

Abstract
Excessive oxidative damage to DNA leads to activation of poly(ADP-ribose) polymerase-1 (PARP-1), accumulation of poly(ADP-ribose) (PAR) polymers, translocation of apoptosis-inducing factor (AIF) from mitochondria to the nucleus, and cell death. Here, we compared the effect of gene deletion of PARP-1 and PARP-2, enzymes activated by DNA oxidative damage, in male mice subjected to 2 h of focal cerebral ischemia. Infarct volume at 3 days of reperfusion was markedly decreased to a similar extent in PARP-1- and PARP-2-null mice. The ischemia-induced increase in nuclear AIF accumulation was largely suppressed in both knockout genotypes. The transient increase in PAR during early reperfusion was nearly blocked in PARP-1-null mice, but only moderately decreased at 1-h reperfusion in PARP-2-null mice. Differences in the tissue volume at risk, as assessed by arterial casts and autoradiographic analysis of regional blood flow, did not fully account for the large reductions in AIF translocation and infarct volume in both PARP null mice. Cell death was attenuated in PARP-2-null neurons exposed to a submaximal concentration of 100 micromolar NMDA for 5 minutes, but not in those exposed to a near-maximal toxic concentration of 500 micromolar NMDA. We conclude that PARP-2 contributes substantially to nuclear translocation of AIF and infarct size after transient focal cerebral ischemia in male mice, but that protection is disproportionate to the attenuation of overall PARP activity.
Keywords: cerebral blood flow, iodoantipyrine, middle cerebral artery, NMDA, poly(ADP-ribose) polymers, stroke
Introduction
Oxidative damage to DNA leads to activation of poly(ADP-ribose) polymerase (PARP), which participates in DNA repair. Within the nucleus, PARP-1 and PARP-2 are the major enzymes that contribute to overall PARP activity induced by DNA damage (Ame et al. 1999), and they appear to have specific functions in the DNA repair process (Schreiber et al. 2006; Yelamos et al. 2008). With extensive oxidative damage, as occurs with cerebral ischemia, PARP activity increases substantially, and poly(ADP-ribose) (PAR) polymers accumulate on a variety of proteins (Li et al. 2007). Interestingly, inhibition of PARP activity decreases infarct volume in models of experimental stroke (Abdelkarim et al. 2001; Park et al. 2004; Takahashi et al. 1997). Studies in cell systems demonstrated that over-activation of PARP produces cell death by a mechanism that involves translocation of apoptosis-inducing factor (AIF) from mitochondria to the nucleus (Yu et al. 2002). Furthermore, delivery of PAR polymer of sufficient size can trigger release of AIF from the mitochondria (Andrabi et al. 2006; Yu et al. 2006). Focal cerebral ischemia is associated with an increase in PAR polymer accumulation and translocation of AIF to the nucleus (Eliasson et al. 1997; Li et al. 2007; Endres et al. 1997; Plesnila et al. 2004). Pharmacological inhibition of PARP activity decreases AIF translocation and infarct size (Culmsee et al. 2005), and infarct size is inversely related to the expression of poly(ADP-ribose) glycohydrolase, the enzyme that catabolizes PAR polymers (Andrabi et al. 2006). Thus, a major downstream pathway of focal ischemic damage appears to involve activation of PARP and subsequent PAR-dependent translocation of AIF.
PARP-1-null (PARP-1-/-) neurons exhibit less AIF translocation and cell death in response to NMDA than do wild-type (WT) neurons, and male PARP-1-/- mice have robust neuroprotection from NMDA and focal cerebral ischemia in association with decreased AIF nuclear translocation (Wang et al. 2004; Yuan et al. 2009). However, PARP-2-/- mice also exhibit significant reductions in infarct volume after transient middle cerebral artery (MCA) occlusion (Kofler et al. 2006). This reduction was somewhat unexpected because the contribution of PARP-2 to overall PARP activity is considerably less than that of PARP-1 (Schreiber et al. 2002). The role of PARP-2 in PAR accumulation and AIF translocation after focal ischemia has not been evaluated. Moreover, NMDA excitotoxicity plays a significant role in focal ischemic injury, and PARP-1 contributes to NMDA excitotoxicity (Eliasson et al. 1997; Mandir et al. 2000). However, the contribution of PARP-2 to NMDA excitotoxicity has not been investigated.
In the present study, we tested the hypothesis that the reduction in infarct volume in PARP-1-/- and PARP-2-/- male mice is associated with reductions in PAR accumulation and AIF translocation and that PARP-2-/- neurons are resistant to NMDA toxicity. Because reperfusion initiates rapid formation of PAR polymers, we used a transient focal cerebral ischemia model to examine the time course of PAR formation. We also carried out Western immunoblotting to provide quantitative assessments of PAR and nuclear AIF accumulation. The study was restricted to male mice because female mice treated with a PARP inhibitor and female PARP-1-/- mice are not further protected from cerebral ischemia beyond the effects of gender (Hagberg et al. 2004; McCullough et al. 2005) despite decreases in AIF translocation (Yuan et al. 2009). In previous work that showed protection from focal ischemia in male PARP-2-/-, infarct volume was measured at 1 day of reperfusion (Kofler et al. 2006). In the present study, we extended reperfusion to 3 days to determine if protection was sustained. Lastly, the location of the border region between the MCA and the anterior cerebral artery (ACA) was delineated as a measure of the tissue at risk, and regional cerebral blood flow (CBF) during focal ischemia was quantified to determine if differences in infarct volume could be attributed to differences in perfusion between PARP-null mice and their respective WT controls.
Methods
Transient focal cerebral ischemia model
All experiments were conducted in accordance with the guidelines of the National Institutes of Health guide for the care and use of animals in research and were approved by the Johns Hopkins University Animal Care and Use Committee. PARP-1-/- mice were bred on a Sv129 mouse background, and PARP-2-/- mice were backcrossed with C57Bl/6 mice (Eliasson et al. 1997; Kofler et al. 2006; Mandir et al. 2000). Adult male mice (20 to 28 g) were anesthetized with 1.5–2% isoflurane. Body temperature was maintained at 37°C by a feedback-controlled heating system. Focal cerebral ischemia was induced with the intraluminal filament technique and Laser-Doppler flow (LDF) was monitored over the lateral parietal cortex. (Li et al. 2007; Mito et al. 2009). A small incision was made in the scalp, and a 1-mm LDF probe was secured against the lateral temporal bone to assess the adequacy of vascular occlusion during the first 10 min of ischemia. To produce focal ischemia, a midline ventral incision was made in the neck, the common carotid artery was occluded with a 6-0 silk suture, the external carotid artery was ligated and cut, and a 7-0 monofilament was passed through the cut stump of the external carotid and into the internal carotid artery. The filament tip was enlarged by coating with silicone glue. The filament tip was advanced until stable reductions in LDF were obtained (approximately 6 mm past the bifurcation of the internal carotid and pterygopalantine arteries). Incisions were closed with suture and isoflurane was discontinued. In most experiments, the mice were briefly re-anesthetized with isoflurane after 2 hours of ischemia and the neck incision was re-opened. To allow reperfusion, the filament in the internal carotid artery was withdrawn and the suture around the common carotid artery was untied. The incision was then closed with suture, and isoflurane was discontinued. Infarct volume was evaluated 3 days after 2-h of ischemia by standard volumetric analysis of five 1-mm thick coronal sections stained with 1.5% 2,3,5-triphenyltetrazolium chloride, with correction for swelling. Infarct volume was measured on 9 WT and 10 PARP-1-/- brains and on 12 WT and 10 PARP-2-/- brains.
Location of MCA–ACA border region
To visualize the line of anastomoses between the MCA and ACA distribution regions, mice were anesthetized with pentobarbital (50 mg/kg, ip) and perfused with a black latex suspension (Maeda et al. 1998). A midline incision was made, and papaverine hydrochloride (50 mg/kg) was injected into the liver to produce maximal vasodilation and to minimize cerebrovascular resistance. A 20-gauge intracatheter was inserted through the left ventricular wall with the tip placed into the aorta, and the right atrium was incised to allow outflow. A solution of warmed 0.9% NaCl was infused first to remove blood, and then a warmed black latex suspension was infused until the suspension was observed to flow freely from the right atrium. Next, the mouse was placed in ice for 15 min and decapitated. Finally, the brain was removed, fixed in 10% formalin, and photographed. The line of anastomoses between the MCA and ACA territories was delineated, and the distance of this line from the midline was measured at 1-mm intervals from the frontal pole. Measurements were made on 6 WT and 6 PARP-1-/- brains and on 8 WT and 7 PARP-2-/- brains.
Regional CBF
At 1.5 h of ischemia, mice were anesthetized with isoflurane, the femoral artery was catheterized with a 15-cm length of PE-10 tubing and the femoral vein was catheterized with a 10-cm length of PE-10 tubing. Arterial blood pressure was monitored and an arterial blood sample was obtained for analysis of blood gases, pH and hemoglobin concentration. At 2 h of ischemia, regional CBF was determined by infusing 4 μCi of [14C]-iodoantipyrine intravenously at a rate of 108 μL/min and sampling arterial blood at 5-s intervals to obtain the arterial input function. The brain was rapidly harvested after 45 s of infusion and was frozen for later cutting on a cryostat into 20-μm coronal sections for autoradiography. Optical density measurements were made on triplicate sections at 1-mm increments from +2 through −3 mm from bregma and regional CBF was calculated (Sakurada et al. 1978) using image analysis software (Inquiry Image Analysis, Loats Associates, Westminister, MD). Measurements of regional CBF and the volume of tissue with different ranges of CBF (Goto et al. 2003; Mito et al. 2009) were made on 6 WT and 7 PARP-1-/- brains and on 7 WT and 5 PARP-2-/- brains. Regions of interest included dorsal, dorsolateral, and lateral cortex corresponding to cortical tissue approximately 10%, 33%, and 100%, respectively, along the perimeter of the dorsolateral quadrant of cerebral cortex.
Nuclear fractionation
Brains were rapidly harvested 24 h after sham surgery or focal ischemia. After removing the anterior and posterior poles at approximately +2.2 mm and −4.9 mm from bregma, hemispheres (including cortex and striatum) from two mice were pooled together to obtain a sufficient highly purified nuclear pellet. A sucrose density-gradient centrifugation method was used to separate nuclear and mitochondrial fractions as described (Li et al. 2007). Briefly, freshly harvested tissue was homogenized in a handheld Teflon-coated glass homogenizer on ice with 10 ml of ice-cold buffer A (250 mM sucrose, 10 mM HEPES at pH 7.4, 1 mg/mL bovine serum albumin, 0.5 mM EDTA, 0.5 mM EGTA), and then centrifuged at 2,000 g for 3 min at 4°C. The loose pellet was homogenized with a Dounce homogenizer in buffer A followed by suspension in 4 mL of buffer B (2 M sucrose, 5 mM Mg-acetate, 0.1 mM EDTA, 10 mM Tris-HCl at pH 8.0, 1 mM dithiothreitol). This suspension was placed in an ultracentrifuge tube on top of 4.4 mL of buffer B, followed by the addition of 2–3 mL of buffer C [320 mM sucrose, 3 mM CaCl2, 2 mM Mg-acetate, 0.1 mM EDTA, 10 mM Tris-HCl at pH 8.0, 1 mM dithiothreitol, and 0.5% octylphenoxypolyethoxyethanol (IGEPAL® CA-630)] for balance. The sample underwent ultracentrifugation at 30,000 g for 45 min at 4°C. The pellet containing the nuclear fraction was resuspended in glycerol storage buffer and stored at −80°C.
Immunoblotting
Standard Western immunoblotting techniques were performed. Proteins were separated by 4–12% Tris-glycine gels, transferred to nitrocellulose membranes, and revealed with 62 kDa rabbit anti-AIF, rabbit anti-Mn superoxide dismutase (SOD) (Yu et al. 2002), sheep anti-histones (US-Biological, Swampscott MA, USA), mouse anti-actin (Sigma-Aldrich, St. Louis MO, USA), anti-PARP-1 antibody (Cell Signaling, Beverly, MA, USA), anti-PARP-2 antibody (Alexis, Lausen, Switzerland), and rabbit anti-poly(ADPribose) polyclonal antibody to poly(ADP-ribose) (LP96-10; BD Biosciences Pharmingen); horseradish peroxidase-conjugated anti-rabbit (BIO-RAD, Hercules CA, USA) and anti-sheep (Pierce, Rockford IL, USA) secondary antibodies were incubated for 1 h at room temperature and detected with ECL reagents. After detection, the films were quantified with Imagequant software (Molecular Dynamics, Sunnyvale CA, USA).
For immunoblots of PAR, striatal samples were dissected from mice at 20 min, 1 h, 4 h, or 24 h of reperfusion after 2 h of ischemia. Samples were homogenized in 200 μl NuPAGE LDS Sample Buffer (Invitrogen, Carlsbad CA, USA). Equal samples were loaded onto the gels for Western immunoblotting. Anti-human β–III tubulin antibody (Chemicon, Temecula CA, USA) was used as a protein loading control. For each of the four genotypes, samples from different mice were used on five independent gels probed for PAR at each of the four time points after ischemia and at 24 h after sham surgery and anesthesia. Thus, 25 mice of each genotype were used to obtain 5 lanes (5 time points) for 5 gels, and all of the PAR data required a total of 100 mice.
For AIF immunoblots, nuclear protein from two hemispheres were pooled for each lane, and samples from different mice were used on four or five independent gels to determine nuclear AIF at 24 h after sham surgery and ischemia. Four gels were used for the WT and PARP-1-/- comparisons, which represents the results from 16 WT and 16 PARP-1-/- mice (2 mice/lane × 2 time points × 4 gels). Five gels were used for the WT and PARP-2-/- comparisons, which represents the results from 20 WT and 20 PARP-1-/- mice (2 mice/lane × 2 time points × 5 gels). Thus, another 72 mice were used to generate the AIF data.
To test for altered expression of PARP-1 and PARP-2 in each genotype, immunoblots were performed on nuclear fractions from non-ischemic brains from 4 WT and 4 PARP-1-/- brains and from 4 WT and 4 PARP-2-/- brains.
NMDA toxicity in primary neuronal cultures
Primary cortical cell cultures were prepared from PARP-2 WT, PARP-2-/-, and PARP-1-/- mouse embryos at gestational day 15 as previously described (Gonzalez-Zulueta et al. 1998). Experiments were carried out at 14 days in vitro. Under these conditions, neurons represent 90% of the cells in the culture. Mature neurons were treated with NMDA (100 or 500 μM for 5 min) and 10 μM glycine in control salt solution (CSS) containing 120 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 25 mM Tris-HCl (pH 7.4) and 15 mM D-glucose. Control cultures received CSS alone for 5 min. Following 5 min of NMDA or CSS treatment, the cells were washed with CSS and re-supplemented growth medium. At 24 h after CSS/NMDA exposure, cell death was assessed with 5 μM Hoechst 33342 and 2 μM propidium iodide (Invitrogen) to stain live and dead cells, respectively. Cell death was quantified with a mechanized stage Zeiss microscope (Axiovert 200M) coupled with automated computer-assisted counting software Axiovision (Carl Zeiss, Jena, Germany). Percent cell death was determined as the ratio of live-to-dead cells compared with the percent cell death in control wells. Glial nuclei fluoresce at a lower intensity than neuronal nuclei and were gated out. Thus, the procedure largely eliminates glial cell death, which is not distinguished in the commonly used lactate dehydrogenase assay.
Statistical analysis
Comparisons of PAR and AIF optical densities between null mice and their respective WT controls at different times were made by ANOVA and the Newman-Kuels multiple range test. Comparisons of infarct volume, blood flow, and the line of anastomoses location were made between PARP-null mice and their respective WT controls by t-test. Comparisons of the percent cell death after NMDA exposure between WT neurons and PARP-1-/- and PARP-2-/- neurons at each dose were made by ANOVA and the Newman-Kuels multiple range test. Data are presented as mean ± SD.
Results
PARP-1 and PARP-2 expression
To test if gene deletion of PARP-2 upregulated PARP-1 expression or if gene deletion of PARP-1 upregulated PARP-2 expression, immunoblots were performed on the nuclear fractions of WT and null mouse brains. Expression of PARP-1 was similar in WT and PARP-2-/- brains (Fig.1). Likewise, expression of PARP-2 was similar in WT and PARP-1-/- brains. Thus, gene deletion of one isoform did not result in a substantial upregulation of the other isoform.
Fig. 1.

Western immunoblot for PARP-1, PARP-2, and histones in nuclear fractions from brains of 4 PARP-1-/- and 4 PARP-1 WT mice and of 4 PARP-2-/- and 4 PARP-2 WT mice. No major differences were apparent in PARP-1 expression between WT and PARP-2-/- brains or in PARP-2 expression between WT and PARP-1-/- brains.
MCA distribution
Latex casts of pial artery anatomy were used to visualize the border of the MCA distribution territory. The distance from the midline to the line of anastomoses was measured (Fig. 2). No significant difference was evident in the distance at any coronal level between PARP-1-/- or PARP-2-/- and its WT counterpart.
Fig. 2.
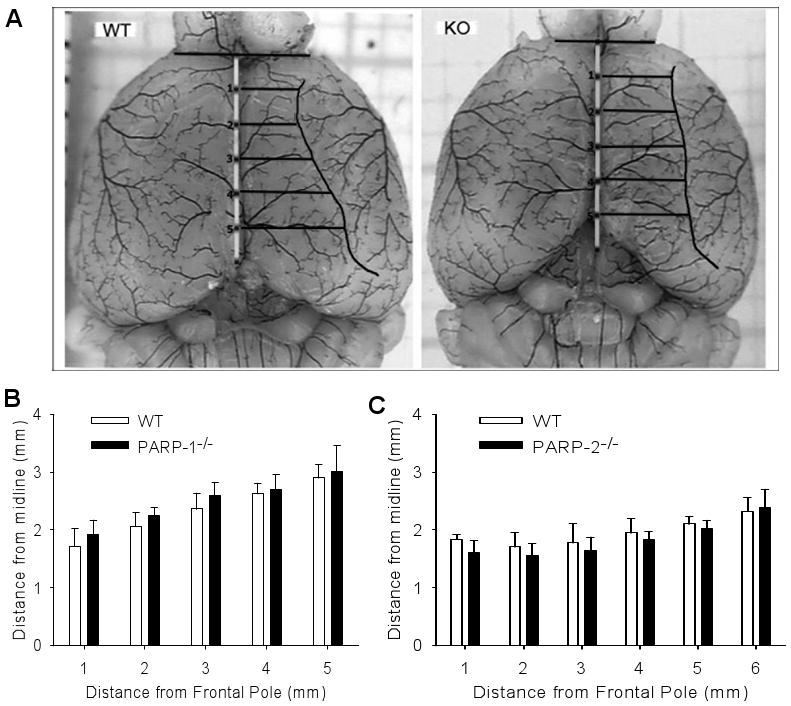
A. Example of latex-perfused WT and PARP-1-/- (KO) brains for delineating distance from midline to site of anastomoses between middle cerebral artery and anterior cerebral artery distribution regions. To pool data among mice, distances were interpolated at 1-mm increments caudal from the frontal pole. B. Mean distance (± SD) to the line of anastomoses in 6 WT and 6 PARP-1-/- brains. C. Mean distance to the line of anastomoses in 8 WT and 7 PARP-2-/- brains. No significant differences were seen between PARP-null mice and the respective WT mice.
Regional CBF
Autoradiograms revealed widespread reductions of regional CBF at 2 h of ischemia in all genotypes (Fig. 3). Analysis of CBF in dorsal cortex (ACA territory), dorsolateral cortex (MCA-ACA border region), dorsolateral tissue underlying cortex, lateral cortex (cortical ischemic core), and ventrolateral striatum did not show significant differences between WT and PARP-1-/- mice or between WT and PARP-2-/- mice in the hemisphere ipsilateral to the occlusion (Fig. 4). CBF averaged over the entire non-ischemic contralateral hemisphere was similar in PARP-null mice and the corresponding WT mice. Interestingly, contralateral values in PARP-1-/- mice backcrossed with Sv129 mice were 59% greater than those in PARP-2-/- mice backcrossed with C57Bl/6 mice. Likewise, contralateral values in PARP-1 WT mice were 52% greater than those in PARP-2 WT mice. Arterial blood gases, pH, and blood pressure were similar between WT and PARP-1-/- mice and between WT and PARP-2-/- mice (Table 1).
Fig. 3.
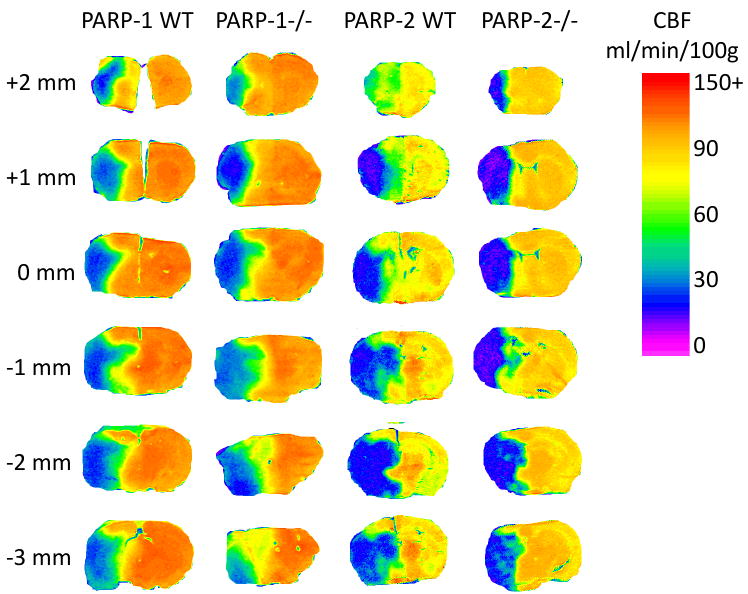
Representative pseudo-color images of cerebral blood flow (CBF) at 6 coronal levels relative to bregma for PARP-1 WT and null brains and for PARP-2 WT and null brains at 2 h of ischemia.
Fig. 4.
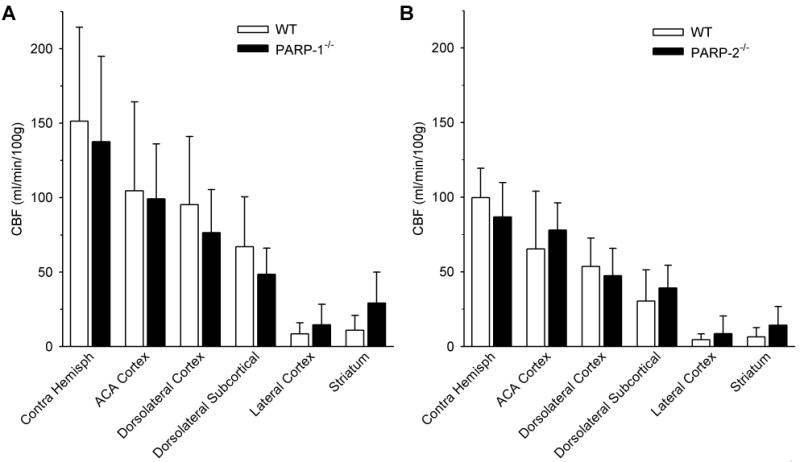
Mean regional cerebral blood flow (CBF; ±SD) at 2 h of ischemia in 6 WT and 7 PARP-1-/- mice (A) and in 7 WT and 5 PARP-2-/- mice (B). Values from dorsal cortex, dorsolateral cortex, dorsolateral subcortical tissue, lateral cortex, striatum, and entire contralateral hemisphere (Contra Hemisph) were not significantly different between PARP-null mice and the corresponding WT mice.
Table 1.
Physiological parameters prior to regional cerebral blood flow measurements during ischemia
| Parameter | PARP-1 WT | PARP-1-/- | PARP-2 WT | PARP-2-/- |
|---|---|---|---|---|
| MABP (mmHg) | 78±5 | 75±6 | 80±4 | 81±9 |
| Temperature (°C) | 36.9±0.4 | 36.9±0.3 | 37.0±0.3 | 36.9±0.3 |
| pH | 7.37±0.04 | 7.31±0.06 | 7.33±0.04 | 7.30±0.07 |
| PCO2 (mmHg) | 51±4 | 56±8 | 49±5 | 53±5 |
| PO2 (mmHg) | 143±10 | 156±34 | 149±16 | 157±39 |
| Hemoglobin (g/dL) | 13.1±1.1 | 14.2±2.1 | 13.4±1.6 | 14.4±1.4 |
MABP, mean arterial blood pressure
The distribution of intraischemic CBF also was analyzed by calculating the volume of tissue with different ranges of CBF summed over coronal sections between +2 and −3 mm from bregma (Fig. 5A and B). Two-way ANOVA did not indicate a significant interaction between the volume of tissue with different ranges of CBF and genotype for WT and PARP-1-/- mice. However, a significant interaction was detected between WT and PARP-2-/- mice. PARP-2-/- mice had a somewhat greater volume of tissue in the CBF ranges of 50–60 and 60–70 mL/min/100 g (P < 0.05). As expected, the volumetric distribution of CBF in the hemisphere ipsilateral to the occlusion differed markedly from the distribution in the contralateral hemisphere for each genotype (Fig. 5C and D). However, the contralateral flow distribution was similar in WT and PARP-1-/- mice and in WT and PARP-2-/- mice.
Fig. 5.
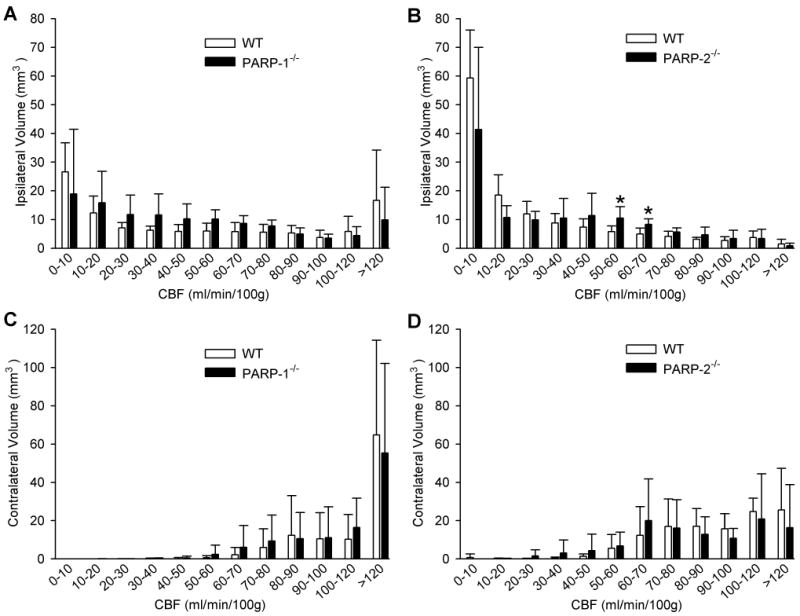
Mean volume of tissue (±SD) between +2 mm and −3 mm from bregma with CBF in discrete ranges in PARP-1 WT and PARP-1-/- mice ipsilateral (A) and contralateral (C) to arterial occlusion and in PARP-2 WT and PARP-2-/- mice ipsilateral (B) and contralateral (D) to arterial occlusion. * P < 0.05 from WT.
Infarct volume
At 10 min of occlusion, LDF measured over lateral cortex was decreased to 20 ± 5% of baseline in WT mice and to 22 ± 9% of baseline in PARP-1-/- mice. After discontinuing anesthesia, neurologic deficits were scored on a 0–4 scale (Zeynalov et al. 2006). The deficits were similar in WT (2.4 ± 0.4) and PARP-1-/- (2.4 ± 0.2) mice during occlusion, consistent with a similar ischemic insult between groups. After 2 h of ischemia and 3 days of reperfusion, infarct volume was 78% smaller in cerebral cortex, 75% smaller in striatum, and 74% smaller in the cerebral hemisphere of PARP-1-/- mice than in their WT counterparts (Fig. 6C). The decrease in infarct volume was significant at all coronal levels except the most posterior level where infarction was small and variable (Fig. 6A).
Fig. 6.
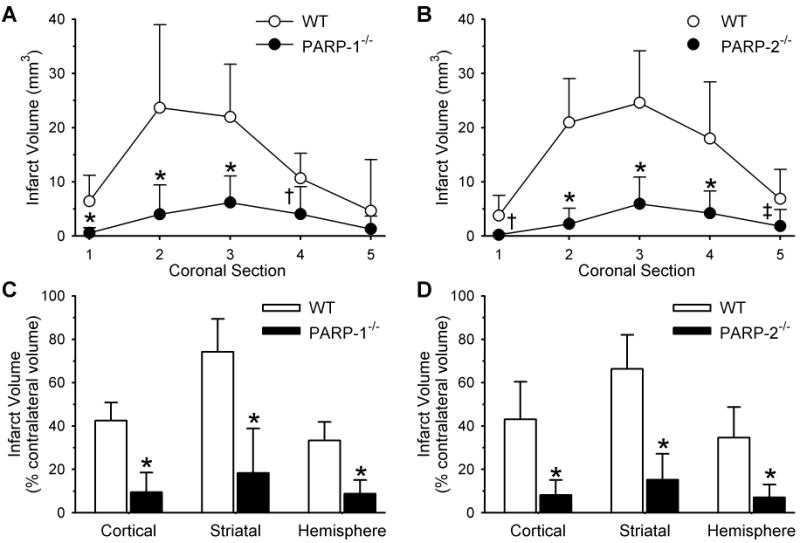
Infarct volume (mm3; mean±SD) of individual coronal sections (1 anterior, 5 posterior) in 9 WT and 10 PARP-1-/- mice (A) and in 12 WT and 10 PARP-2-/- mice (B). Infarct volume (% of contralateral structure) in cerebral cortex, striatum, and the entire hemisphere for WT and PARP-1-/- mice (C) and WT and PARP-2-/- mice (D). * P < 0.001 from WT; † P < 0.01 from WT; ‡ P < 0.02 from WT.
Likewise, LDF was decreased to a similar extent in WT (23 ± 9% of baseline) and PARP-2-/- (27 ± 5% of baseline) mice and the neurological deficit scores were not different (2.5 ± 0.2 for both genotypes). Furthermore, PARP-2-/- mice had infarct volumes that were 81% smaller in cerebral cortex, 77% smaller in striatum, and 80% smaller in the hemisphere than those of WT mice (Fig. 6D). The reduction in infarct volume was consistent at all coronal levels (Fig. 6B).
PAR accumulation
As in previous work (Li et al. 2007), Western blots showed that PAR was increased in PARP-1 WT striatum at 20 min and 1 h of reperfusion and gradually recovered to baseline levels from 4 to 24 h of reperfusion (Fig. 7). In PARP-1-/- mice, PAR expression remained close to values obtained in sham-operated animals. Values in PARP-1-/- mice were significantly less than those in WT mice throughout 24 h of reperfusion. In PARP-2 WT mice, robust increases in PAR occurred at 20 min and 1 h of reperfusion. In PARP-2-/- mice, PAR increased during reperfusion, but the increase was significantly less than that in WT mice at 1 h of reperfusion.
Fig. 7.
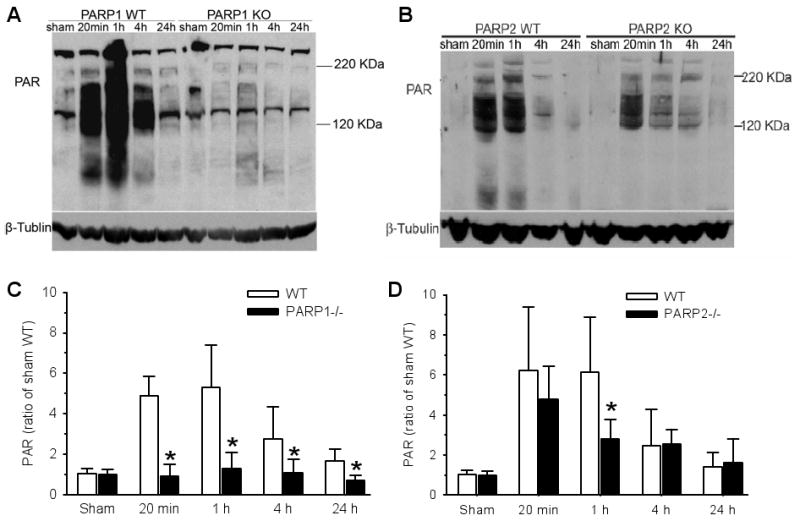
Examples of PAR immunoblots from WT and PARP-1-/- striatum (A) and WT and PARP-2-/- striatum (B) after sham surgery or 2 h ischemia + 20 min, 1, 4, or 24 h of reperfusion. The increase in PAR during early reperfusion was largely attenuated in PARP-1-/- mice (C; ±SD; n=5 independent gels) and moderately attenuated at 1 h of reperfusion in PARP-2-/- mice (D; n=5). Separate mice were used for each gel. * P < 0.05 from WT.
Nuclear AIF accumulation
Previous work showed that AIF progressively increased in the nucleus over a 24-h period of reperfusion (Li et al. 2007). Accordingly, we made comparisons between groups at 24 h in the current study. Western blots of the nuclear fraction indicated an increase in AIF immunoreactivity at 24 h of reperfusion in both WT groups (Fig. 8). The increase in nuclear AIF was significantly reduced in both PARP-1-/- and PARP-2-/- mice compared to their WT counterparts.
Fig. 8.
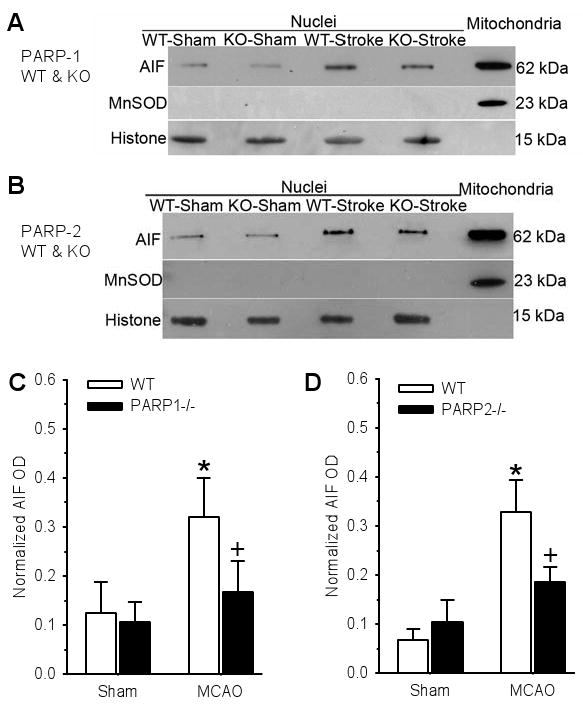
Example of Western blots of AIF, the mitochondrial marker Mn superoxide dismutase (MnSOD), and histone (loading control) from WT and PARP-1 knockout (KO) mice (A) and from WT and PARP-2 KO mice (B). Lanes 1–4 are nuclear fractions obtained from two cerebral hemispheres of sham-operated mice and from two ischemic hemispheres of WT and PARP-1-/- (KO) mice subjected to 2-h ischemia and 1-day reperfusion. Lane 5 is the mitochondrial fraction from naïve mouse brain used as an internal standard for AIF on each gel. Normalized nuclear AIF (±SD) from WT and PARP-1-/- mice (C; n=4 gels) and from WT and PARP-2-/- mice (D; n=5 gels) showed significant reduction in nuclear AIF accumulation at 24 h after middle cerebral artery occlusion (MCAO) in both PARP-1-/- and PARP-2-/- post-ischemic brains. Separate mice were used for each gel. * P < 0.05 from sham; + P < 0.05 from WT.
NMDA toxicity
Exposure of primary cortical neurons to NMDA produced dose-dependent cell death in PARP-2 WT neurons (Fig. 9). The magnitude of cell death was significantly lower in PARP-1-/- neurons in response to 100 and 500 μM NMDA and was significantly lower in PARP-2-/- neurons in response to 100 μM NMDA (P = 0.012). Protection in PARP-2-/- neurons was lost at the higher concentration of 500 μM NMDA.
Fig. 9.
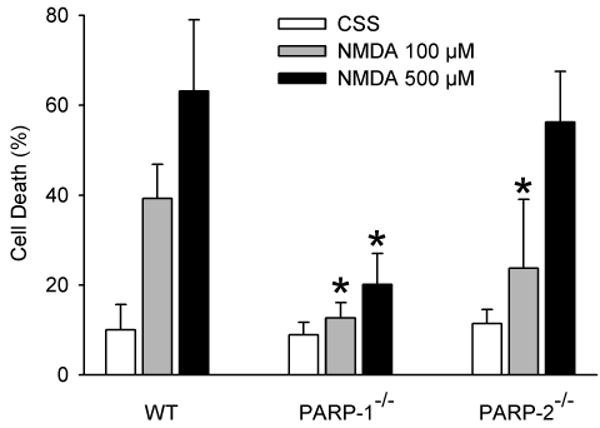
Percent of dead cells in primary neuronal cultures (±SD; n = 5 independent culture experiments) of PARP-2 WT, PARP-1-/-, and PARP-2-/- neurons 24 h after a 5-min exposure to controlled saline solution (CSS), 100 μM NMDA, or 500 μM NMDA. * P < 0.05 from WT at same NMDA concentration by ANOVA and the Newman-Keuls test.
Discussion
Previous studies have shown that infarct volume is reduced at 1 day of reperfusion in PARP-1-/- and PARP-2-/- mice compared with WT mice (Eliasson et al. 1997; Goto et al. 2002; Kofler et al. 2006). Our current work extends these findings by demonstrating that the reduction in infarct volume remains robust at 3 days of reperfusion without major differences in the MCA distribution territory or intraischemic blood flow between each of the PARP isoform-null mice and their respective WT mice. The principal new findings are 1) that PAR accumulation is largely suppressed throughout 24 h of reperfusion in PARP-1-/- mice but only moderately attenuated at 1 h of reperfusion in PARP-2-/- mice, and 2) that AIF accumulation in the nucleus is substantially lower in both PARP-1-/- and PARP-2-/- mice than in WT mice.
The large suppression of PAR accumulation in PARP-1-/- mice and the modest suppression in PARP-2-/- mice are consistent with the concept that PARP-1 comprises the majority of PARP activity (Schreiber et al. 2002). Earlier work showed that activation of PARP activity by direct damage to DNA within an alkylating agent or indirectly by NMDA-induced excitotoxicity produces cell death that is dependent on AIF translocation from the mitochondria to the nucleus (Wang et al. 2004; Yu et al. 2002). Subsequent work revealed that PAR was a critical factor in mediating the release of AIF from the mitochondria (Andrabi et al. 2006; Yu et al. 2006). Thus, the results in PARP-2-/- mice were somewhat unexpected in that the reduction in nuclear AIF and infarct volume were disproportionately greater than the reduction in PAR accumulation.
Several points need to be considered for understanding this apparent nonlinear relationship between PAR accumulation and AIF translocation. PARP activity generates polymers of different sizes and branching complexity. The PAR antibody does not discriminate the size or complexity of the polymer. Previously it was reported that large, complex polymers are the most potent for stimulating release of AIF (Andrabi et al. 2006). In addition, a variety of proteins are poly-ADP-ribosylated by PARP-1 and PARP-2, and these target proteins do not appear to be identical for each PARP isoform (Yelamos et al. 2008). It is likely that only one or a select subset of these proteins acts as a key PAR binding partner in signaling AIF release. Our analysis of PAR blots, which integrated optical density over a wide range of molecular weights, did not discriminate the molecular weight of the individual PAR polymer or binding partner. For these reasons, the amount of AIF translocation integrated over 24 h may not be strictly proportional to the total PAR immunoreactivity measured at discrete time points.
Another interesting finding is that the reductions in AIF translocation and infarct volume in PARP-2-/- mice were approximately as great as those seen in PARP-1-/- mice. The fact that the reduction was greater than 50% in both PARP-1-/- and PARP-2-/- mice implies that each isoform exerts a non-additive interaction on AIF translocation and infarct volume. PARP-1 and PARP-2 form catalytically active homodimers and heterodimers (Schreiber et al. 2002), and colocalization of PARP-1 and PARP-2 is particularly evident in the nucleolus (Meder et al. 2005). One possibility is that PARP-1 and PARP-2 heterodimers poly-ADP-ribosylate a specific binding partner with a large-size polymer that is selective for triggering AIF release. Another consideration is that PARP-1 and PARP-2 can selectively interact with specific proteins (Bai et al. 2007) and play a role in chromatin modifications (El Ramy et al. 2009; Quenet et al. 2009). Thus, it is conceivable that PARP-2 could influence cell death by interactions independent of PAR generation. Consequently, several explanations can be offered for why infarct volume and nuclear AIF are markedly reduced in PARP-2-/- mice with only a moderate decrease in overall PAR availability and why PARP-1 and PARP-2 appear to act in a non-additive fashion during focal cerebral ischemia.
It should also be appreciated that because the PARP-1-/- mice were bred on a Sv129 background and the PARP-2-/- mice were bred on a C57Bl/6 background, quantitative comparisons of infarct volume between PARP-1-/- and PARP-2-/- mice can not strictly be made. Moreover, the Western blot experiments were designed to compare null mice with their respective WT and not to directly compare null mice with each other. Nevertheless, the large reduction in infarct volume and AIF translocation in each null genotype compared to their respective WT indicates that each PARP isoform makes a major contribution. We did not test a PARP inhibitor for residual effects of PARP activity in the null mice because the reductions were already large and statistically detecting further reductions in infarct volume and nuclear AIF would have required a very large sample size. Furthermore, combined gene deletion of PARP-1 and PARP-2 is embryonically lethal (Menissier et al. 2003). PARP-1 and PARP-2 are the two major isoforms that respond to DNA strand breaks. Because we did not observe a compensatory increase in the expression of the other isoform in either the PARP-1-/- or PARP-2-/- brain, the difference in PAR accumulation between WT and null mice reflects the loss of that isoform rather than a change in expression of the other isoform. Another point to consider is that a small increase in AIF translocation in PARP-1-/- mice observed on some gels despite the marked reduction in PAR accumulation could be due to non-PAR–dependent triggering mechanisms of AIF release.
We found that the distance from the midline to the line of MCA-ACA anastomoses did not differ between WT and knockout mice. This finding suggests that the volume at risk was not influenced by the loss of each PARP isoform. This line of anastomoses shifts further from the midline in SV129 mice than in C57Bl/6 mice (Maeda et al. 1998). Because the PARP-1-/- mice were bred on an SV129 background, the finding that PARP-1-/- mice had a greater distance to the line of anastomoses than did the PARP-2-/- mice, which were backcrossed with C57BL/6 mice, was expected.
The level of CBF did not significantly differ between the PARP-null mice and the respective WT mice at 2 h of ischemia in lateral cortex or striatum, where ischemia was severe, or in dorsolateral cortex or subcortex, where ischemia was moderate. Although there was some variability in the CBF measurements among mice in each cohort that may have prevented small differences in CBF from being detected, large differences in CBF that would have accounted for the large reduction in infarct volume in each of the PARP-null mice should have been detectable with this technique. Moreover, levels of CBF <50 mL/min/100 g are associated with impaired protein synthesis and eventual infarction in rats (Jacewicz et al. 1992; Mies et al. 1991). Mice, which have normal CBF at least as great as rats, would be expected to have an ischemic threshold in approximately the same range as rats. Volumetric analysis did not reveal significant differences in the amount of tissue with CBF in any of the ranges below 50 mL/min/100 g during ischemia. Nevertheless, there was a tendency for the amount of tissue with CBF <20 ml/min/100g to be smaller in PARP-2-/- mice than in WT mice, whereas the amount of tissue with levels of CBF in the 50–60 and 60–70 mL/min/100 g range was statistically greater in PARP-2-/- mice than in WT mice. However, the statistically significant difference in tissue volume over the 50–70 ml/min/100g range amounted to a total of only 8 mm3, which is only a fraction of the 50 mm3 difference in infarct volume between WT and PARP-2-/- mice. Although we cannot completely exclude improved perfusion at 2 h of occlusion in PARP-2-/- mice as one explanation for the reduced infarct volume, improved perfusion is unlikely to be the major reason for tissue protection.
Comparison of CBF between PARP-1-/- and PARP-2-/- mice was not a primary endpoint. However, CBF in the non-ischemic hemispheres was noted to be 52% higher in PARP-1 WT than in PARP-2 WT mice and 58% higher in PARP-1-/- mice than in PARP-2-/- mice. Interestingly, the contralateral CBF in the PARP-1 WT on the Sv129 background is similar to the 166 ml/min/100g value previously obtained in C57Bl/6 mice not subjected to ischemia (Mito et al. 2009). Thus, one possible explanation for the low CBF in the hemisphere contalateral to the vascular occlusion of the PARP-2 WT mice on a C57Bl/6 background is that contralateral CBF is more greatly reduced by transcallosal diaschisis in C57Bl/6 mice.
NMDA excitotoxicity is a major factor that contributes to focal ischemic injury and PARP-dependent neuronal cell death. NMDA is known to exert less toxicity in PARP-1-/- neurons than in PARP-1 WT neurons (Eliasson et al. 1997; Wang et al. 2004). Although PARP-1-/- neurons were not directly compared to their own WT neurons in the present experiment, we have found no differences in NMDA toxicity among neurons from these different background strains, and the degree of injury obtained in PARP-2 WT neurons in the present study is similar to that previously obtained in PARP-1 WT neurons (Mandir et al. 2000). Moreover, the low number of dead PARP-1-/- neurons exposed to 500 μM NMDA for 5 min is similar to that previously observed (Eliasson et al. 1997; Wang et al. 2004). Thus, the results are consistent with PARP-1 acting as a principal contributor to NMDA excitotoxicity. The present study demonstrated that NMDA toxicity also is attenuated in PARP-2-/- neurons compared to their WT neurons at a concentration of 100 μM NMDA, which exerts moderate toxicity. However, protection was lost at the higher concentration of 500 μM, which exerts near maximal toxicity. These observations indicate that PARP-2 has the potential to contribute directly to moderate excitotoxic neuronal injury in vivo.
In conclusion, this study provides further evidence that PARP activation and translocation of AIF from the mitochondria to the nucleus comprise a major pathway of injury in transient focal cerebral ischemia in male subjects. Despite the smaller contribution of PARP-2 to overall PARP activity, PARP-2 plays a role in the injury process that is comparable to PARP-1. This role for PARP-2 may be specific for focal ischemia because, unlike PARP-1, the hippocampus in PARP-2-/- mice remains vulnerable to delayed cell death from global cerebral ischemia (Kofler et al. 2006). Because calpain appears to play a role in causing AIF release from the mitochondria in hippocampus after global ischemia (Cao et al. 2007) but not focal ischemia (Wang et al. 2009), the precise mechanisms for triggering AIF release may depend on the nature of the ischemic stimulus.
Acknowledgments
This work was supported by grants from the National Institute of Neurological Diseases and Stroke (NS039148 and NS067525). Xiaoling Li was the recipient of a Scientist Development Grant from the American Heart Association. Shaida A. Andrabi was the recipient of a Postdoctoral Fellowship Grant from the American Heart Association. The authors are grateful to Jennifer Mytar for her technical assistance and Claire Levine for her editorial assistance.
Abbreviations used
- ACA
anterior cerebral artery
- AIF
apoptosis-inducing factor
- CBF
cerebral blood flow
- CSS
control salt solution
- LDF
laser-Doppler flow
- MCA
middle cerebral artery
- PAR
poly(ADP-ribose) polymer
- PARP
poly(ADP-ribose) polymerase
- WT
wild-type
Footnotes
The authors have no competing financial interests.
References
- Abdelkarim GE, Gertz K, Harms C, Katchanov J, Dirnagl U, Szabo C, Endres M. Protective effects of PJ34, a novel, potent inhibitor of poly(ADP-ribose) polymerase (PARP) in in vitro and in vivo models of stroke. Int J Mol Med. 2001;7:255–260. [PubMed] [Google Scholar]
- Ame JC, et al. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J Biol Chem. 1999;274:17860–17868. doi: 10.1074/jbc.274.25.17860. [DOI] [PubMed] [Google Scholar]
- Andrabi SA, et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A. 2006;103:18308–18313. doi: 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai P, Houten SM, Huber A, Schreiber V, Watanabe M, Kiss B, de Murcia G, Auwerx J, Menissier-de Murcia J. Poly(ADP-ribose) polymerase-2 controls adipocyte differentiation and adipose tissue function through the regulation of the activity of the retinoid X receptor/peroxisome proliferator-activated receptor-gamma heterodimer. J Biol Chem. 2007;282:37738–37746. doi: 10.1074/jbc.M701021200. [DOI] [PubMed] [Google Scholar]
- Cao G, Xing J, Xiao X, Liou AK, Gao Y, Yin XM, Clark RS, Graham SH, Chen J. Critical role of calpain I in mitochondrial release of apoptosis-inducing factor in ischemic neuronal injury. J Neurosci. 2007;27:9278–9293. doi: 10.1523/JNEUROSCI.2826-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culmsee C, Zhu C, Landshamer S, Becattini B, Wagner E, Pellechia M, Blomgren K, Plesnila N. Apoptosis-inducing factor triggered by poly(ADP-Ribose) polymerase and bid mediates neuronal cell death after oxygen-glucose deprivation and focal cerebral ischemia. J Neurosci. 2005;25:10262–10272. doi: 10.1523/JNEUROSCI.2818-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Ramy R, et al. Functional interplay between Parp-1 and SirT1 in genome integrity and chromatin-based processes. Cell Mol Life Sci. 2009;66:3219–3234. doi: 10.1007/s00018-009-0105-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliasson MJL, et al. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nature Med. 1997;3:1089–1095. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- Endres M, Wang ZQ, Namura S, Waeber C, Moskowitz MA. Ischemic brain injury is mediated by the activation of poly(ADP- ribose)polymerase. J Cereb Blood Flow Metab. 1997;17:1143–1151. doi: 10.1097/00004647-199711000-00002. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Zulueta M, Ensz LM, Mukhina G, Lebovitz RM, Zwacka RM, Engelhardt JF, Oberley LW, Dawson VL, Dawson TM. Manganese superoxide dismutase protects nNOS neurons from NMDA and nitric oxide-mediated neurotoxicity. J Neurosci. 1998;18:2040–2055. doi: 10.1523/JNEUROSCI.18-06-02040.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto S, Sampei K, Alkayed NJ, Doré S, Koehler RC. Characterization of a new double-filament model of focal cerebral ischemia in heme oxygenase-2-deficient mice. Am J Physiol Regul Integr Comp Physiol. 2003;285:R222–R230. doi: 10.1152/ajpregu.00067.2003. [DOI] [PubMed] [Google Scholar]
- Goto S, et al. Poly (ADP-ribose) polymerase (PARP-1) impairs early and long-term experimental stroke therapy. Stroke. 2002;33:1101–1106. doi: 10.1161/01.str.0000014203.65693.1e. [DOI] [PubMed] [Google Scholar]
- Hagberg H, et al. PARP-1 gene disruption in mice preferentially protects males from perinatal brain injury. J Neurochem. 2004;90:1068–1075. doi: 10.1111/j.1471-4159.2004.02547.x. [DOI] [PubMed] [Google Scholar]
- Jacewicz M, Tanabe J, Pulsinelli WA. The CBF threshold and dynamics for focal cerebral infarction in spontaneously hypertensive rats. J Cereb Blood Flow Metab. 1992;12:359–370. doi: 10.1038/jcbfm.1992.53. [DOI] [PubMed] [Google Scholar]
- Kofler J, et al. Differential effect of PARP-2 deletion on brain injury after focal and global cerebral ischemia. J Cereb Blood Flow Metab. 2006;26:135–141. doi: 10.1038/sj.jcbfm.9600173. [DOI] [PubMed] [Google Scholar]
- Li X, et al. Influence of duration of focal cerebral ischemia and neuronal nitric oxide synthase on translocation of apoptosis-inducing factor to the nucleus. Neuroscience. 2007;144:56–65. doi: 10.1016/j.neuroscience.2006.08.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Hata R, Hossmann KA. Differences in the cerebrovascular anatomy of C57black/6 and SV129 mice. Neuroreport. 1998;9:1317–1319. doi: 10.1097/00001756-199805110-00012. [DOI] [PubMed] [Google Scholar]
- Mandir AS, et al. NMDA but not non-NMDA excitotoxicity is mediated by Poly(ADP-ribose) polymerase. J Neurosci. 2000;20:8005–8011. doi: 10.1523/JNEUROSCI.20-21-08005.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough LD, Zeng Z, Blizzard KK, Debchoudhury I, Hurn PD. Ischemic nitric oxide and poly (ADP-ribose) polymerase-1 in cerebral ischemia: male toxicity, female protection. J Cereb Blood Flow Metab. 2005;25:502–512. doi: 10.1038/sj.jcbfm.9600059. [DOI] [PubMed] [Google Scholar]
- Meder VS, Boeglin M, de Murcia G, Schreiber V. PARP-1 and PARP-2 interact with nucleophosmin/B23 and accumulate in transcriptionally active nucleoli. J Cell Sci. 2005;118:211–222. doi: 10.1242/jcs.01606. [DOI] [PubMed] [Google Scholar]
- Menissier dM, et al. Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J. 2003;22:2255–2263. doi: 10.1093/emboj/cdg206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mies G, Ishimaru S, Xie Y, Seo K, Hossmann KA. Ischemic thresholds of cerebral protein synthesis and energy state following middle cerebral artery occlusion in rat. J Cereb Blood Flow Metab. 1991;11:753–761. doi: 10.1038/jcbfm.1991.132. [DOI] [PubMed] [Google Scholar]
- Mito T, Nemoto M, Kwansa H, Sampei K, Habeeb M, Murphy SJ, Bucci E, Koehler RC. Decreased damage from transient focal cerebral ischemia by transfusion of zero-link hemoglobin polymers in mouse. Stroke. 2009;40:278–284. doi: 10.1161/STROKEAHA.108.526731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EM, Cho S, Frys K, Racchumi G, Zhou P, Anrather J, Iadecola C. Interaction between inducible nitric oxide synthase and poly(ADP-ribose) polymerase in focal ischemic brain injury. Stroke. 2004;35:2896–2901. doi: 10.1161/01.STR.0000147042.53659.6c. [DOI] [PubMed] [Google Scholar]
- Plesnila N, Zhu C, Culmsee C, Groger M, Moskowitz MA, Blomgren K. Nuclear translocation of apoptosis-inducing factor after focal cerebral ischemia. J Cereb Blood Flow Metab. 2004;24:458–466. doi: 10.1097/00004647-200404000-00011. [DOI] [PubMed] [Google Scholar]
- Quenet D, El Ramy R, Schreiber V, Dantzer F. The role of poly(ADP-ribosyl)ation in epigenetic events. Int J Biochem Cell Biol. 2009;41:60–65. doi: 10.1016/j.biocel.2008.07.023. [DOI] [PubMed] [Google Scholar]
- Sakurada O, Kennedy C, Jehle J, Brown JD, Carbin GL, Sokoloff L. Measurement of local cerebral blood flow with iodo [14C] antipyrine. Am J Physiol. 1978;234:H59–H66. doi: 10.1152/ajpheart.1978.234.1.H59. [DOI] [PubMed] [Google Scholar]
- Schreiber V, Ame JC, Dolle P, Schultz I, Rinaldi B, Fraulob V, Menissier-de Murcia J, de Murcia G. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J Biol Chem. 2002;277:23028–23036. doi: 10.1074/jbc.M202390200. [DOI] [PubMed] [Google Scholar]
- Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Greenberg JH, Jackson P, Maclin K, Zhang J. Neuroprotective effects of inhibiting poly(ADP-ribose) synthetase on focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 1997;17:1137–1142. doi: 10.1097/00004647-199711000-00001. [DOI] [PubMed] [Google Scholar]
- Wang H, et al. Apoptosis-inducing factor substitutes for caspase executioners in NMDA-triggered excitotoxic neuronal death. J Neurosci. 2004;24:10963–10973. doi: 10.1523/JNEUROSCI.3461-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kim NS, Li X, Greer PA, Koehler RC, Dawson VL, Dawson TM. Calpain activation is not required for AIF translocation in PARP-1-dependent cell death (parthanatos) J Neurochem. 2009;110:687–696. doi: 10.1111/j.1471-4159.2009.06167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelamos J, Schreiber V, Dantzer F. Toward specific functions of poly(ADP-ribose) polymerase-2. Trends Mol Med. 2008;14:169–178. doi: 10.1016/j.molmed.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, Dawson VL. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci U S A. 2006;103:18314–18319. doi: 10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- Yuan M, Siegel C, Zeng Z, Li J, Liu F, McCullough LD. Sex differences in the response to activation of the poly (ADP-ribose) polymerase pathway after experimental stroke. Exp Neurol. 2009;217:210–218. doi: 10.1016/j.expneurol.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeynalov E, Nemoto M, Hurn PD, Koehler RC, Bhardwaj A. Neuroprotective effect of selective kappa opioid receptor agonist is gender specific and linked to reduced neuronal nitric oxide. J Cereb Blood Flow Metab. 2006;26:414–420. doi: 10.1038/sj.jcbfm.9600196. [DOI] [PubMed] [Google Scholar]