. Sleep. 2003;6:953–959. doi: 10.1093/sleep/26.8.953. [DOI] [PubMed] [Google Scholar]
- Gill A. Idiopathic and traumatic narcolepsy. Lancet. 1941:474–9. [Google Scholar]
- Hallmayer J, Faraco J, Lin L, Hesselson S, Winkelmann J, Kawashima M, Mayer G, Plazzi G, Nevsimalova S, Bourgin P, Hong SS, Honda Y, Honda M, Hogl B, Longstreth WT, Jr, Montplaisir J, et al. Narcolepsy is strongly associated with the T-cell receptor alpha locus. Nat Genet. 2009 doi: 10.1038/ng.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohjoh H, Nakayama T, Ohashi J, Miyagawa T, Tanaka H, Akaza T, Honda Y, Juji T, Tokunaga K. Significant association of a single nucleotide polymorphism in the tumor necrosis factor-alpha (TNF-alpha) gene promoter with human narcolepsy. Tissue Antigens. 1999;54:138–45. doi: 10.1034/j.1399-0039.1999.540204.x. [DOI] [PubMed] [Google Scholar]
- Hohjoh H, Terada N, Kawashima M, Honda Y, Tokunaga K. Significant association of the tumor necrosis factor receptor 2 (TNFR2) gene with human narcolepsy. Tissue Antigens. 2000;56:446–8. doi: 10.1034/j.1399-0039.2000.560508.x. [DOI] [PubMed] [Google Scholar]
- Honda M, Eriksson KS, Zhang S, Tanaka S, Lin L, Salehi A, Hesla PE, Maehlen J, Gaus SE, Yanagisawa M, Sakurai T, Taheri S, Tsuchiya K, Honda Y, Mignot E. IGFBP3 colocalizes with and regulates hypocretin (orexin) PLoS ONE. 2009;4:e4254. doi: 10.1371/journal.pone.0004254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda Y. Clinical features of Narcolepsy: Japanese Experience. In: HONDA Y, JUJI T, editors. HLA in Narcolepsy. New York: Springer-Verlag; 1988. [Google Scholar]
- ICSD-2. Diagnostic and coding manual, Westchester. 2. Illinois: American Academy of Sleep Medicine; 2005. ICSD-2-International classification of sleep disorders. [Google Scholar]
- Juji T, Satake M, Honda Y, Doi Y. HLA antigens in Japanese patients with narcolepsy. All the patients were DR2 positive. Tissue Antigens. 1984;24:316–319. doi: 10.1111/j.1399-0039.1984.tb02144.x. [DOI] [PubMed] [Google Scholar]
- Kanbayashi T, Inoue Y, Chiba S, Aizawa R, Saito Y, Tsukamoto H, Fujii Y, Nishino S, Shimizu T. CSF hypocretin-1 (orexin-A) concentrations in narcolepsy with and without cataplexy and idiopathic hypersomnia. J Sleep Res. 2002;11:91–3. doi: 10.1046/j.1365-2869.2002.00284.x. [DOI] [PubMed] [Google Scholar]
- Kanbayashi T, Shimohata T, Nakashima I, Yaguchi H, Yabe I, Shimizu T, Nishino S. Symptomatic narcolepsy in MS and NMO patients; new neurochemical and immunological implications. Arch Neurol. 2009 doi: 10.1001/archneurol.2009.264. in press. [DOI] [PubMed] [Google Scholar]
- Kato T, Kanbayashi T, Yamamoto K, Nakano T, Shimizu T, Hashimoto T, Ikeda S. Hypersomnia and low CSF hypocretin-1 (orexin-A) concentration in a patient with multiple sclerosis showing bilateral hypothalamic lesions. Intern Med. 2003;42:743–5. doi: 10.2169/internalmedicine.42.743. [DOI] [PubMed] [Google Scholar]
- Kawashima M, Tamiya G, Oka A, Hohjoh H, Juji T, Ebisawa T, Honda Y, Inoko H, Tokunaga K. Genomewide association analysis of human narcolepsy and a new resistance gene. Am J Hum Genet. 2006;79:252–63. doi: 10.1086/505539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–7. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lhermitte J, Roques A. A. Narcolepsie et Cataplexie post-encephalitiques. Rev Neurol. 1928;II:264–267. [Google Scholar]
- Lieberman AP, Pitha PM, Shin HS, Shin ML. Production of tumor necrosis factor and other cytokines by astrocytes stimulated with lipopolysaccharide or a neurotropic virus. Proc Natl Acad Sci U S A. 1989;86:6348–52. doi: 10.1073/pnas.86.16.6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X, Qiu X, de Jong PJ, Nishino S, Mignot E. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98:365–76. doi: 10.1016/s0092-8674(00)81965-0. [DOI] [PubMed] [Google Scholar]
- Mamelak M. A perspective on narcolepsy. Encephale. 1992;18:347–51. [PubMed] [Google Scholar]
- Marcus CL, Trescher WH, Halbower AC, Lutz J. Secondary narcolepsy in children with brain tumors. Sleep. 2002;25:435–9. [PubMed] [Google Scholar]
- Matsuki K, Juji T, Honda Y. Immunological features of Narcolepsy in Japan. In: HONDA Y, JUJI T, editors. HLA in Narcolepsy. New York: Springer-Verlag; 1988. [Google Scholar]
- Mieda M, Willie JT, Hara J, Sinton CM, Sakurai T, Yanagisawa M. Orexin peptides prevent cataplexy and improve wakefulness in an orexin neuron-ablated model of narcolepsy in mice. Proc Natl Acad Sci U S A. 2004;101:4649–4654. doi: 10.1073/pnas.0400590101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignot E. Genetic and familial aspects of narcolepsy. Neurology. 1998;50:S16–S22. doi: 10.1212/wnl.50.2_suppl_1.s16. [DOI] [PubMed] [Google Scholar]
- Mignot E, Guilleminault C, Grumet FC, Dement WC. Is narcolepsy an autoimmune disease?. In: SMIRNE S, FRANCESCI M, FERINI-STRAMBI L, ZUCCONI M, editors. Sleep, Hormones, and the Immune System; Proceedings of the Third Milano International Symposium; September 18–19; Milan: Masson; 1992. [Google Scholar]
- Mignot E, Hayduk R, Grumet FC, Black J, Guilleminault C. HLA DQB1*0602 is associated with cataplexy in 509 narcoleptic patients. Sleep. 1997;20:1012–1020. [PubMed] [Google Scholar]
- Mignot E, Lammers GJ, Ripley B, Okun M, Nevsimalova S, Overeem S, Vankova J, Black J, Harsh J, Bassetti C, Schrader H, Nishino S. The role of cerebrospinal fluid hypocretin measurement in the diagnosis of narcolepsy and other hypersomnias. Arch Neurol. 2002;59:1553–62. doi: 10.1001/archneur.59.10.1553. [DOI] [PubMed] [Google Scholar]
- Mishima K, Fujiki N, Yoshida Y, Sakurai T, Honda M, Mignot E, Nishino S. Hypocretin receptor expression in canine and murine narcolepsy models and in hypocretin-ligand deficient human narcolepsy. Sleep. 2008;31:1119–26. [PMC free article] [PubMed] [Google Scholar]
- Miyagawa T, Kawashima M, Nishida N, Ohashi J, Kimura R, Fujimoto A, Shimada M, Morishita S, Shigeta T, Lin L, Hong SC, Faraco J, Shin YK, Jeong JH, Okazaki Y, Tsuji S, et al. Variant between CPT1B and CHKB associated with susceptibility to narcolepsy. Nat Genet. 2008;40:1324–8. doi: 10.1038/ng.231. [DOI] [PubMed] [Google Scholar]
- Montplaisir J, Poirier G, Lapierre O, Montplaisir S. Streptococcal antibodies in Narcolepsy and idiopathic hypersomnia. Sleep Res. 1989;18:271. [Google Scholar]
- Nakamura M, Nishii M, Maki S, Nakamuara M, Suenaga T. A MS cses with EDS and bilateral hypothlamic lesions. Clinical Neurology. 2005;45:187. in Japanese. [Google Scholar]
- Nishino S. The hypocretin/orexin system in health and disease. Biol Psychiatry. 2003;54:87–95. doi: 10.1016/s0006-3223(03)00349-4. [DOI] [PubMed] [Google Scholar]
- Nishino S. Clinical and neurobiological aspects of narcolepsy. Sleep Med. 2007;8:373–99. doi: 10.1016/j.sleep.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino S, Kanbayashi T. Symptomatic narcolepsy, cataplexy and hypersomnia, and their implications In the hypothalamic hypocretin/orexin system. Sleep Med Rev. 2005;9:269–310. doi: 10.1016/j.smrv.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Nishino S, Kanbayashi T, Fujiki N, Uchino M, Ripley B, Watanabe M, Lammers GJ, Ishiguro H, Shoji S, Nishida Y, Overeem S, Toyoshima I, Yoshida Y, Shimizu T, Taheri S, Mignot E. CSF hypocretin levels in Guillain-Barre syndrome and other inflammatory neuropathies. Neurology. 2003;61:823–5. doi: 10.1212/01.wnl.0000081049.14098.50. [DOI] [PubMed] [Google Scholar]
- Nishino S, Mignot E. Pharmacological aspects of human and canine narcolepsy. Prog Neurobiol. 1997;52:27–78. doi: 10.1016/s0301-0082(96)00070-6. [DOI] [PubMed] [Google Scholar]
- Nishino S, Riehl J, Hong J, Kwan M, Reid M, Mignot E. Is narcolepsy REM sleep disorder? Analysis of sleep abnormalities in narcoleptic Dobermans. Neuroscience Research. 2000a;38:437–446. doi: 10.1016/s0168-0102(00)00195-4. [DOI] [PubMed] [Google Scholar]
- Nishino S, Ripley B, Overeem S, Lammers G, Mignot E. Hypocretin (orexin) deficiency in human narcolepsy. Lancet. 2000b;355:39–40. doi: 10.1016/S0140-6736(99)05582-8. [DOI] [PubMed] [Google Scholar]
- Nishino S, Ripley B, Overeem S, Lammers GJ, Mignot E. Hypocretin (orexin) deficiency in human narcolepsy. Lancet. 2000c;355:39–40. doi: 10.1016/S0140-6736(99)05582-8. [DOI] [PubMed] [Google Scholar]
- Nishino S, Ripley B, Overeem S, Nevsimalova S, Lammers GJ, Vankova J, Okun M, Rogers W, Brooks S, Mignot E. Low CSF hypocretin (orexin) and altered energy homeostasis in human narcolepsy. Ann Neurol. 2001;50:381–388. doi: 10.1002/ana.1130. [DOI] [PubMed] [Google Scholar]
- Nishino S, Taheri S, Black J, Nofzinger E, Mignot E. The neurobiology of sleep in relation to mental illness. In: CHARNEY DSNE, editor. Neurobiology of Mental Illness. New York: Oxford University Press; 2004. [Google Scholar]
- Nozaki H, Shimohata T, Kanbayashi T, Sagawa Y, Katada SI, Satoh M, Onodera O, Tanaka K, Nishizawa M. A patient with anti-aquaporin 4 antibody who presented with recurrent hypersomnia, reduced orexin (hypocretin) level, and symmetrical hypothalamic lesions. Sleep Med. 2008 doi: 10.1016/j.sleep.2007.11.022. [DOI] [PubMed] [Google Scholar]
- Oka Y, Kanbayashi T, Mezaki T, Iseki K, Matsubayashi J, Murakami G, Matsui M, Shimizu T, Shibasaki H. Low CSF hypocretin-1/orexin-A associated with hypersomnia secondary to hypothalamic lesion in a case of multiple sclerosis. J Neurol. 2004;251:885–6. doi: 10.1007/s00415-004-0442-z. [DOI] [PubMed] [Google Scholar]
- Overeem S, Dalmau J, Bataller L, Nishino S, Mignot E, Vershuuren J, Lammers GJ. Secondary narcolepsy in patients with praneoplastic anti-Ma2 antibodies is associated with hypocretin deficiency. J Sleep Res. 2001;11 (suppl 1):166–167. [Google Scholar]
- Park MH, Roh EY, Park H, Kim SJ, Jeong DU. Association of TNF-alpha gene promoter polymorphism with narcolepsy-cataplexy in Koreans. Human Immunology. 2004;65:S110. [Google Scholar]
- Peyron C, Faraco J, Rogers W, Ripley B, Overeem S, Charnay Y, Nevsimalova S, Aldrich M, Reynolds D, Albin R, Li R, Hungs M, Pedrazzoli M, Padigaru M, Kucherlapati M, Fan J, et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med. 2000a;6:991–7. doi: 10.1038/79690. [DOI] [PubMed] [Google Scholar]
- Peyron C, Faraco J, Rogers W, Ripley B, Overeem S, Charnay Y, Nevsimalova S, Aldrich M, Reynolds D, Albin R, Li R, Hungs M, Pedrazzoli M, Padigaru M, Kucherlapati M, Fan J, et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med. 2000b;6:991–7. doi: 10.1038/79690. [DOI] [PubMed] [Google Scholar]
- Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittock SJ, Weinshenker BG, Lucchinetti CF, Wingerchuk DM, Corboy JR, Lennon VA. Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol. 2006;63:964–8. doi: 10.1001/archneur.63.7.964. [DOI] [PubMed] [Google Scholar]
- Poirier G, Montplaisir J, Dumont M, Duquette P, Decary F, Pleines J, Lamoureux G. Clinical and sleep laboratory study of narcoleptic symptoms in multiple sclerosis. Neurology. 1987;37:693–5. doi: 10.1212/wnl.37.4.693. [DOI] [PubMed] [Google Scholar]
- Rao KV, Qureshi IA. Decompensation of hepatic and cerebral acyl-CoA metabolism in BALB/cByJ mice by chronic riboflavin deficiency: restoration by acetyl-L-carnitine. Can J Physiol Pharmacol. 1997;75:423–30. [PubMed] [Google Scholar]
- Ripley B, Fujiki N, Okura M, Mignot E, Nishino S. Hypocretin levels in sporadic and familial cases of canine narcolepsy. Neurobiol of Dis. 2001a;8:525–534. doi: 10.1006/nbdi.2001.0389. [DOI] [PubMed] [Google Scholar]
- Ripley B, Overeem S, Fujiki N, Nevsimalova S, Uchino M, Yesavage J, Di Monte D, Dohi K, Melberg A, Lammers GJ, Nishida Y, Roelandse FW, Hungs M, Mignot E, Nishino S. CSF hypocretin/orexin levels in narcolepsy and other neurological conditions. Neurology. 2001b;57:2253–8. doi: 10.1212/wnl.57.12.2253. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, Arch JRS, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–585. doi: 10.1016/s0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–63. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- Scammell T, Nishino S, Mignot E, Saper C. Narcolepsy and low CSF orexin (hypocretin) concentration after a diencephalic stroke. Neurology. 2001;56:1751–3. doi: 10.1212/wnl.56.12.1751. [DOI] [PubMed] [Google Scholar]
- Scammell TE. The frustrating and mostly fruitless search for an autoimmune cause of narcolepsy. Sleep. 2006;29:601–2. doi: 10.1093/sleep/29.5.601. [DOI] [PubMed] [Google Scholar]
- Schatzberg SJ, Barrett J, Cutter Kl, Ling L, Mignot E. Case Study: Effect of hypocretin replacement therapy in a 3-year-old Weimaraner with narcolepsy. J Vet Internal Med. 2004;18:586–588. doi: 10.1892/0891-6640(2004)18<586:teohrt>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Siegel J. Brainstem Mechanisms Generating REM Sleep. In: KRYGER MR, TDEMENT WC, editors. Principles and Practice of Sleep Medicine. 3. Philadelphia: W.B. Saunders Company; 2000. [Google Scholar]
- Smith C, Hamilton J. Psychological factors in the narcolepsy-cataplexy syndrome. Psychosom Med. 1959;21:40–9. doi: 10.1097/00006842-195901000-00006. [DOI] [PubMed] [Google Scholar]
- Stiefler G. Narkolepsie nach Enzephalitis Lethargica. Wien Klin. 1924;37:1044–6. [Google Scholar]
- Tachibana N, Howard RS, Hirsch NP, Miller DH, Moseley IF, Fish D. Sleep problems in multiple sclerosis. Eur Neurol. 1994;34:320–3. doi: 10.1159/000117070. [DOI] [PubMed] [Google Scholar]
- Tafti M, Petit B, Chollet D, Neidhart E, de Bilbao F, Kiss JZ, Wood PA, Franken P. Deficiency in short-chain fatty acid beta-oxidation affects theta oscillations during sleep. Nat Genet. 2003;34:320–5. doi: 10.1038/ng1174. [DOI] [PubMed] [Google Scholar]
- Takeuchi T, Miyasita A, Sasaki Y, Inugami M, Fukuda K. Isolated sleep paralysis elicited by sleep interruption. Sleep. 1992;15:217–225. doi: 10.1093/sleep/15.3.217. [DOI] [PubMed] [Google Scholar]
- Thannickal T, Moore R, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, Cornford M, Siegel J. Reduced number of hypocretin neurons in human narcolepsy. Neuron. 2000a;27:469–74. doi: 10.1016/s0896-6273(00)00058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, Cornford M, Siegel JM. Reduced number of hypocretin neurons in human narcolepsy. Neuron. 2000b;27:469–74. doi: 10.1016/s0896-6273(00)00058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma A, Anand V, Verma NP. Sleep disorders in chronic traumatic brain injury. J Clin Sleep Med. 2007;3:357–62. [PMC free article] [PubMed] [Google Scholar]
- von Economo C. Sleep as a problem of localization. J Nerv Ment Dis. 1930;71:249–59. [Google Scholar]
- von Economo C. Encephalitis lethargica: its sequelae and treatment 1931 [Google Scholar]
- Wieczorek S, Gencik M, Rujescu D, Tonn P, Giegling I, Epplen JT, Dahmen N. TNFA promoter polymorphisms and narcolepsy. Tissue Antigens. 2003;61:437–42. doi: 10.1034/j.1399-0039.2003.00068.x. [DOI] [PubMed] [Google Scholar]