

Abstract
This study examines the role of the p12 subunit in the function of the human DNA polymerase δ (Pol δ) holoenzyme by comparing the kinetics of DNA synthesis and degradation catalyzed by the four subunit complex, the three subunit complex lacking p12, and site directed mutants of each lacking proofreading exonuclease activity. Results show that p12 modulates the rate and fidelity of DNA synthesis by Pol δ. All four complexes synthesize DNA in a rapid burst phase and a slower, more linear phase. In the presence of p12, the burst rates of DNA synthesis are about five times faster, while the affinity of the enzyme for its DNA and dNTP substrates appears unchanged. The p12 subunit alters Pol δ fidelity by modulating the proofreading 3′ to 5′ exonuclease activity. In the absence of p12, Pol δ is more likely to proofread DNA synthesis because it cleaves single-stranded DNA twice as fast and transfers mismatched DNA from the polymerase to the exonuclease sites 9 times faster. Pol δ also extends mismatched primers 3 times more slowly in the absence of p12. Taken together, the changes that p12 exerts on Pol δ are ones that can modulate its fidelity of DNA synthesis. The loss of p12, which occurs in cells upon exposure to DNA damaging agents, converts Pol δ to a form that has an increased capacity for proofreading.
Keywords: DNA replication, genome stability, mutations, exonuclease, proofreading
Each diploid cell in the human body contains over 6 billion base pairs of DNA, which must be accurately copied before a cell can divide. The macromolecular machine that replicates human DNA must therefore function astonishingly fast, yet at the same time, this DNA ‘replisome’ must be amazingly accurate (1). Thus, replicative DNA polymerases copy DNA with very low error rates. Extensive structural and kinetic studies have provided insights into how this fidelity is achieved. All DNA polymerases possess a common polymerase fold that resembles a right hand, which is formed by fingers, palm and thumb domains. The finger and thumb regions undergo conformational changes from an open to closed formation upon binding of a dNTP,1 providing for an extremely high level of selection for a correctly based paired incoming nucleotide as well as an appropriately based paired primer terminus (2-6). In addition, proofreading polymerases possess a 3′ to 5′ exonuclease domain (exo) that is distant from the polymerase site (pol), which further adds to the fidelity of DNA synthesis. The maintenance of genomic integrity also depends on a complex network of cellular responses to DNA damage that orchestrate cell cycle check points (7) and the repair of DNA damage (8).
Three DNA polymerases participate in eukaryotic chromosomal DNA replication. DNA polymerase δ (Pol δ) and ε are proofreading enzymes that possess pol and exo active sites, while polymerase a/primase synthesizes the primers on the lagging strand that are extended by the proofreading polymerases. Studies in budding yeast have provided evidence that Pol ε synthesizes most of the DNA on the leading strand template, while Pol δ synthesizes most of the DNA on the lagging strand template (9, 10). However, the extent to which this division of labor takes place in the replication of the more complex genome of mammalian cells is still uncertain (9, 10). Pol δ also plays a role in gap filling during DNA repair processes (11).
Human Pol δ has four subunits, p125, p50, p68, and p12 (12, 13). The largest subunit, p125, is the catalytic subunit and its catalytic domain is conserved with other members of the B-family of DNA polymerases (14-16). The p125 subunit interacts with p50 to form the core enzyme, while p68 interacts with p50, and p12 forms a bridge between p125 and p50 (17, 18). The subunit composition of eukaryotic Pol δ is conserved between human and S. pombe, but the most extensively studied form of Pol δ that of S. cerevisiae, is a three-subunit enzyme, which lacks a homolog of p12 (11). The human Pol δ p125/p50/p68 trimer (Pol δ3) is active, in contrast to the less active p125/p50 dimer (18). The crystal structure of S. cerevisiae Pol δ (19) reveals extensive similarity between Pol δ and the monomeric bacteriophage RB69 polymerase, RB69 gp43, for which a number of structures have been obtained in different conformational states (2, 16, 20-24). RB69 gp43 shares homology with T4 polymerase, which has been more extensively studied (25).
Pol δ activity is involved in a number of DNA transactions that include not only DNA replication but also gap filling during DNA repair processes. Pol δ4 is converted into Pol δ3 by the depletion of the p12 subunit when cells are subjected to genotoxic stress by DNA damaging agents such as ultraviolet light and methyl methanesulfonate, or by replication stress induced by treatment with hydroxyurea or aphidicolin (26). These findings raise a number of questions of how the conversion of Pol δ4 to Pol δ3 might contribute to the DNA damage response. One way to gain insights into this question is to compare the properties of Pol δ3 with that of its progenitor. Comparison of the activities of Pol δ3 and Pol δ4 on damaged DNA templates reveal Pol δ3 is less able to bypass templates containing base lesions (O6-MeG, 8-oxoG), produces more exonucleolytic products, and has a decreased tendency for inserting wrong nucleotides and extending mismatched primers (27). Thus, Pol δ3 appears to display a classic ‘antimutator’ phenotype (28), whereby an increase in exonucleolytic ability relative to the polymerase function enhances proofreading and fidelity.
In this study, we examined the kinetics of DNA synthesis and degradation catalyzed by Pol δ3 and Pol δ4 and their exonuclease-deficient mutants to provide insights into the nature of their functional differences. The rates of DNA synthesis were examined by pre-steady state kinetic analysis and reveal that the loss of p12 decreases kpol. Analysis of the rates of exonuclease activity reveals that the rate of translocation of DNA from pol to exo catalytic sites is increased when p12 is absent. Both changes are consistent with the exhibition of an antimutator phenotype for Pol δ3, in which Pol δ4 is less likely to proofread and more likely to extend mismatched primers. Thus, loss of p12 modulates both the rate and fidelity of DNA synthesis by facilitating a more rapid and frequent transfer of the DNA primer from the polymerase to the exonuclease active center. These findings provide novel insights into the role of p12 in human Pol δ function at the mechanistic level and into the potential cellular consequences of the in vivo conversion of Pol δ4 to Pol δ3.
Experimental procedures
Materials
Calf thymus DNA was obtained from Sigma-Aldrich (St. Louis, MO), dNTPs were obtained from GE Healthcare (Piscataway, NJ), and dGTP-α-thiotriphosphate (dGTP-αS) was obtained from Glen Research (Sterling, VA).
Recombinant human PCNA, recombinant unmodified Pol δ4, Pol δ3, and the D402A site-directed mutants of each (Pol δ4exo- and Pol δ3exo-) were expressed in insect cells and purified as previously described (27, 29). The Pol δ4 and Pol δ3 enzymes exhibited four and three major protein bands on SDS-PAGE. Specific activities of the enzymes were similar to those previously reported (27). Pol δ enzyme concentrations for these studies were expressed as concentrations of the p125 subunit. This was determined by SDS-PAGE in which the purified Pol δ3 or Pol δ4 complexes were separated together with known amounts of catalase and aldolase (GE Healthcare) to generate standard curves after densitometry of the gels. Digital images of the stained gels were analyzed with AlphaEaseFC software (Alpha Innotech, San Leandro, CA). Recombinant human PCNA was prepared as previously described (29). PCNA (400 nM) was included in all reactions. Such a concentration is at least 10 times more than is necessary to stimulate Pol δ to its maximal activity in any of the assays used in this study (Zhou, Meng, and Lee, unpublished data).
Polymerase Substrates
All DNA oligonucleotides were synthesized and PAGE-purified by Integrated DNA Technologies, Inc. (Coralville, IA). The sequences of the primers (25mer, 26merC, 26merT) and the template (40mer) are as follows: 5′ - GCC ACT ACA GCA CCT TGA CAG CCA G - 3′ (25mer), 5′ - GCC ACT ACA GCA CCT TGA CAG CCA G T- 3′ (26merT), 5′ - GCC ACT ACA GCA CCT TGA CAG CCA G C- 3′ (26merC), 5′- TCA TCG GTC GCA TCG CTG GCT GTC AAG GTG CTG TAG TGGC - 3′ (40mer). The primers were [5′-32P]labeled using [γ-32P]ATP (5000 Ci/mmol, MP Biochemicals) and T4 polynucleotide kinase (New England Biolabs, MA). Labeled primers were purified using QIAquick Spin Columns (Qiagen), and concentrations were determined from absorbance at 260 nm using extinction coefficients provided by Integrated DNA Technologies. Each primer was separately mixed with the template (40mer) in a ratio of 1:1.2 in 10 mM Tris-HCl pH 7.5, incubated at 90 °C for 3 minutes, and gradually cooled to room temperature. All substrates were analyzed using non-denaturing PAGE to ensure that all primers were annealed.
Pre-steady state DNA Polymerase assays
Reactions were carried out at 24 °C using a KinTek RQF-3 Rapid Quench Flow apparatus (KinTek Corp., Austin, TX). Pol δ preparations were first diluted in enzyme dilution buffer [8 μM PCNA, 50 mM Bis(2-hydroxyethyl)-imino-tris(hydroxymethyl)-methane (Bis-Tris) pH 6.5, 5 mM dithiothreitol (DTT), 500 μg/ml bovine serum albumin (BSA) and 10% glycerol (v/v)] so that the concentration of p125 and PCNA were 10 times the concentration to be loaded into syringe A. Syringe A contained 2× final concentrations both of Pol δ, and DNA substrate, 800 nM PCNA (based on a trimeric molecular weight), 100 mM Bis-Tris pH 6.5, 10 mM DTT, 400 μg/ml BSA, and 60 mM NaCl. After loading syringe A, solutions were incubated at 24 °C for approximately 5 minutes. During this time, syringe B was loaded with 20 mM MgCl2 and 2× final concentrations of dNTPs. Syringe C was loaded with 500 mM EDTA pH 8.
Reactions were initiated by rapidly mixing 20 μl from syringe A (Pol δ*PCNA*DNA complexes), with 20 μl from syringe B [Mg(dNTP)2- complexes] such that final reaction mixtures contained 50 mM Bis-Tris pH 6.5, 5 mM DTT, 200 μg/ml BSA, 30 mM NaCl, 10 mM MgCl2, 400 nM PCNA, dNTP, DNA, and Pol δ. After indicated times, reactions were rapidly quenched by adding 80 μl from syringe C (EDTA) such that the final concentration of EDTA was 330 mM. To analyze remaining substrate and products, 20 μl of loading dye (95% formamide, 0.01% bromophenol blue and 0.01% xylene cyanol) was added to 10 μl of each quenched reaction. After heating these samples to 90 °C for 3 min, 4 μl of each was loaded on a 16% polyacrylamide (acrylamide/bisacrylamide 19:1) 8 M urea denaturing gel. After electrophoresis, [5′-32P]DNA was visualized with a Molecular Dynamics Storm Phosphorimaging system and quantified with ImageQuant software (Amersham Biosciences, NJ).
Transient state time courses (products vs. time) of polymerase reactions were fit to equation 1 to determine burst amplitude [ED], which is a measurement of catalytically competent enzyme substrate present at the start of the reaction; a first order rate constant describing the burst phase, kobs, and a velocity of the linear phase of the reaction, v. GraphPad Prism 4.0 software (La Jolla, CA) was used for non-linear regression.
| (1) |
To determine the Kd for the incoming dNTP and kpol, the kobs values of reactions performed at various dNTP concentrations were fit to equation 2.
| (2) |
Steady-state kcat was calculated by equation 3.
| (3) |
In Eq. 3, [dNTP] is the final dNTP concentration in reaction, Et is Pol δ amount determined by active site titration, v is velocity from equation 1, and Km is the Michaelis-Menten constant.
Single turnover polymerase assays
Single turnover reactions for determining burst amplitudes were performed in the rapid quench apparatus using the same procedure as described above except that 1 mg/ml calf-thymus DNA was also added to syringe B solutions so that the final concentration was 0.5 mg/ml. When 0.5 mg/ml calf-thymus DNA was added to syringe A before mixing, no reaction was observed, confirming that this amount of enzyme trap is sufficient to force single turnover conditions. In the presence of an enzyme trap, both Pol δ4exo- and Pol δ3exo- appear to catalyze only a single round of synthesis with the same amplitude as that observed in the burst phase of reactions without trap. We therefore determined amplitudes by simply stopping “single-turnover” reactions after the burst phase was complete. Typically, such reactions were stopped after 1 s, so that the amplitude of the single turnover could be measured.
To determine the equilibrium dissociation constant (KDNA, Scheme 1) describing binding of Pol δ and DNA, the products formed under single turnover conditions, which reflect [ED], were fit to equation 4.
Scheme 1. Reactions involved in Pol δ-catalyzed DNA synthesis and editing.
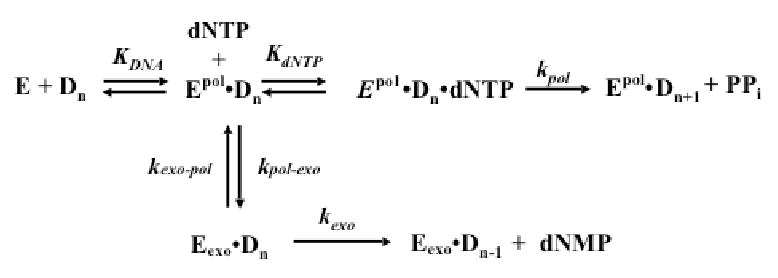
Epol and Eexo indicate DNA binding to pol site or exo sites, respectively.
| (4) |
In equation 4, Et is the total active site concentration, Dt is the total DNA concentration, and KDNA is a dissociation constant describing the affinity of the enzyme for DNA.
Pre-steady state exonuclease assays
Exonuclease assays were performed in the rapid quench apparatus as described above for the polymerase assays except that no dNTPs (or calf thymus DNA) were included in syringe B, 2× Pol δ solutions containing 100 nM of p125 and 100 nM of each DNA substrate was added to syringe A. The final reactions contained 50 mM Bis-Tris pH 6.5, 5 mM DTT, 200 μg/ml BSA, 30 mM NaCl, 10 mM MgCl2, 400 nM PCNA, 50 nM DNA, and 100 nM Pol δ. Assays were performed with the 25mer primer as a single stranded DNA substrate, with end-labeled 26merC/40mer as a double stranded DNA and with 26merT/40mer as a substrate with a mispaired primer terminus.
Pseudo-first order rate constants describing exonuclease reactions were determined by fitting time courses of substrate remaining to equation 5. In equation 5, [S] is substrate remaining at each time, t. [S]0 is substrate concentration at the beginning of the reaction, and kobs is a first order rate constant describing either cleavage of DNA (kexo, Scheme 1) or transfer of DNA from the polymerase to exonuclease sites (kpol-exo, Scheme 1)
| (Eq. 5) |
Steady-state polymerase/exonuclease assays
Assays monitoring the simultaneous action of the polymerase and exonuclease were performed at 37 °C. Pol δ (2 nM p125) was first incubated with 800 nM PCNA, 500 nM DNA substrate (25mer/40mer in Fig. 5A, 26merT/40mer in reaction in Fig. 5B) in 10 μl. Reactions were initiated by adding 10 μl of a 2× final solution which has 20 mM MgCl2 and twice the amounts of various dNTPa as indicated, such that final reactions contained 50 mM Bis-Tris pH 6.5, 5 mM DTT, 200 μg/ml BSA, 30 mM NaCl, 10 mM MgCl2, 400 nM PCNA, dNTP, 250 nM DNA, and Pol δ (2 nM μg/ml p125). After 3 minutes at 37°C, reactions were stopped by removing 6 μl and adding it to 12 μl loading buffer (25 mM EDTA in 95% formamide, 0.01% bromophenol blue and 0.01% xylene cyanol). Reaction products were analyzed on gels as described above. Products of the polymerase reaction [Ppol] and products of the exonuclease reaction [Pexo] were measured in each gel, and percent of DNA edited was calculated using equation 6.
FIGURE 5.
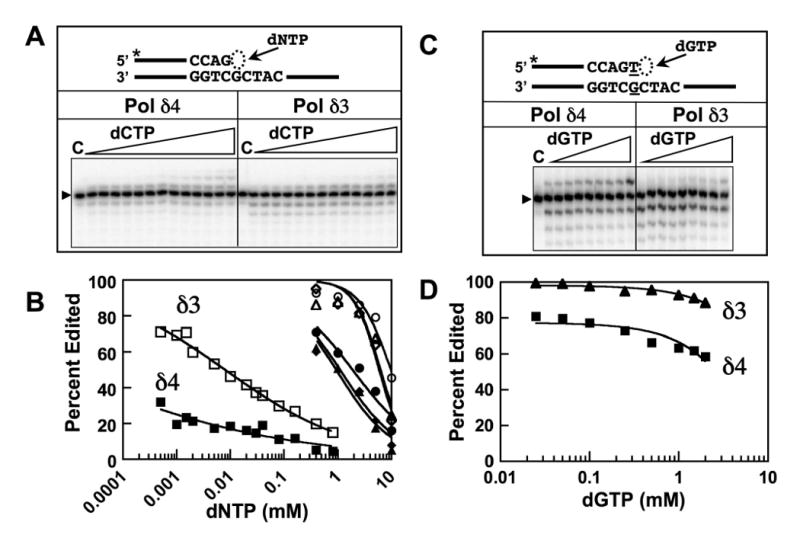
Steady-state rates of Pol δ polymerase and exonuclease activities in the presence of varying concentrations of the correct or an incorrect nucleotide. Reactions contained catalytic amounts (2 nM p125) of wild type Pol δ4 or Pol δ3, 250 nM of the indicated templates, and individual dNTPs as indicated (Experimental Procedures). (A) Product formation by Pol δ4 or Pol δ3 on a matched primer (25mer/40mer, inset). The gel shows the product formation when the reactions were performed with increasing concentrations of the correct nucleotide, dCTP (0.5, 1, 1.5, 2, 5,10, 20, 30, 40, 80, 160, 400, and 800 mM). Lane C is a control with no enzyme added, and the arrowhead indicates the position of the 25mer primer. (B) Reactions with dCTP or the individual incorrect nucleotides were performed as in panel A. Percentages of substrate that were edited by Pol δ were calculated as the percentage of the total exonucleolytic products over the sum of the exonucleolytic and extension products (equation 6), and plotted as Percent Edited vs log[dNTP]. Data for dCTP, squares; dATP, triangles; dTTP, circles; dGTP, diamonds. Data for Pol δ4 are shown as solid symbols and that for Pol δ3 as open symbols. (C) Product formation by Pol δ4 or Pol δ3 on a mismatched primer (26merT/40mer, inset). Reactions were performed as in panel A, and the products formed with dGTP, the next correct nucleotide are shown in the gel. Concentrations of dGTP used were 0.025, 0.05, 0.10, 0.25, 0.5, 1.0, 1.5, and 2.0 mM). Data for Pol δ4 are shown as squares and that for Pol δ3 as circles. Lane C is a control with no enzyme added, and the arrowhead indicates the position of the 26merT primer. (D) Percentages of substrate that were edited by Pol δ4 (solid squares) and Pol δ3 (solid triangles) were calculated as for Panel B and plotted against log[dGTP]. No extension was observed with either Pol δ4 or Pol δ3 in the presence of dATP, dTTP, or dGTP.
| (Eq. 6) |
Results
The abilities of Pol δ4 and Pol δ3 to synthesize DNA have been compared previously only by examining the ability of catalytic amounts of each complex to extend various primers annealed to various templates (26, 27). In all these prior assays, Pol δ4 synthesized more DNA than Pol δ3, but it is not clear from those results whether Pol δ4 is a faster DNA polymerase than Pol δ3. It has long been established that the rate-limiting step in standard polymerase assays, where the enzyme is allowed to catalyze multiple reactions, is not DNA synthesis but rather template release (30, 31). Similarly, prior assays indicating that Pol δ3 degrades DNA faster than Pol δ4 were done with limiting amounts of enzyme, so observed rates could reflect differences in the amounts of active enzyme in each preparation rather than different exonuclease functions per se (27). The goal of this study is to compare the polymerase and exonuclease functions of Pol δ4 and Pol δ3 under conditions where rates of DNA synthesis and degradation can be more accurately measured.
Human DNA Pol δ synthesizes DNA faster in the presence of p12
To understand how p12 influences Pol δ-catalyzed DNA synthesis, we monitored reactions catalyzed by Pol δ4 and Pol δ3 in the presence of a 5′-end labeled 25mer primer annealed to a 40mer template and a single complementary nucleotide, dCTP. Reactions were performed for very short incubation times (5 ms – 5 s) in a rapid quench apparatus, and 26mer products representing the addition of a single nucleotide were analyzed by polyacrylamide gel electrophoresis under denaturing conditions (Experimental Procedures). Both Pol δ4 and Pol δ3 synthesized DNA in a rapid burst phase followed by a slower, more linear phase that is characteristic of DNA polymerases-catalyzed DNA synthesis (Fig. 1). The data were fit to a model typically used to analyze pre-steady state DNA polymerase reactions (Eq. 1), in which the initial burst phase is described by a first order rate constant and a linear velocity describes the second phase. For the experiment shown in Fig. 1A, values for the kobs describing the fast phase of the reaction for Pol δ4 and Pol δ3 were 73 s-1 and 17 s-1, respectively, showing that Pol δ4 was ca. 4 times faster than Pol δ3. The amplitudes of the burst phases were calculated to be 27 nM and 19 nM respectively, for Pol δ4 and Pol δ3-catalyzed reactions; these amplitudes likely reflect the concentrations of catalytically competent polymerase-substrate complexes present at the start of the reactions, which each contained 50 nM enzyme based on p125 protein content. Calculation of the rates of the linear portions of the reactions corrected for the concentration of active sites gave turnover numbers of 0.16 s-1 and 0.17 s-1 for Pol δ3 and Pol δ4, respectively.
FIGURE 1.

Pre-steady state kinetics of DNA synthesis catalyzed by human Pol δ4 and Pol δ3. (A) Reactions with wildtype Pol δ4 and Pol δ3 were performed as described in Experimental Procedures using a rapid quench apparatus and the reaction products representing addition of a single nucleotide to the primer were determined. Reactions contained Pol δ4 or Pol δ3 (50 nM p125), 500 nM [5′-32P]25mer/40mer (see inset), and 500 μM dCTP. Primers extended by one nucleotide ([5′-32P]26mer, [P]) are plotted vs. time. Data are fit to equation 1 with the following parameters: Pol δ4 (solid squares) [ED] = 26.2 nM, kobs = 72.7 s-1, v= 4.6 nM/s; Pol δ3 (open squares), [ED] = 18.6 nM, kobs = 16.7 s-1, v= 3.0 nM/s. (B) Pre-steady state kinetics of DNA synthesis catalyzed by the exonuclease deficient forms of Pol δ4 and Pol δ3. Reactions contained Pol δ4exo- or Pol δ3exo- (40 nM p125), 250 nM [5′-32P]26merC/40mer (see inset), and 500 μM dGTP. Data are fit to equation 1 with the following constants: Pol δ4exo- (solid circles), [ED] = 19.3 nM, kobs = 54.7 s-1, v= 6.4 nM/s; Pol δ3exo (open circles), [ED] = 10.0 nM, kobs = 13.3 s-1, and v= 9.6 nM/s. (C) Pre-steady state kinetics of DNA synthesis catalyzed by Pol δ4exo- or Pol δ3exo- in the presence of a mismatched DNA substrate. 40 nM of p125, 200 nM mismatched [5′-32P]26merT/40mer (see inset; the T:G mismatch is underlined), and 500 μM dGTP. Data are fit to equation 1 using the following constants: Pol δ4exo- : kobs = 7.6 s-1, [ED] = 8.3 nM, v=1.9 nM/s; Pol δ3exo- :kobs = 2.5 s-1. [ED]= 4.4 nM, v= 0.9 nM/s. Data for Pol δ4 and Pol δ3 are shown as solid and open squares, respectively, and for Pol δ4exo- and Pol δ3exo- as solid and open circles, respectively.
In the gels that were used to analyzed the experiments in Fig. 1A, we observed that small but significant amounts of exonucleolytic products were present in the reactions; these were more pronounced with Pol δ3 and were more prominent in the later linear phases of the reactions, so that it is possible that the exonuclease activity of Pol δ3 may have caused reductions in the amount of product observed. Therefore, the same experiments were repeated using exonuclease-deficient forms of Pol δ. Pol δ4exo- and Pol δ3exo- each possess an alanine substitution for Asp402 in p125, a mutation that disrupts Pol δ exonuclease activity without affecting its polymerase function (27, 32). The time courses of product formation for Pol δ4exo- and Pol δ3exo- are shown in Fig. 1B. In this experiment, values of kobs of 54 s-1 and 13 s-1 were obtained for Pol δ4exo- and Pol δ3exo, respectively, a four fold difference. As with the wild type enzymes, Pol δ4exo- is four times faster than Pol δ3exo-. The rates of the linear (steady-state) phases divided by reaction amplitudes give turnover numbers for the reactions for Pol δ4exo- and Pol δ3exo- of 0.34 s-1 and 0.5 s-1 respectively (Fig. 1B). Thus, rates seen in the fast phase of the reaction were similar while rates in the slow phase were slightly faster than when reactions were performed with wild type Pol δ4 or Pol δ3, indicating that the exonuclease masks part of the polymerase activity in the native forms in the steady state (slow) phase of the reactions.
Having shown that Pol δ4 extends a correctly based paired primer faster than Pol δ3, we next examined their abilities to extend a primer with a T:G mismatch at the primer terminus under the same conditions (Fig. 1C). The rate constants for Pol δ4exo- and Pol δ3exo- were 7.6 s-1 and 2.5 s-1 respectively (Fig. 1C, Table 1). Both enzymes extended the mismatched primer more slowly than the matched primer, but Pol δ3exo- was still three times slower than Pol δ4exo-.
TABLE 1.
Estimates of rate and dissociation constants describing Pol δ catalyzed reactions
| Pol δ4 | Pol δ3 | δ4/δ3 a | |
|---|---|---|---|
| kcat b | 0.24 ± 0.08 s-1 | 0.18 ± 0.03 s-1 | 1.3 |
| kpol c | 87 ± 5.7 s-1 | 19 ± 2.9 s-1 | 4.6 |
| KdNTP c | 3.6 ± 0.8 μM | 3.2 ± 1.6 μM | 1.0 |
| KDNA d | 34 ± 5.4 nM | 35 ± 6.5 nM | 1.0 |
| kpol (mismatch)e | 7.6 ± 2.4 s-1 | 2.5 ± 0.6 s-1 | 3.0 |
| kexo f | 0.8 ± 0.2 s-1 | 1.9 ± 0.4 s-1 | 0.42 |
| kpol-exo g | 0.003 ± 0.0004 s-1 | 0.026 ± 0.003 s-1 | 0.12 |
| kpol-exo (mismatch) h | 0.062 ± 0.006 s-1 | 0.29 ± 0.03 s-1 | 0.21 |
| kpol-exo : kpol i | 1 : 29,000 | 1 : 730 | 40 |
| kpol-exo(mismatch) : kpol(mismatch) i | 1 : 123 | 1 : 8.6 | 14 |
Calculated by dividing the value observed with Pol δ4 by the value observed with Pol δ3.
Steady state initial velocities of incorporation performed at various dNTP concentration were fit to the Michaelis-Menten equation (equation 3) to determine a Vmax (Fig. 3H). The relationship Vmax/Et (active sites, Fig. 3G), was use to determine a steady state kcat. Uncertainties are standard errors from three independent reaction sets.
Values were obtained by fitting equations 1 and 2 to time courses obtained at various dNTP concentrations with Pol δ4exo- or Pol δ 3exo- (Fig. 3F). Errors are the standard errors from non-linear regression.
Values represent the average KDNA obtained when equation 4 is used to fit three independent titrations with DNA (Fig. 2B). Errors are standard deviations between three separate fits.
The observed first order rate constant describing the burst phase for extension of a mismatched primer (Fig. 1C). Errors are the standard errors from non-linear regression.
The observed first order rate constant describing the degradation of single-stranded DNA (Fig. 4A). Errors are the standard errors from non-linear regression.
The observed first order rate constant describing the degradation of matched duplex DNA (Fig. 4B). Errors are the standard errors from non-linear regression.
The observed first order rate constants describing the degradation of mismatched duplex DNA (Fig. 4C). Errors are the standard errors from non-linear regression.
The apparent affinities of human Pol δ for DNA and dNTPs are not influenced by p12
To probe whether the above differences result from the possibility that p12 enhances the affinity of Pol δ for any of it substrates, we repeated the above experiments at several different substrate concentrations. We used the exonuclease deficient mutants of Pol δ4 and Pol δ3 for these experiments to avoid potential losses of product formation by the exonuclease activity.
The affinities of Pol δ4 exo- and Pol δ3 exo- for DNA were estimated by examining the kinetics of DNA synthesis at various DNA concentrations. The goal was to relate amplitude of the burst phase of the reaction, which is commonly assumed to represent the number of catalytically competent enzyme-DNA complexes (active sites), with DNA concentration. To first demonstrate that the fast phase of the reaction represents a single turnover, we repeated time courses in the presence of an enzyme trap (0.5 mg/ml calf-thymus DNA) (Fig. 2A). Addition of an enzyme trap did not significantly alter the burst amplitude, but it eliminated the slow phase of the reaction. Such results support the notion that amplitude of the burst phase depends on the amount of enzyme-DNA complex present at the start of the reaction. To estimate the affinity of Pol δexo- for DNA, therefore, single-turnover reactions were performed in triplicate at various DNA concentrations (Fig. 2B). The data were then fit to a one-site binding equation (equation 4), from which dissociation constants KDNA of ca. 35 nM were found for both for Pol δ4exo- and Pol δ3exo– (Table 1). Thus, p12 does not appear to affect the binding of DNA to Pol δ.
FIGURE 2.

Effect of DNA primer/template concentration on the amplitude of the burst phase of DNA synthesis catalyzed by Pol δ4exo- or Pol δ3exo-. (A) Effect of an enzyme trap. The reaction shown in Fig. 1B (x′s) was repeated in the presence of an enzyme trap (0.5 mg/ml calf-thymus DNA). The trap abolished the slow phase of the reaction catalyzed by Pol δ4exo- (solid circles), and data was fit to equation 1 with the following parameters [ED] = 19.8 nM, kobs = 94 s-1, v= 0 nM/s (B) Single turnover reactions (see Experimental Procedures) containing Pol δ4exo- or Pol δ3exo- (20 nM p125), 500 μM dGTP, 0.5 mg/ml calf thymus DNA and the indicated amounts of [5′-32P]26merC/40mer. Product concentration ([P]) at 1 s, which reflects the burst amplitude ([ED]) of the reaction, is plotted versus DNA concentration and fit to equation 3 with a KDNA of 34 ± 5.4 nM for Pol δ4exo- and 35 ± 6.5 nM for Pol δ3exo-. Data for Pol δ4exo- and Pol δ3exo- are shown as filled and solid circles, respectively.
To examine how p12 affects the affinity of Pol δ for its dNTP substrates, burst assays were repeated with saturating levels of DNA (250 nM) in the presence of various dGTP concentrations for Pol δ4exo- and Pol δ3exo- as shown in Fig. 3, panels A-E. Under these conditions, the rates of the fast phase of the reaction (Fig. 3F) and the rates of the slow phase increased (Fig. 2H) with dGTP, as one would expect. Surprisingly, however, the amplitude of the fast phase of the reaction also increased (Fig. 3G), implying that the concentration of catalytically competent complexes also varies with dGTP. Nevertheless, each factor describing the reactions increased similarly with dGTP regardless of whether Pol δ4exo- and Pol δ3exo- was present. We therefore chose to use the relationship between kobs and dGTP concentration (Fig. 3F) to estimate the maximum rate constant for DNA synthesis (kpol, scheme 1) and the concentration of dGTP leading to half this rate (KdNTP) by fitting the data to a one site binding equation (equation 2), as is common practice (33).
FIGURE 3.

Effect of dNTP concentration on the pre-steady state kinetics of Pol δ4 and Pol δ3 catalyzed DNA synthesis. Reactions contained Pol δ4exo- (filled circles) or Pol δ3exo- (open circles), each at 20 nM p125, 250 nM [5′-32P]26C/40mer template (see inset, Fig. 1), and varying concentrations of dGTP ((A), 1 μM; (B), 2 μM; (C), 5 μM; (D), 10 μM; (E) 100 μM). Data were fit to equation 1. (F) kobs values from fits in panels A-E as a function of dGTP concentration. Data are fit to equation 2 with a kpol of 87 ± 12 s-1 and a Kd dGTP of 3.6 ± 0.8 μM for Pol δ4exo-, and kpol of 19 ± 2.8 s-1 and a KdGTP of 3.2 ± 1.6 μM for Pol δ3exo-. (G) Reaction amplitudes ([ED]) from the fits in panels A-E as function of dGTP concentration. Data are fit to a one site binding equation with a Bmax of 13 ± 3 nM and a Kd of 1.6 ± 0.6 μM for Pol δ4exo-, and Bmax of 8 ± 4 nM and a Kd of 1.3 ± 0.9 μM for Pol δ4exo- for Pol δ3exo-. (H) Slow phase velocities (v) from fits in panels A-E as function of dGTP concentration. Data are fit to equation 3 with a Vmax of 3.1 ± 2.0 nM/s μM for Pol δ4exo-, and Vmax of 1.4 ± 0.6 nM/s for Pol δ4exo- and a Km of 0.8 ± 0.5 for Pol δ3exo-. Data in (A-F) are from single reaction sets, but two additional assay sets were performed prior, to determine appropriate time and concentration ranges. Fits to the other datasets yielded constants within the stated errors. Error bars and uncertainties reflect the standard error for the curve fits.
The values of kpol for Pol δ4 and Pol δ3 determined from these experiments were 87 ± 5.7 s-1 and 19 ± 2.9 s-1, respectively. Values for KdNTP for Pol δ4exo- and Pol δ3exo- for the incorporation of dGTP obtained in the same analysis for Pol δ4exo- and Pol δ3exo- were 3.6 ± 0.8 and 3.2 ± 1.6 μM, respectively, showing that binding affinities for dNTP are not affected by p12. Care should be exercised so as not to over-interpret these estimates since the reactions were clearly not performed under pseudo-first order conditions despite the fact that best attempts were made to saturate the enzyme with all substrates besides dGTP. We also attempted to use these data to determine the Michaelis constants for Pol δ3exo- and Pol δ4exo-, but because of errors in the rates of the slow phases, we were not able to determine an accurate Km using the Michaelis-Menten equation for Pol δ4exo-. However, a Vmax could be estimated to be 3.2 nM/s and 1.5 nM/s for Pol δ4exo- and Pol δ3exo-, respectively. Adjusted for the maximal active sites determined in Fig. 3G, kcat values were estimated at 0.24 s-1 and 0.18 s-1 for Pol δ4exo- and Pol δ3exo-, respectively (Table 1).
p12 modulates the rate of DNA transfer between the polymerase and exonuclease active sites
We have previously examined the activities of Pol δ4 and Pol δ3 on oligonucleotide substrates, and observed that Pol δ3 generated more exonucleolytic products, particularly when template lesions are encountered. Measurements of the exonuclease activities of Pol δ4 and Pol δ3 under steady state conditions indicated that Pol δ3 might degrade DNA faster than Pol δ4 (27). However, in order to determine if p12 affects the true rates of the 3′ to 5′ exonuclease activity, we monitored rates of Pol δ4 and Pol δ3-catalyzed DNA cleavage in a rapid quench device under single turnover conditions where the amount of enzyme exceeded the amount of DNA. The goal was to use pseudo-first order reactions to estimate rate constants for DNA cleavage (kexo, Scheme 1), and for the transfer of the primer (kpol-exo, Scheme 1), from the polymerase to the exonuclease site.
Kinetic studies (34) as well as structural studies have shown that during DNA synthesis, duplex DNA lies in the pol site, while single-stranded DNA directly binds to the exo site (2, 20, 21). Three to four nucleotides of the primer must be melted so that the frayed primer can enter the exonuclease site, which typically only can accommodate ssDNA (16, 20, 34). Measurement of the rate of ssDNA hydrolysis, therefore, provides a determination of kexo, the first order rate constant describing the cleavage of a single nucleotide from the 3′ end of DNA (Scheme 1). Because duplex DNA binds to the pol site, kexo measured with duplex DNA provides values for kpol-exo, since switching of the primer to the exonuclease site is rate-limiting in such reactions (3).
Reactions containing Pol δ4 or Pol δ3 and a single-stranded oligonucleotide were quenched at various times (Fig. 4A). The substrate remaining was fit to a first order exponential decay equation (equation 5) to obtain the rate constants (kexo) describing hydrolytic cleavage. The value of kexo obtained for Pol δ4 (0.8 s-1) was about half that obtained for Pol δ3 (1.9 s-1) (Fig. 4A, Table 1). The values of kexo for Pol δ have not been previously reported, and are roughly 40-fold lower than values reported for RB69 polymerase (23).
FIGURE 4.

Determination of the rates of exonuclease cleavage of ssDNA and dsDNA by Pol δ4 and Pol δ3. Reactions were performed as described in Experimental Procedures using a rapid quench apparatus under single turnover conditions. The final reactions contained Pol δ3 or Pol δ4 (100 nM p125) and 50 nM DNA substrate. Reactions were initiated by addition of 10 mM Mg2+, and quenched at various times (from 0.05s to 150s). The remaining 26mer was determined and the data were fit into an exponential decay equation (Equation 5), and yield observed rates of excision. (A) Time course of hydrolysis of a 26mer ssDNA DNA, [5′-32P]26merT, by Pol δ4 (squares) and Pol δ3 (triangles); values for the rates are given as kexo in Table 1. (B) Time course of hydrolysis of a 26mer/40mer duplex DNA, [5′-32P]26merC/40mer, by Pol δ4 (squares) and Pol δ3 (triangles); values for the rates are given in Table 1 as the rates for switching of the primer terminus from the pol to the exo site (kpol-exo, Scheme I). (C) Time course of hydrolysis of a 26mer/40mer duplex DNA containing a mismatched primer terminus, [5′-32P]26merT/40mer, by Pol δ4 (squares) and Pol δ3 (triangles); values for the rates are given in Table 1 as the rates for switching of the primer terminus from the pol to the exo site (kpol-exo, mismatch).
Next, we determined the rate constants when similar reactions were performed with a 26mer/40mer duplex DNA (Fig. 4B). This rate constant (kpol-exo) for Pol δ3 was 0.026 s-1, ca. 9 times faster than that for Pol δ4 (0.003 s-1) (Fig. 2B, Table 1). When a 26mer/40mer with a T:G mismatch at the primer terminus was used, the switching rates for Pol δ4 and Pol δ3 were increased 20 and 10 fold respectively, but the rate constant for Pol δ3 (0.29 s-1) was still 5 fold faster than that for Pol δ4 (Fig 4C, Table 1).
These findings show that p12 exerts a large negative effect on the rate at which the primer terminus is switched from the polymerase to the exonuclease site and only a small effect (ca. 2 fold) on kexo. Thus, Pol δ3 would be expected to exhibit an enhanced proofreading ability by comparison with Pol δ4. Taken with the negative effect on kpol when p12 is absent, it is apparent that these two effects both lead to an increased capacity of Pol δ3 for proofreading. Changes in kpol or kpol-exo have been well established to alter the proofreading or fidelity of DNA polymerases, and to generate mutator or antimutator phenotypes (3, 35-37). To understand the combined effects of alterations in kpol-exo and kpol it is useful to examine the kpol-exo to kpol ratio, as an indication of the probability of excision of the primer terminus, and thus the proofreading activity. For Pol δ4 the ratio is 1:29,000 and 1:730 for Pol δ3 on an unmodified template (Table 1). This ca. 40 fold difference indicates that Pol δ3 is more active in proofreading than Pol δ4. When assayed on a template with a mismatched primer terminus, the ratios for Pol δ4 and Pol δ3 were 1:123 and 1:8.6, respectively (Table 1). The increase in the ratios for either enzyme when compared to the normal template reflects the increased probabilities for the excision of the mismatch. Comparison of the ratios for the mismatched primer terminus shows that Pol δ3 is 14 times more likely to remove a mismatched primer terminus than Pol δ4.
Taken together, our findings show that removal of p12 leads to reciprocal changes in kpol and the switching rate, kpol-exo, that are well established to result in increased proofreading, that are associated with a capacity for increased fidelity of DNA synthesis.
In the absence of p12, human Pol δ is more likely to proofread errors
The above data clearly show that in the presence of p12, human Pol δ synthesizes DNA more rapidly and degrades DNA more slowly. Slower DNA cleavage in the presence of p12 seems to arise not from an intrinsically slower exonuclease but rather from a slower transfer of the DNA from the polymerase to the exonuclease site. To examine how these differences might impact the fidelity of DNA synthesis, we examined Pol δ action under conditions that might force the enzyme to introduce a mutation. Pol δ4 and Pol δ3 were incubated with the 25mer/40mer in the presence of concentrations ranging from 5 μM to 10 mM of correct next nucleotide, dCTP, or the dNTPs that do not base pair with the template. Products formed under steady-state conditions were examined to determine the percentage of DNA that was edited, which was defined as the amount of exonucleolytic products as a percentage of the sum of exonucleolytic (<26mer) and extension products (>26mer) (equation 6, Experimental Procedures). In the presence of dCTP, editing is suppressed with increasing dCTP concentration since the insertion of dCTP suppresses the appearance of exonucleolytic products; when plotted against log[dNTP], a linear relationship is observed. It is apparent that Pol δ3 is far more active than Pol δ4 in editing (Fig. 5). When the incorrectly based paired dNTPs are present, very high concentrations are required to suppress editing, but nevertheless, it is Pol δ3 that again exhibits greater editing activity than Pol δ4. We performed a similar analysis in an experiment with a substrate in which the primer contained a T:G mismatch, and only the next correct nucleotide, dGTP, was added in increasing concentrations. Once the terminal mismatch is removed, the dGTP represents a mismatched dNTP, and thus editing activity prevails. This is seen for Pol δ3, where inhibition of editing dominates and essentially no primer extension can be observed until ca. 1 mM dGTP concentration. Pol δ4 exhibited a lower extent of editing initially, and this could be suppressed at much lower dGTP concentrations than with Pol δ3. While the conditions used are forcing, in that they require high concentrations of dNTPs, these experiments provide a demonstration of that Pol δ3 exhibits a greater degree of fidelity than Pol δ4, since this editing assay monitors in tandem their different propensities for insertion of a wrong nucleotide and for excision of a mismatched primer terminus.
Discussion
We have investigated the kinetic properties of human Pol δ4 and Pol δ3 with two goals in mind, first, to understand the role of the p12 subunit in Pol δ catalysis, and second, to gain insights into a rationale for the removal of the p12 subunit during DNA damage, based on their altered properties. We examined the polymerase and exonuclease functions of Pol δ4 and Pol δ3 using pre-steady state kinetic methods to obtain values for kinetic parameters that might provide more information on how p12 alters the catalytic functions of Pol δ. Pre-steady state kinetic analyses have provided powerful tools for understanding the kinetic mechanisms for DNA polymerase functions, and have complemented the structural studies that have lead to current models for the mechanisms whereby the exquisite fidelity of DNA polymerases is achieved (3, 4).
The above experiments have provided estimates the constants in scheme 1: KDNA, KdNTP, kpol, kpol-exo and kexo for Pol δ4 and Pol δ3 (Table 1). Note that scheme 1 is simpler than minimal kinetic mechanisms that have been determined for other DNA polymerases like those encoded by bacteriophages or prokaryotes (25, 38) in that the various steps in DNA synthesis are not delineated. In other words, kpol in scheme 1 may include more than one sub-steps, such a conformational change and chemical step. Scheme 1 is also intended to mainly highlight the fact that because the exonuclease active site (exo) is spatially separated from the polymerase active site (pol) (6), the primer terminus has to be translocated, or switched, to the exo site for proofreading to take place (3, 34). Kinetic analyses of polymerase mutants have provided significant insights into understanding how alterations in the rates of these parameters affect the fidelity of DNA polymerases. Alterations in the switching rates from pol to exo sites (kpol-exo) affect the efficiency of proofreading, while reductions in kpol that occur by mutation or by encounter of a mismatched terminus lead to a kinetic barrier that favors translocation of the DNA from the pol to the exo site (3, 35). Our findings show that two key rate constants, kpol and kpol-exo, are affected by the absence or presence of p12 in the Pol δ enzyme.
The difference in kpol implies that p12 accelerates the rate of DNA synthesis by about 4.6-fold. The kpol of 87 s-1 for Pol δ4 is higher than the value of 21 s-1 previously reported for the p125/p50 dimer calf thymus Pol δ (39), which is closer to the kpol of 19 s-1 for Pol δ3 (Table 1). However, since the calf thymus experiments were performed at a higher temperature (37 °C) than our studies (24 °C) such a comparison might not be meaningful. Nevertheless, these findings show that p12 has a crucial role in modulating Pol δ catalysis, in which it is able to modulate the rate-limiting step(s) involved in the catalysis of phosphodiester bond formation. This capacity has important implications for the changes that take place when p12 is removed; decreases in kpol have been well established to form a kinetic barrier that increases the probability of switching of the primer terminus from the pol to the exo site, thereby providing a basis for increased proofreading (3). Thus, p12 acts as a gearshift that regulates kpol in a manner that will alter the proofreading capacity of Pol δ.
The second step altered by p12 is kpol-exo, a rate constant describing the switching of the primer terminus from the pol to the exo sites, a step which plays a critical role in proofreading. This alteration has significant implications for the effects of p12 on the capacity of Pol δ to perform proofreading. The alterations of the balance between polymerase and exonuclease activities that we observed are ones that are well established to alter the proofreading of DNA polymerases, and to generate mutator or antimutator phenotypes in related enzymes that reduce the mutation frequency during replication (3, 35-37). We have performed experiments which assess the degree of editing by Pol δ4 and Pol δ3 on oligonucleotide substrates and observed the expected increase in editing (Fig. 5). The characteristics displayed by Pol δ3 allow for a prediction that it would display greater fidelity when synthesizing DNA in vitro, and possibly in vivo. Future studies will be needed to obtain quantitative assessments of alterations in the fidelity of DNA synthesis by Pol δ4 and Pol δ3.
Our findings are illustrated in the diagram in Fig. 6A, which shows the directional shifts in kpol, kpol-exo, and the ratio kpol-exo/kpol when p12 is absent or present. The functions of p12 can be viewed as that of a gearshift. The presence of p12 increases the rate of polymerization, and slows the switching rate. When p12 is removed from the Pol δ complex, there is a downshift in the rate of phosphodiester bond formation. This will facilitate proofreading via the kinetic barrier effect, in which reduction of kpol favors the probability of primer terminus translocation from the pol to exo sites. Coupled with this, removal of p12 increases the rates of translocation of primer from the pol to exo site, a change that further enhances proofreading. Thus, p12 functions as a gearshift whose effect on the catalytic properties of Pol δ changes two of the key rate constants that govern proofreading efficiency. Taken together, our data provide a mechanistic basis for the earlier observations that Pol δ3 exhibits behaviors consistent with an increased level of proofreading (27).
FIGURE 6.
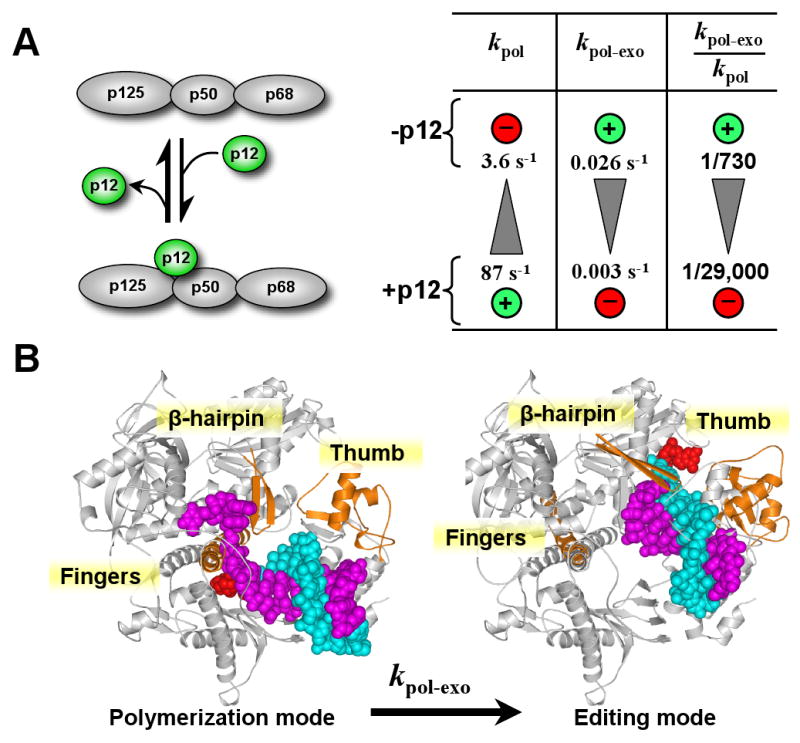
The p12 subunit of Pol δ acts as a gearshift that alters the rates of polymerization and primer switching. (A) Diagram of the effects of p12 on Pol δ. The first two columns show the effects of p12 on the kinetic constants for polymerization (kpol) and translocation of the primer terminus from the pol to the exo site (kpol-exo), respectively. The third column shows the effects on proofreading efficiency, expressed as the ratio kpol-exo:kpol. The shaded triangles indicate the direction of change the direction of change of the rate constants from low to high, and the circled plus and minus signs indicate the effects of the presence and absence of p12 on the rate constants. (B) Models of the switch of the DNA from the pol to the exo sites of Pol δ The model on the left is the recently published structure of the yeast Pol δ catalytic subunit (19) in the polymerization mode, viz., in a ternary complex bound to a primer (cyan), template (magenta) and nucleoside triphosphate (red) (protein data bank (PDB) file 1IAY). The right model shows a putative structure of yeast Pol δ in the editing mode, which was made by aligning the yeast Pol δ structure (1IAY) and a structure of the related RB69 polymerase (PDB file 1CLQ) with DNA bound in the in the exonuclease site (20). The RB69 and Pol δ structures superimpose except along one helix in the fingers domain, the tip of thumb domain, and a β-hairpin. These three regions of RB69 are shown (orange) to point out the conformational changes needed to transfer the primer between the pol and exo sites. The fingers, thumb and b-hairpin structures of yeast Pol δ are highlighted (orange) in the left model. The rest of the RB69 protein is not shown for clarity, but the primer (cyan), the primer 3′ terminus (red), and template (magenta) bound to RB69 are shown to mark the exonuclease site of Pol δ (grey).
The structural basis for the influence of p12 on both pol and exo functions is unknown, and at this point one can only speculate on how p12 might exert its effects on Pol δ. Comparison of structures of RB69 polymerase with yeast Pol δ (19) can provide some insights into the mechanisms by which a primer terminus is switched from the pol to the exo sites (2, 16, 20-24, 40). These studies have shown that there are large DNA movements as well as protein conformational changes that take place upon switching between the editing and polymerization modes (Fig. 6B). In particular, movement of the tip of the thumb domain (21) and a β hairpin loop (23, 40) must occur in order for DNA to migrate from the pol to the exo site for editing to take place. Notably, in contrast to analogous RB69 structures, the recent yeast Pol δ structure (19) reveals this β-hairpin (β16-β17) in a conformation where it directly interacts with the template strand, consistent with an involvement with primer switching between pol and exo sites. Thumb movement, however, might be even more critical as evidenced by the fact that some of the first antimutator alleles discovered map to this region of the related T4 DNA polymerase (41) and the mutations have now been shown to clearly affect primer switching in the T4 enzyme (37, 42). Effects of p12 on the reduction of the rate of switching of the primer from the pol to the exo site (kpol-exo) could be exerted by an interaction with such regions of Pol δ. p12 could exert effects on conformation changes, cause steric interference of the movement of the primer terminus, or modify access to the exonuclease site (23, 43). Alternatively, p12 might impede primer movement in a channel crossed by the primer terminus during primer shuttling between pol and exo sites (44).
Our findings also provide insights into how loss of p12 might alter the biological function of the human replisome. Agents that damage DNA cause p12 to rapidly disappear in cells (26), and the results above show that this change causes Pol δ to shift to a form that is capable of greater proofreading ability, and which has previously been shown to exhibit reduced capacity for lesion bypass synthesis (26). More proofreading would be needed when the genome is damaged, especially if such damage leads to base modifications that cause nucleotides to ambiguously base pair. As has been previously discussed, loss of the p12 subunit could affect a number of the DNA transactions in which Pol δ is engaged in vivo, e.g., its ability to interact with other proteins involved in DNA damage, or its ability to participate in DNA repair processes (26). This study provides significant new mechanistic information that may assist in evaluating the role of Pol δ3 in cells undergoing DNA damage.
Acknowledgments
This work was supported by grants from the NIH National Institutes of Health (GM31973 and ES014737 to M.Y.W.T.L.; AI052395 to D.N.F.).
Footnotes
Abbreviations: dNTP, deoxynucleoside triphosphate; Exo, exonuclease active site, Pol, polymerase active site, Pol δ, human DNA polymerase δ; Pol δ3, Pol δ p125/p50/p68 trimer; Pol δ4, Pol δ p125/p50/p68 p125/p50/p68/p12 tetramer.
References
- 1.Nachman MW, Crowell SL. Estimate of the mutation rate per nucleotide in humans. Genetics. 2000;156:297–304. doi: 10.1093/genetics/156.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steitz TA. DNA polymerases: structural diversity and common mechanisms. J Biol Chem. 1999;274:17395–17398. doi: 10.1074/jbc.274.25.17395. [DOI] [PubMed] [Google Scholar]
- 3.Johnson KA. Conformational coupling in DNA polymerase fidelity. Annu Rev Biochem. 1993;62:685–713. doi: 10.1146/annurev.bi.62.070193.003345. [DOI] [PubMed] [Google Scholar]
- 4.Kunkel TA, Bebenek K. DNA replication fidelity. Annu Rev Biochem. 2000;69:497–529. doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]
- 5.Doublie S, Sawaya MR, Ellenberger T. An open and closed case for all polymerases. Structure. 1999;7:R31–5. doi: 10.1016/S0969-2126(99)80017-3. [DOI] [PubMed] [Google Scholar]
- 6.Steitz TA, Yin YW. Accuracy, lesion bypass, strand displacement and translocation by DNA polymerases. Philos Trans R Soc Lond B Biol Sci. 2004;359:17–23. doi: 10.1098/rstb.2003.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Branzei D, Foiani M. Interplay of replication checkpoints and repair proteins at stalled replication forks. DNA Repair (Amst) 2007;6:994–1003. doi: 10.1016/j.dnarep.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 8.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 9.Kunkel TA, Burgers PM. Dividing the workload at a eukaryotic replication fork. Trends Cell Biol. 2008;18:521–527. doi: 10.1016/j.tcb.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burgers PM. Polymerase dynamics at the eukaryotic DNA replication fork. J Biol Chem. 2009;284:4041–4045. doi: 10.1074/jbc.R800062200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garg P, Burgers PM. DNA polymerases that propagate the eukaryotic DNA replication fork. Crit Rev Biochem Mol Biol. 2005;40:115–128. doi: 10.1080/10409230590935433. [DOI] [PubMed] [Google Scholar]
- 12.Liu L, Mo J, Rodriguez-Belmonte EM, Lee MY. Identification of a fourth subunit of mammalian DNA polymerase delta. J Biol Chem. 2000;275:18739–18744. doi: 10.1074/jbc.M001217200. [DOI] [PubMed] [Google Scholar]
- 13.Mo J, Liu L, Leon A, Mazloum N, Lee MY. Evidence that DNA polymerase delta isolated by immunoaffinity chromatography exhibits high-molecular weight characteristics and is associated with the KIAA0039 protein and RPA. Biochemistry. 2000;39:7245–7254. doi: 10.1021/bi0000871. [DOI] [PubMed] [Google Scholar]
- 14.Yang CL, Chang LS, Zhang P, Hao H, Zhu L, Toomey NL, Lee MY. Molecular cloning of the cDNA for the catalytic subunit of human DNA polymerase delta. Nucleic Acids Res. 1992;20:735–745. doi: 10.1093/nar/20.4.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hao H, Jiang Y, Zhang SJ, Zhang P, Zeng RX, Lee MY. Structural and functional relationships of human DNA polymerases. Chromosoma. 1992;102:S121–7. doi: 10.1007/BF02451795. [DOI] [PubMed] [Google Scholar]
- 16.Wang J, Sattar AK, Wang CC, Karam JD, Konigsberg WH, Steitz TA. Crystal structure of a pol alpha family replication DNA polymerase from bacteriophage RB69. Cell. 1997;89:1087–1099. doi: 10.1016/s0092-8674(00)80296-2. [DOI] [PubMed] [Google Scholar]
- 17.Xie B, Mazloum N, Liu L, Rahmeh A, Li H, Lee MY. Reconstitution and characterization of the human DNA polymerase delta four-subunit holoenzyme. Biochemistry. 2002;41:13133–13142. doi: 10.1021/bi0262707. [DOI] [PubMed] [Google Scholar]
- 18.Li H, Xie B, Zhou Y, Rahmeh A, Trusa S, Zhang S, Gao Y, Lee EY, Lee MY. Functional roles of p12, the fourth subunit of human DNA polymerase delta. J Biol Chem. 2006;281:14748–14755. doi: 10.1074/jbc.M600322200. [DOI] [PubMed] [Google Scholar]
- 19.Swan MK, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Structural basis of high-fidelity DNA synthesis by yeast DNA polymerase delta. Nat Struct Mol Biol. 2009;16:979–986. doi: 10.1038/nsmb.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shamoo Y, Steitz TA. Building a replisome from interacting pieces: sliding clamp complexed to a peptide from DNA polymerase and a polymerase editing complex. Cell. 1999;99:155–166. doi: 10.1016/s0092-8674(00)81647-5. [DOI] [PubMed] [Google Scholar]
- 21.Franklin MC, Wang J, Steitz TA. Structure of the replicating complex of a pol alpha family DNA polymerase. Cell. 2001;105:657–667. doi: 10.1016/s0092-8674(01)00367-1. [DOI] [PubMed] [Google Scholar]
- 22.Hogg M, Wallace SS, Doublie S. Crystallographic snapshots of a replicative DNA polymerase encountering an abasic site. EMBO J. 2004;23:1483–1493. doi: 10.1038/sj.emboj.7600150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hogg M, Aller P, Konigsberg W, Wallace SS, Doublie S. Structural and biochemical investigation of the role in proofreading of a beta hairpin loop found in the exonuclease domain of a replicative DNA polymerase of the B family. J Biol Chem. 2007;282:1432–1444. doi: 10.1074/jbc.M605675200. [DOI] [PubMed] [Google Scholar]
- 24.Aller P, Rould MA, Hogg M, Wallace SS, Doublie S. A structural rationale for stalling of a replicative DNA polymerase at the most common oxidative thymine lesion, thymine glycol. Proc Natl Acad Sci U S A. 2007;104:814–818. doi: 10.1073/pnas.0606648104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karam JD, Konigsberg WH. DNA polymerase of the T4-related bacteriophages. Prog Nucleic Acid Res Mol Biol. 2000;64:65–96. doi: 10.1016/s0079-6603(00)64002-3. [DOI] [PubMed] [Google Scholar]
- 26.Zhang S, Zhou Y, Trusa S, Meng X, Lee EY, Lee MY. A novel DNA damage response: rapid degradation of the p12 subunit of dna polymerase delta. J Biol Chem. 2007;282:15330–15340. doi: 10.1074/jbc.M610356200. [DOI] [PubMed] [Google Scholar]
- 27.Meng X, Zhou Y, Zhang S, Lee EY, Frick DN, Lee MY. DNA damage alters DNA polymerase delta to a form that exhibits increased discrimination against modified template bases and mismatched primers. Nucleic Acids Res. 2009;37:647–657. doi: 10.1093/nar/gkn1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muzyczka N, Poland RL, Bessman MJ. Studies on the biochemical basis of spontaneous mutation. I. A comparison of the deoxyribonucleic acid polymerases of mutator, antimutator, and wild type strains of bacteriophage T4. J Biol Chem. 1972;247:7116–7122. [PubMed] [Google Scholar]
- 29.Zhang P, Zhang SJ, Zhang Z, Woessner JFJ, Lee MY. Expression and physicochemical characterization of human proliferating cell nuclear antigen. Biochemistry. 1995;34:10703–10712. doi: 10.1021/bi00034a002. [DOI] [PubMed] [Google Scholar]
- 30.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 31.Byrnes JJ, Downey KM, Que BG, Lee MY, Black VL, So AG. Selective inhibition of the 3′ to 5′ exonuclease activity associated with DNA polymerases: a mechanism of mutagenesis. Biochemistry. 1977;16:3740–3746. doi: 10.1021/bi00636a002. [DOI] [PubMed] [Google Scholar]
- 32.Goldsby RE, Hays LE, Chen X, Olmsted EA, Slayton WB, Spangrude GJ, Preston BD. High incidence of epithelial cancers in mice deficient for DNA polymerase delta proofreading. Proc Natl Acad Sci USA. 2002;99:15560–15565. doi: 10.1073/pnas.232340999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson KA. Rapid quench kinetic analysis of polymerases, adenosinetriphosphatases, and enzyme intermediates. Methods Enzymol. 1995;249:38–61. doi: 10.1016/0076-6879(95)49030-2. [DOI] [PubMed] [Google Scholar]
- 34.Donlin MJ, Patel SS, Johnson KA. Kinetic partitioning between the exonuclease and polymerase sites in DNA error correction. Biochemistry. 1991;30:538–546. doi: 10.1021/bi00216a031. [DOI] [PubMed] [Google Scholar]
- 35.Khare V, Eckert KA. The proofreading 3′-->5′ exonuclease activity of DNA polymerases: a kinetic barrier to translesion DNA synthesis. Mutat Res. 2002;510:45–54. doi: 10.1016/s0027-5107(02)00251-8. [DOI] [PubMed] [Google Scholar]
- 36.Reha-Krantz LJ. Learning about DNA polymerase function by studying antimutator DNA polymerases. Trends Biochem Sci. 1995;20:136–140. doi: 10.1016/s0968-0004(00)88987-2. [DOI] [PubMed] [Google Scholar]
- 37.Reha-Krantz LJ. Regulation of DNA polymerase exonucleolytic proofreading activity: studies of bacteriophage T4 “antimutator” DNA polymerases. Genetics. 1998;148:1551–1557. doi: 10.1093/genetics/148.4.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Capson TL, Peliska JA, Kaboord BF, Frey MW, Lively C, Dahlberg M, Benkovic SJ. Kinetic characterization of the polymerase and exonuclease activities of the gene 43 protein of bacteriophage T4. Biochemistry. 1992;31:10984–10994. doi: 10.1021/bi00160a007. [DOI] [PubMed] [Google Scholar]
- 39.Einolf HJ, Guengerich FP. Kinetic analysis of nucleotide incorporation by mammalian DNA polymerase delta. J Biol Chem. 2000;275:16316–16322. doi: 10.1074/jbc.M001291200. [DOI] [PubMed] [Google Scholar]
- 40.Subuddhi U, Hogg M, Reha-Krantz LJ. Use of 2-aminopurine fluorescence to study the role of the beta hairpin in the proofreading pathway catalyzed by the phage T4 and RB69 DNA polymerases. Biochemistry. 2008;47:6130–6137. doi: 10.1021/bi800211f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reha-Krantz LJ. Locations of amino acid substitutions in bacteriophage T4 tsL56 DNA polymerase predict an N-terminal exonuclease domain. J Virol. 1989;63:4762–4766. doi: 10.1128/jvi.63.11.4762-4766.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stocki SA, Nonay RL, Reha-Krantz LJ. Dynamics of bacteriophage T4 DNA polymerase function: identification of amino acid residues that affect switching between polymerase and 3′ --> 5′ exonuclease activities. J Mol Biol. 1995;254:15–28. doi: 10.1006/jmbi.1995.0595. [DOI] [PubMed] [Google Scholar]
- 43.Hadjimarcou MI, Kokoska RJ, Petes TD, Reha-Krantz LJ. Identification of a mutant DNA polymerase delta in Saccharomyces cerevisiae with an antimutator phenotype for frameshift mutations. Genetics. 2001;158:177–186. doi: 10.1093/genetics/158.1.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu P, Nossal N, Benkovic SJ. Kinetic characterization of a bacteriophage T4 antimutator DNA polymerase. Biochemistry. 1998;37:14748–14755. doi: 10.1021/bi980835a. [DOI] [PubMed] [Google Scholar]
- 45.Nick McElhinny SA, Stith CM, Burgers PM, Kunkel TA. Inefficient proofreading and biased error rates during inaccurate DNA synthesis by a mutant derivative of Saccharomyces cerevisiae DNA polymerase delta. J Biol Chem. 2007;282:2324–2332. doi: 10.1074/jbc.M609591200. [DOI] [PMC free article] [PubMed] [Google Scholar]