

Abstract
Microglia, the innate immune cells in the brain, can become chronically activated in response to dopaminergic neuron death, fuelling a self-renewing cycle of microglial activation followed by further neuron damage (reactive microgliosis), which is implicated in the progressive nature of Parkinson’s disease. Here, we use an in vitro approach to separate neuron injury factors from the cellular actors of reactive microgliosis and discover molecular signals responsible for chronic and toxic microglial activation. Upon injury with the dopaminergic neurotoxin 1-methyl-4-phenylpyridinium, N27 cells (dopaminergic neuron cell line) released soluble neuron injury factors that activated microglia and were selectively toxic to dopaminergic neurons in mixed mesencephalic neuron-glia cultures through nicotinamide adenine dinucleotide phosphate oxidase. μ-Calpain was identified as a key signal released from damaged neurons, causing selective dopaminergic neuron death through activation of microglial nicotinamide adenine dinucleotide phosphate oxidase and superoxide production. These findings suggest that dopaminergic neurons may be inherently susceptible to the pro-inflammatory effects of neuron damage, i.e. reactive microgliosis, providing much needed insight into the chronic nature of Parkinson’s disease.
Keywords: microglia, inflammation-mediated neurodegeneration, oxidative stress, chronic neurotoxicity, extracellular μ-calpain, reactive microgliosis, Parkinson’s disease
Introduction
Parkinson’s disease is a devastating movement disorder characterized by selective and progressive loss of dopaminergic neurons in the substantia nigra. An active pathological process is implicated in Parkinson’s disease (McGeer et al., 1988a), but the mechanisms responsible for the chronic and progressive neuron damage are unclear. Microglia, the resident innate immune cells in the brain, are activated in Parkinson’s disease (McGeer et al., 1988b). Recent evidence points to microglial activation and the production of toxic factors (i.e. interleukin-1β, tumour necrosis factor-α, prostaglandin E2, nitric oxide and superoxide) as key contributors to dopaminergic neuron damage implicated in Parkinson’s disease (Block et al., 2007). The mechanisms fuelling this chronic and toxic microglial response, however, are largely unknown.
Microglial activation in response to neurodegeneration or neuron injury was initially perceived as a transient event (Streit et al., 1999). However, recent reports indicate that the microglial response to neuronal damage can be toxic, long-lived and self-propelling (Gao et al., 2003b; Huh et al., 2003; McGeer et al., 2003). In fact, dying/damaged neurons themselves are implicated as potential triggers of toxic microglial activation (Gao et al., 2003b). This repeating cycle of neurotoxic microglial activation in response to neuron injury is commonly referred to as reactive microgliosis, which may be culpable in persistent inflammation and chronic neuron damage associated with Parkinson’s disease (Block et al., 2007).
This premise is supported by studies investigating the effects of the selective dopaminergic neurotoxin, 1-methyl 4-phenyl 1,2,3,6-tetrahydropyridine (MPTP), an illicit drug contaminant linked to human Parkinsonism cases (Langston et al., 1983). To damage dopaminergic neurons, MPTP must be metabolized to 1-methyl-4-phenylpyridinium (MPP+), which is then selectively taken up by the dopamine transporter, resulting in inhibition of the mitochondrial electron transport chain complex I (Przedborski et al., 2004). In addition to this mechanism of direct neurotoxicity, MPTP-induced neurotoxicity is also clearly linked with microglial activation in vivo and in vitro (Gao et al., 2003b; McGeer et al., 2003; Wu et al., 2003). While in vitro studies show that MPTP directly damages dopaminergic neurons, both MPTP and MPP+ fail to activate microglia directly (Gao et al., 2003b). Rather, microglial activation in response to MPTP or MPP+ occurs only when neurons are present and this response takes time (days) to accumulate (Gao et al., 2003b). Furthermore, the addition of microglia to enriched neuron cultures greatly enhances MPTP-induced dopaminergic toxicity (Gao et al., 2003b), demonstrating that microglia cause dopaminergic neuron damage in addition to the direct toxic effects of MPTP/MPP+ on the neuron.
In vivo studies also emphasize the important role of inflammation as a toxic component of MPTP/MPP+ neurotoxicity (Wu et al., 2002), where dopaminergic neuron damage in response to MPTP is significantly reduced in mutant mice with deficient production of pro-inflammatory factors, such as nitric oxide (Liberatore et al., 1999), superoxide (Wu et al., 2003; Zhang et al., 2004), prostaglandins (Feng et al., 2002; Teismann et al., 2003) and tumour necrosis factor-α (Sriram et al., 2002). Interestingly, chronic neuroinflammation and neuron damage is reported to continue years after MPTP exposure in humans (Langston et al., 1999) and primates (McGeer et al., 2003), indicating an active pathology that persists long after the initial toxic insult has been metabolized and eliminated. Thus, several lines of evidence suggest that microglial activation initiated by neuronal damage may be toxic and persistent, continuing long after the initiating damaging/toxic stimulus is gone. At present, how neuronal damage results in microglial activation is poorly understood.
In the current study, we begin to address two fundamental questions regarding reactive microgliosis: (i) why the microglial response to neuron damage persists, and (ii) why this response is toxic. First, using an in vitro MPP+ model, we examined whether dopaminergic neuron damage/death causes the release of soluble factors that are selectively toxic to neighbouring/additional dopaminergic neurons through the activation of microglia. Second, we identify a key neuron injury signal driving the toxic component of reactive microgliosis. More specifically, we determined that μ-calpain, a cytosolic calcium-dependent cysteine protease, is released extracellularly upon dopaminergic neuron damage with MPP+, activating microglia to produce superoxide, which is selectively toxic to dopaminergic neurons.
Methods
Animals
Timed-pregnant (gestational day 14) adult female Fisher 344 rats were purchased from Charles River Laboratories (Raleigh, NC). Eight-week-old (25–30 g) male and female B6.129S6-Cybbtm1Din (PHOX−/−) and C57BL/6J (PHOX+/+) mice were purchased from Jackson Laboratories (Bar Harbor, Maine) and maintained in a strict pathogen-free environment. The PHOX−/− mice lack the functional catalytic subunit of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex, gp91. NADPH oxidase is an inducible electron transport system in phagocytic cells that is responsible for the generation of the respiratory burst. PHOX−/− mice are unable to generate extracellular superoxide in response to lipopolysaccharide or other immunological stimulus. Breeding of the mice was designed to achieve accurate timed-pregnancy ± 0.5 days. Because the PHOX−/− mutation is maintained in the C57BL/6J background, the C57BL/6J (PHOX+/+) mice were used as control animals. Housing, breeding and experimental use of the animals were performed in strict accordance with the National Institutes of Health guidelines.
Reagents
Lipopolysaccharide (strain O111:B4), μ-calpain and the polyclonal anti-μ-calpain antibody were purchased from EMD Chemicals (Gibbstown, NJ). Cell culture reagents were obtained from Invitrogen (Carlsbad, CA). [3H] Dopamine (28 Ci/mmol) and [2,3-H3] GABA (81 Ci/mmol) were purchased from NEN Life Science (Boston, MA). The polyclonal antibody against tyrosine hydroxylase was purchased from Protos Immunoresearch (Burlingame, CA). The neuron-specific nuclear protein (NeuN) monoclonal antibody was obtained from Millipore (Billerica, MA). The polyclonal antibody against the ionized calcium-binding adaptor molecule-1 microglial marker was purchased from Wako (Richmond, VA). The biotinylated horse anti-mouse and goat anti-rabbit secondary antibodies were purchased from Vector Laboratories (Burlingame, CA). 2-(4-lodophenyl)-3-(4-nitrophenyl)-5-(2,4,-disulphophenyl)-2H-tetrazolium, monosodium salt (WST-1) was purchased from Dojindo Laboratories (Gaithersburg, MD). Tumour necrosis factor-α enzyme-linked immunosorbent assay (ELISA) kits were purchased from R&D Systems (Minneapolis, MN). Prostaglandin E2 ELISA kits were purchased from Cayman (Ann Arbor, MI). All other reagents were procured from Sigma-Aldrich (St. Louis, MO).
Primary cultures
Rat and mouse ventral mesencephalic neuron-glia cultures from day 14 Fisher 344 rat embryos or day 14 mouse embryos (PHOX+/+ or PHOX−/−) were prepared as previously described (Liu et al., 2001). Cultures were treated 7 days after seeding.
Primary enriched microglia cultures were prepared from the whole brains of a day old Fisher 344 rat pups, as previously described (Block et al., 2004). Cells were treated 24 h after seeding the enriched microglia. Immunocytochemistry revealed <1% astrocyte or neuron contamination.
Primary microglia-depleted cultures were prepared according to Wang et al. (2006) by adding 1 μM leucine methyl ester into the primary neuron-glia cultures 24 h after the initial seeding. Seven-day-old cultures were used for treatment. At the time of treatment, immunocytochemistry analysis indicated that the microglial composition was <0.1%.
N27 cells
N27 cells are T-antigen immortalized rat mesencephalic dopaminergic neuron cells (Zhou et al., 2000). N27 cells were grown in Roswell Park Memorial Institute medium 1640 supplemented with 10% foetal bovine serum, penicillin (100 U/ml), streptomycin (100 U/ml) and 2 mM l-glutamine. Cells were maintained at 37°C in a 5% CO2 humidified atmosphere.
Uptake assays (dopamine and GABA)
Cells were incubated in Krebs-ringer buffer (16 mM NaH2PO4, 16 mM NaH2PO4, 1.2 mM MgSO4, 1.3 mM EDTA, 4.7 nM KCL, 16 mM Na2HPO4) for 15 min at 37°C with either 5 μM [3H] GABA or 1 μM [3H] dopamine. Non-specific uptake was blocked for GABA with 10 μM nitric oxide-711 and 1 mM β alanine. Non-specific uptake was blocked for dopamine with 10 μM mazindole. After incubation, cells were lysed and radioactivity was measured, where specific [3H] GABA or [3H] dopamine uptake was calculated by subtracting the mazindole or the nitric oxide-711 and β-alanine counts from the wells without the uptake inhibitors.
Soluble neuron injury factors-N27-conditioned medium
At 24 h post-seeding, N27 cells (5 × 106 cells/well) were exposed to treatment medium alone or MPP+ (5 or 10 μM) for 24 h. Cells were then washed three times with 1 ml/well of warm treatment medium to remove MPP+. Next, 1 ml of fresh treatment medium was added to each well and soluble factors were allowed to accumulate (0 or 6 h). The conditioned medium was then transferred to primary mixed neuron-glia cultures prepared from the mesencephalon of embryonic day 14 rats, PHOX+/+ mice, or PHOX−/−.
Immunostaining
Cells were fixed in 3.7% formaldehyde, washed twice, and followed by treatment with 1% hydrogen peroxide. Cultures were then washed three times and incubated with a blocking solution [phosphate buffered saline (PBS) containing 1% bovine serum albumin, 0.4% Triton X-100 and 4% serum] and incubated overnight (4°C) with the primary antibody diluted in Dako antibody diluent. Cultures were then incubated with an appropriate biotinylated secondary antibody. After washing three times, the cultures were incubated with Vectastain ABC Kit reagents according to the manufacturer’s instructions. Images were captured with an Axio Cam MRc5 imaging system (Carl Zeiss MicroImaging Thornwood, NY). To quantify cell numbers, nine representative areas were counted per well in the 24 well plate, at 100 × magnification by two individuals. The average of these scores is reported.
Superoxide assay
Extracellular superoxide 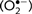 production from microglia was determined as reported previously by measuring the superoxide dismutase inhibitable reduction of WST-1 (Block et al., 2006). Cell-free experiments with and without μ-calpain were conducted to determine that μ-calpain did not alter absorbance by itself (data not shown). The amount of superoxide dismutase-inhibitable superoxide was calculated and expressed as percent of vehicle-treated control cultures.
production from microglia was determined as reported previously by measuring the superoxide dismutase inhibitable reduction of WST-1 (Block et al., 2006). Cell-free experiments with and without μ-calpain were conducted to determine that μ-calpain did not alter absorbance by itself (data not shown). The amount of superoxide dismutase-inhibitable superoxide was calculated and expressed as percent of vehicle-treated control cultures.
Confocal microscopy
Microglia were seeded on Matek (Sussex, UK) confocal chambers (2 × 106 cells/chamber) for 24 h and then treated with either medium alone or medium containing 3.8 μg/ml μ-calpain. At 12 h post-treatment, cells were washed, fixed [3.7% paraformaldehyde (w/w)/0.2% glutaraldehyde (v/v)] and permeabilized with 0.1% surfact-amps X-100. Following blocking (1% bovine serum albumin), fixed cells were incubated with anti-ionized calcium binding adaptor molecule-1 (1:200), followed by an Alexa-Fluor 488 secondary antibody (1:500). Microglia morphology was visualized using a laser scanning confocal microscope (LSM 510 mounted on an Axiovert 100 M microscope, Carl Zeiss, Inc.). The fluorescence and differential interference contrast images were captured simultaneously using the 488 nm line from an Argon laser and a Zeiss C-Apo 40 × N.A. = 1.2 water immersion objective lens.
Tumour necrosis factor-α and prostaglandin E2 ELISA
The production and release of tumour necrosis factor-α into the medium was measured with a commercial ELISA kit from R&D Systems (Minneapolis, MN), as described previously (Liu et al., 2001). The prostaglandin E2 release was measured with a commercial ELISA kit from Cayman Chemical Company (Ann Arbor, MI), as described previously (Liu et al., 2001).
Nitrite assay
As an indicator of nitric oxide production, the amount of nitrite accumulated in the culture supernatant was determined with a colorimetric assay using Griess reagent [1% sulphanilamide, 2.5% H3PO4, 0.1% N-(1-naphthyl) ethylenediamine dihydrochloride] as previously reported (Block et al., 2004). The sample nitrite concentration was determined from a sodium nitrite standard curve, with a lower limit of detection of 1.2 μM.
Immunoblotting
Conditioned medium (6 h) was collected from treated N27 cells with a serum-free treatment medium. Serum was omitted from the medium for these studies as it confounds the western blot analysis. The absence of serum did not affect culture viability, as determined by lactate dehydrogenase release from the cells determined using the Promega LDH Kit according to the manufacturer’s instructions (data not shown). Complete protease inhibitor (Roche Applied Sciences, Indianapolis, IN) was added to pooled samples from each treatment and the samples were concentrated using Amicon Ultra-15 centrifugal filter units with a 3 kDa cut-off (Millipore, Billerica, MA). Concentrated samples were electrophorezed on Nu-PAGE® 4–12% Bis–Tris electrophoresis gels (Invitrogen, Carlsbad, CA), transferred to nitrocellulose membranes, blocked with 5% non-fat milk for 1 h, followed by incubation overnight with a rabbit anti-μ-calpain antibody (1:1000) at 4°C. Blots were then incubated with horseradish peroxidase-linked mouse anti-rabbit (1:5000) for 1 h and bands were visualized with enhanced chemiluminescence and Plus reagents (Amersham, Piscataway, NJ).
Calpain activity
Calpain activity was determined using the InnoZyme™ Calpain 1 and 2 Activity Kit (EMD Chemicals, Gibbstown, NJ), per manufacturer instructions. N27 cell lysate was acquired using Cytobuster (EMD Chemicals, Gibbstown, NJ) lysis buffer and 100 μg of total protein was assayed. A conditioned medium was collected before cell lysis and 100 μl from each treatment was measured with the kit. Data are reported as relative fluorescence units, as defined by manufacturer instructions.
μ-Calpain ELISA
We developed a sandwich ELISA to quantitate rat μ-calpain in a conditioned medium. Nunc-immuno™ 96 well plates were coated with 100 μl (5 μg/ml) of anti-μ-calpain capture antibody (H-65, Santa Cruz Biotechnology, Santa Cruz, CA) in PBS at 24°C overnight and washed three times with 0.1% Tween in PBS. The plate was then blocked for 1 h with 300 μl of the blocking buffer (1% bovine serum albumin and 5% sucrose in PBS). Conditioned medium samples were diluted 1:2 in reagent diluent (1% bovine serum albumin in PBS) containing protease inhibitors (Halt Protease Inhibitor Cocktail, Thermo Scientific, Rockford, IL). Human μ-calpain (4.0–0.125 μg/ml, EMD Chemicals, Gibbstown, NJ) suspended in a 1:2 dilution of reagent diluent and the neuron-glia treatment medium was used as a standard. The treatment medium diluted 1:2 with reagent diluent was used as a blank. After blocking, 100 μl of either sample or standard was added to the plate in duplicates, at room temperature for 2 h and washed three times with 0.1% Tween in PBS. Next, 100 μl (10 μg/ml) of detection antibody (N-19, Santa Cruz Biotechnology, Santa Cruz, California) in reagent diluent was added for 1 h and washed three times with 0.1% Tween in PBS. The capture (H-65) and detection (N-12) antibodies recognize both human and rat μ-calpain. Then, 100 μl (1 μg/ml) of anti-goat horseradish peroxidase-conjugated antibody (Vector Laboratories, Burlingame, CA) in reagent diluent was added for 1 h and washed three times with 0.1% Tween in PBS. 3,3′,5,5′-Tetramethylbenzidine solution was added (100 μl, Sigma Aldrich Chemical Company, St. Louis, MO) for 30 min. Finally, 50 μl of stop solution was added (1N H2SO4) and the microplate was read at an absorbance of 450. The ELISA has a lower detection limit of 1 μg/ml of μ-calpain. Protein quantitation is considered relative in that the standards are derived from human μ-calpain.
Lactate dehydrogenase release
Cell viability was determined by lactate dehydrogenase release using the CytoTox 96 Non-Radioactive Cytotoxicity Assay Kit (Promega, Madison, WI), according to the manufacturer’s instructions.
Statistical analysis
Data are expressed either as raw values, the percentage of control or the difference from control, where control values were set to either 100% or 0 accordingly. The treatment groups are expressed as the mean ± SEM and statistical significance was assessed with an analysis of variance followed by Bonferroni’s t-test. In cases where only two means could be compared, an independent t-test was used. A value of P < 0.05 was considered statistically significant.
Results
Soluble neuron injury factors accumulate in a conditioned medium and are toxic to dopaminergic neurons
The ability of damaged neurons to release soluble signals into a conditioned medium, which then propagates additional dopaminergic neuron damage, was tested using the N27 cell line as a means to generate soluble neuron injury signals upon MPP+ exposure and primary neuron-glia cultures to test the effects of the N27-derived conditioned medium on dopaminergic neuron survival. N27 cells were exposed to either vehicle (Control) or MPP+ (5 and 10 μM) for 24 h. Consistent with other reports (Drechsel et al., 2007), approximately 35% of N27 cells released lactate dehydrogenase in response to 10 μM MPP+ at 24 h post-treatment and cells remained adherent to the cell culture plate (Supplementary Fig. S1). N27 cells provided a subtle model of MPP+ toxicity that allowed damage to accumulate over 30 h. Specifically, N27 cells express lower amounts of dopamine transporter, making them more resistant to MPP+ when compared to primary dopaminergic neurons (Supplementary Fig. S2A). After washing MPP+ from N27 dopaminergic neuron cells three times with 1 ml of medium, the conditioned medium was collected from N27 cells at either 0 or 6 h following the final wash. Fresh medium alone (unconditioned) or the conditioned medium collected from washed N27 cells was then added to mixed neuron-glia cultures.
To discern the effect of these injury factors in the conditioned medium on dopaminergic neuron function, the ability of cells to uptake [3H] dopamine was measured. As expected, the conditioned medium taken immediately after the wash (0 h), allowing no time for factors to accumulate, failed to result in any significant loss of dopaminergic neuron function or tyrosine hydroxylase-immunoreactive cells, supporting that MPP+ was not present in the conditioned medium (P < 0.05) (Fig. 1A and B). However, the conditioned medium collected from MPP+-treated N27 cells after 6 h (allowing time for soluble factors to be secreted) resulted in a significant and dose-dependent reduction in dopaminergic uptake (P < 0.05) (Fig. 1A). That is, the conditioned medium from N27 cells treated with MPP+ (10 μM) and MPP+ (5 μM) resulted in a 74% and 57% loss of dopaminergic neuron cell function (P < 0.05) (Fig. 1A), when compared to the conditioned medium from N27 cells exposed to the medium containing no MPP+ or fresh unconditioned medium controls, respectively. Neuron-glia cultures were also stained with the tyrosine hydroxylase antibody, where the number of tyrosine hydroxylase-positive neurons was counted 9 days post-treatment. Only the medium conditioned at 6 h from MPP+ (5 and 10 μM)-treated N27 cells reduced the number of tyrosine hydroxylase-immunoreactive neurons significantly (P < 0.05) (Fig. 1B), where the conditioned medium from N27 cells treated with MPP+ (10 μM) produced a 52% loss of dopaminergic neurons. Thus, further analyses only included 6 h conditioned medium. Figure 1C demonstrates that 6 h conditioned medium from N27 cells treated with MPP+ (10 μM) also results in changes in dopaminergic neuron morphology indicating damage, such as loss of dendrites and axons. In contrast, the conditioned medium from N27 cells that received only medium failed to affect dopaminergic uptake or cell loss, indicating that neuron damage had to occur for the conditioned medium to be toxic. Thus, the conditioned medium from MPP+-damaged N27 cells contained soluble factors that accumulated with time to cause further dopaminergic neuron damage in primary neuron-glia cultures.
Figure 1.

Dopaminergic neurons are selectively vulnerable to soluble neuron injury signals. Mesencephalic neuron-glia cultures were treated with medium alone (unconditioned medium, −/−) or conditioned medium (CM) from N27 dopaminergic (DA) cells exposed to MPP+ to determine if signals released from damaged neurons propagate further dopaminergic neuronal damage. N27 cells were treated with either medium alone (+/−) or MPP+ (5 or 10 μM) for 24 h (N27 MPP+ TRT). After washing MPP+ from N27 dopaminergic neuron cells three times with 1 ml of medium, the conditioned medium was collected from N27 cells at either 0 or 6 h following the final wash. The fresh medium alone (−/−) or the conditioned medium was added to mixed neuron-glia cultures. (A) Soluble neuron-injury signals take time to accumulate in the conditioned medium. Loss of dopaminergic neuron function was measured 9 days later with the [3H] dopamine uptake assay. (B) Loss of dopaminergic neurons was determined 9 days later by counting the number of tyrosine hydroxylase (TH)-immunoreactive (IR) neurons. (C) Tyrosine hydroxylase staining demonstrates the ability of 6 h conditioned medium (from N27 cells treated with 10 μM MPP+) to cause morphological damage to dopaminergic neurons. The arrow denotes the damaged dopaminergic neuron and the scale bar indicates 50 μm. Representative images are from three separate experiments. (D) Loss of dopaminergic neurons (tyrosine hydroxylase-immunoreactive neurons) and total neurons (NeuN-immunoreactive neurons) was determined 9 days later by cell count. Graphs show the results expressed as percentage of the control cultures (unconditioned medium, −/−) and are the mean ± standard error of mean from three independent experiments in triplicate. *P < 0.05, control compared to treatment, #P < 0.05 indicates significant differences due to either time (0 versus 6 h) or selective neurotoxicity (tyrosine hydroxylase versus NeuN).
Soluble neuron injury factors are selectively toxic to dopaminergic neurons
In an effort to determine whether dopaminergic neurons are selectively vulnerable to the toxic effects of reactive microgliosis, neuron-glia cultures were exposed to the conditioned medium from damaged N27 cells and stained for tyrosine hydroxylase and NeuN 9 days after treatment. Figure 1D indicates that while 6 h conditioned medium from N27 cells treated with MPP+ (10 μM and 5 μM) caused significant loss of tyrosine hydroxylase-immunoreactive neurons (P < 0.05), there was no overall effect on the neuron number, indicating that dopaminergic neurons are selectively vulnerable to the toxic effects of dopaminergic neuron (N27 cells) damage that is transferred in the conditioned medium. Immunohistochemical staining revealed that the total amount of dopaminergic neurons present in untreated neuron-glia cultures represent <1% of the total neurons, as previously reported (Liu and Hong, 2003a). Thus, consistent with our previous results, robust and selective dopaminergic neurotoxicity does not significantly impact overall neuron number in the neuron-glia culture system (Block et al., 2004, 2006).
Soluble neuron injury factors toxically activate microglia
Neuron-glia cultures were also stained for ionized calcium-binding adaptor molecule-1 at 12 h post-treatment to observe changes in microglia morphology. Figure 2A depicts microglial activation in response to 6 h conditioned medium from N27 cells treated with MPP+ (10 μM) when compared to controls. Activated microglia cells are larger and display a more irregular, amoeboid morphology. MPP+ itself does not activate microglia (Supplementary Fig. S2B). Previous studies have shown that damaged dopaminergic neurons must be present for microglial activation to occur in response to MPP+ (Gao et al., 2003c). To discern the role of activated microglia as a mechanism through which the conditioned medium was causing dopaminergic neuron death, neuron-glia cultures depleted of microglia (<0.1% microglia present) were exposed to the conditioned medium from N27 cells treated with MPP+ (10 μM and 5 μM) and dopaminergic neurotoxicity was abolished (Fig. 2B) (P < 0.05). Thus, not only do damaged neurons secrete soluble signals that activate microglia, but this microglial activation is responsible for the further toxicity of soluble neuron injury factors. In addition, the lack of any toxicity for the conditioned medium treatments in the absence of microglia confirms that MPP+ is not present in the conditioned medium, as MPP+ is well known to kill at least some dopaminergic neurons directly in the absence of microglia (Gao et al., 2003c).
Figure 2.

Soluble neuron injury signals are toxic to dopaminergic neurons through microglial activation and NADPH oxidase. Mesencephalic neuron-glia cultures were treated with medium alone (−/−) or conditioned medium (CM) from N27 dopaminergic (DA) cells exposed to MPP+ to determine if signals released from damaged neurons propagate further dopaminergic neuron damage. N27 cells were treated with either medium alone (+/−) or MPP+ (5 or 10 μM) for 24 h (N27 MPP+ TRT). After washing MPP+ from N27 dopaminergic neuron cells three times with 1 ml of medium, the conditioned medium was collected from N27 cells at 6 h following the final wash. Fresh unconditioned medium (−/−) or the conditioned medium was added to mixed neuron-glia cultures. (A) At 12 h post-treatment, neuron-glia cultures were stained for ionized calcium-binding adaptor molecule-1. The arrows denote examples of activated microglia and the scale bar indicates 50 μm. Microglial activation in response to the conditioned medium from N27 cells damaged with MPP+ is depicted by an increase in the number of stained cells, enlarged size of stained cells and irregular amoeboid morphology. Representative images are from three separate experiments. (B) Dopamine neurotoxicity was measured 9 days later with the [3H] dopamine uptake assay in neuron-glia cultures (N/G: containing microglia, astrocytes and neurons) and microglia-depleted cultures (MG-depleted: containing astrocytes and neurons). The conditioned medium was only toxic in the presence of microglia. (C) Dopamine neurotoxicity was measured 9 days later with the [3H] dopamine uptake assay in neuron-glia cultures from mice missing functional NADPH oxidase (PHOX−/−) and control strains (PHOX+/+). The conditioned medium was only toxic in the presence of NADPH oxidase. Graphs show the results expressed as percentage of the control cultures and are the mean ± SEM from three independent experiments in triplicate. *P < 0.05, control compared to treatment, #P < 0.05 indicates significant differences due to microglia (N/G versus microglia) or mouse strain (PHOX+/+ versus PHOX−/−).
NADPH oxidase is a key for conditioned medium-induced dopaminergic neurotoxicity
NADPH oxidase is the enzyme complex responsible for the respiratory burst in phagocytes. Activation of this enzyme in microglia induces the production of extracellular superoxide, resulting in selective dopaminergic neurotoxicity (Qin et al., 2004). Mesencephalic neuron-glia cultures from PHOX−/− mice are resistant to MPTP and MPP+-induced dopaminergic neurotoxicity, when compared to PHOX+/+ mice (Gao et al., 2003c; Wu et al., 2003). Here, we show that this is probably due to an insensitivity to the soluble pro-inflammatory signals released by damaged neurons, as PHOX−/− mice are resistant to 6 h conditioned medium from N27 cells treated with MPP+ (10 μM and 5 μM) (Fig. 2C) (P < 0.05). These results confirm that NADPH oxidase is critical to the toxic effects of reactive microgliosis and demonstrate for the first time that the factors released into the conditioned medium upon neuron damage are essential to this process.
μ-Calpain is released by MPP+-treated N27 cells
Next we began to address the identity of the factors present in the conditioned medium that were responsible for the selectively toxic aspects of reactive microgliosis. We began by concentrating the pooled conditioned medium from N27 cells exposed to medium alone (Control) or MPP+ (10 μM and 5 μM). The western blot analysis of concentrated samples showed that μ-calpain was present as a 78 kDa band that increased in the conditioned medium in response to increasing concentrations of MPP+ treatment and consequent N27 cell damage (Fig. 3A). Densitometry analysis averaged over five separate experiments demonstrated that these changes in extracellular μ-calpain were significant from control values (Fig. 3B) (P < 0.05). In a separate study, 2.7 μg/ml μ-calpain was measured by ELISA in a conditioned medium of N27 cells treated with 10 μM MPP+ (Fig. 3C). This is further supported by the enzyme activity assay, where relative μ-calpain levels in the conditioned medium from N27 cells treated with 10 μM MPP+ were predicted to be ∼3.13 μg/ml (Supplementary Table S2). Notably, this concentration of calpain is less than extracellular calpain concentrations predicted to play a role in progressive liver damage (Limaye et al., 2003; Mehendale and Limaye, 2005).
Figure 3.

μ-Calpain is a soluble neuron injury factor released by damaged dopaminergic neurons. N27 dopaminergic neuron cells were treated with either medium alone or MPP+ (5 or 10 μM) for 24 h (N27 MPP+ TRT). After washing MPP+ from N27 dopaminergic neuron cells three times with 1 ml of medium, conditioned medium (CM, serum-free medium) from N27 cells at 6 h following the final wash. Samples were concentrated from 15 ml to 100 μl and ran out on a sodium dodecyl sulphate polyacrylamide gel electrophoresis gel. (A) Representative image of western blot analysis. Blots of concentrated conditioned medium probed with an anti-μ-calpain antibody reveals a 78 kDa protein band that increases with the concentration the MPP+ exposure (i.e. increases with enhanced neuron damage). (B) The densitometry graph shows the results expressed as percentage of the control and are the mean ± SEM from five independent experiments. (C) The relative amount of extracellular μ-calpain present in the unconcentrated N27 conditioned medium, as determined by ELISA. The results are from six separate experiments. *P < 0.05, control compared to treatment.
Extracellular μ-calpain is toxic to dopaminergic neurons
While intracellular μ-calpain is known to play a key role in how cells die and are damaged (Hara and Snyder, 2007), the role of this enzyme once outside the cell is unknown. Here, we compared the neurotoxic effect of extracellular μ-calpain on dopaminergic neurons in rat mesencephalic neuron-glia cultures. To discern the effect of μ-calpain on dopaminergic neuron function, the ability of cells to uptake [3H] dopamine was measured. Addition of μ-calpain to neuron-glia cultures resulted in a significant dose-dependent reduction in dopamine uptake (P < 0.05) (Fig. 4A) at 9 days post-treatment. The highest dose of 3.8 μg/ml calpain produced a 49% loss of dopaminergic neuron cell function that was abolished when the enzyme was boiled, indicating that enzymatic function was essential for toxicity. Lipopolysaccharide was used as a positive control for microglia-mediated neurotoxicity and the loss of dopaminergic neuron function seen with lipopolysaccharide (10 ng/ml) was comparable to calpain (3.8 μg/ml). Notably, boiled calpain (3.8 μg/ml) had no effect. The same assay performed at 3 days post-treatment showed no significant changes in [3H] dopamine uptake (Supplementary Fig. S3), indicating that damage took time (days) to accumulate. This delayed dopaminergic neurotoxicity is consistent with previously reported mechanisms of microglia-mediated neurotoxicity, such as the case with lipopolysaccharide (Gao et al., 2002).
Figure 4.

Extracellular μ-calpain selectively kills dopaminergic neurons. Rat mesencephalic neuron-glia cultures were treated with either medium alone (control), lipopolysaccharide (LPS; 10 ng/ml) or μ-Calpain (3.8 μg/ml, 1.9 μg/ml or boiled). (A) Loss of dopaminergic neuron function was measured 9 days later with the [3H] dopamine uptake assay. (B) Loss of dopaminergic neurons was determined 9 days later by counting the number of tyrosine hydroxylase (TH)-immunoreactive (IR) neurons. (C) Tyrosine hydroxylase staining demonstrates μ-calpain-induced morphological damage to dopaminergic neurons. The arrow denotes the damaged dopaminergic neuron and the scale bar indicates 50 μm. Representative images are from three separate experiments. (D) Dopamine neurotoxicity (DA) and GABA neurotoxicity (GABA) was determined 9 days later by [3H] dopamine or GABA uptake assay. (E) General loss of total neurons (NeuN-immunoreactive neurons) was determined 9 days later by the cell count. Graphs show the results expressed as percentage of the control cultures and are the mean ± SEM from three independent experiments in triplicate. *P < 0.05, control compared to treatment; #P < 0.05 indicates significant differences in selective neurotoxicity (tyrosine hydroxylase versus GABA).
To quantitate the dopaminergic neuron cell loss associated with μ-calpain treatment, neuron-glia cultures were exposed to μ-calpain and stained with the tyrosine hydroxylase antibody, where the number of tyrosine hydroxylase-positive neurons were counted. μ-Calpain reduced the number of tyrosine hydroxylase-immunoreactive neurons by 31% and this was abolished by boiling the enzyme (P < 0.05) (Fig. 4B). μ-Calpain also caused morphological damage to dopaminergic neurons, as evidenced by loss of dendrites and axons (Fig. 4C).
Extracellular μ-calpain neurotoxicity is selective for dopaminergic neurons
Neuron-glia cultures exposed to μ-calpain were compared for the ability to uptake [3H] dopamine and [3H] GABA. Figure 4D shows that dopaminergic uptake was reduced by the addition of μ-calpain to only neuron-glia cultures (P < 0.05), while GABA uptake remained unchanged. The number of NeuN immunoreactive neurons in neuron-glia cultures was also counted to determine the toxic effect of μ-calpain on the overall neuron number (Fig. 4E). These data showed that μ-calpain had no effect on the number of NeuN-positive cells present in neuron-glia cultures. Together, the lack of μ-calpain effect on both GABA uptake and NeuN cell count in neuron-glia cultures supports the specificity of μ-calpain-induced dopaminergic neurotoxicity.
Extracellular μ-calpain activates microglia
It has been well established that the activation of microglia, and the consequent production of pro-inflammatory factors, has been linked to dopaminergic neurotoxicity (Liu and Hong, 2003b; Qin et al., 2004). The supernatant from μ-calpain-treated neuron-glia cultures was tested for the presence of several pro-inflammatory factors. μ-Calpain added to cultures was lipopolysaccharide- free, per manufacturer specifications. Analysis of supernatant collected at 3, 6, 12 and 24 h, and 4, 6, 8 and 9 days post-μ-calpain treatment in neuron-glia cultures revealed that prostaglandin E2, tumour necrosis factor-α and nitrite (indicative of nitric oxide production) were not produced (Supplementary Table S1). However, immunocytochemistry staining with the ionized calcium-binding adaptor molecule-1 antibody for microglia at 12 h post-μ-calpain treatment revealed activated microglia morphology (Fig. 5A). The activated microglia were identified by the altered amoeboid morphology when compared to the control group. This early evidence of microglial activation occurred eight days before the determination of dopaminergic neurotoxicity and supported the role of microglia as the triggering event of μ-calpain-induced dopaminergic neurotoxicity.
Figure 5.

Extracellular μ-calpain is neurotoxic due to activation of microglial NADPH oxidase. (A) Confocal images of neuron-glia cultures stained for ionized calcium-binding adaptor molecule-1. Cells were treated with either vehicle (medium alone) or μ-calpain (3.8 μg/ml) for 12 h at 37°C. (1) Background fluorescence (no secondary antibody added); (2), transmitted light (Differential Interference Contrast) image; (3) untreated control cells; (4) μ-calpain-treated cells. The fluorescence micrographs depict representative changes in morphology caused by μ-calpain that indicates microglial activation. Activated microglial cells are amoeboid with multiple extended processes (4). The scale bar indicates 50 μm. (B) Neuron-glia cultures (N/G—containing microglia, astrocytes, and neurons) and microglia-depleted cultures (MG-depleted—containing astrocytes and neurons) were treated with medium alone (Control), lipopolysaccharide (LPS) 10 ng/ml (positive control for microglia-mediated neurotoxicity) or μ-calpain (3.8 or 1.9 μg/ml). Dopamine neurotoxicity was measured 9 days later with the [3H] dopamine uptake assay. μ-Calpain was only toxic in the presence of microglia. (C) Enriched microglia cultures were treated with medium alone (Control), μ-calpain (3.8 μg/ml), calpeptin (1 μM, a specific calpain inhibitor) or calpeptin + μ-calpain. The production of extracellular superoxide was measured by the superoxide dismutase-inhibitable reduction of tetrazolium salt, WST-1 at 30 min post-treatment. Results are mean ± SEM. Data are from four separate experiments. *P < 0.05, compared with control cultures. (D) Mesencephalic midbrain neuron-glia cultures from PHOX+/+ and PHOX−/− mice were treated with medium alone (Control), μ-calpain (3.8 μg/ml) or boiled μ-calpain (3.8 μg/ml). Graphs show the results expressed as percentage of the control cultures and are the mean ± SEM from three independent experiments in triplicate. *P < 0.05, control compared to treatment; #P < 0.05 indicates significant differences due to microglia (N/G versus microglia), superoxide reduction (calpeptin reduction of μ-calpain) or mouse strain (PHOX+/+ versus PHOX−/−).
Microglia mediate dopaminergic neurotoxicity caused by extracellular μ-calpain
To investigate the role of microglia in μ-calpain-induced dopaminergic neurotoxicity, [3H] dopamine uptake was compared in microglia-depleted cultures versus neuron-glia cultures. While μ-calpain remained toxic to dopaminergic neurons in the neuron-glia culture (P < 0.05), the dopamine uptake in microglia-depleted cultures was not affected (Fig. 5B), demonstrating that μ-calpain concentrations of 3.8 μg/ml and below were not directly toxic to dopaminergic neurons.
Extracellular μ-calpain causes superoxide production in microglia
The production of reactive oxygen species from activated microglia has also been strongly linked to dopaminergic neurotoxicity (Qin et al., 2002, 2004; Gao et al., 2003a, 2003b). The results presented in Fig. 5C demonstrate that the addition of μ-calpain to enriched-microglial cultures caused a significant increase in extracellular superoxide from control, which was inhibited by the calpain-specific inhibitor calpeptin (1 μM) (P < 0.05).
Extracellular μ-calpain-induced neurotoxicity is mediated by NADPH oxidase
To test the importance of extracellular superoxide for μ-calpain-induced dopaminergic neurotoxicity, dopamine uptake in PHOX−/− mouse mesencephalic neuron-glia cultures was compared to PHOX+/+ mouse cultures. PHOX−/− mice lack the functional gp91 protein, the catalytic subunit of the NADPH oxidase complex and thus have no phagocytic respiratory burst (production of extracellular superoxide). Dopamine uptake at 9 days post-treatment was measured to determine the μ-calpain neurotoxicity. While μ-calpain was toxic to dopaminergic neurons in the PHOX+/+ culture (P < 0.05), the dopamine uptake in PHOX−/− cultures was unaffected by μ-calpain (Fig. 5D), demonstrating the pivotal role of NADPH oxidase generated reactive oxygen species in extracellular μ-calpain-induced dopaminergic neurotoxicity.
E64 attenuates MPP+ neurotoxicity in neuron-glia cultures
To confirm that extracellular μ-calpain played a role in MPP+-induced dopaminergic neurotoxicity in primary cultures, rat mesencephalic neuron-glia cultures were pre-treated with E64, a μ-calpain inhibitor that is not cell-permeable (Limaye et al., 2003; Mehendale and Limaye, 2005) and exposed to either conditioned medium (MPP+ 10 μM) or MPP+ directly. Figure 6A demonstrates that E64 was not cell-permeable in our in vitro system, as cell lysates from N27 cells treated with medium, E64 (1 μM), MPP+ (10 μM) or E64 combined with MPP+ for 24 h show no significant differences in calpain enzyme activity (P > 0.05). μ-Calpain is ubiquitously expressed and activity is regulated by calcium influx. When cells are lysed and added to the activation buffer (containing calcium) provided with the calpain activity kit, differences in activation due to treatment (i.e. cellular calcium currents) are lost, as depicted in Fig. 6A. Specifically, the increase in intracellular N27 calpain activity due to MPP+ treatment is masked by the nature of the activity assay, where calcium is added to total cell protein. Cell-permeable inhibitors present in the cell and contained in the lysate would still be able to reduce activity. While E64 does not modulate intracellular calpain activity in N27 cells (Fig. 6A, P > 0.05), E64 does inhibit extracellular calpain activity in the conditioned medium in response to MPP+ (10 μM) treatment (Fig. 6B, P < 0.05). Furthermore, pre-treatment with E64 (1 μM) significantly protected dopaminergic neurons from the conditioned medium (MPP+ 10 μM)-induced dopaminergic neurotoxicity, bringing dopaminergic neuron survival from 23.9% to near control levels at 84.3% (Fig. 6C, P < 0.05).
Figure 6.

Inhibiting extracellular μ-calpain protects dopaminergic neurons. (A) E64 fails to inhibit intracellular calpain activity in N27 cells. N27 cells were pre-treated with medium alone or E64 (1 μM, non-permeable, extracellular only calpain inhibitor) for 30 min prior to treatment with either medium alone or 10 μM MPP+. After 24 h, the conditioned medium was removed, cells were lysed and intracellular calpain activity was calculated with a commercially available kit. Data are presented as relative fluorescence units (RFU) and are the mean ± SEM from three independent experiments performed with duplicate samples. (B) E64 inhibits extracellular calpain activity. The conditioned medium from the N27 cells above was tested for extracellular calpain activity using the commercially available kit. Data are presented as relative fluorescence units and are the mean ± SEM from three independent experiments performed with duplicate samples. (C) E64 protects against dopaminergic neurotoxicity caused by soluble neuron-injury factors (conditioned medium). Rat mesencephalic neuron-glia cultures were pre-treated with medium alone or E64 (1 μM, non-permeable, extracellular only calpain inhibitor) for 30 min prior to treatment with either medium alone or the conditioned medium (CM N27 10 μM MPP+). [3H] Dopamine (DA) uptake assay was performed at 9 days following the MPP+ treatment. The data are expressed as the percent of the control cultures and are the mean ± SEM from three independent experiments performed with triplicate samples. (D) E64 protects against dopamine neurotoxicity caused by soluble neuron-injury factors (MPP+). Rat mesencephalic neuron-glia cultures were pre-treated with medium alone or E64 (1 μM, non-permeable, extracellular only calpain inhibitor) for 30 min prior to treatment with either medium alone or 0.1 μM MPP+. [3H] Dopamine uptake assay was performed at 9 days following the MPP+ treatment. The data are expressed as the percent of the control cultures and are the mean ± SEM from three independent experiments performed with triplicate samples. *P < 0.05, compared to control-treated cultures. #P < 0.05, compared to MPP+ treatment.
To confirm that E64 neuroprotection extends to reactive microgliosis that occurs in the presence of MPP+ toxin, rat mesencephalic neuron-glia cultures were pre-treated with E64 for 30 min, followed by MPP+ treatment (0.1 μM). The MPP+ concentration of 0.1 μM was chosen for moderate direct toxicity at 9 days post-treatment. Primary dopaminergic neurons are more sensitive to MPP+ and higher concentrations of 5–10 μM result in immediate and severe direct neurotoxicity of 70% of dopaminergic neurons (Supplementary Fig. S2A), allowing little room for reactive microgliosis to occur. Figure 6D indicates that E64 can inhibit a portion of MPP+-induced dopaminergic neurotoxicity at 9 days post-treatment (P < 0.05), where MPP+ (0.1 μM) was toxic to all but 19% of dopaminergic neurons and E64 treatment boosted dopaminergic neuron survival to 44%. Notably, complete inhibition of dopaminergic neurotoxicity is not achieved, as extracellular inhibition of μ-calpain is only able to inhibit the microglia-mediated component of the damage, rather than the intracellular processes related to the direct toxicity of the MPP+ on the dopaminergic neuron itself.
Discussion
When dopaminergic neurons are damaged, chronic microglial activation and the persistent selective loss of dopaminergic neurons can occur, long after the initial toxic insult has abated. This persistent cellular response called reactive microgliosis is widely believed to be a predominant mechanism underlying progressive neuron damage in neurodegenerative diseases. Here we specifically addressed why reactive microgliosis in response to dopaminergic neuron damage is chronic and toxic.
We used an in vitro model to separate the soluble signals released by damaged dopaminergic neurons from the toxicant causing the damage (MPP+). This approach allowed analysis of the dopaminergic neuron survival in response to only the neuron injury signals, revealing that dopaminergic neurons are inherently more susceptible to soluble neuron injury signals when compared to other neuronal-sub-types and that microglia and NADPH oxidase are key to the mechanism of damage. These data strongly support that dopaminergic neurons may be inherently susceptible to reactive microgliosis and relentless propagation of the chronic and neurotoxic response continuing on in the absence of the initial toxic stimulus. Given that the substantia nigra contains both the population of dopaminergic neurons selectively lost in Parkinson’s disease and a disproportionately high concentration of microglia (4.5 times as many as other regions of the brain) (Kim et al., 2000), our findings indicate that damaged neurons themselves are culpable in propagating further neurotoxicity with pro-inflammatory signals to microglia (Fig. 7), providing a key insight into the chronic and selective nature of Parkinson’s disease.
Figure 7.

μ-Calpain is a key factor driving the progressive nature of dopaminergic neuron damage. Dopaminergic (DA) neuron damage is chronic in part because damaged cells release soluble factors that accumulate over time to active resident microglia, driving further toxicity (reactive microgliosis). μ-Calpain is externalized in the process of dopamine neuron damage and is a fundamental soluble neuron injury factor responsible for the toxic aspects of reactive microgliosis. Specifically, extracellular μ-calpain activates microglial NADPH oxidase, producing superoxide to damage neighbouring dopaminergic neurons selectively and propagate neurotoxicity. This feed-forward cycle provides much needed insight into the progressive nature of dopaminergic neuron damage.
After isolating the soluble-neuron injury factors released by damaged dopaminergic neurons and confirming that they were selectively toxic through microglial activation and the super-oxide producing enzyme, NADPH oxidase, we then began to identify which soluble neuron injury factors were responsible for microglial activation and consequent dopaminergic neuron damage. In the present study, we chose a hypothesis-directed approach and investigated whether μ-calpain is a key neuron injury signal causing reactive microgliosis and consequent chronic dopaminergic neuron damage.
Calpain is a family of calcium-dependent cysteine proteases that have been implicated in chronic cellular damage, e.g. extracellular calpain in the liver (Limaye et al., 2003; Mehendale and Limaye, 2005). While calpain is traditionally viewed as an intracellular protease, it is found extracellularly in human disease and tissue damage, such as arthritis (Fushimi et al., 2004) and liver damage (Limaye et al., 2003; Mehendale and Limaye, 2005). In fact, extracellular calpain is present in the supernatant from damaged cortical neurons in vitro (Siman et al., 2004), supporting a potential role for this protease as a neuron injury signal. Notably, while the mechanisms remain unclear, calpain has been linked to intracellular process of dopaminergic neuron death (apoptosis and necrosis) (Crocker et al., 2003), microglial activation (Shields et al., 2000), and Parkinson’s disease (Mouatt-Prigent et al., 1996; Crocker et al., 2003). Intracellularly, calpain activity is shown to increase in dopaminergic neurons damage by MPP+ and MPTP in vitro and in vivo, where calpain inhibitors can attenuate MPP+-induced dopamine cell loss (Crocker et al., 2003). Calpain is also upregulated in post-mortem Parkinson’s disease brain, verifying that calpain is present in Parkinson’s disease brain and is upregulated upon dopaminergic neuron damage (Crocker et al., 2003). However, until the present study, the role of extracellular calpain in dopaminergic neuron damage and its effect on chronic and toxic microglia activation were unknown.
Calpain has multiple isoforms, with μ- and m-calpain ubiquitously distributed in the cytoplasm of all cells. Intracellular calpain is involved in numerous cellular functions, including membrane trafficking, receptor signalling, inflammatory signalling, apoptosis and necrosis (Franco and Huttenlocher, 2005). Calpain’s intracellular functions are critical for both survival and death, making the tight regulation of calpain activity essential. The dysregulation of calpain has been associated with numerous diseases, such as arthritis (Fushimi et al., 2004), Alzheimer’s disease (Zatz and Starling, 2005), multiple sclerosis (Sloane et al., 2003; Guyton et al., 2005), optic neuritis (Shields and Banik, 1998) and Parkinson’s disease (Crocker et al., 2003).
While most studies have focused on the intracellular role of calpain in cell death, extracellular calpain has also been documented (Nishihara et al., 2001; Xu and Deng, 2004) and implicated in disease. For example, calpain is found in the synovial fluid of arthritis patients (Fushimi et al., 2004) and has been associated with degeneration of the myelin sheath (Shields et al., 1999; Sloane et al., 2003). Calpain is reported to be released extracellularly in the case of tissue damage in liver (Limaye et al., 2003; Mehendale and Limaye, 2005) and cortical neuron damage (Siman et al., 2004). Interestingly, calpain and the breakdown products of its substrates have been found in the cerebral spinal fluid after acute ischaemia and are proposed to be markers for neuronal damage (Siman et al., 2005). Once in the extracellular space, calpain is presumed to be highly active and unregulated, as extracellular calcium concentrations (1.3 mM) (Limaye et al., 2003) are higher than the μM (μ-calpain) and the mM (m-calpain) required for activation. In the case of liver injury, once outside of the cell, calpain is believed to attack the membrane of surrounding cells to result in propagation of tissue damage (Limaye et al., 2003; Mehendale and Limaye, 2005). Alternatively, several extracellular proteases are reported to be pro-inflammatory when extracellular, which may result in neuronal death (Choi et al., 2005a, b; Kim et al., 2005), but the mechanisms remain poorly described.
In this study we show that μ-calpain is a key soluble neuron injury factor driving reactive microgliosis. μ-Calpain is present in the conditioned medium from MPP+-damaged dopaminergic neurons (N27 cells), where this conditioned medium was shown to be selectively toxic to dopaminergic neurons through microglial NADPH oxidase. Extracellular μ-calpain added to cultures activated microglia, as evidenced by changes in morphology and superoxide production. Furthermore, μ-calpain-induced neurotoxicity was selective for dopaminergic neurons and occurred only in the presence of microglia. Finally, μ-calpain only exerted dopaminergic neurotoxicity in cultures with functioning NADPH oxidase, indicating that superoxide production was the predominant mechanism of μ-calpain-induced neuron damage. Thus, we show that μ-calpain is a soluble neuron injury factor that is selectively toxic to dopaminergic neurons through microglia-generated oxidative insult, revealing key mechanisms of chronic neurodegenerative pathology (Fig. 7), as reactive microgliosis may contribute to neuronal damage in diverse neuronal diseases (Block and Hong, 2005, 2007; Block et al., 2007).
While μ-calpain may be a key signal that damaged dopaminergic neurons release to activate microglia and propagate damage, reactive microgliosis is a complex phenomenon and there are probably multiple factors released that contribute to the toxic microglial response. In fact, previous studies have reported other damage signals implicated in toxic reactive microgliosis, such as α-synuclein and neuromelanin (Block and Hong, 2007). However, μ-calpain is ubiquitously expressed in all cell types and previous reports indicate that it is also released from damaged cortical neurons (Siman et al., 2004), suggesting that μ-calpain may be a general factor of reactive microgliosis. The dopamine selective toxicity of μ-calpain and the conditioned medium that we report here is likely to be due to the characteristic selective vulnerability of dopaminergic neurons to microglial activation and consequent production of reactive oxygen species, rather than a dopaminergic neuron-specific signal. For example, previous reports have shown that multiple other toxins are selectively toxic to dopaminergic neurons through microglial reactive oxygen species production, such α-synuclein, neuromelanin, lipopolysaccharide, paraquat, air pollution, rotenone and substance P (Block and Hong, 2007). Our current results indicate that μ-calpain and the soluble neuron-injury signals contained in the conditioned medium from damaged dopaminergic neurons converge on this basic mechanism of selective dopaminergic neurotoxicity. Thus, while reactive microgliosis may underlie diverse neurodegenerative diseases, our study suggests that dopaminergic neurons are more likely to be negatively affected than other cell types, providing much needed insight into the progressive nature of Parkinson’s disease. Indeed, this study supports the premise that dopaminergic neurons may be particularly vulnerable to the chronic effects of a single neurotoxic insult that propagates because of the microglial response to neuronal injury, which then becomes the driving force of persistent and progressive dopaminergic neuron damage.
Supplementary material
Supplementary material is available at Brain online.
Supplementary Material
Funding
National Institute of Environmental Health Sciences/National Institute of Health Pathway to Independence Award (R00ES01549) and in part by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences, in fulfilment of the Pathway to Independence Award (K99ES015409-01).
Glossary
Abbreviations
- ELISA
enzyme-linked immunosorbent assay
- MPP+
1-methyl-4-phenylpyridinium
- MPTP
1-methyl 4-phenyl 1,2,3,6-tetrahydropyridine
- NADPH
nicotinamide adenine dinucleotide phosphate
- NeuN
neuron-specific nuclear protein
- PBS
phosphate buffered saline
References
- Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol. 2005;76:77–8. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Block ML, Hong JS. Chronic microglial activation and progressive dopaminergic neurotoxicity. Biochem Soc Trans. 2007;35:1127–32. doi: 10.1042/BST0351127. [DOI] [PubMed] [Google Scholar]
- Block ML, Li G, Qin L, Wu X, Pei Z, Wang T, et al. Potent regulation of microglia-derived oxidative stress and dopaminergic neuron survival: substance P vs. dynorphin. Faseb J. 2006;20:251–8. doi: 10.1096/fj.05-4553com. [DOI] [PubMed] [Google Scholar]
- Block ML, Wu X, Pei Z, Li G, Wang T, Qin L, et al. Nanometer size diesel exhaust particles are selectively toxic to dopaminergic neurons: the role of microglia, phagocytosis, and NADPH oxidase. Faseb J. 2004;18:1618–20. doi: 10.1096/fj.04-1945fje. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Choi SH, Lee da Y, Chung ES, Hong YB, Kim SU, Jin BK. Inhibition of thrombin-induced microglial activation and NADPH oxidase by minocycline protects dopaminergic neurons in the substantia nigra in vivo. J Neurochem. 2005a;95:1755–65. doi: 10.1111/j.1471-4159.2005.03503.x. [DOI] [PubMed] [Google Scholar]
- Choi SH, Lee da Y, Kim SU, Jin BK. Thrombin-induced oxidative stress contributes to the death of hippocampal neurons in vivo: role of microglial NADPH oxidase. J Neurosci. 2005b;25:4082–90. doi: 10.1523/JNEUROSCI.4306-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker SJ, Smith PD, Jackson-Lewis V, Lamba WR, Hayley SP, Grimm E, et al. Inhibition of calpains prevents neuronal and behavioral deficits in an MPTP mouse model of Parkinson’s disease. J Neurosci. 2003;23:4081–91. doi: 10.1523/JNEUROSCI.23-10-04081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drechsel DA, Liang LP, Patel M. 1-Methyl-4-phenylpyridinium-induced alterations of glutathione status in immortalized rat dopaminergic neurons. Toxicol Appl Pharmacol. 2007;220:341–8. doi: 10.1016/j.taap.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng ZH, Wang TG, Li DD, Fung P, Wilson BC, Liu B, et al. Cyclooxygenase-2-deficient mice are resistant to 1-methyl-4-phenyl1, 2, 3, 6-tetrahydropyridine-induced damage of dopaminergic neurons in the substantia nigra. Neurosci Lett. 2002;329:354–8. doi: 10.1016/s0304-3940(02)00704-8. [DOI] [PubMed] [Google Scholar]
- Franco SJ, Huttenlocher A. Regulating cell migration: calpains make the cut. J Cell Sci. 2005;118:3829–38. doi: 10.1242/jcs.02562. [DOI] [PubMed] [Google Scholar]
- Fushimi K, Nakashima S, Banno Y, Akaike A, Takigawa M, Shimizu K. Implication of prostaglandin E(2) in TNF-alpha-induced release of m-calpain from HCS-2/8 chondrocytes. Inhibition of m-calpain release by NSAIDs. Osteoarthritis Cartilage. 2004;12:895–903. doi: 10.1016/j.joca.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Gao HM, Jiang J, Wilson B, Zhang W, Hong JS, Liu B. Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: relevance to Parkinson’s disease. J Neurochem. 2002;81:1285–97. doi: 10.1046/j.1471-4159.2002.00928.x. [DOI] [PubMed] [Google Scholar]
- Gao HM, Liu B, Hong JS. Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopaminergic neurons. J Neurosci. 2003a;23:6181–7. doi: 10.1523/JNEUROSCI.23-15-06181.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Liu B, Zhang W, Hong JS. Critical role of microglial NADPH oxidase-derived free radicals in the in vitro MPTP model of Parkinson’s disease. Faseb J. 2003b;17:1954–6. doi: 10.1096/fj.03-0109fje. [DOI] [PubMed] [Google Scholar]
- Gao HM, Liu B, Zhang W, Hong JS. Critical role of microglial NADPH oxidase-derived free radicals in the in vitro MPTP model of Parkinson’s disease. Faseb J. 2003c;17:1954–6. doi: 10.1096/fj.03-0109fje. [DOI] [PubMed] [Google Scholar]
- Guyton MK, Wingrave JM, Yallapragada AV, Wilford GG, Sribnick EA, Matzelle DD, et al. Upregulation of calpain correlates with increased neurodegeneration in acute experimental auto-immune encephalomyelitis. J Neurosci Res. 2005;81:53–61. doi: 10.1002/jnr.20470. [DOI] [PubMed] [Google Scholar]
- Hara MR, Snyder SH. Cell signaling and neuronal death. Annu Rev Pharmacol Toxicol. 2007;47:117–41. doi: 10.1146/annurev.pharmtox.47.120505.105311. [DOI] [PubMed] [Google Scholar]
- Huh Y, Jung JW, Park C, Ryu JR, Shin CY, Kim WK, et al. Microglial activation and tyrosine hydroxylase immunoreactivity in the substantia nigral region following transient focal ischemia in rats. Neurosci Lett. 2003;349:63–7. doi: 10.1016/s0304-3940(03)00743-2. [DOI] [PubMed] [Google Scholar]
- Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J Neurosci. 2000;20:6309–16. doi: 10.1523/JNEUROSCI.20-16-06309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YS, Kim SS, Cho JJ, Choi DH, Hwang O, Shin DH, et al. Matrix metalloproteinase-3: a novel signaling proteinase from apoptotic neuronal cells that activates microglia. J Neurosci. 2005;25:3701–11. doi: 10.1523/JNEUROSCI.4346-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–80. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- Langston JW, Forno LS, Tetrud J, Reeves AG, Kaplan JA, Karluk D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann Neurol. 1999;46:598–605. doi: 10.1002/1531-8249(199910)46:4<598::aid-ana7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, et al. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med. 1999;5:1403–9. doi: 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- Limaye PB, Apte UM, Shankar K, Bucci TJ, Warbritton A, Mehendale HM. Calpain released from dying hepatocytes mediates progression of acute liver injury induced by model hepatotoxicants. Toxicol Appl Pharmacol. 2003;191:211–26. doi: 10.1016/s0041-008x(03)00250-3. [DOI] [PubMed] [Google Scholar]
- Liu B, Hong JS. Primary rat mesencephalic neuron-glia, neuron-enriched, microglia-enriched, and astroglia-enriched cultures. Methods Mol Med. 2003a;79:387–95. doi: 10.1385/1-59259-358-5:387. [DOI] [PubMed] [Google Scholar]
- Liu B, Hong JS. Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther. 2003b;304:1–7. doi: 10.1124/jpet.102.035048. [DOI] [PubMed] [Google Scholar]
- Liu B, Wang K, Gao HM, Mandavilli B, Wang JY, Hong JS. Molecular consequences of activated microglia in the brain: overactivation induces apoptosis. J Neurochem. 2001;77:182–9. doi: 10.1046/j.1471-4159.2001.t01-1-00216.x. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Akiyama H, McGeer EG. Rate of cell death in parkinsonism indicates active neuropathological process. Ann Neurol. 1988a;24:574–6. doi: 10.1002/ana.410240415. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988b;38:1285–91. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Schwab C, Parent A, Doudet D. Presence of reactive microglia in monkey substantia nigra years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administration. Ann Neurol. 2003;54:599–604. doi: 10.1002/ana.10728. [DOI] [PubMed] [Google Scholar]
- Mehendale HM, Limaye PB. Calpain: a death protein that mediates progression of liver injury. Trends Pharmacol Sci. 2005;26:232–6. doi: 10.1016/j.tips.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Mouatt-Prigent A, Karlsson JO, Agid Y, Hirsch EC. Increased M-calpain expression in the mesencephalon of patients with Parkinson’s disease but not in other neurodegenerative disorders involving the mesencephalon: a role in nerve cell death? Neuroscience. 1996;73:979–87. doi: 10.1016/0306-4522(96)00100-5. [DOI] [PubMed] [Google Scholar]
- Nishihara H, Nakagawa Y, Ishikawa H, Ohba M, Shimizu K, Nakamura T. Matrix vesicles and media vesicles as nonclassical pathways for the secretion of m-Calpain from MC3T3-E1 cells. Biochem Biophys Res Commun. 2001;285:845–53. doi: 10.1006/bbrc.2001.5242. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Tieu K, Perier C, Vila M. MPTP as a mitochondrial neurotoxic model of Parkinson’s disease. J Bioenerg Biomembr. 2004;36:375–9. doi: 10.1023/B:JOBB.0000041771.66775.d5. [DOI] [PubMed] [Google Scholar]
- Qin L, Liu Y, Cooper C, Liu B, Wilson B, Hong JS. Microglia enhance beta-amyloid peptide-induced toxicity in cortical and mesencephalic neurons by producing reactive oxygen species. J Neurochem. 2002;83:973–83. doi: 10.1046/j.1471-4159.2002.01210.x. [DOI] [PubMed] [Google Scholar]
- Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, et al. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem. 2004;279:1415–21. doi: 10.1074/jbc.M307657200. [DOI] [PubMed] [Google Scholar]
- Shields DC, Banik NL. Putative role of calpain in the pathophysiology of experimental optic neuritis. Exp Eye Res. 1998;67:403–10. doi: 10.1006/exer.1998.0537. [DOI] [PubMed] [Google Scholar]
- Shields DC, Schaecher KE, Hogan EL, Banik NL. Calpain activity and expression increased in activated glial and inflammatory cells in penumbra of spinal cord injury lesion. J Neurosci Res. 2000;61:146–50. doi: 10.1002/1097-4547(20000715)61:2<146::AID-JNR5>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Shields DC, Schaecher KE, Saido TC, Banik NL. A putative mechanism of demyelination in multiple sclerosis by a proteolytic enzyme, calpain. Proc Natl Acad Sci USA. 1999;96:11486–91. doi: 10.1073/pnas.96.20.11486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siman R, McIntosh TK, Soltesz KM, Chen Z, Neumar RW, Roberts VL. Proteins released from degenerating neurons are surrogate markers for acute brain damage. Neurobiol Dis. 2004;16:311–20. doi: 10.1016/j.nbd.2004.03.016. [DOI] [PubMed] [Google Scholar]
- Siman R, Zhang C, Roberts VL, Pitts-Kiefer A, Neumar RW. Novel surrogate markers for acute brain damage: cerebrospinal fluid levels corrrelate with severity of ischemic neurodegeneration in the rat. J Cereb Blood Flow Metab. 2005;25:1433–44. doi: 10.1038/sj.jcbfm.9600138. [DOI] [PubMed] [Google Scholar]
- Sloane JA, Hinman JD, Lubonia M, Hollander W, Abraham CR. Age-dependent myelin degeneration and proteolysis of oligodendrocyte proteins is associated with the activation of calpain-1 in the rhesus monkey. J Neurochem. 2003;84:157–68. doi: 10.1046/j.1471-4159.2003.01541.x. [DOI] [PubMed] [Google Scholar]
- Sriram K, Matheson JM, Benkovic SA, Miller DB, Luster MI, O’Callaghan JP. Mice deficient in TNF receptors are protected against dopaminergic neurotoxicity: implications for Parkinson’s disease. Faseb J. 2002;16:1474–6. doi: 10.1096/fj.02-0216fje. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Walter SA, Pennell NA. Reactive microgliosis. Prog Neurobiol. 1999;57:563–81. doi: 10.1016/s0301-0082(98)00069-0. [DOI] [PubMed] [Google Scholar]
- Teismann P, Vila M, Choi DK, Tieu K, Wu DC, Jackson-Lewis V, et al. COX-2 and neurodegeneration in Parkinson’s disease. Ann NY Acad Sci. 2003;991:272–7. doi: 10.1111/j.1749-6632.2003.tb07482.x. [DOI] [PubMed] [Google Scholar]
- Wang T, Zhang W, Pei Z, Block M, Wilson B, Reece JM, et al. Reactive microgliosis participates in MPP+-induced dopaminergic neurodegeneration: role of 67 kDa laminin receptor. Faseb J. 2006;20:906–15. doi: 10.1096/fj.05-5053com. [DOI] [PubMed] [Google Scholar]
- Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, et al. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci. 2002;22:1763–71. doi: 10.1523/JNEUROSCI.22-05-01763.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Teismann P, Tieu K, Vila M, Jackson-Lewis V, Ischiropoulos H, et al. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Proc Natl Acad Sci USA. 2003;100:6145–50. doi: 10.1073/pnas.0937239100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Deng X. Tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone induces phosphorylation of μ- and m-calpain in association with increased secretion, cell migration, and invasion. J Biol Chem. 2004;279:53683–90. doi: 10.1074/jbc.M409889200. [DOI] [PubMed] [Google Scholar]
- Zatz M, Starling A. Calpains and disease. N Engl J Med. 2005;352:2413–23. doi: 10.1056/NEJMra043361. [DOI] [PubMed] [Google Scholar]
- Zhang W, Wang T, Qin L, Gao HM, Wilson B, Ali SF, et al. Neuroprotective effect of dextromethorphan in the MPTP Parkinson’s disease model: role of NADPH oxidase. Faseb J. 2004;18:589–91. doi: 10.1096/fj.03-0983fje. [DOI] [PubMed] [Google Scholar]
- Zhou W, Hurlbert MS, Schaack J, Prasad KN, Freed CR. Overexpression of human alpha-synuclein causes dopamine neuron death in rat primary culture and immortalized mesencephalon-derived cells. Brain Res. 2000;866:33–43. doi: 10.1016/s0006-8993(00)02215-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.